
Epoxy and aziridinyl enolsilanes in diastereoselective inter- and intramolecular Friedel–Crafts alkylations†
Jesse
Ling
ab,
Sze Kui
Lam
a,
Brian
Lo
a,
Sarah
Lam
ab,
Wing-Tak
Wong‡
a,
Jian
Sun
a,
Guanhua
Chen
a and
Pauline
Chiu
*ab
aDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
bThe State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: pchiu@hku.hk
First published on 12th February 2016
Abstract
The Friedel–Crafts alkylations of epoxy enolsilanes with arenes occur under silyl triflate catalysis at low temperatures. Intermolecular alkylations with optically-enriched epoxides occur with conservation of ee. Intramolecular reactions give rise to cyclized products with high diastereoselectivity in good yields. Aziridinyl enolsilanes also undergo selective inter- and intramolecular Friedel–Crafts reactions in a similar manner.
Introduction
The reactions of epoxides and aziridines with π-nucleophiles have been a topic of a longstanding synthetic interest.1 There are numerous examples of such reactions with heteroaromatic compounds such as indoles, which are more electron-rich and allow such reactions to proceed under milder conditions.2 In comparison, there are far fewer examples of intermolecular reactions of epoxides/aziridines with arenes.3 This latter group of Friedel–Crafts reactions has been promoted by strong Lewis and protic acids, and proceeds in moderate to good yields. It is a useful strategy to alkylate and install functionality and chirality proximate to arenes, and there are more examples of the intramolecular version of this reaction than the intermolecular.4 In addition to the traditional strong Lewis and protic acids for Friedel–Crafts reactions, this reaction has also been promoted with H-mordenite,5 Sc(OTf)3,3a AuCl3/AgOTf,6 Ag(PF6),7 InCl3,4e B(C6F5)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and silica gel,9 in solvents including hexafluoroisopropanol4d and water,4d with a range of stereoselectivities. Cyclizations to generate 5- to 8-membered rings have been observed.
8 and silica gel,9 in solvents including hexafluoroisopropanol4d and water,4d with a range of stereoselectivities. Cyclizations to generate 5- to 8-membered rings have been observed.
With respect to the electrophiles, many of these reactions involve the more reactive, aryl-substituted epoxides and aziridines, which are attacked at the benzylic position via SN1-like transition states. The reactions of vinyl epoxides are complicated by nucleophilic attack via SN2 and SN2′ pathways. Epoxides bearing electron-withdrawing substituents are opened at the distal position by nucleophiles. Varying degrees of inversion have been recorded for optically-enriched epoxides, depending on the substituents on the epoxide.10
The conversion of an epoxyketone to an epoxy enolsilane has the overall effect of transforming the electron-withdrawing carbonyl group into a nucleophilic moiety, promoting carbon–carbon bond formation at the allylic position. Recently, a related umpolung strategy has been employed by MacMillan et al. to induce nucleophilic attack of α-haloketones under basic and ionizing conditions, by heteroatomic nucleophiles and indoles via the intermediacy of oxyallyl cations.11
We have investigated extensively the reactions of epoxy enolsilanes with π-nucleophiles such as furans and dienes, which, under silyl triflate catalysis, are predominantly (4 + 3) cycloadditions.12 Side products arising from an arrested cycloaddition, undergoing deprotonation instead of a second carbon–carbon bond formation, and culminating in a Friedel–Crafts process, have been observed as minor products. Our computational studies showed that furan forms carbon–carbon bonds with the activated epoxide via a backside attack, and not through the putative oxyallyl cation intermediate of typical (4 + 3) cycloadditions.13 This explains why the cycloadditions proceeded with high levels of inversion and conservation of ee. It occurred to us that we could also engage the activated epoxide in Friedel–Crafts chemistry, to further exploit the high fidelity of the transfer of chiral information conferred by this intermediate.
Results and discussion
As expected, with arenes as π-nucleophiles, the Friedel–Crafts pathway predominated. Under silyl triflate catalysis at very low temperatures (the same conditions optimized for (4 + 3) cycloadditions with dienes), electron-rich 1,3,5-trimethoxybenzene underwent an SN2-like ring opening of rac-1 to give rise to β-hydroxy-α-arylketone rac-2a as the major product (Scheme 1). Doubly arylated product 3a was also isolated as a minor product in 13% yield, which was deduced to have been obtained from an initial SN2′ addition of 1 by the arene. Benzene was apparently not electron-rich enough to react efficiently as the nucleophile (0% yield), and even mono-activated arenes such as t-butylbenzene and anisole reacted to give Friedel–Crafts products in ≤20% yield.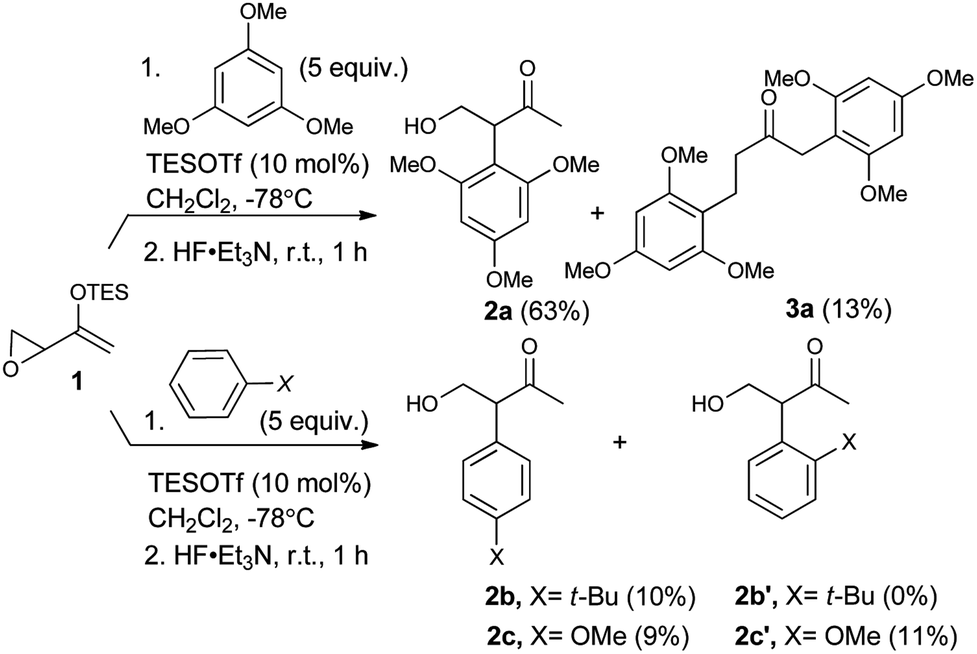 | ||
| Scheme 1 | ||
We continued to examine this reaction for optically-enriched 1 with electron-rich arenes (Table 1). Indeed, the yields of the intermolecular Friedel–Crafts alkylation increased with increasing electron density of the arenes. In all cases, the Friedel–Crafts reaction proceeded to give 2 with high enantiomeric purity, inferring that a similar activated epoxide was the electrophile, and that the arenes underwent an SN2-like, backside attack.14 It should be noted that asymmetric aldol reactions of arylated ketones with formaldehyde as a strategy to synthesize similar arrays remains a challenge.
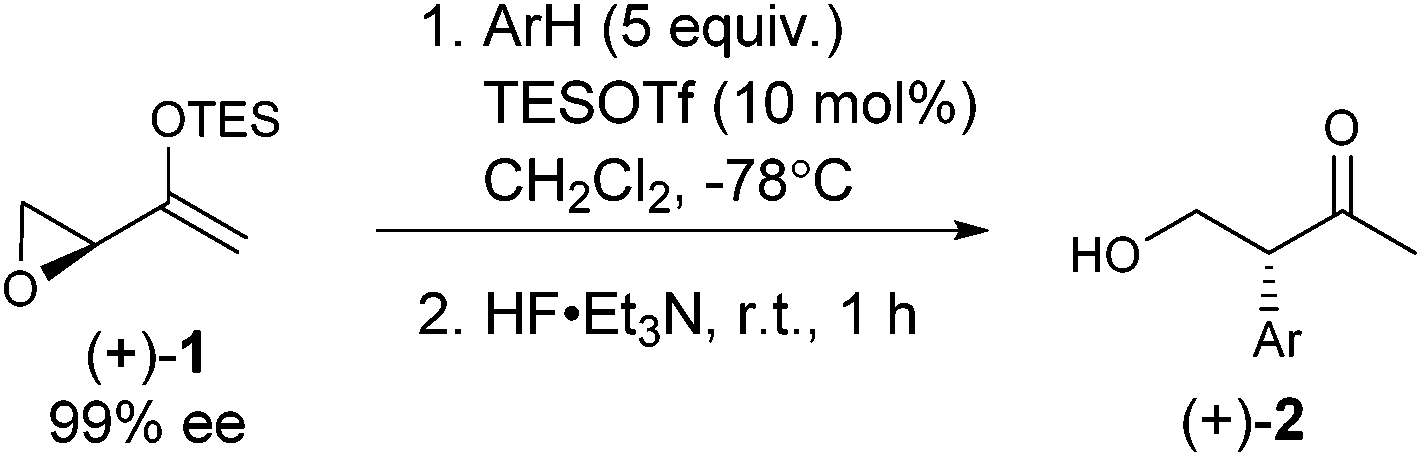
We proceeded to investigate the intramolecular version of this alkylation (Table 2). A series of substrates 4a–k, each with an arene coupled to an epoxy enolsilane through all-carbon tethers were synthesized. Gratifyingly, substrates that cyclize by a (π-endo)-endo epoxide reaction to generate six-membered rings and yield octalin derivatives, proceeded with high diastereoselectivity. The pseudoaxial methine proton alpha to the acyl group had 3J = 8–9 Hz by coupling with the methine proton at the secondary hydroxyl group, which is consistent with a trans-relationship of the protons. The relative stereochemistry was further confirmed by X-ray crystallographic analysis of 5a (Fig. 1).15 This stereochemical outcome again inferred an SN2-like attack on an activated epoxide intermediate, rather than an SN1-like transition state resembling a siloxyallyl cation. It follows then, that the reaction of optically pure (+)-4b generated a single diastereomer of 5b with high enantiomeric purity also (Scheme 3).14
 | (1) |
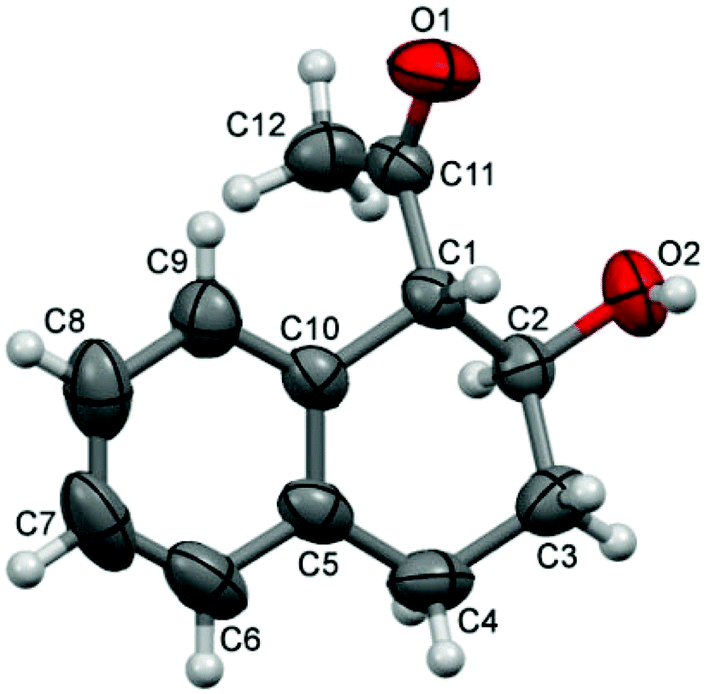 | ||
| Fig. 1 X-ray crystallographic structure of 5a. | ||
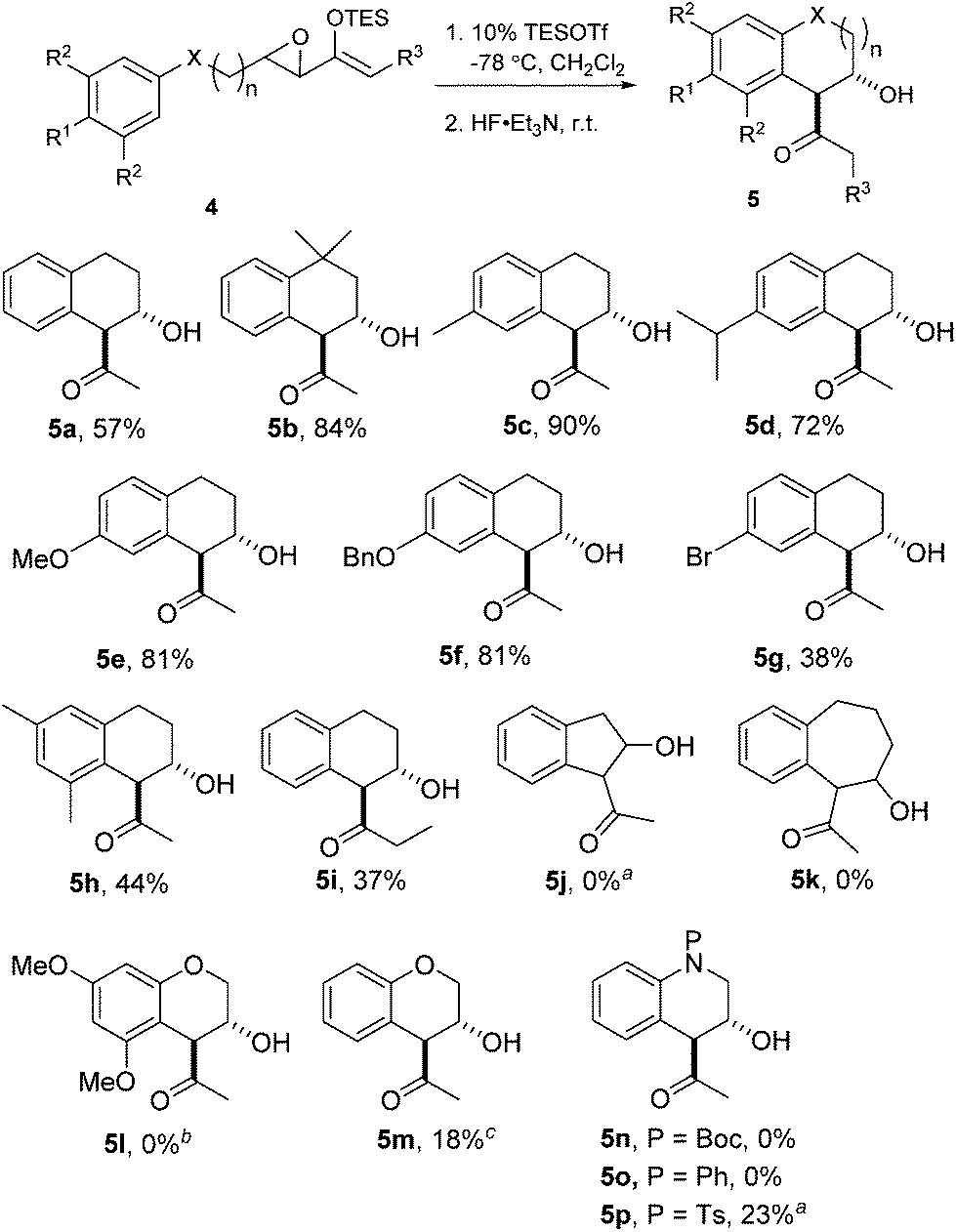
gem-Dimethyl effects in 4 facilitated the cyclization process to give 5 significantly (e.g.4avs.4b). Electron-rich arenes underwent cyclizations with up to 90% yield, whereas the less electron-rich substrates cyclized with moderate yields (e.g.4g). The para-disubstituted arenes such as 4c–f underwent cyclization efficiently, whereas cyclizations of meta-substituted arenes such as 5g were less facile due to steric congestion. When 4i was substituted at the enolsilane (R3 ≠ H), the Friedel–Crafts cyclization was also less efficient. The low yield of 5i may be due to the formation of side products arising from increased oxyallyl cation characteristic in the intermediate.
Substrates 4j–k were synthesized to explore the variations in the length of the tether, but neither reacted significantly in Friedel–Crafts cyclizations.4a In fact, 4j was completely unreactive under the typical conditions and could be essentially completely recovered.
Surprisingly, the oxygen-tethered substrates 4l, m, as well as the nitrogen-tethered 4n–p, which are rather electron-rich arene nuclei, failed to undergo any Friedel–Crafts cyclization under the typical reaction conditions. Only under more forcing conditions were some Friedel–Crafts products detected. In the case of 4p, only when one equivalent of TESOTf was used, and the reaction proceeded to give a modest yield of 5p. The addition of 2,6-lutidine as a proton scavenger allowed some of the reactions to be conducted at elevated temperatures and over longer reaction times, because trace amounts of Brønsted acid promoted the desilylation of the enolsilanes. When the reaction of 4m was heated in the presence of 2,6-lutidine, a modest 18% yield of 5m was isolated.
For those substrates that failed to cyclize, it was not because the epoxy enolsilane was unreactive: in the presence of 5 equivalents of 1,3,5-trimethoxybenzene, the intermolecular Friedel–Craft alkylation product 6 was obtained at −78 °C, while no 5m was observed (Scheme 2).
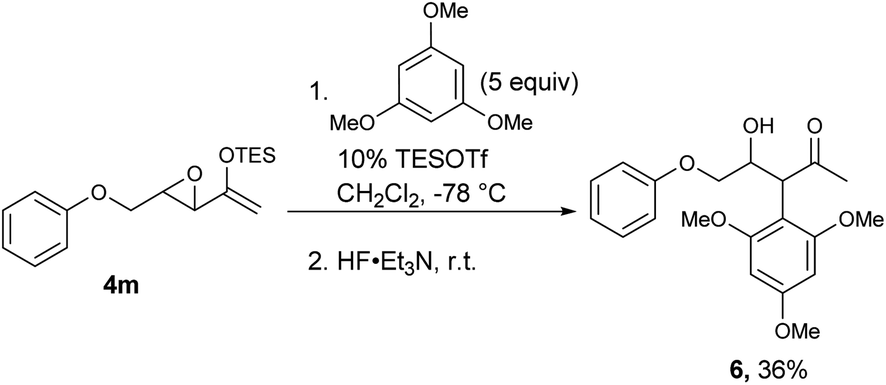 | ||
| Scheme 2 | ||
On the other hand, we noted in the literature that there were examples of related arene substrates in which epoxides were similarly tethered via a phenolic oxygen atom, that underwent Friedel–Crafts reactions successfully, albeit at higher reaction temperatures than those used in our reactions (Scheme 3).6,16
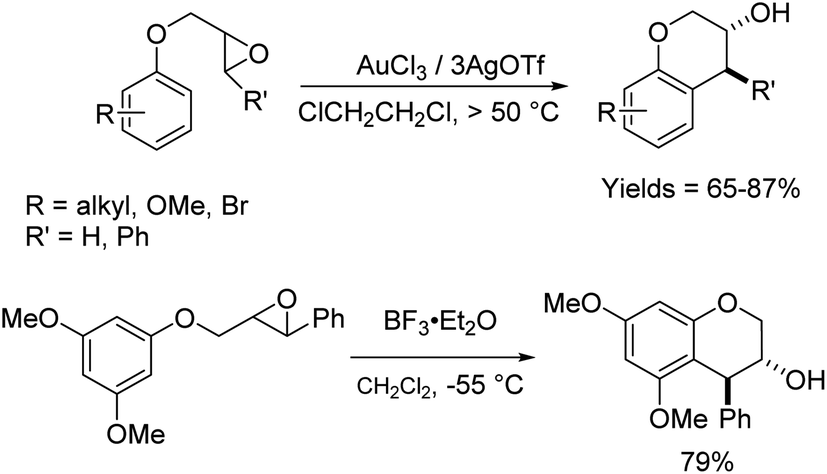 | ||
| Scheme 3 | ||
A rudimentary computational study was carried out to sample the ground state conformations of 4a and 4m using the MMFF method.17Fig. 2 shows the structures of the lowest energy conformers. For 4a, the two carbons (C1, C2) that would eventually need to approach at a bonding distance were quite close to each other at about 3.8 Å, whereas for the lowest energy conformer of 4m, the two carbons were much farther apart (5.2 Å). We found that the population of the two substrates also differed significantly (Tables S1 and S2, see the ESI†). The majority of the conformers of 4a were found to have bent carbon tethers, which bring the arene close to the epoxide and place C1 and C2 at ≤4.0 Å proximity. In contrast, the conformers of 4m possessed tethers that were more linear and directed the epoxide away from the aromatic ring, such that at 298 K, fewer than 2% of the sampled population had C1 and C2 within 5.0 Å of each other. Based on this, it can be reasoned qualitatively that the transition state of the Friedel–Crafts reaction which has C1 and C2 at a bonding distance would be lower in energy for 4a than 4m. However, this scenario does not preclude the cyclization of 4m at higher temperatures. Probably the more extended conformations of the tether of 4m are due to electron donation from the oxygen atom into the arene ring.
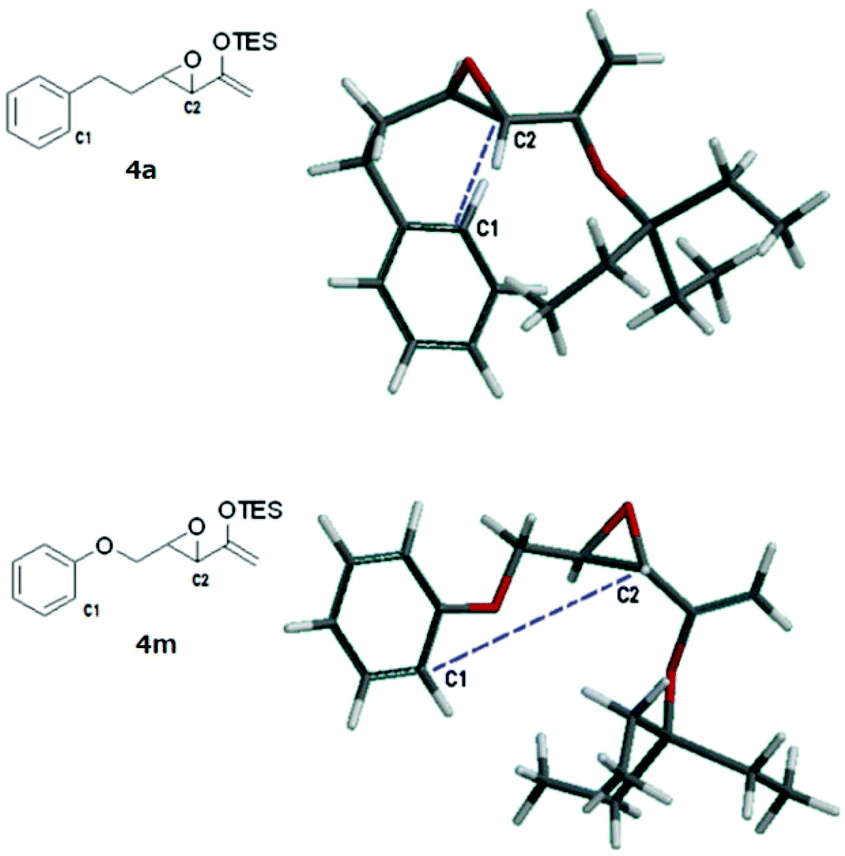 | ||
| Fig. 2 The lowest energy conformers of 4a and 4m. | ||
We also examined the Friedel–Crafts reaction of the related aziridinyl enolsilanes. Under acid or Lewis acid activation, they reacted in a similar fashion as the epoxy enolsilanes, to afford the corresponding aminoalkylated arenes. The reaction of optically pure 7 with 1,3,5-trimethoxybenzene produced β-aminoketone 8 in good yield and with the conservation of ee, along with some SN2′ product 9 (Scheme 4). The Friedel–Crafts cyclization of 10 proceeded to afford a single diastereomer of β-aminoketone 11, along with a carbamate ring expansion product 12 as the major side product.18 Replacing the carbamate protecting group with a tosyl group in 13 obviated the side reaction and improved the yield of β-aminoketone 14 significantly.
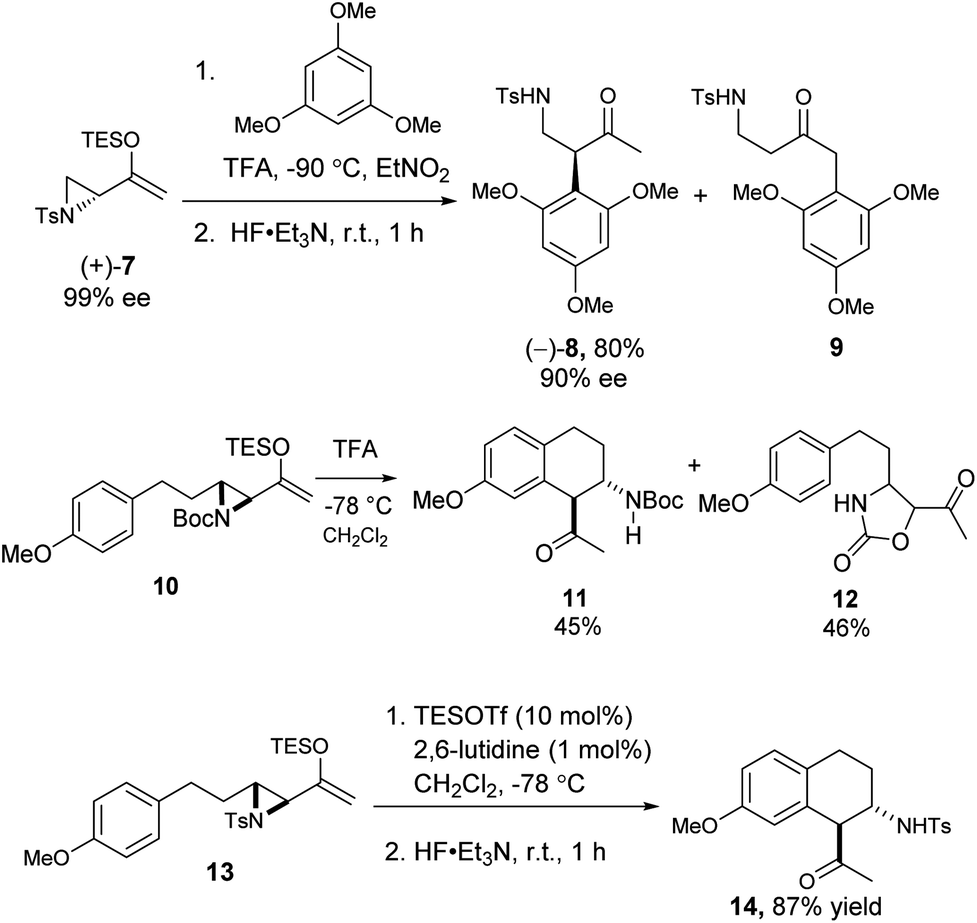 | ||
| Scheme 4 | ||
The mechanism of the intramolecular Friedel–Crafts alkylation is proposed as depicted in Scheme 5. Epoxide 1 reacted with silyl triflate to give activated epoxide 1*, which is subsequently attacked by the arene from the back in an SN2-like manner. The aromaticity of the arene is restored by deprotonation, while the enolsilane could react as a proton acceptor to form the activated oxonium species that could then silylate 1 and propagate the reaction. The reaction involving aziridinyl enolsilanes would be expected to be very similar.
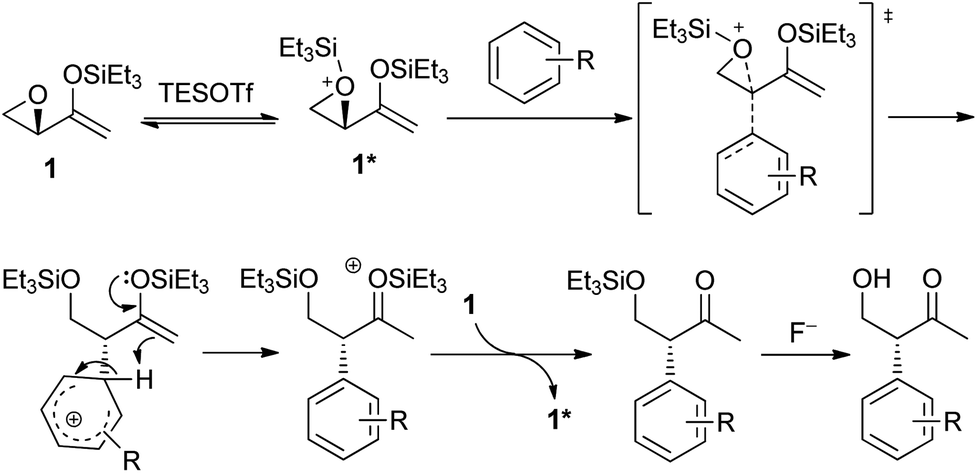 | ||
| Scheme 5 Proposed mechanism of the Friedel–Crafts reaction of 1. | ||
Conclusions
In summary, the scope of intermolecular and intramolecular Friedel–Crafts cyclization of epoxy enolsilanes with arenes has been explored. The reaction occurs with high stereoselectivity through the intermediacy of a stereochemically defined chiral activated epoxide, rather than an oxyallyl cation, to give β-hydroxy-α-arylketones in enantiomerically pure forms. Electron-rich arenes underwent the reaction with moderate to high yields. Aziridinyl enolsilanes undergo a similar stereoselective reaction to afford β-amino-α-arylketones. This reaction may offer us a strategy to tackle the synthesis of the differently functionalized anthracenone hemispheres in a natural product such as setomimycin (Fig. 3).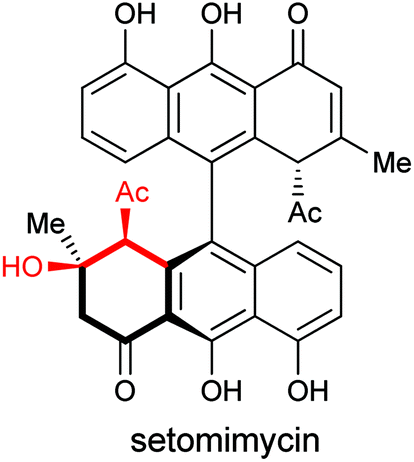 | ||
| Fig. 3 Natural products containing a β-hydroxy-α-arylketone substructure. | ||
Acknowledgements
We thank the University of Hong Kong, The State Key Laboratory of Synthetic Chemistry, and the Hong Kong Research Grants Council General Research Fund (HKU 7016/12P) for financial support. We also thank Dr L. Szeto for collecting the X-ray diffraction data. J. L. thanks the University of Hong Kong for a conference grant.References
- S. H. Krake and S. C. Bergmeier, Tetrahedron, 2010, 66, 7337 CrossRef CAS.
- (a) H. Kotsuki, K. Hayashida, T. Shimanouchi and H. Nishizawa, J. Org. Chem., 1996, 61, 984 CrossRef CAS; (b) M. Bandini, M. Fagioli, A. Melloni and A. Umani-Ronchi, Adv. Synth. Catal., 2004, 346, 573 CrossRef CAS; (c) K. Tabatabaeian, M. Mamaghani, N. O. Mahmoodi and A. Khorshidi, Tetrahedron Lett., 2008, 49, 1450 CrossRef CAS; (d) M. Westermaier and H. Mayr, Chem. – Eur. J., 2008, 14, 1638 CrossRef CAS PubMed; (e) B. V. Subba Reddy, B. Bramha Reddy, K. V. Raghavendra Rao and J. S. Yadav, Tetrahedron Lett., 2012, 53, 2500 CrossRef; (f) C. Huo, X. Xu, J. An, X. Jia, X. Wang and C. Wang, J. Org. Chem., 2012, 77, 8310 CrossRef CAS PubMed.
- (a) D. Stadler, F. Mühlthau, P. Rubenbauer, E. Herdtweck and T. Bach, Synlett, 2006, 2573 CAS; (b) K.-G. Ji, X.-Z. Shu, J. Chen, S.-C. Zhao, Z.-J. Zheng, X.-Y. Liu and Y.-M. Liang, Org. Biomol. Chem., 2009, 7, 2501 Search PubMed; (c) Y.-H. Liu, Q.-S. Liu and Z.-H. Zhang, Tetrahedron Lett., 2009, 50, 916 CrossRef CAS; (d) B. Thirupathi, R. Srinivas, A. N. Prasad, J. K. Prashanth Kumar and B. M. Reddy, Org. Process Res. Dev., 2010, 14, 1457 CrossRef CAS; (e) M. Hosseini-Sarvari and G. Parhizgar, Green Chem. Lett. Rev., 2012, 5, 439 CrossRef CAS.
- (a) S. K. Taylor, G. H. Hockerman, G. L. Karrick, S. B. Lyle and S. B. Schramm, J. Org. Chem., 1983, 48, 2449 CrossRef CAS; (b) S. K. Taylor, C. L. Blankespoor, S. M. Harvey and L. J. Richardson, J. Org. Chem., 1988, 53, 3309 CrossRef CAS; (c) S. Nagumo, T. Miura, M. Mizukami, I. Miyoshi, M. Imai, N. Kawahara and H. Akita, Tetrahedron, 2009, 65, 9884 CrossRef CAS; (d) G.-X. Li and J. Qu, Chem. Commun., 2010, 46, 2653 RSC; (e) N. Ahmed, B. Venkata Babu and H. Kumar, Synthesis, 2011, 2471 CrossRef CAS; (f) L. Albrecht, L. K. Ransborg, V. Lauridsen, M. Overgaard, T. Zweifel and K. A. Jorgensen, Angew. Chem., Int. Ed., 2011, 50, 12496 CrossRef CAS PubMed; (g) M. Mizukami, K. Wada, G. Sato, Y. Ishii, N. Kawahara and S. Nagumo, Tetrahedron, 2013, 69, 4120 CrossRef CAS; (h) D. Naduthambi, S. Bhor, M. B. Elbaum and N. J. Zondlo, Org. Lett., 2013, 15, 4892 CrossRef CAS PubMed.
- J. A. Elings, R. S. Downing and R. A. Sheldon, Eur. J. Org. Chem., 1999, 837 CrossRef CAS.
- Z. Shi and C. He, J. Am. Chem. Soc., 2004, 126, 5964 CrossRef CAS PubMed.
- H. Stamm, A. Onistschenko, B. Buchholz and T. Mall, J. Org. Chem., 1989, 54, 193 CrossRef CAS.
- A. B. Pulipaka and S. C. Bergmeier, Synthesis, 2008, 1420 CAS.
- J. Collins, M. Drouin, X. Sun, U. Rinner and T. Hudlicky, Org. Lett., 2008, 10, 361 CrossRef CAS PubMed.
- Examples of incomplete inversion: (a) S. K. Taylor, W. C. Haberkamp, D. W. Brooks and D. N. Whittern, J. Heterocycl. Chem., 1983, 20, 1745–1747 CrossRef CAS; (b) Ref. 3a .
- M. N. Vander Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075 RSC.
- (a) W. K. Chung, S. K. Lam, B. Lo, L. L. Liu, W.-T. Wong and P. Chiu, J. Am. Chem. Soc., 2009, 131, 4556 CrossRef CAS PubMed; (b) B. Lo and P. Chiu, Org. Lett., 2011, 13, 864 CrossRef CAS PubMed; (c) S. Lam, B. Lo, W.-T. Wong and P. Chiu, Asian J. Org. Chem., 2012, 1, 30 CrossRef CAS; (d) B. Lo, S. Lam, W.-T. Wong and P. Chiu, Angew. Chem., Int. Ed., 2012, 51, 12120 CrossRef CAS PubMed.
- E. H. Krenske, S. Lam, J. P. L. Ng, B. Lo, S. K. Lam, P. Chiu and K. N. Houk, Angew. Chem., Int. Ed., 2015, 54, 7422 CrossRef CAS PubMed.
- The absolute stereochemistry is assumed to be as shown, based on the mechanism of backside attack of activated epoxide, ref. 13.
- Crystal data for 5a: C12H14O2, MW = 190.23, monoclinic, space group P21/c (#14), a = 9.8964(16) Å, b = 7.7982(13) Å, c = 13.421(2) Å, β = 91.267(2)°, V = 1035.5(3) Å3, Z = 4, Dx = 1.220 Mg m−3, μ(Mo Kα) = 0.08 mm−1, F(000) = 408, T = 301 K; crystal dimensions: 0.50 mm × 0.35 mm × 0.32 mm. Of the 5495 reflections that were collected, 1813 reflections were unique. (Rint = 0.015), equivalent reflections were merged. All non-H atoms were refined anisotropically. R[F2 > 2σ(F2)] = 0.040, wR(F2) = 0.116. Full crystallographic data for 5a (CCDC 1055230).
- (a) R. Marcos, C. Rodríguez-Escrich, C. I. Herrerías and M. A. Pericàs, J. Am. Chem. Soc., 2008, 130, 16838 CrossRef CAS PubMed; (b) R. Devi, T. Kalita and S. K. Das, RSC Adv., 2015, 5, 39692 RSC.
- Spartan'14, Wavefunction, Inc., Irvine, CA, 2013 Search PubMed. See the ESI.†.
- C. Tomasini and A. Vecchione, Org. Lett., 1999, 1, 2153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for synthesis, full characterization of all new compounds, and details of computations. CCDC 1055230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00333d |
| ‡ Author responsible for X-ray diffraction analyses. |
| This journal is © the Partner Organisations 2016 |