DOI:
10.1039/C5QO00315F
(Research Article)
Org. Chem. Front., 2016,
3, 330-334
Access to bicyclic hydroxamate macrocycles via intramolecular aza-(4 + 3) cyloaddition reactions of aza-oxyallylic cation intermediates†
Received
13th October 2015
, Accepted 27th December 2015
First published on 28th December 2015
Abstract
The intramolecular aza-(4 + 3) cycloaddition reactions of in situ generated aza-oxyallylic cations and furans have been reported for the construction of medium sized hydroxamate macrocycles. This method provides direct access to 12–18 membered bicyclic macrocycles. The highly functionalized macrocycles have been transformed easily in to a wide range of highly functionalized heterocyclic scaffolds including lactones and lactams that could serve the synthesis of complex macrocyclic natural products and pharmaceuticals.
Macrocyclic motifs have demonstrated profound pharmacological properties. The balance between conformational flexibility and rigidity demonstrated by macrocyclic molecules often leads to specific and potent activity against a variety of biological targets.1 Among the various classes of macrocycles, nitrogen-containing heterocycles have drawn considerable interest in chemical synthesis as they are widely represented in natural products and pharmaceuticals.
There are various methods reported for the construction of nitrogen containing macrocycles. Lactonization and lactimization reactions are most commonly used methods.2–6 Other methods are cantered on constructing C–C bonds via nucleophilic substitution,5 ring-closing metathesis7–12 Pd-catalysed reactions,13,14 Diels–Alder reactions,3,15 cycloaddition reactions,16,17 photochemical reactions,18 among others. Aside from these approaches, multicomponent reactions have also been applied for the preparation of macrocyles.19–23 Despite the promising biological activity of various macrocycles only a few reports have demonstrated the synthesis of five to eight membered cyclic hydroxamates from corresponding lactones.24 We report herein, the development of a direct method of medium size hydroxamates using an aza-(4 + 3) cycloaddition reaction of furans and aza-oxyallylic cations. Additionally, we report further reactions of these highly functionalized macrocycles that provide access to products with unique polycyclic architectures.
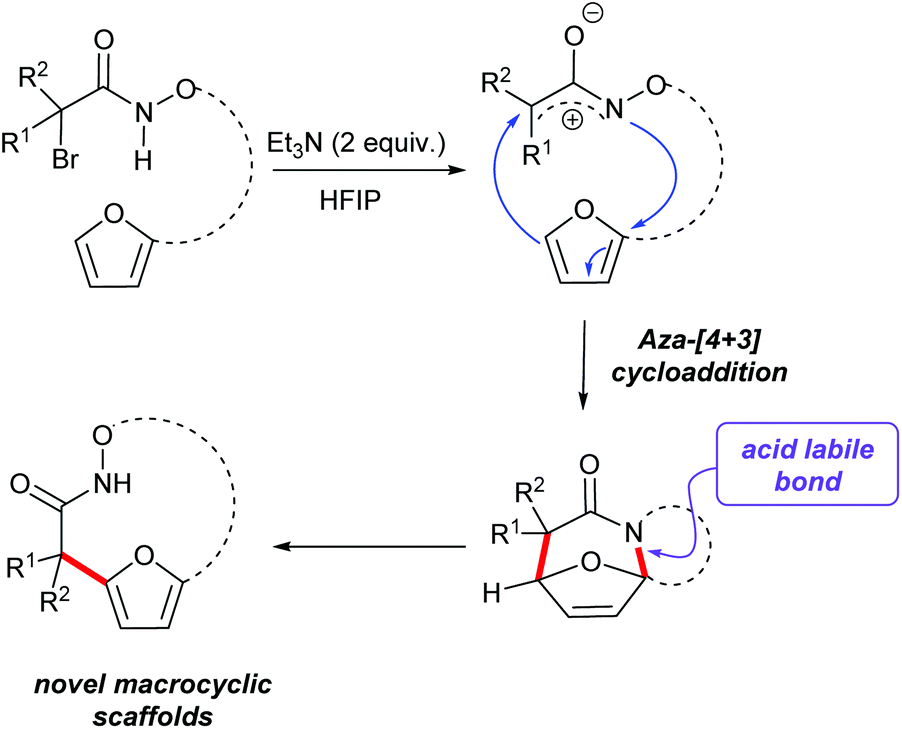
(4 + 3)-Cycloaddition reactions of oxyallyl cations have been reported as efficient methods to construct seven membered carbocycles and natural products.25–28 As part of our interest in exploring the reactivity of electrophilic nitrogen species, we have reported the aza-(4 + 3) reaction of a transient aza- and diaza-oxyallylic cation with cyclic dienes to construct seven membered N-heterocycles.29–32 Recently our group and Wu and co-workers have reported a (3 + 2) cycloaddition of aza-oxyallyl cationic intermediate and 1,3-disubstituted indoles to form pyrroloindolines.33 In the course of studying the intramolecular cycloaddition reactions of α-halohydroxamates to afford tricyclic lactams, we observed further conversion of some of the initial cycloadducts to interesting macrocyclic products. We found that acid-catalyzed ring opening of polycyclic cycloadducts efficiently provided macrocyclic products via rupture of the aminal bond and rearomatization to the furan (Scheme 1).31 These observations prompted us to further explore this strategy as a method for the construction of hydroxamate macrocycles. Herein, our efforts to expand the reactivity of in situ generated aza-oxyallyl cationic intermediates by the dehydrohalogenation reaction of α-halohydroxamates to access medium rings bicyclic hydroxamates macrocycles are described.
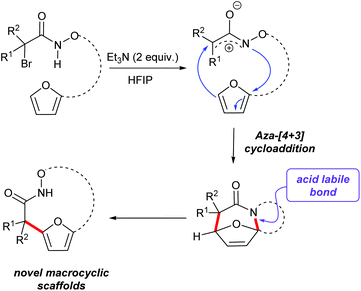 |
| | Scheme 1 An aza-(4 + 3) cycloaddition of a stabilized aza-oxyallylic to form macrocyclic scaffolds. | |
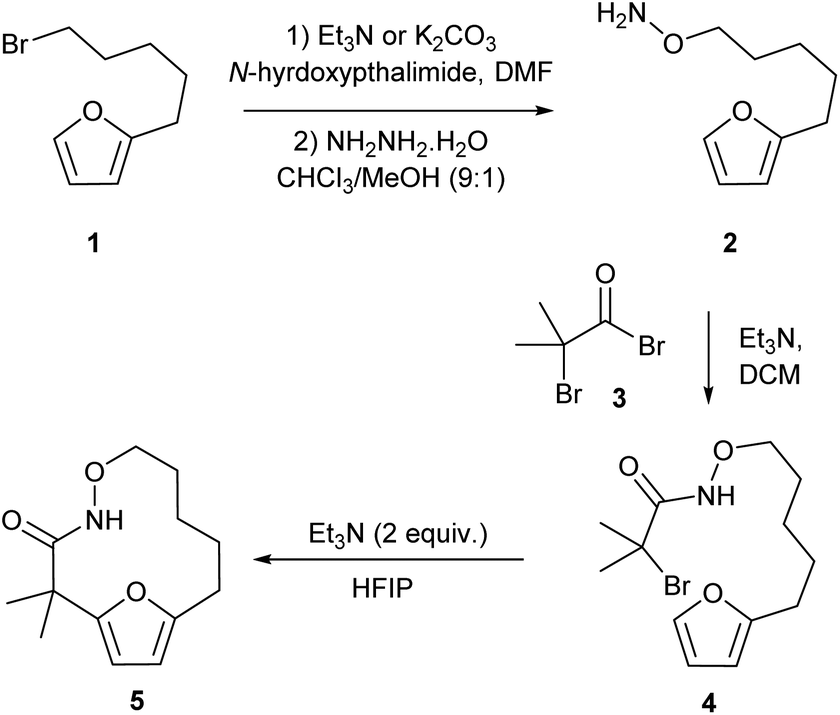
Our studies began by preparing the hydroxylamine 2 in two steps from the 5-furylpentanyl bromide 1. Acylation of the hydroxylamine with 2-bromo-2-methyl-propanoylbromide 3 provided the cycloaddition precursor 4 (Scheme 2). Initial attempts to construct macrocycles via intramolecular (4 + 3)-cycloaddition reaction of in situ generated aza-oxyallyl cation intermediate from halohydroxamates 4 using triethylamine as a base and trifluoroethanol (TFE) as a solvent34,35 exclusively provided solvolysis product of the α-halohydroxamate by TFE along with 15% desired macrocycle.29 (Table 1, entry 1). Use of the more bulky and less nucleophilic solvent, hexafluoroisopropanol (HFIP), was found to be effective in suppressing solvolysis and increasing the yield of the macrocyclic product. Optimization of the reaction conditions were explored varying the concentration of α-halohydroxamate, the base and the order of addition. We found that slow addition of triethylamine to a solution of the halohydroxamate in HFIP (0.1 M) to be optimal reaction conditions providing the desired macrocyclization product in 58% yield (Table 1, entry 8). The reaction conditions were further optimized by examination of the effects of temperature and solvent (Table 1).
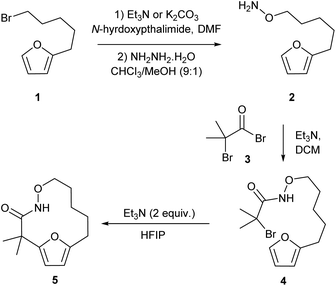 |
| | Scheme 2 Synthesis of the cycloaddition precursor and preliminary studies of the macrocyclization reaction. | |
Table 1 Optimization of the reaction conditions for substrate 5a
| Entry |
Base |
Solvent |
Conc.b |
Time |
Yieldc (%) |
|
All reactions were conducted by adding the base to a solution of the α-haloamide in HFIP and stirred until complete consumption of starting material was observed.
(M).
Isolated yield.
Refluxed.
Slow addition of TEA in HFIP over 2 h at room temperature.
Slow addition of SM in HFIP over 2 h at room temperature.
|
| 1 |
TEA |
TFE |
0.25 |
0.5 h |
15 |
| 2d |
TEA, LiClO4 |
Ether |
0.25 |
3 h |
ND |
| 3 |
TEA |
HFIP |
0.25 |
1 h |
36 |
| 4 |
DIPEA |
HFIP |
0.25 |
3 h |
35 |
| 5 |
DBU |
HFIP |
0.25 |
0.5 h |
50 |
| 6 |
TEA |
HFIP |
0.1 |
1 h |
48 |
| 7 |
TEA |
HFIP |
0.05 |
2 h |
45 |
| 8e |
TEA |
HFIP |
0.1 |
2.5 h |
58 |
| 9f |
TEA |
HFIP |
0.1 |
2.5 h |
36 |
| 10 |
Proton sponge |
HFIP |
0.25 |
3 h |
47 |
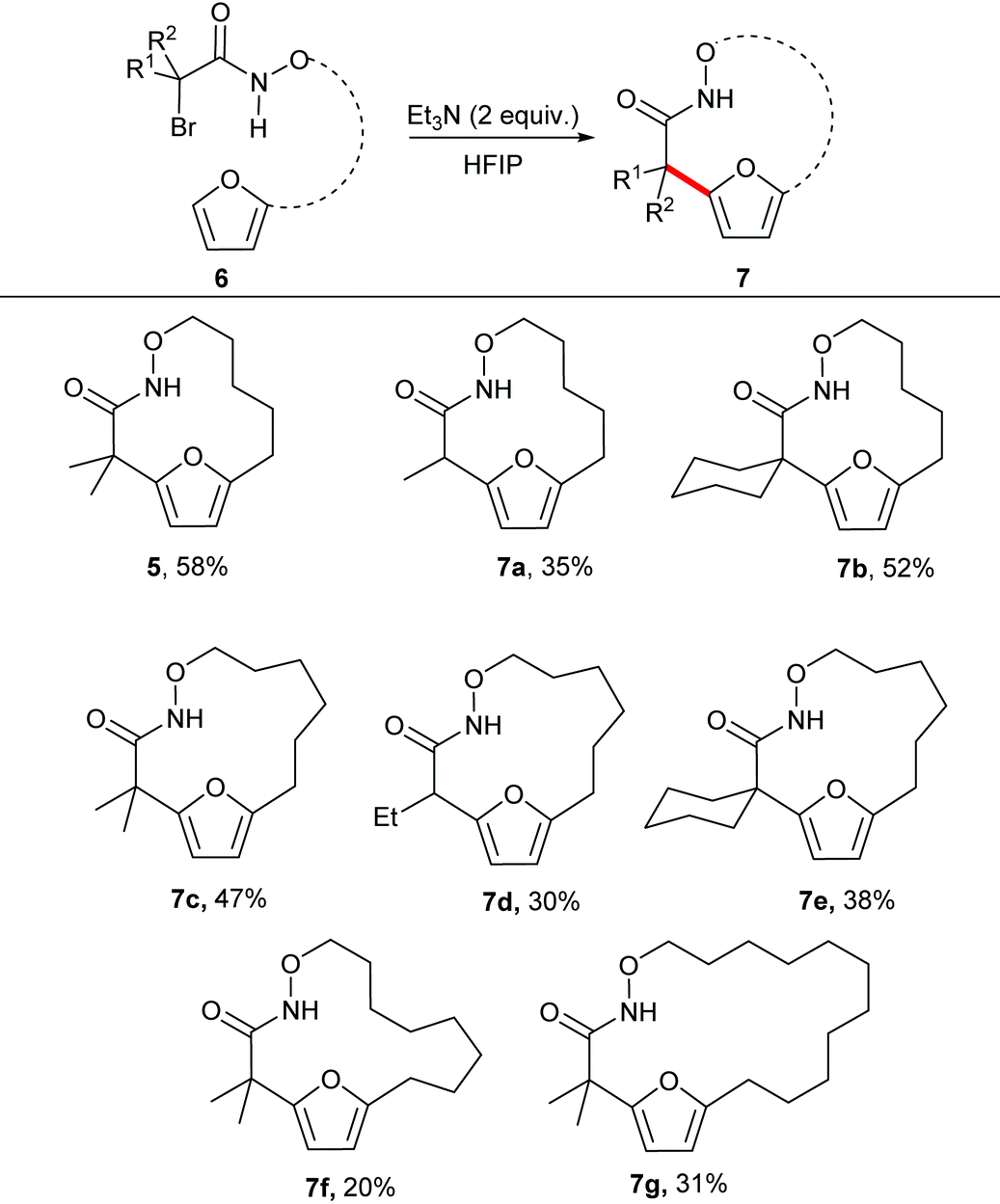
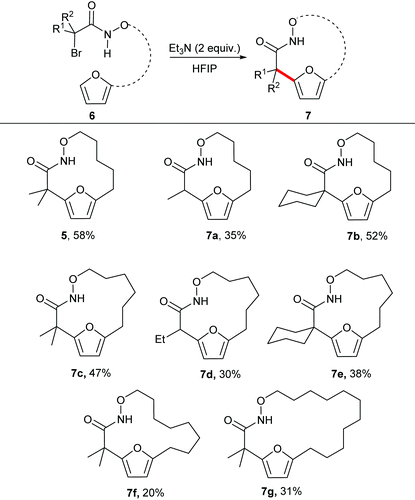
With the optimal reaction conditions in hand, we explored the substrate scope for the synthesis of medium sized macrocycles (Table 2). In general all the reactions proceeded smoothly to afford the desired product in satisfactory yield. Substitution at the 2-position of the substrate was found to influence the overall rate of reaction, with tertiary halides undergoing the reaction at a faster rate than the corresponding secondary halides. It is hypothesized that tertiary carbon helps to stabilize the aza-oxyallyl cation as compared to secondary carbon, thereby accelerating the overall rate of the reaction.
Table 2 Evaluation of the effect of tether length on macrocyclization reactions of α-halohydroxamatesa
|
All reactions were conducted by adding the base to a solution of the α-haloamide in HFIP and stirred until complete consumption of starting material was observed (see ESI).
|
|
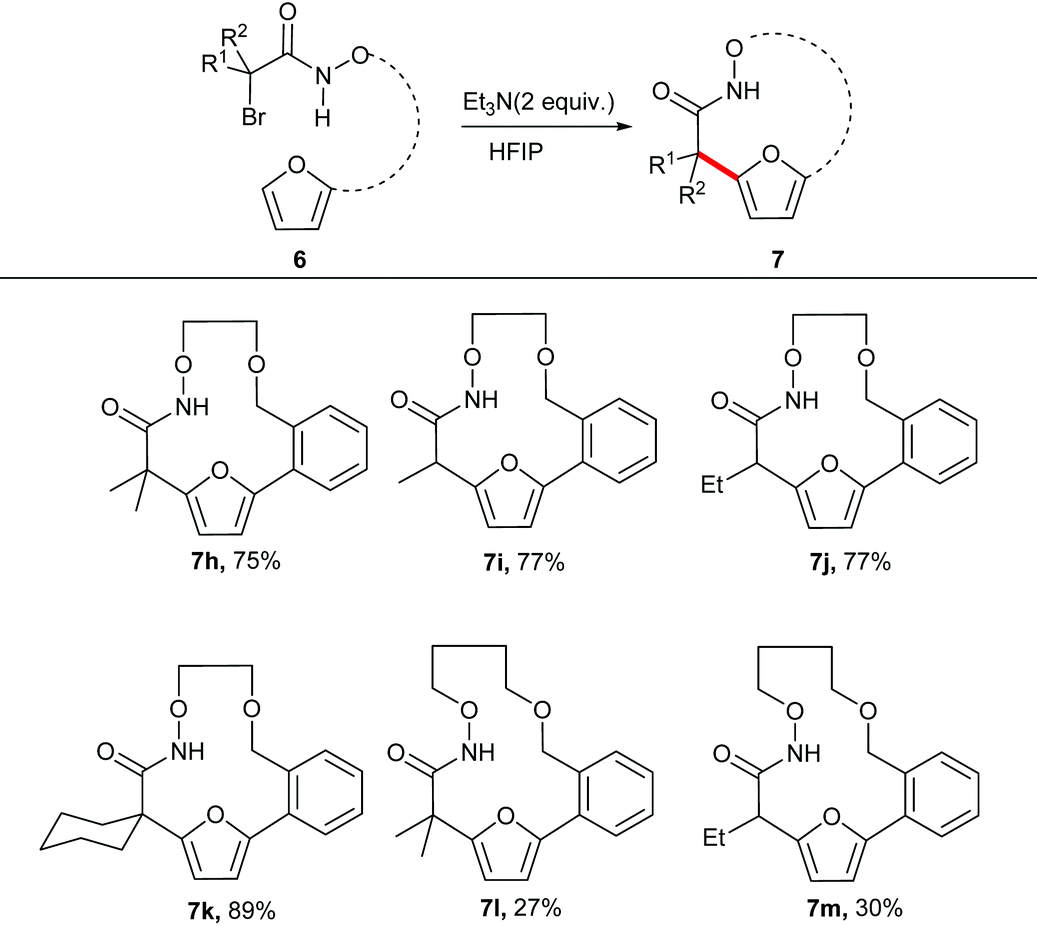
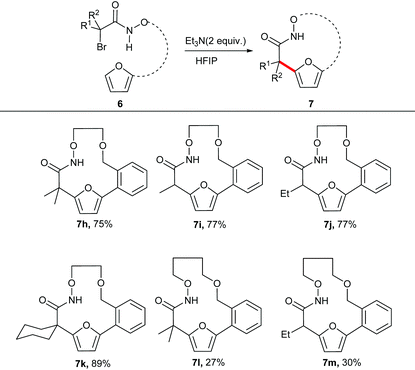
The unsubstituted α-halohydroxamate was found to be unreactive under the reaction conditions. The effect of tether length between both reacting termini was next explored. Increasing the tether length from five carbons to six carbons substantially diminished yield of desired products (7c–e) along with a complex mixture of side products. Extension of the tether further diminished the yield of the macrocyles, however still provided the large fifteen and eighteen membered macrocycles (7f and 7g). We hypothesized that rigidifying the tether could enhance preference for cyclization by buttressing the reactive termini through the Thorpe–Ingold effect. To explore this hypothesis, substrates incorporating an aromatic backbone within the tether were prepared and their macrocyclization using the optimal reaction conditions was explored (Table 3). These substrates were found to be more effective than the less rigid precursors and provided desired macrocycle in good yield (7h–j). However, increasing tether length with the backbone again diminished the yield of desired macrocycles (7k–m).
Table 3 Evaluation of the effect of variable tether length having rigid backbone on macrocyclization reactions of α-halohydroxamatesa
|
All reactions were conducted by adding the base to a solution of the α-haloamide in HFIP and stirred until complete consumption of starting material (see ESI).
|
|
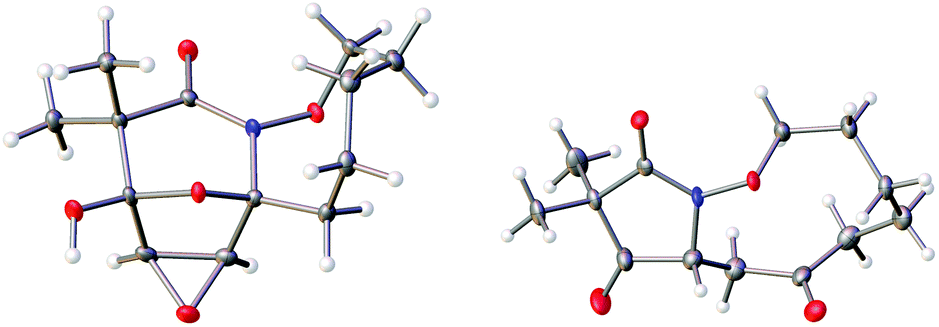
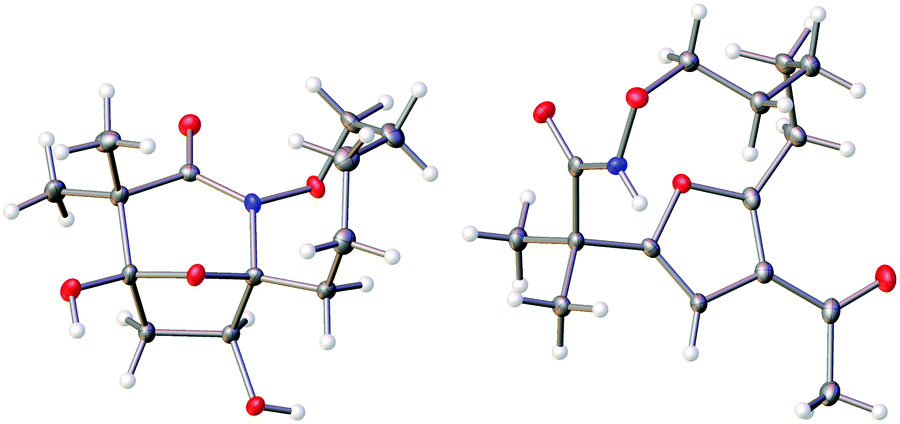
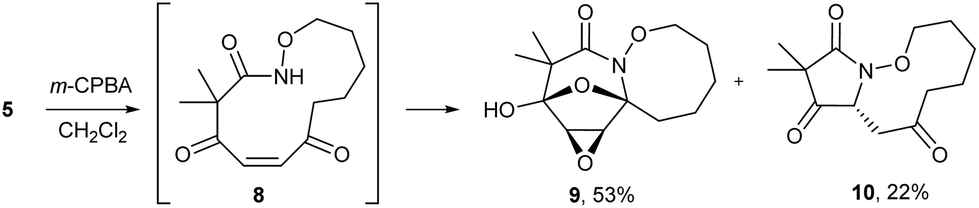
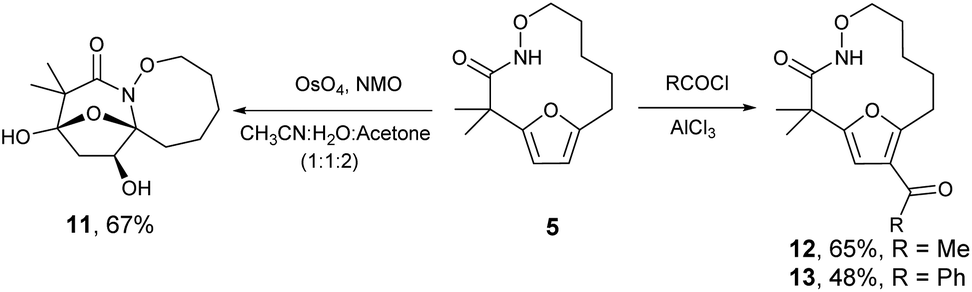
The highly functionalized macrocyclic products provide ample opportunity for the synthesis of biologically active macrocycles. The aromatic furan ring present in macrocycles 5, underwent efficient oxidation with m-CPBA to afford epoxy lactam 9 and pyrrolidinedione 10 (Scheme 3 and Fig. 1). The introduction of epoxy group in 9 and diketone group in 10 suggests that the reaction proceeds via ring opening of furan to provide diketone intermediate 8, which was observed in crude NMR analysis during the reaction. An OsO4 oxidation of 5 provides the interesting bicyclic products 11 in good yield, now with the incorporation of a hydroxylated tetrahydrofuran ring (Scheme 4). Friedel–Crafts acylation of the macrocyclic furan was found to be regioselective affording 12 and 13 in good yield (Scheme 4 and Fig. 2).
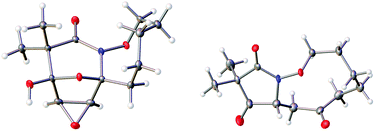 |
| | Fig. 1 Thermal ellipsoid plot of 9 and 10 at 50% probability showing relative stereochemistry. Hydrogen atoms are represented as spheres of arbitrary radius. Grey = carbon, red = oxygen, blue = nitrogen. | |
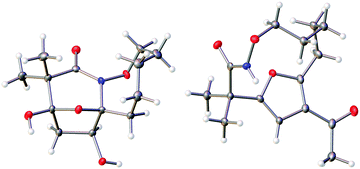 |
| | Fig. 2 Thermal ellipsoid plot of 11 and 12 at 50% probability showing relative stereochemistry. Hydrogen atoms are represented as spheres of arbitrary radius. Grey = carbon, red = oxygen, blue = nitrogen. | |
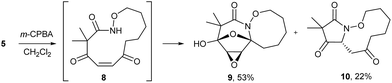 |
| | Scheme 3 Oxidation of macrocycle 5 with m-CPBA. | |
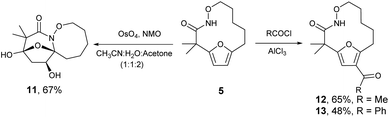 |
| | Scheme 4 Osmium tetroxide oxidation and Friedel–Craft acylation of macrocycle 5. | |
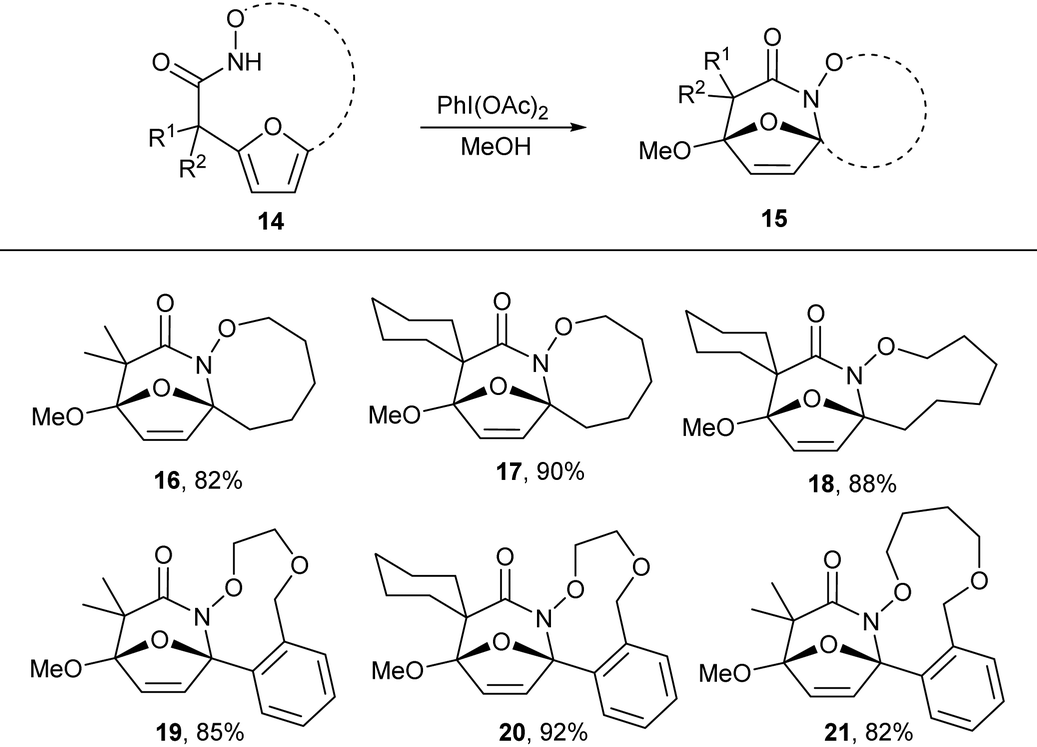
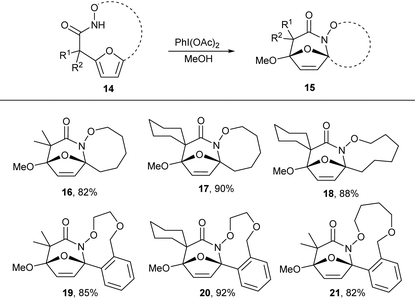
N-Acyl hydroxamates are established to be efficiently oxidized to electrophilic N-acyl nitrenium ions using hypervalent iodide oxidants. We envisioned that oxidation of the macrocyclic hydroxamates to the nitrenium ion would promote intramolecular electrophilic aromatic substitution by the pendant furan to form and oxidized tricyclic lactams. Treatment of the macrocyclic products 5 with PhI(OAc)2 in methanol afforded tricyclic lactam 16 in good yield. We further explored this reactivity in other macrocycles 7b,e,h,k and 7l, which all provided desired tricyclic products in excellent yield (Table 4). These tricyclic lactams provide excellent opportunity for the construction of biologically active classes of heterocycles including nucleoside mimics and polyhydroxylated azepanes.
Table 4 Substrate scope for the oxidative lactamization of macrocyclesa
|
All reactions were conducted by adding PhI(OAc)2 in to the solution of macrocycles in methanol.
|
|
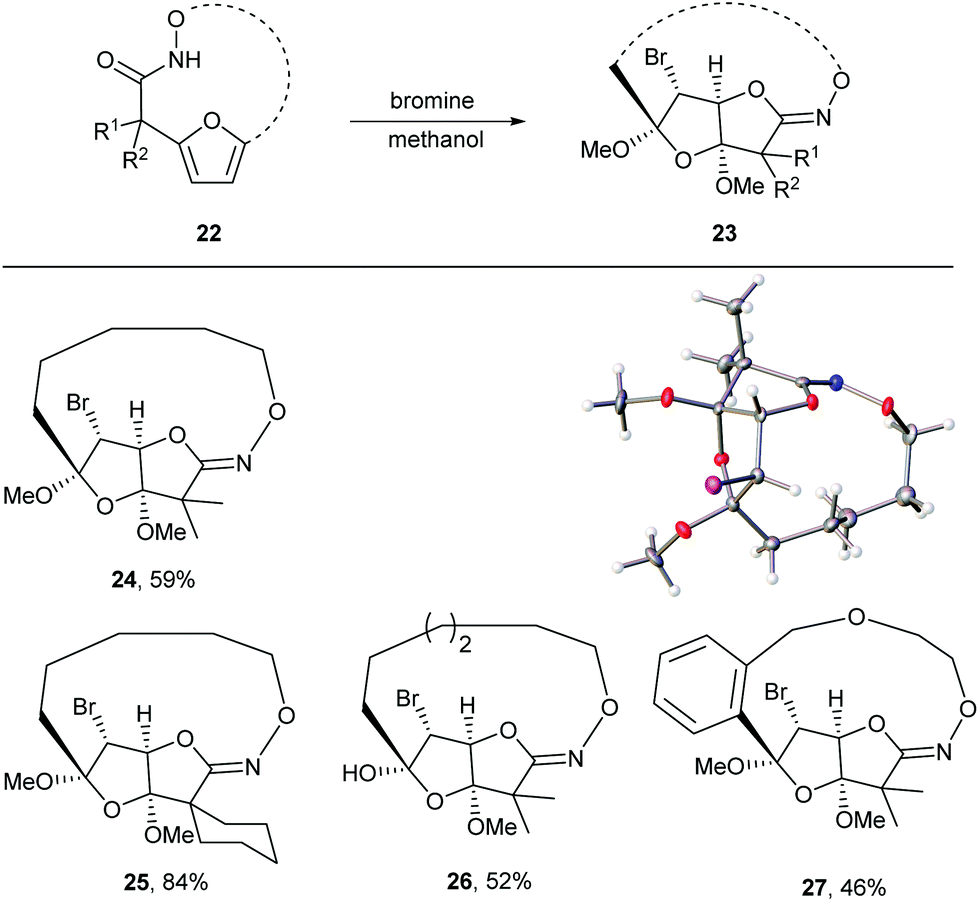
Given the oxidative susceptibility of the furan ring, we pursued the bromination reaction of a few selected macrocycles (5, 7b, 7c and 7h). Treatment of the macrocycles with bromine in methanol/ether provided the unique tricyclic imidate products (Fig. 3) in good yield and as a single diastereoisomer. The stereochemistry of the product 24 was confirmed by X-ray crystallography. These highly functionalized products represent important building blocks that could be used to prepare a number of lactone natural products including ascorbigen,36–38 microphynolides,39 amarusine,40 dichotomains,41 helioscopin.42
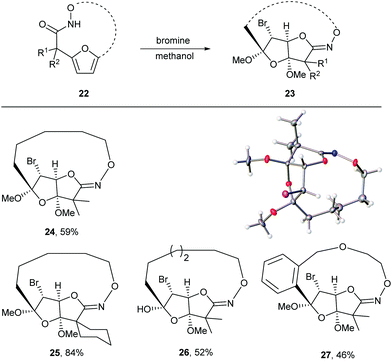 |
| | Fig. 3 Bromination of macrocycles and thermal ellipsoid plot of 24 at 50% probability showing relative stereochemistry. Hydrogen atoms are represented as spheres of arbitrary radius. Grey = carbon, red = oxygen, blue = nitrogen, purple = bromine. | |
Conclusions
In summary, we have developed a novel macrocyclization method for the preparation of aza-macrocyclic compounds in fair to good yield. Intramolecular (4 + 3)-cycloaddition reactions of in situ generated aza-oxyallylic cation intermediates followed by ring opening of bicyclic products provide medium sized macrocycles. The imbedded furan core undergoes a variety of different functionalization reactions, including regioselective Friedel–Crafts acylation/benzoylation, and a diversity of oxidative cyclizations using m-CPBA, osmium tetroxide, PhI(OAc)2 and Br2/MeOH. These methods provide access to a wide range of interesting architectures including polycyclic lactams, polyhydroxylated caprolactam, oxazines, and lactones. Further application of these strategies in total synthesis of the natural products is in under investigation in our group.
Crystal structure determination of compounds 9–12 and 20
Crystal data for 9 (CCDC no. 1429958).
C13H19NO5, M = 269.29, monoclinic, a = 10.9816(5), b = 9.2222(4), c = 13.0751(6) Å, U = 1261.57(10) Å3, T = 100.15 K, space group P21/n (no. 14), Z = 4, 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 reflections measured, 2898 unique (Rint = 0.0452) which were used in all calculations. The final wR(F2) was 0.1382 (all data).
450 reflections measured, 2898 unique (Rint = 0.0452) which were used in all calculations. The final wR(F2) was 0.1382 (all data).
Crystal data for 10 (CCDC no. 1429956).
C13H19NO4, M = 253.29, monoclinic, a = 11.3134(5), b = 9.4290(4), c = 12.7345(6) Å, U = 1268.29(10) Å3, T = 100 K, space group P21/n (no. 14), Z = 4, 21397 reflections measured, 2947 unique (Rint = 0.0433) which were used in all calculations. The final wR(F2) was 0.2774 (all data).
Crystal data for 11 (CCDC no. 1429955).
C14H23Cl2NO5, M = 356.23, monoclinic, a = 11.7775(8), b = 8.7429(6), c = 16.8595(12) Å, U = 1670.0(2) Å3, T = 100(2) K, space group P21/n (no. 14), Z = 4, 23957 reflections measured, 3102 unique (Rint = 0.0652) which were used in all calculations. The final wR(F2) was 0.1240 (all data).
Crystal data for 12 (CCDC no. 1429957).
C15H21NO4, M = 558.65, monoclinic, a = 17.0197(5), b = 16.2710(5), c = 10.1414(3) Å, U = 2773.26(14) Å3, T = 100 K, space group P21/c (no. 14), Z = 4, 41363 reflections measured, 5170 unique (Rint = 0.0413) which were used in all calculations. The final wR(F2) was 0.1275 (all data).
Crystal data for 24 (CCDC no. 1429954).
C15H24BrNO5, M = 378.26, triclinic, a = 8.8508(8), b = 10.0352(8), c = 10.7879(9) Å, U = 811.83(12) Å3, T = 99.65 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 2, 14678 reflections measured, 3029 unique (Rint = 0.0794) which were used in all calculations. The final wR(F2) was 0.0722 (all data).
(no. 2), Z = 2, 14678 reflections measured, 3029 unique (Rint = 0.0794) which were used in all calculations. The final wR(F2) was 0.0722 (all data).
Acknowledgements
This work was supported through an ACS-PRF grant (51442-DNI) and through start-up funding from the University of Nevada, Reno.
Notes and references
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed
 .
.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef CAS PubMed .
- X. F. Yu and D. Q. Sun, Molecules, 2013, 18, 6230 CrossRef CAS PubMed .
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS PubMed .
- V. M. Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736 CrossRef PubMed .
- W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn and D. P. Fairlie, Chem. Rev., 2004, 104, 6085 CrossRef CAS PubMed .
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed .
- C. Obradors, D. Leboeuf, J. Aydin and A. M. Echavarren, Org. Lett., 2013, 15, 1576 CrossRef CAS PubMed .
- J. Prunet, Eur. J. Org. Chem., 2011, 3634 CrossRef CAS .
- M. Ibrahim-Ouali, J. Zoubir and E. Romero, Tetrahedron Lett., 2011, 52, 7128 CrossRef CAS .
- M. Yu, C. B. Wang, A. F. Kyle, P. Jakubec, D. J. Dixon, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 479, 88 CrossRef CAS PubMed .
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94 CrossRef CAS PubMed .
- E. A. Crane and K. A. Scheidt, Angew. Chem., Int. Ed., 2010, 49, 8316 CrossRef CAS PubMed .
- S. P. Breazzano, Y. B. Poudel and D. L. Boger, J. Am. Chem. Soc., 2013, 135, 1600 CrossRef CAS PubMed .
- C. W. Zapf, B. A. Harrison, C. Drahl and E. J. Sorensen, Angew. Chem., Int. Ed., 2005, 44, 6533 CrossRef CAS PubMed .
- D. E. Lonsdale and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4496 CrossRef CAS .
- D. E. Lonsdale, C. A. Bell and M. J. Monteiro, Macromolecules, 2010, 43, 3331 CrossRef CAS .
- K. Nishikawa, Y. Yoshimi, K. Maeda, T. Morita, I. Takahashi, T. Itou, S. Inagaki and M. Hatanaka, J. Org. Chem., 2013, 78, 582 CrossRef CAS PubMed .
- L. A. Wessjohann, D. G. Rivera and O. E. Vercillo, Chem. Rev., 2009, 109, 796 CrossRef CAS PubMed .
- L. A. Wessjohann and E. Ruijter, Mol. Diversity, 2005, 9, 159 CrossRef CAS PubMed .
- D. G. Rivera and L. A. Wessjohann, J. Am. Chem. Soc., 2009, 131, 3721 CrossRef CAS PubMed .
- D. G. Rivera and L. A. Wessjohann, J. Am. Chem. Soc., 2006, 128, 7122 CrossRef CAS PubMed .
- J. M. Knapp, J. C. Fettinger and M. J. Kurth, Org. Lett., 2011, 13, 4732 CrossRef CAS PubMed .
- S. Wolfe, M. C. Wilson, M. H. Cheng, G. V. Shustov and C. I. Akuche, Can. J. Chem., 2003, 81, 937 CrossRef CAS .
- J. Mann, Tetrahedron, 1986, 42, 4611 CrossRef CAS .
- J. K. Cha and J. Oh, Curr. Org. Chem., 1998, 2, 217 CAS .
- R. Noyori, S. Makino, T. Okita and Y. Hayakawa, J. Org. Chem., 1975, 40, 806 CrossRef CAS .
- H. M. Hoffmann, Angew. Chem., Int. Ed., 1973, 12, 819 CrossRef .
- C. S. Jeffrey, K. L. Barnes, J. A. Eickhoff and C. R. Carson, J. Am. Chem. Soc., 2011, 133, 7688 CrossRef CAS PubMed ; K. L. Barnes, A. K. Koster and C. S. Jeffrey, Tetrahedron Lett., 2014, 55, 4690 CrossRef .
- C. S. Jeffrey, D. Anumandla and C. R. Carson, Org. Lett., 2012, 14, 5764 CrossRef CAS PubMed .
- A. Acharya, J. A. Eickhoff and C. S. Jeffrey, Synthesis, 2013, 1825 CAS .
- D. Anumandla, R. Littlefield and C. S. Jeffrey, Org. Lett., 2014, 16, 5112 CrossRef CAS PubMed .
-
(a) A. Acharya, D. Anumandla and C. S. Jeffrey, J. Am. Chem. Soc., 2015, 137, 14858 CrossRef CAS PubMed ;
(b) M. C. DiPoto, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2015, 137, 14861 CrossRef CAS PubMed .
- B. Föhlisch, E. Gehrlach and R. Herter, Angew. Chem., Int. Ed. Engl., 1982, 21, 137 CrossRef .
- R. Herter and B. Föhlisch, Synthesis, 1982, 976 CrossRef CAS .
- A. Tai, K. Fukunaga, A. Ohno and H. Ito, Biosci., Biotechnol., Biochem., 2014, 78, 1723 CrossRef CAS PubMed .
- M. Opietnik, S. N. B. S. Jaafar, M. Becker, S. Boehmdorfer, A. Hofinger and T. Rosenau, Mini-Rev. Org. Chem., 2012, 9, 411–417 CrossRef CAS .
- C. H. Wang, H. S. Huang, N. T. Dai, M. J. Sheu and D. M. Chang, Phytother. Res., 2013, 27, 1863 CrossRef CAS PubMed .
- H. Ghanem, H. Haba, L. Marcourt, M. Benkhaled and J. L. Wolfender, Nat. Prod. Res., 2014, 28, 1800 CrossRef CAS PubMed .
- J. Sun, P. Zhang, Q. Wei, H. Xun, F. Tang, Y. Yue, L. Li, X. Guo and R. Zhang, Tetrahedron Lett., 2014, 55, 4529 CrossRef CAS .
- X. L. Li, X. Cheng, L. M. Yang, R. R. Wang, Y. T. Zheng, W. L. Xiao, Y. Zhao, G. Xu, Y. Lu, Y. Chang, Q. T. Zheng, Q. S. Zhao and H. D. Sun, Org. Lett., 2006, 8, 1937 CrossRef CAS PubMed .
- S. H. Lee, M. Jun, J. Y. Choi, E. J. Yang, J. M. Hur, K. Bae, Y. H. Seong, T. L. Huh and K. S. Song, Arch. Pharm. Res., 2007, 30, 827 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1429954–1429958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00315f |
| ‡ Current address: Department of Chemistry, University of California, Santa Barbara, CA 93106 United States. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a