
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Endogenous Cu in the central nervous system fails to satiate the elevated requirement for Cu in a mutant SOD1 mouse model of ALS
J. B.
Hilton
a,
A. R.
White
ab and
P. J.
Crouch
*ab
aDepartment of Pathology, the University of Melbourne, Victoria 3010, Australia. E-mail: pjcrouch@unimelb.edu.au
bFlorey Institute of Neuroscience and Mental Health, the University of Melbourne, Victoria 3010, Australia
First published on 22nd June 2016
Abstract
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease, a fatal degenerative disorder in which motor neurons in the central nervous system (CNS) progressively deteriorate. Most cases of ALS are sporadic, but 10% are familial and mutations affecting the copper (Cu)-dependent antioxidant Cu/Zn-superoxide dismutase (SOD1) are the most common familial cause. Cu malfunction is evident in CNS tissue from transgenic mice that over-express mutant SOD1 and modulating Cu bioavailability in the CNS provides positive therapeutic outcomes. In the present study we assessed levels of Cu and Zn, SOD activity, and SOD1 protein levels in CNS and non-CNS tissue from transgenic mutant SOD1 mice (SOD1G37R) and non-transgenic controls. Physiological SOD1 binds one structural Zn and one catalytic Cu per subunit. Due to over-expression of the transgene, SOD activity and SOD1 protein levels are elevated in all tissues examined from the SOD1G37R mice and a commensurate increase in Zn is evident. There is a comparable increase in Cu in non-CNS tissue, but the increase in Cu in the SOD1G37R mouse brain is limited and there is no increase in Cu in the spinal cord. The limited change in CNS Cu is associated with a strong disparity between SOD1 protein and SOD activity in the brain and spinal cord. We hypothesise that the limited capacity for CNS tissue to respond to an increased requirement for bioavailable Cu contributes to CNS vulnerability in ALS.
Significance to metallomicsPerturbations to copper are evident in the central nervous system (CNS) of mouse models of amyotrophic lateral sclerosis, the most common form of motor neuron disease, and targeted deletion of the copper transporter ATP7A induces a disease phenotype. Despite this apparent role for copper malfunction in the disease, explanations for why the CNS is primarily affected, particularly in those cases caused by ubiquitous expression of a disease-causing mutation, have remained elusive. In the present study we hypothesise that the CNS is primarily affected because, unlike other tissues, it has a relatively limited capacity to satiate an increased requirement for copper. |
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease, an adult-onset degenerative disorder in which the motor neurons that relay signals between the brain and muscles progressively deteriorate. Initial symptoms are as innocuous as weakness in a hand or slurred speech, but inevitably and relentlessly the symptoms escalate; people with ALS become paralysed, lose the ability to breathe, speak and swallow, and due to the absence of an effective treatment most will die within 5 years of diagnosis.Most cases of ALS are sporadic but 10% are familial and the heritable basis for these cases has been ascribed to mutations in over 20 different genes. Mutations in the Cu-dependent antioxidant Cu/Zn-superoxide dismutase (SOD1) were the first described genetic cause of ALS.1 Mutant SOD1 is the most common cause of familial ALS and transgenic mice expressing mutant SOD1 provide a robust animal model of the disease.2,3 Underscoring the clear role for mutant SOD1 in the ALS-like phenotype of these mice, higher levels of mutant SOD1 protein cause earlier symptom onset, faster progression of symptoms, and shorter lifespan.2,4 Conversely, spontaneous loss of transgene copy number or silencing transgene expression improves the ALS-like phenotype and extends survival.5,6
But despite the clear role for mutant SOD1 in familial ALS (and its apparent role in some cases of sporadic ALS7) the mechanisms by which mutant SOD1 selectively causes deterioration in the CNS remain incompletely understood. Many mechanisms have been proposed for mutant SOD1-mediated neurodegeneration in ALS, including: aberrant pro-oxidant gain of function, altered proteostasis and protein aggregation, protein misfolding and prion-like propagation, mitochondrial dysfunction, and loss of calcium regulation (see ref. 8–11 for recent reviews). And of these mechanisms, several provide plausible explanations for why ubiquitous expression of mutant SOD1 would impair motor neurons in the CNS yet leave other tissues unaffected (e.g. high metabolic demand of motor neurons making them more vulnerable to mitochondrial dysfunction).
An alternate mechanism, supported by therapeutic intervention studies, is that the metal state of SOD1 is a significant contributing factor. Physiological SOD1 functions as a homo-dimer in which each subunit binds one Zn and one Cu; the Zn serves a structural role whereas the redox properties of Cu are harnessed to confer catalytic activity to the enzyme. But in the CNS of ALS model mice mutant SOD1 accumulates in a Cu-deficient form.12–14 Moreover, treating these mice with the Cu-containing compound diacetyl-bis(4-methylthiosemicarbazonato)copperII [CuII(atsm)] converts the Cu-deficient SOD1 to holo-SOD1 via a Cu-delivery mechanism of action.13 This is associated with an improvement in the animals’ ALS-like phenotype, preservation of spinal cord motor neurons, and an increase in the animals’ overall survival13,14 (by 1.7 years in one study14).
Significantly, treating with CuII(atsm) results in an overall increase in mutant SOD1 protein levels in the CNS,13,14 thereby demonstrating that severity of disease symptoms in SOD1 transgenic mice is not strictly proportional to levels of the expressed SOD1 protein, and that the Cu state of the protein may be a greater determinant of its role in ALS. In the present study we assessed levels of Cu and Zn, SOD activity, and SOD1 protein levels in transgenic mutant SOD1 mice and non-transgenic controls. By comparing various CNS and non-CNS tissues, our aim was to assess whether limited Cu presentation to SOD1 may be involved in the selective vulnerability of CNS tissue to ubiquitously expressed mutant SOD1.
Experimental methods
ALS mice
All research involving mice was approved by a University of Melbourne Animal Experimentation Ethics Committee (clearance #1513556) and conformed with guidelines of the Australian Health and Medical Research Council. Hemizygous mice expressing a transgene for human SOD1 containing the G37R substitution mutation (SOD1G37R) on the congenic C57Bl/6 background were used as an ALS model.3 To collect tissues for biochemical analysis, SOD1G37R mice and non-transgenic (non-Tg) littermates were killed at 25 weeks of age. At this age the SOD1G37R mice display a substantial ALS-like phenotype, including loss of spinal cord motor neurons, loss of locomotive function, and hind-limb paralysis.3,13 Mice were anaesthetised by intraperitoneal injection of ketamine (120 mg kg−1) and xylazine (16 mg kg−1) in PBS, then perfused transcardially with PBS containing 0.25% (v/v) phosphatase inhibitor cocktail 2 (Sigma), 1% (v/v) Complete EDTA-free protease inhibitor (Roche), and 20 U mL−1 heparin (Sigma). Following perfusion, tissues were excised, snap frozen on dry ice, then stored at −80 °C.SDS-PAGE and western blotting
Frozen tissue samples were homogenised in PBS supplemented with 0.5% (v/v) phosphatase inhibitor cocktail 2 (Sigma), 2% (v/v) Complete EDTA-free protease inhibitor (Roche), and 5% (v/v) DNAse. PBS-soluble material was collected by centrifugation (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 30 min, 4 °C) then the protein content determined using the BCA Assay (Thermo Scientific). All samples were normalised to a consistent protein concentration using PBS homogenising buffer. PBS-soluble samples were prepared in reducing and denaturing sample buffer containing 62.2 mM Tris, 5% (v/v) glycerol, 2% (w/v) SDS, 0.5% (v/v) β-mercaptoethanol, and 0.0025% (w/v) bromophenol blue prior to loading onto 4–12% NuPAGE Novex Bis-Tris Midi gels (Life Technologies) and electrophoresis at 200 V for 40 minutes in MES SDS running buffer (Life Technologies).
000 × g, 30 min, 4 °C) then the protein content determined using the BCA Assay (Thermo Scientific). All samples were normalised to a consistent protein concentration using PBS homogenising buffer. PBS-soluble samples were prepared in reducing and denaturing sample buffer containing 62.2 mM Tris, 5% (v/v) glycerol, 2% (w/v) SDS, 0.5% (v/v) β-mercaptoethanol, and 0.0025% (w/v) bromophenol blue prior to loading onto 4–12% NuPAGE Novex Bis-Tris Midi gels (Life Technologies) and electrophoresis at 200 V for 40 minutes in MES SDS running buffer (Life Technologies).
Resolved proteins were transferred to PVDF membranes using iBlot gel transfer stacks (Life Technologies). Membranes were blocked for 1 hour in PBS supplemented with 0.05% (v/v) Tween-20 (Chemsupply) and 4% (w/v) skim milk powder prior to incubation with primary antibodies in blocking buffer, overnight at 4 °C. The primary antibody used to detect overall levels of SOD1 in the non-Tg and SOD1G37R tissues was chosen based on its ability to react with endogenous mouse SOD1 (present in the non-Tg and SOD1G37R mice) and with the human mutant SOD1 expressed in the SOD1G37R mice (Abcam ab16831; 1:2000) thereby enabling an assessment of changes to SOD1 protein in the SOD1G37R mice relative to SOD1 protein levels in the non-Tg controls. A primary antibody to GAPDH was used a loading control for all western blot analyses (cell signaling 2118; 1:5000). Membranes were subsequently probed using a horseradish peroxidase-conjugated anti-rabbit secondary antibody (cell signaling 7074S; 1:5000). Immunoreactive protein bands were visualised by adding Enhanced Chemiluminescence (ECL Advance, GE Healthcare) to membranes and detecting luminescence using a DNR Bio-Imaging Systems MicroChemi imager. Quantitation of immunoreactivity was performed using ImageJ software on TIFF file images.
SOD activity
SOD activity in tissue samples was assessed via a pyrogallol assay based on published procedure.15,16 This activity assay does not discriminate between mouse SOD and human SOD and was therefore used to measure total SOD activity in the non-Tg mice and SOD1G37R mice (i.e. mouse SOD1 only in the non-Tg mice and mouse plus human SOD1 in the SOD1G37R mice). Pyrogallol (Sigma) was added to a reaction buffer (50 mM Tris, 1 mM EGTA, pH 7.4) to a final concentration of 200 μM and allowed to equilibrate for 1 minute. PBS-soluble tissue extracts were added then reaction mixtures monitored at 325 nm. Reaction mixtures supplemented with 10 mM KCN were used to determine the KCN-sensitive activity attributable to SOD. SOD activity was determined by calculating the rate of change through the linear phase of reaction and by comparison to a known range of purified bovine holo SOD1 (Sigma) concentrations. Reactions mixtures containing equivalent volumes of PBS homogenising buffer ±KCN were included as additional controls.Inductively coupled plasma mass spectrometry (ICP-MS) bulk metal analysis
Sections of frozen tissue were weighed then analysed for total copper, zinc, magnesium, phosphorous, and potassium levels following protocols described previously.17 Briefly, the samples were digested using concentrated nitric acid then analysed for metal content using an Agilent 7700 Series ICP-MS with a helium reaction cell.Statistical analyses
All data sets contained 5–7 replicate samples whereby each replicate represents tissue from an individual mouse. Statistically significant outliers were identified using the Grubb's test (Extreme Studentised Deviate). Statistical significance of differences between SOD1G37R and non-Tg mice was determined using the non-parametric Mann–Whitney test. Statistically significant P values (P < 0.05) are indicated in all figures and represent two-tailed P values. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. All statistical analyses were performed using GraphPad Prism Version 6.Results
SOD1 activity and protein levels vary across tissues and activity is increased in the SOD1G37R mice
Assessing SOD activity in CNS tissue (brain, spinal cord) and non-CNS tissue (kidney, liver, quadriceps) from SOD1G37R mice and non-Tg controls revealed considerable tissue-specific variability in overall activity (Fig. 1A). Given the G37R mutation does not prevent SOD1 dismutase activity,18 overall SOD activity was higher in all tissues from the SOD1G37R mice, as expected, due to over-expression of the transgene. In other words, SOD activity in the non-Tg mice is due to the endogenous mouse SOD1 whereas SOD activity in the SOD1G37R mice is the sum of the endogenous mouse SOD1 plus the over-expressed human SOD1. When expressed relative to the amount of SOD activity within each tissue, the overall increase in SOD activity (Δ SOD activity) in tissues from the SOD1G37R ranged from a modest 1.5-fold increase in the brain up to a 4.7-fold increase in the quadriceps (Fig. 1B).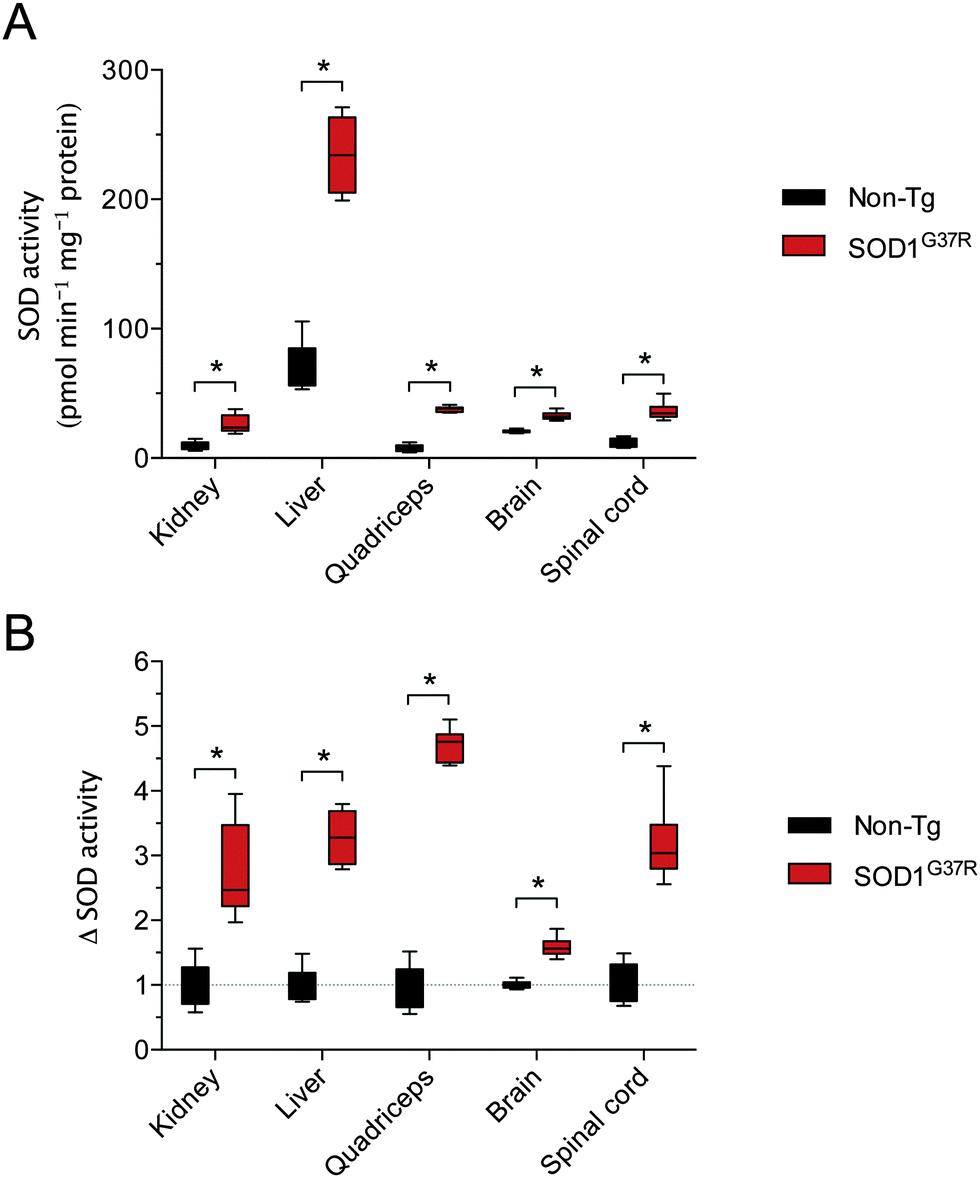 | ||
| Fig. 1 Tissue-specific SOD activity in ALS model SOD1G37R mice and non-transgenic (non-Tg) controls. (A) SOD activity in soluble tissue extracts expressed as the amount of pyrogallol oxidation prevented per minute per mg protein in tissue extracts. (B) SOD activity in SOD1G37R mouse tissues from (A) expressed relative to SOD activity in corresponding tissues from non-Tg control mice. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. *P < 0.05, two-tailed Mann Whitney test, n = 6–7. | ||
As per SOD activity, abundance of the SOD1 protein was consistently higher in the SOD1G37R mice when compared to the non-Tg controls (Fig. 2A and B). In contrast to antibodies that specifically detect mouse or human forms of the SOD1 protein, the antibody we used to monitor changes to total SOD1 in the SOD1G37R mice detects both endogenous mouse SOD1 and the over-expressed human SOD1 (Fig. 2C). Thus, as per the SOD activity assay, our western blot methodology enabled a comparison of total SOD1 in the non-Tg mice (endogenous mouse SOD1 only) to total SOD1 in the SOD1G37R mice (endogenous mouse SOD1 plus the over-expressed human SOD1).
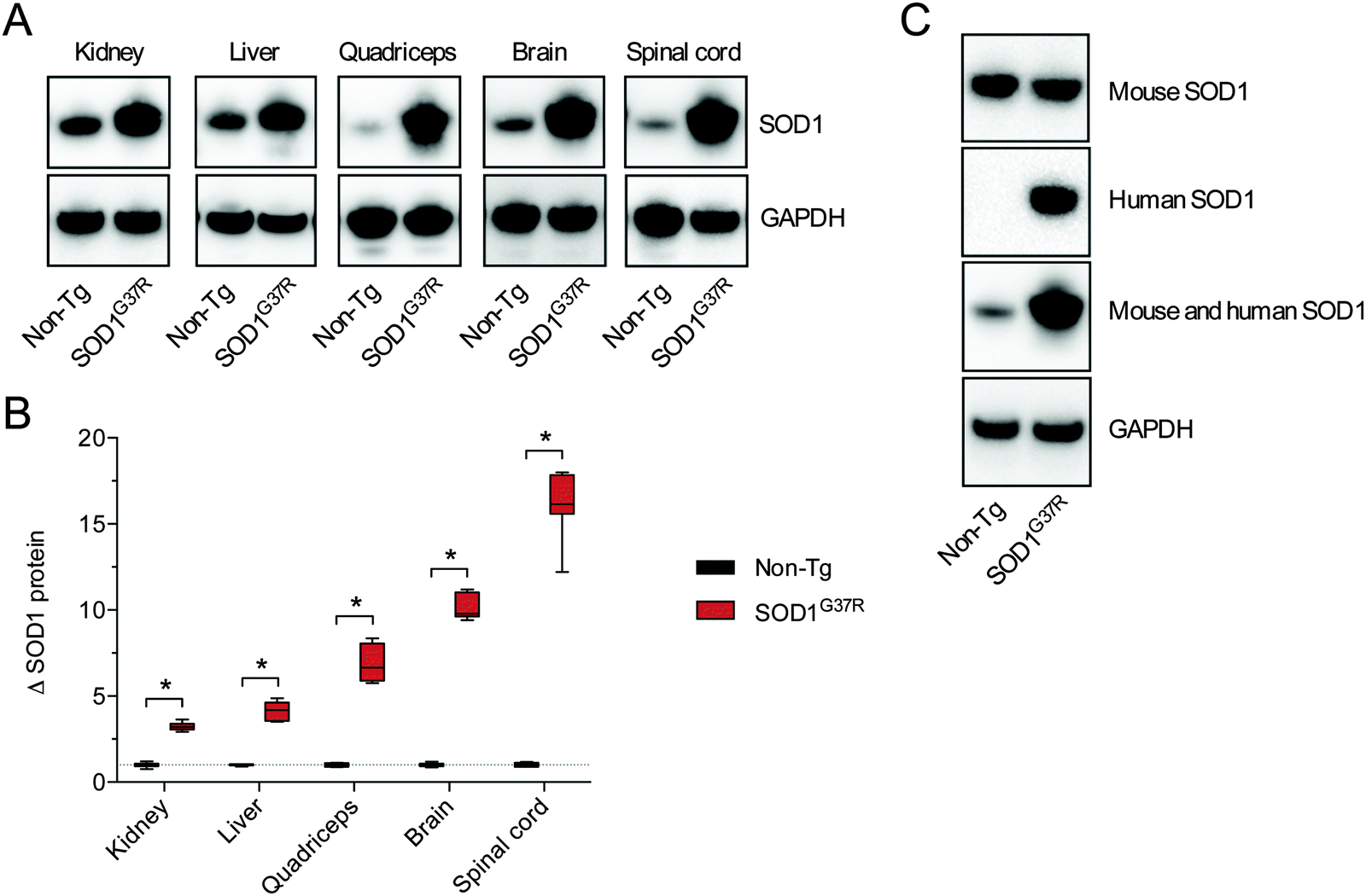 | ||
| Fig. 2 Tissue-specific SOD1 protein levels in ALS model SOD1G37R mice and non-transgenic (non-Tg) controls. (A) Representative western blots showing total SOD1 protein in SOD1G37R and non-Tg mouse tissues. Total SOD1 was detected using an anti-SOD1 antibody that reacts with human and mouse SOD1, thus enabling detection of endogenous SOD1 in the non-Tg mice as well as endogenous SOD1 and the over-expressed mutant SOD1 in the SOD1G37R mice. GAPDH immunoreactivity is used as a loading control. (B) Tissue-specific total SOD1 protein levels in SOD1G37R mice expressed relative to total SOD1 in corresponding tissues from non-Tg control mice. SOD1 levels were normalised to the loading control GAPDH before expressing relative to SOD1 in non-Tg mice. (C) Representative western blots showing selective immunoreactivity of different SOD1 antibodies to the endogenous mouse SOD1 (expressed in non-Tg and SOD1G37R mice) and the human SOD1 (expressed only in the SOD1G37R mice) in spinal cord tissue. The antibody used for analyses presented in this study (Abcam ab16831) detects mouse and human SOD1, enabling comparative assessment of total SOD1 in SOD1G37R and non-Tg mice. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. *P < 0.05, two-tailed Mann Whitney test, n = 5–7. | ||
Disparity between SOD1 protein and activity in SOD1G37R mice
A comparison of tissue-specific Δ SOD activity (Fig. 1B) to tissue-specific Δ SOD1 protein (Fig. 2B) indicates a strong disparity between the two; the Δ SOD1 protein results span a broader range (Fig. 2B) when compared to the Δ SOD activity results (Fig. 1B). Most obvious is that the relatively large Δ SOD1 protein in the CNS tissues is not matched by a comparable Δ SOD activity. To assess the nature of this disparity we calculated the ratio of SOD activity to SOD1 protein in all tissues. This was achieved using overall SOD activity data (from Fig. 1A) and expressing it relative to overall SOD1 protein levels in each tissue (from Fig. 2B but also accounting for differences in SOD1 protein across the different tissues). These calculations revealed considerable differences in the activity:protein ratio across the tissues examined, but more noticeably, also revealed that while the activity:protein ratio is sustained in the kidney and liver of SOD1G37R mice it is significantly decreased in quadriceps, brain and spinal cord, with the greater decrease observed in the two CNS tissues (Fig. 3).
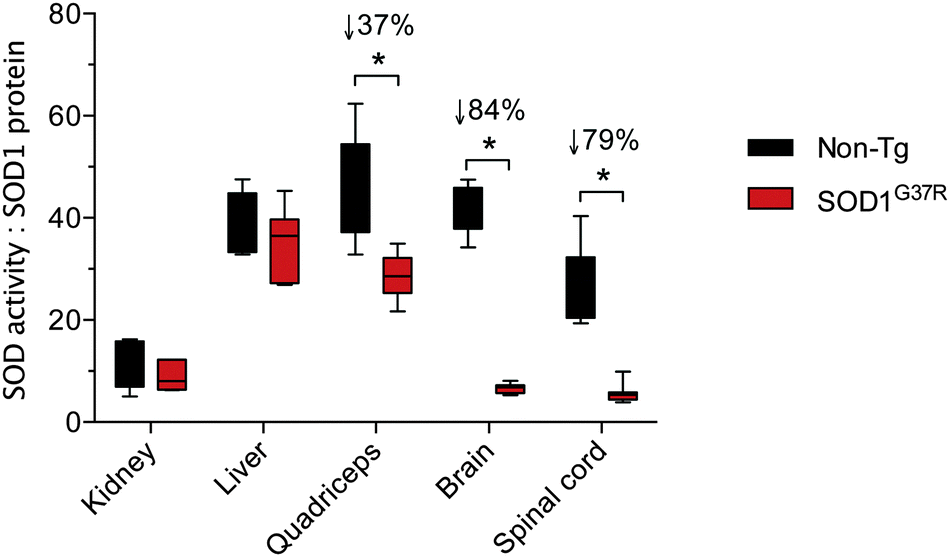 | ||
| Fig. 3 Tissue-specific ratios for SOD activity to SOD1 protein in ALS model SOD1G37R mice and non-transgenic (non-Tg) controls. The activity:protein ratio for each tissue represents overall SOD activity data (from Fig. 1A) expressed relative to overall SOD1 protein levels in each tissue (from Fig. 2B, but also accounting for differences in SOD1 protein across the different tissues). All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. *P < 0.05, two-tailed Mann Whitney test, n = 5–7. % values above statistically different data sets represent the difference in means for the SOD1G37R mice compared to the non-Tg controls. | ||
In order to take into account the native differences in the activity:protein observed across the different tissue types, we further assessed the apparent disparity between SOD activity and SOD1 protein in the SOD1G37R mice by directly expressing Δ SOD activity relative to Δ SOD1 protein. These calculations revealed the ratio of Δ SOD activity to Δ SOD1 protein is approximately 0.7–0.8 for the non-CNS tissues compared to approximately 0.2 for the brain and spinal cord (Fig. 4).
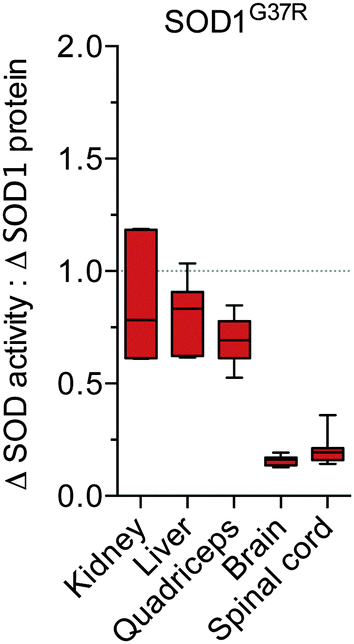 | ||
| Fig. 4 Tissue-specific disparity between SOD1 protein and activity in SOD1G37R mice. The tissue-specific change in SOD activity in SOD1G37R mice (Δ SOD activity from Fig. 1B) is expressed relative to the tissue-specific change in SOD1 protein in the SOD1G37R mice (Δ SOD1 protein from Fig. 2B). A value of 1 (grey dotted line) represents a proportional increase in SOD activity and SOD1 protein in the SOD1G37R mice. Values below 1 indicate the increase in SOD activity in the SOD1G37R mice is disproportionate to (less than) the increase in SOD1 protein in the SOD1G37R mice. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots, n = 7. | ||
Given that our analysis of SOD1 protein utilised reducing conditions in which all SOD1 is detected as monomeric SOD1, and given that physiological SOD1 functions as a dimer, the Δ SOD activity:Δ SOD1 protein ratio would be expected to be 0.5 at most if all of the additional SOD1 produced in the SOD1G37R mice was converted to metal-replete, dimeric holo-SOD1. In other words, a one unit increase in SOD activity for every two unit increase in SOD1 monomer. The observed variance from the expected maximal 0.5 value is likely due to the quantitative limitations of western blot. Alternatively, additional factors may be involved, such as the formation of hetero-dimers or the pre-existence of Cu-deficient SOD1 in the non-Tg mice. Regardless, the salient outcome remains: Δ SOD activity and Δ SOD1 protein both increase in all tissues of the SOD1G37R mice due to over-expression of the transgene, but in the CNS tissue Δ SOD activity is substantially less than could be expected when expressed relative to Δ SOD1 protein.
The increased requirement for copper in the central nervous system of SOD1G37R mice is not satiated
Given that SOD1 requires Cu for its catalytic activity, and based on evidence for Cu-deficient SOD1 in CNS tissue from transgenic mutant SOD1 mice,12–14 we hypothesised that the disparity between Δ SOD activity and Δ SOD1 protein in CNS tissue of the SOD1G37R mice may be related to limited Cu availability. An assessment of Cu in all tissues confirmed that in response to SOD1 over-expression Cu levels increased substantially in all non-CNS tissues from the SOD1G37R mice (Fig. 5A). By contrast, there was only a marginal increase in Cu in the brain, and Cu levels in the spinal cord were unchanged (Fig. 5A). Consistent with the additional requirement for Zn in SOD1, Zn levels were significantly increased in all CNS and non-CNS tissues (Fig. 5B). Others elements examined (Mg, P, and K) were not increased (Fig. 5C and D) indicating the observed changes to Cu and Zn were primarily occurring in response to over-expression of the Cu and Zn binding SOD1.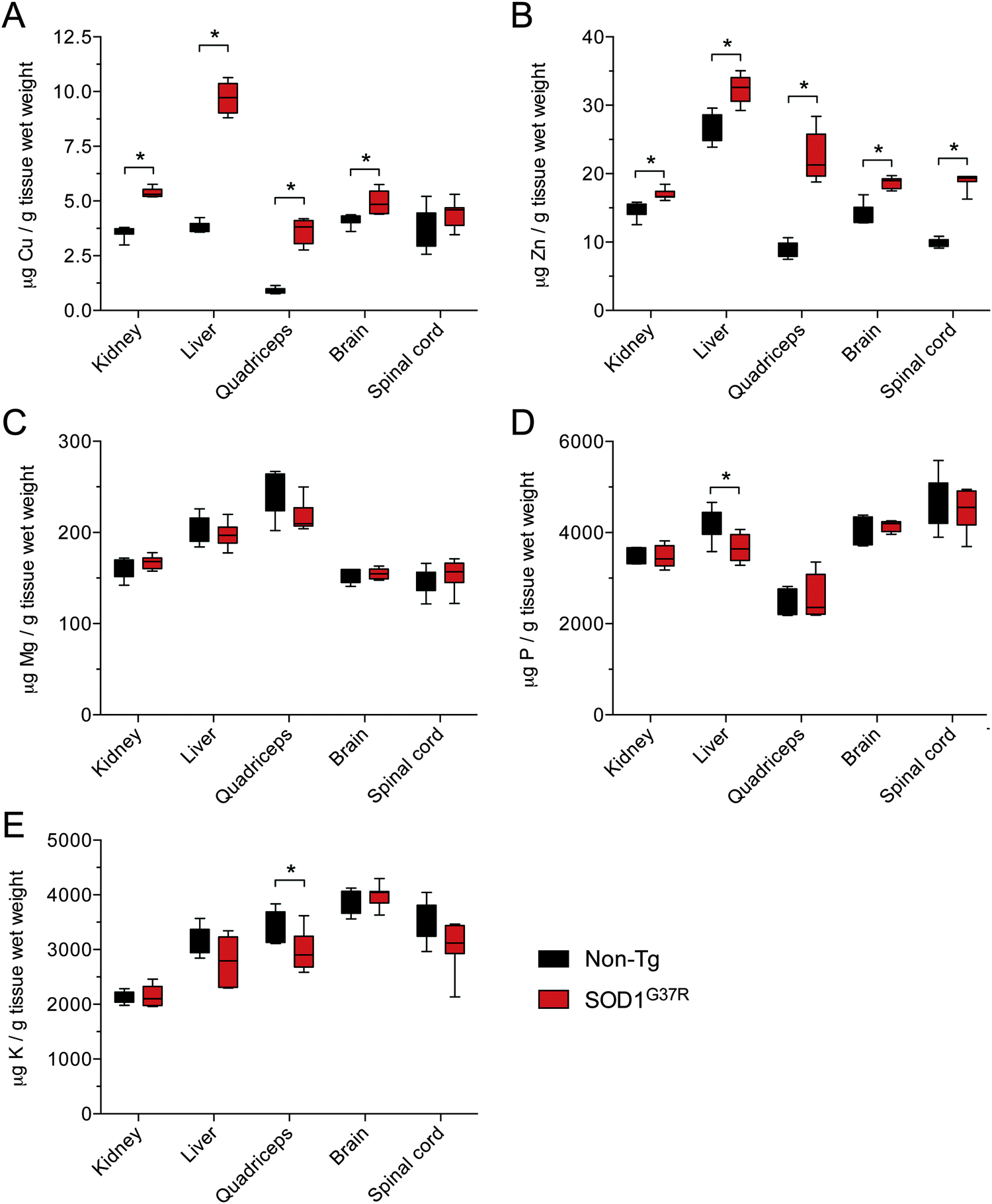 | ||
| Fig. 5 Metal content of tissues from SOD1G37R mice and non-transgenic (non-Tg) controls. The amount of copper (A), zinc (B), magnesium (C), phosphorous (D) and potassium (E) per g tissue wet weight in kidney, liver, quadriceps, brain and spinal cord tissue collected from SOD1G37R mice and non-Tg controls. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. *P < 0.05, two-tailed Mann Whitney test, n = 6–7. | ||
Considering that physiological SOD1 binds Cu and Zn in a molar ratio of 1:1, we assessed the changes to Cu and Zn in the SOD1G37R mice from the perspective of a molar increase above non-Tg levels (Δ μmol Cu and Δ μmol Zn). These data again show that Zn is increased in all tissues in response to over-expression of SOD1 in the SOD1G37R mice but that changes to Cu in the CNS tissue are either marginal (brain) or not significant (spinal cord) (Fig. 6A and B). Moreover, when Δ μmol Cu in the SOD1G37R mouse tissues is expressed relative to Δ μmol Zn, it becomes evident that a 1:1 molar increase predicted to ensure the increased expression of SOD1 is satiated for its Cu and Zn requirement, the 1:1 requirement is not met in the CNS tissue (Fig. 6C) due to a limited increase in Cu. The same Δ μmol Cu:Δ μmol Zn analysis indicates the increased requirement for Cu is also unmet in the quadriceps of SOD1G37R mice (Fig. 6C), but this appears driven by a disproportionate increase in Zn in the quadriceps (Fig. 5B), and not limited availability of Cu. Data to support Δ SOD1 protein in the quadriceps is satiated for its increased Cu requirement is also supported by a relatively proportionate increase in Δ SOD activity in the quadriceps in SOD1G37R mice (Fig. 1B and 4). The alternate calculation for overall SOD1 activity:protein in each tissue however, indicates activity in the quadriceps may be impaired to some extent (Fig. 3).
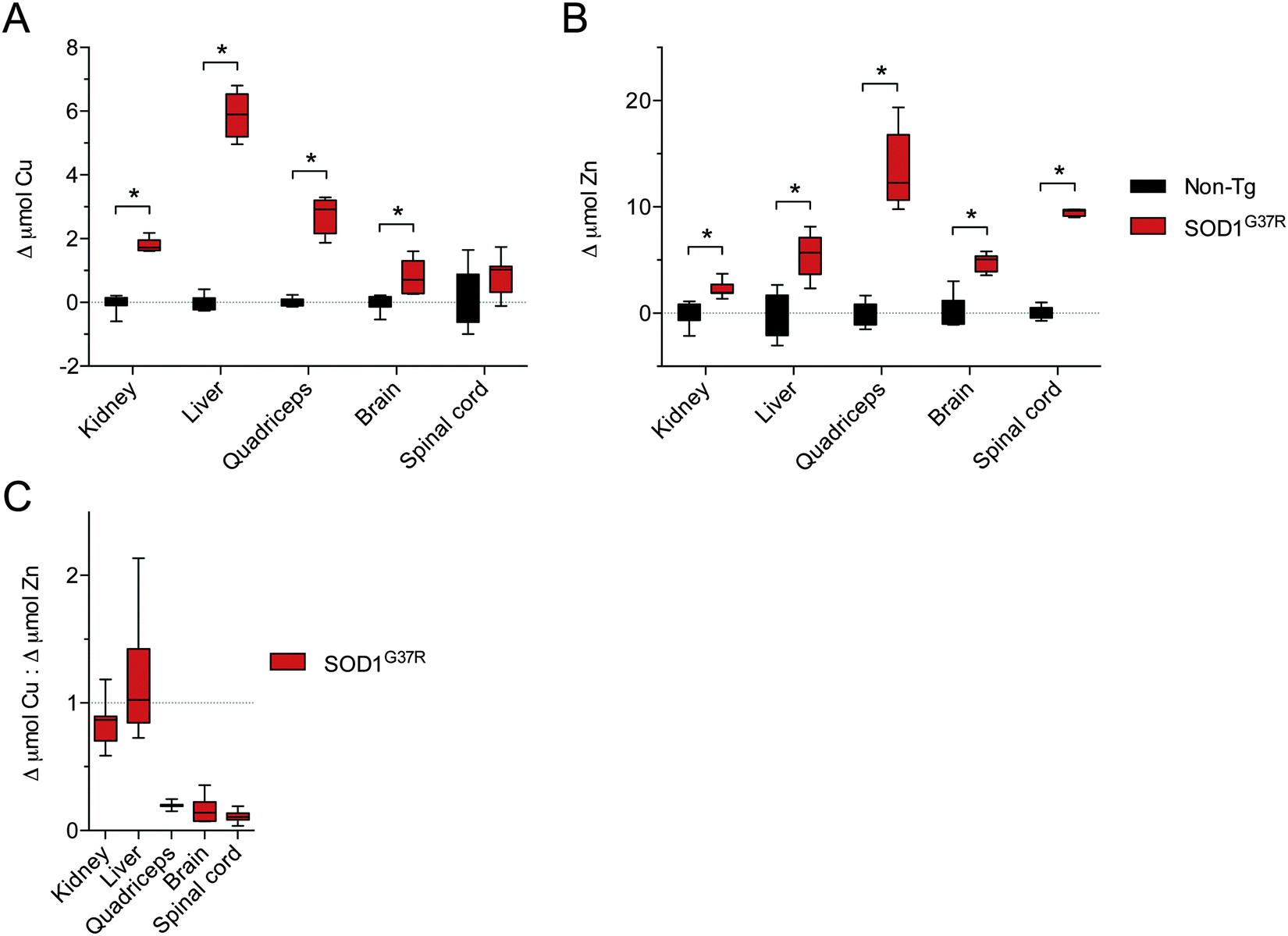 | ||
| Fig. 6 Disproportionate changes to Cu and Zn in tissues from SOD1G37R mice relative to non-transgenic (Non-Tg) controls. (A) The increase in Cu in various tissues from SOD1G37R mice above the mean for Cu in each tissue from non-Tg controls. (B) The increase in Zn in various tissues from SOD1G37R mice above the mean for Zn in each tissue from non-Tg controls. Data in (A) and (B) are calculated from raw data presented in Fig. 5A and B, respectively. (C) Ratio of the increase in Cu in different tissues of SOD1G37R mice to the increase in Zn in the same tissues. A value of 1 (grey dotted line) represents a proportional molar increase in Cu and Zn in the SOD1G37R mice. Values below 1 indicate the increase in Cu in the SOD1G37R mice is disproportionate to (less than) the increase in Zn in the SOD1G37R mice. All data are presented as box (median ±95% CI) and whisker (maximum and minimum) plots. *P < 0.05, two-tailed Mann Whitney test, n = 6–7. | ||
Our calculations for Δ μmol Cu:Δ μmol Zn and the deviation from a predicted 1:1 change in response to the over-expression of SOD1 in the SOD1G37R mice places considerable emphasis on the assumption that the observed changes to Cu and Zn reflect metal bound to SOD1. To partly address the veracity of this, we calculated the overall concentrations for Cu and Zn in the spinal cord tissue and related these to the overall concentration of spinal cord SOD1. These calculations are summarised in Table 1. The SOD1 concentration in the spinal cords of SOD1G37R mice was determined in a previous study via mass spectrometry and calculated to be 130 μM for monomeric SOD1.13 Western blot data (Fig. 2B) indicate the concentration of SOD1 in the non-Tg spinal cord is 16-fold lower than in the SOD1G37R mouse spinal cord, an extrapolated difference of 122 μM. Calculating the concentrations for Cu and Zn in the spinal cord tissue revealed the difference between SOD1G37R and non-Tg mice was 14 μM for Cu and 139 μM for Zn (Table 1). These indicate that the increase in Zn detected in the spinal cords of SOD1G37R mice essentially matches the calculated increase in SOD1 protein whereas the increase in Cu falls short. However, using the western blot data to extrapolate the SOD1 concentration in the non-Tg spinal cord is a persistent limitation of these calculations and greater accuracy would be obtained when using a more quantitative method for determining the concentration of SOD1 in the non-Tg spinal.
| μM SOD1 | μM Cu | μM Zn | % Cu bound to SOD1 | |
|---|---|---|---|---|
| a Calculated from direct measurement of SOD1 concentration in spinal cords of SOD1G37R mice13 and the difference between SOD1G37R and non-Tg mice presented in Fig. 2B. b Calculated from data presented in Fig. 5A and B with the assumption that 1 g tissue = 1 mL. N/A: concentration of SOD1 exceeds the concentration of Cu in these mice. | ||||
| Non-Tg | 8.2a | 56.2b | 150.3b | 14.6% |
| SOD1G37R | 130.613 | 70.1b | 288.8b | N/A |
| Difference | 122.4 | 13.9 | 138.3 | |
Discussion
Outcomes from the present study indicate that due to ubiquitous transgenic over-expression of SOD1 the requirement for Cu and Zn is elevated in all CNS and non-CNS tissues in SOD1G37R mice. The elevated requirement for Zn appears satiated across all tissues, but the elevated requirement for Cu appears to be limited, particularly in the CNS tissue. We propose that these outcomes are in part related to the relatively slow natural turnover of Cu in CNS tissue causing Cu bioavailability in the CNS to become a rate-limiting factor in the formation of metal replete holo-SOD1. This possibility is supported by data that indicate the natural turnover rates for copper are 20 to 2000 times slower in the CNS compared to other tissues.19 Thus, if the requirement for copper increases, the CNS will be one of the slowest tissues to respond.Limited Cu bioavailability within specific tissues may explain why more direct measures of the metal state of SOD1 have already shown a large proportion of SOD1 in the CNS of mutant SOD1 mice accumulates in a Cu-deficient form.12–14 But further to this, it may also explain why improving Cu bioavailability via treatment with CuII(atsm) is effective in mutant SOD1 mice; treating mutant SOD1 mice with CuII(atsm) improves the animals’ ALS-like phenotype, protects motor neurons and increases overall survival of the mice.13,14,20,21 Two of these studies have shown that treating with CuII(atsm) decreases the pool of Cu-deficient SOD113,14 and one has shown that Cu from orally administered CuII(atsm) is recovered in SOD1 extracted from the spinal cord tissue of the treated mice.13 These outcomes all indicate that improving Cu bioavailability in the CNS of mutant SOD1 mice is therefore at least part of the protective mechanism of action of CuII(atsm), and this possibility is supported by an additional study which has shown improving Cu bioavailability in the CNS of mutant SOD1 mice via over-expression of the Cu-uptake transporter CTR1 is also protective.13
Insufficient Cu bioavailability leading to the accumulation of Cu-deficient SOD1 could explain some toxic mechanisms in ALS attributed to SOD1. Specifically, the metal state of SOD1 is a key determinant of the protein’s stability and structure22–25 and insufficient presentation of Cu to SOD1 could therefore contribute to the SOD1 mis-folding and aggregation that is a pathological hallmark of sporadic and familial cases of ALS. Evidence for this exists through the reported susceptibility of both mutant and wild-type SOD1 to fibril formation or seed aggregation from its nascent polypeptide stage until dimeric maturity, whereby full metallation and disulphide bond formation is required to overcome this pathological propensity.26 Moreover, a model has been proposed whereby Cu-deficient (Zn-containing) SOD1 contributes differentially to the distinct processes of initiation of SOD1 aggregation and seeded growth of SOD1 amyloid fibrils depending on whether or not the Cu-deficient SOD1 is also disulphide-reduced.26
However, some data already exist to suggest excessive Cu accumulation, not insufficient Cu bioavailability, is problematic in mutant SOD1 mice and that chelating excess Cu is protective. Treating with the Cu-chelating compound tetrathiomolybdate decreased Cu levels and SOD1 activity in the spinal cords of SOD1G93A mice, improved the animals’ ALS-like symptoms and increased their overall survival.27 But these data appear at odds with a more recent study which has demonstrated the obligate requirement for Cu in motor neurons and the direct relationship between limited Cu bioavailability in motor neurons and the development of an ALS-like phenotype; targeted deletion of the copper transporter ATP7A from motor neurons in mice caused a significant decrease in Cu in the spinal cord, decreased innervation of neuromuscular junctions, loss of motor neurons, and a progressive decrease in locomotive function.28 Furthermore, the need to delineate between the chelation activity of compounds such as tetrathiomolybdate and their potential biometal redistribution activity has been articulated.29 However, decreased SOD1 activity in the tetrathiomolybdate treated SOD1G93A mice does suggest the treatment is working via a true chelation mechanism of action in these mice.
It is therefore apparent that a better understanding of the role that Cu plays in the ALS-like phenotype of mutant SOD1 mice is needed to help reconcile these apparent conflicting results; ATP7A deletion decreases total Cu levels and induces an ALS-like phenotype28 and treating with CuII(atsm) or over-expressing CTR1 increases Cu levels and is protective,13,14,20,21 yet treating with tetrathiomolybdate decreases Cu levels and is also protective.27 We propose adequate bioavailability of Cu within a given tissue is more important than bulk changes in Cu content within the tissue. Furthermore, we propose that an overall change in the requirement for Cu is also an insufficient singular factor to consider when attempting to elucidate the role for Cu in ALS. Indeed, over-expression of SOD1 in the mutant SOD1 transgenic mice may explain why the overall requirement for Cu increases, but an increased biological demand for Cu in the CNS does not appear to be the sole reason why mutant SOD1 over-expressing mice develop an ALS-like phenotype. Transgenic mice that over-express wild-type SOD1 protein at levels comparable to SOD1 protein in the mutant SOD1 mice do not develop an ALS-like phenotype,2 indicating that the additional impact that mutant SOD1 has on the expression of Cu chaperones and transporters30,31 may be a significant additional factor. However, homozygous expression of wild-type human SOD1 (at approximately 50 times the level of the endogenous murine SOD1) does induce motor neuron pathology and phenotypic symptoms comparable to those that present in mutant SOD1 expressing mice.32 Only 15% of total SOD1 in the spinal cord of mice homozygous for wild-type human SOD1 was reported to be catalytically active (lower than the brain and substantially lower than the non-disease affected liver tissue), and the addition of exogenous Cu restored SOD activity to extracts from spinal cords of the homozygous wild-type SOD1 over-expressors.32 Thus, a bulk increase in the requirement for Cu driven solely by SOD1 over-expression does appear to cause ALS-like pathology in these mice. But this requires considerably higher expression levels when compared to the mutant protein, suggesting the mutations introduce an additive factor that exacerbates the limited bioavailability of Cu in the CNS. Indeed, further subtlety in the relationship between mutation state and expression levels in the context of pathology is highlighted by the low penetrance L117V SOD1 mutation in ALS.33 Comparable stability and activity levels for the L117V SOD1 and wild-type SOD1 suggest the L117V mutation does not contribute to ALS via a mechanism that can be attributed solely to the SOD1 protein itself, thereby suggesting the mutation causes ALS via other mechanisms. However, these measurements were derived from erythrocytes,33 leaving the possibility that an investigation of disease-affected regions of the CNS may generate alternate outcomes, as proposed here based on the CNS and non-CNS tissue-specific changes observed in the SOD1G37R mice.
Overall, we propose that limited Cu bioavailability is a significant feature of the development of ALS-like symptoms in transgenic mice that ubiquitously over-express mutant SOD1. An elevated requirement for Cu, mutant SOD1-mediated changes to Cu handling processes, and a natural slow turnover of Cu in the CNS all appear to combine to lead to selective degeneration within the CNS.
Conclusions
SOD1 mutations are an unequivocal cause of familial ALS and transgenic mice over-expressing mutant SOD1 are a robust animal model of the disease. But the mechanisms by which mutant SOD1 cause motor neuron degeneration in the CNS remain to be fully elucidated. Better understanding of these mechanisms is needed to expedite development of new therapeutic options. Evidence that improving Cu bioavailability in the CNS is protective in mutant SOD1 mice13,14 indicates a role for Cu. Moreover, new data presented here indicate the role that Cu plays in this disease could partly explain why the CNS is selectively affected, particularly in those cases where the selective degeneration of motor neurons in the CNS is caused by ubiquitous expression of mutant SOD1.Acknowledgements
All ICP-MS analyses were performed at the Biometals Facility, Florey Institute of Neurosciences and Mental Health by Ms Irene Volitakis. The research was supported by funds from the Australian National Health and Medical Research Council (NHMRC Grant 1061550) and the University of Melbourne. JBH is recipient of the Australian Postgraduate Award and the Nancy Frances Curry Scholarship. PJC is recipient of the NHMRC R.D. Wright Biomedical Research Fellowship (CDF2, 1084927).Notes and references
- D. R. Rosen, T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J. P. O'Regan and H. X. Deng, et al., Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis, Nature, 1993, 362, 59 CrossRef CAS PubMed.
- M. E. Gurney, H. Pu, A. Y. Chiu, M. C. Dal Canto, C. Y. Polchow, D. D. Alexander, J. Caliendo, A. Hentati, Y. W. Kwon and H. X. Deng, et al., Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation, Science, 1994, 264, 1772 CrossRef CAS PubMed.
- P. C. Wong, C. A. Pardo, D. R. Borchelt, M. K. Lee, N. G. Copeland, N. A. Jenkins, S. S. Sisodia, D. W. Cleveland and D. L. Price, An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria, Neuron, 1995, 14, 1105 CrossRef CAS PubMed.
- M. C. Dal Canto and M. E. Gurney, A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis, Acta Neuropathol., 1997, 93, 537 CrossRef CAS PubMed.
- G. S. Ralph, P. A. Radcliffe, D. M. Day, J. M. Carthy, M. A. Leroux, D. C. Lee, L. F. Wong, L. G. Bilsland, L. Greensmith, S. M. Kingsman, K. A. Mitrophanous, N. D. Mazarakis and M. Azzouz, Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model, Nat. Med., 2005, 11, 429 CrossRef CAS PubMed.
- A. Henriques, C. Pitzer and A. Schneider, Characterization of a novel SOD-1(G93A) transgenic mouse line with very decelerated disease development, PLoS One, 2010, 5, e15445 CrossRef PubMed.
- P. M. Andersen, K. B. Sims, W. W. Xin, R. Kiely, G. O'Neill, J. Ravits, E. Pioro, Y. Harati, R. D. Brower, J. S. Levine, H. U. Heinicke, W. Seltzer, M. Boss and R. H. Brown Jr, Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes, Amyotrophic Lateral Scler. Other Mot. Neuron Disord., 2003, 4, 62 CrossRef CAS.
- Y. Hayashi, K. Homma and H. Ichijo, SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS, Adv. Biol. Regul., 2016, 60, 95 CrossRef CAS PubMed.
- S. J. Kaur, S. R. McKeown and S. Rashid, Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis, Gene, 2016, 577, 109 CrossRef CAS PubMed.
- S. S. Leal and C. M. Gomes, Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability, Front. Cell. Neurosci., 2015, 9, 225 Search PubMed.
- S. Parakh and J. D. Atkin, Protein Folding Alterations in Amyotrophic Lateral Sclerosis, Brain Res., 2016 DOI:10.1016/j.brainres.2016.04.010.
- H. L. Lelie, A. Liba, M. W. Bourassa, M. Chattopadhyay, P. K. Chan, E. B. Gralla, L. M. Miller, D. R. Borchelt, J. S. Valentine and J. P. Whitelegge, Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice, J. Biol. Chem., 2011, 286, 2795 CrossRef CAS PubMed.
- B. R. Roberts, N. K. Lim, E. J. McAllum, P. S. Donnelly, D. J. Hare, P. A. Doble, B. J. Turner, K. A. Price, S. C. Lim, B. M. Paterson, J. L. Hickey, T. W. Rhoads, J. R. Williams, K. M. Kanninen, L. W. Hung, J. R. Liddell, A. Grubman, J. F. Monty, R. M. Llanos, D. R. Kramer, J. F. Mercer, A. I. Bush, C. L. Masters, J. A. Duce, Q. X. Li, J. S. Beckman, K. J. Barnham, A. R. White and P. J. Crouch, Oral treatment with CuII(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis, J. Neurosci., 2014, 34, 8021 CrossRef CAS PubMed.
- J. R. Williams, E. Trias, P. R. Beilby, N. I. Lopez, E. M. Labut, C. S. Bradford, B. R. Roberts, E. J. McAllum, P. J. Crouch, T. W. Rhoads, C. Pereira, M. Son, J. L. Elliott, M. C. Franco, A. G. Estevez, L. Barbeito and J. S. Beckman, Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD mice co-expressing the Copper-Chaperone-for-SOD, Neurobiol. Dis., 2016, 89, 1 CrossRef CAS PubMed.
- F. Li, N. Y. Calingasan, F. Yu, W. M. Mauck, M. Toidze, C. G. Almeida, R. H. Takahashi, G. A. Carlson, M. Flint Beal, M. T. Lin and G. K. Gouras, Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice, J. Neurochem., 2004, 89, 1308 CrossRef CAS PubMed.
- S. Marklund and G. Marklund, Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase, Eur. J. Biochem., 1974, 47, 469 CrossRef CAS PubMed.
- P. S. Donnelly, J. R. Liddell, S. Lim, B. M. Paterson, M. A. Cater, M. S. Savva, A. I. Mot, J. L. James, I. A. Trounce, A. R. White and P. J. Crouch, An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 47 CrossRef CAS PubMed.
- D. R. Borchelt, M. K. Lee, H. S. Slunt, M. Guarnieri, Z. S. Xu, P. C. Wong, R. H. Brown Jr, D. L. Price, S. S. Sisodia and D. W. Cleveland, Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8292 CrossRef CAS.
- C. W. Levenson and M. Janghorbani, Long-term measurement of organ copper turnover in rats by continuous feeding of a stable isotope, Anal. Biochem., 1994, 221, 243 CrossRef CAS PubMed.
- E. J. McAllum, N. K. H. Lim, J. L. Hickey, B. M. Paterson, P. S. Donnelly, Q. X. Li, K. J. Barnham, A. R. White and P. J. Crouch, Therapeutic effects of CuII(atsm) in the SOD1G37R mouse model of amyotrophic lateral sclerosis, Amyotrophic Lateral Scler. Frontotemporal Degener., 2013, 14, 586 CrossRef CAS PubMed.
- C. P. Soon, P. S. Donnelly, B. J. Turner, L. W. Hung, P. J. Crouch, N. A. Sherratt, J. L. Tan, N. K. Lim, L. Lam, L. Bica, S. Lim, J. L. Hickey, J. Morizzi, A. Powell, D. I. Finkelstein, J. G. Culvenor, C. L. Masters, J. Duce, A. R. White, K. J. Barnham and Q. X. Li, Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model, J. Biol. Chem., 2011, 286, 44035 CrossRef CAS PubMed.
- H. J. Forman and I. Fridovich, On the stability of bovine superoxide dismutase. The effects of metals, J. Biol. Chem., 1973, 248, 2645 CAS.
- S. M. Lynch, S. A. Boswell and W. Colon, Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: implications for ALS, Biochemistry, 2004, 43, 16525 CrossRef CAS PubMed.
- J. A. Rumfeldt, J. R. Lepock and E. M. Meiering, Unfolding and folding kinetics of amyotrophic lateral sclerosis-associated mutant Cu,Zn superoxide dismutases, J. Mol. Biol., 2009, 385, 278 CrossRef CAS PubMed.
- P. Ip, V. K. Mulligan and A. Chakrabartty, ALS-causing SOD1 mutations promote production of copper-deficient misfolded species, J. Mol. Biol., 2011, 409, 839 CrossRef CAS PubMed.
- M. Chattopadhyay, E. Nwadibia, C. D. Strong, E. B. Gralla, J. S. Valentine and J. P. Whitelegge, The Disulfide Bond, but Not Zinc or Dimerization, Controls Initiation and Seeded Growth in Amyotrophic Lateral Sclerosis-linked Cu,Zn Superoxide Dismutase (SOD1) Fibrillation, J. Biol. Chem., 2015, 290, 30624 CrossRef CAS PubMed.
- E. Tokuda, S. Ono, K. Ishige, S. Watanabe, E. Okawa, Y. Ito and T. Suzuki, Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis, Exp. Neurol., 2008, 213, 122 CrossRef CAS PubMed.
- V. L. Hodgkinson, J. M. Dale, M. L. Garcia, G. A. Weisman, J. Lee, J. D. Gitlin and M. J. Petris, X-inked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuon, J. Pathol., 2015, 236, 241 CrossRef CAS PubMed.
- P. J. Crouch and K. J. Barnham, Therapeutic redistribution of metal ions to treat Alzheimer's disease, Acc. Chem. Res., 2012, 45, 1604 CrossRef CAS PubMed.
- E. Tokuda, E. Okawa and S. Ono, Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutase-linked familial amyotrophic lateral sclerosis, J. Neurochem., 2009, 111, 181 CrossRef CAS PubMed.
- E. Tokuda, E. Okawa, S. Watanabe, S. Ono and S. L. Marklund, Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities, Neurobiol. Dis., 2013, 54, 308 CrossRef CAS PubMed.
- K. S. Graffmo, K. Forsberg, J. Bergh, A. Birve, P. Zetterstrom, P. M. Andersen, S. L. Marklund and T. Brannstrom, Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis, Hum. Mol. Genet., 2013, 22, 51 CrossRef CAS PubMed.
- M. Synofzik, D. Ronchi, I. Keskin, A. N. Basak, C. Wilhelm, C. Gobbi, A. Birve, S. Biskup, C. Zecca, R. Fernandez-Santiago, T. Kaugesaar, L. Schols, S. L. Marklund and P. M. Andersen, Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS, Hum. Mol. Genet., 2012, 21, 3568 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |