
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protonation state of F420H2 in the prodrug-activating deazaflavin dependent nitroreductase (Ddn) from Mycobacterium tuberculosis†
A. Elaaf
Mohamed
a,
F. Hafna
Ahmed
a,
Sundaram
Arulmozhiraja
a,
Ching Y.
Lin
a,
Matthew C.
Taylor
b,
Elmars R.
Krausz
a,
Colin J.
Jackson
*a and
Michelle L.
Coote
*ac
aResearch School of Chemistry, The Australian National University, Canberra, ACT 2601, Australia. E-mail: colin.jackson@anu.edu.au; michelle.coote@anu.edu.au
bCSIRO Land and Water, Black Mountain Laboratories, Canberra, ACT 2601, Australia
cARC Centre of Excellence for Electromaterials Science,
First published on 9th February 2016
Abstract
The protonation state of the deazaflavin dependent nitroreductase (Ddn) enzyme bound cofactor F420 was investigated using UV-visible spectroscopy and computational simulations. The reduced cofactor F420H2 was determined to be present in its deprotonated state in the holoenzyme form. The mechanistic implications of these findings are discussed.
The discovery of cofactor F420 (Fig. 1), and a new family of F420-dependent enzymes, in a wide range of Actinobacteria, including pathogenic mycobacteria,1–4 has led to increased interest in F420 dependent oxidoreductases (FDORs). The absence of F420 dependent biochemical reactions in humans, and their prevalence in Mycobacterium tuberculosis (TB), the causative agent of tuberculosis, has made the enzymes that depend on and synthesize this cofactor drug targets for new treatments of the disease.5 Bicyclic nitroimidazoles have recently been discovered to be activated by a class of FDORs called FDORAs,2,4,6 which are thought to have a physiological role in reducing oxidative stress against mycobacteria during infection.7 Bicyclic nitroimidazoles show anti-mycobacterial effects such as respiratory poisoning and the disruption of cell wall biosynthesis.8,9 One of these compounds, pretomanid (formerly PA-824) shown in Fig. 1, is currently in clinical trials and shows promising results against multi-drug resistant strains of TB.10
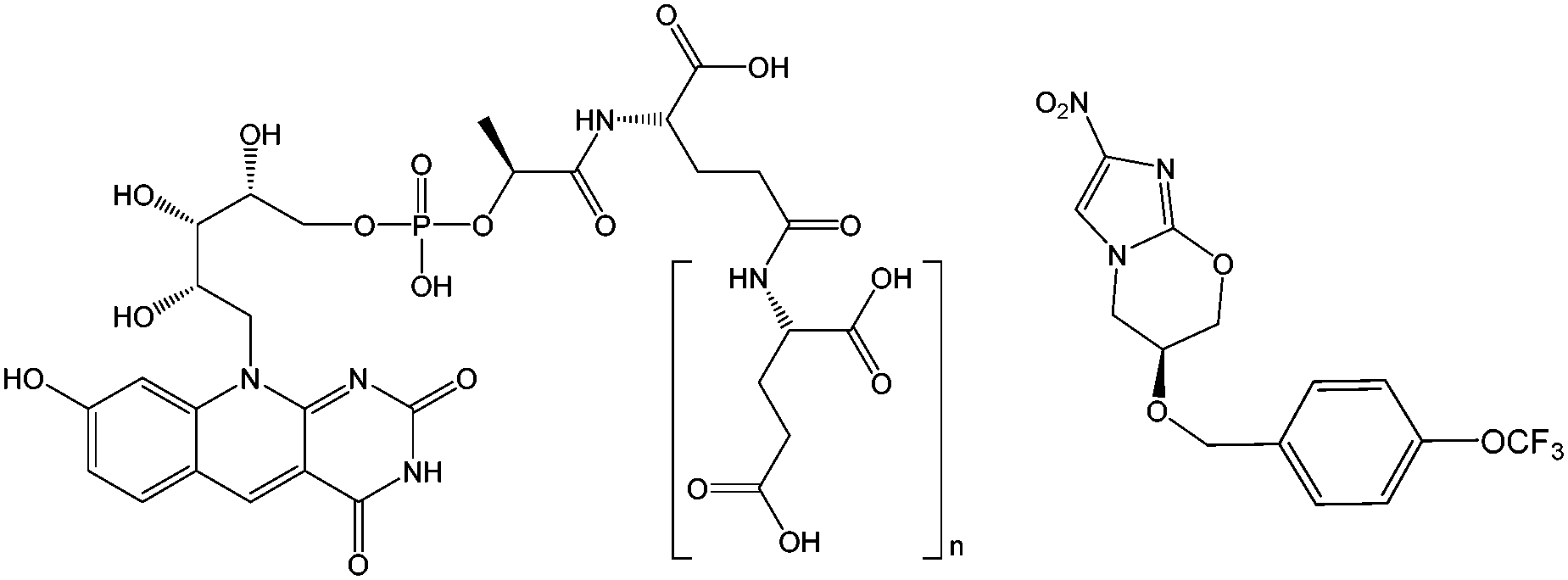 | ||
| Fig. 1 Structures of F420 (left) and pretomanid (right). | ||
In order to exert its anti-mycobacterial activity against M. tuberculosis, pretomanid must initially be reduced by the protein encoded by rv3547, referred to as the Deazaflavin-dependent nitroreductase (Ddn), in concert with reduced cofactor F420 (F420H2).1,6,11 This reaction results in the formation of three stable metabolites, one of which decomposes to nitric oxide (NO) and results in anaerobic anti-mycobacterial activity.1 Although the role of both Ddn and F420 in the activity of pretomanid is known,1,6,8,11 the precise catalytic mechanism is not fully understood. Thus, a better understanding of this reaction and the configuration and protonation state of the reactants within the enzyme's active site is essential to further improving this class of anti-mycobacterial pro-drugs.
While the deazaflavin F420 is structurally analogous to riboflavin based cofactors such as flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), it is functionally closer to nicotinamide (e.g. NAD) cofactors by being involved in redox reactions as a hydride carrier involved in two electron transfer mechanisms.12 Consistent with its involvement in redox reactions, F420 exists in both oxidized (F420) and reduced (F420H2) forms, as shown in Fig. 2. The oxidized cofactor exhibits strong and distinct UV-visible spectroscopic properties, with a characteristic absorbance peak at 420 nm that is different to the spectroscopic properties of the reduced species.13 This allows for the study of F420 dependent reactions using UV/vis spectroscopy to follow changes in the amounts of the oxidized cofactor being formed or consumed.1,4,6,7,11 These spectroscopic properties are also sensitive to pH changes, with the structural changes due to deprotonation of the 8-OH and 1-NH groups in the oxidized and reduced species (Fig. 2), respectively, influencing the absorbance bands.
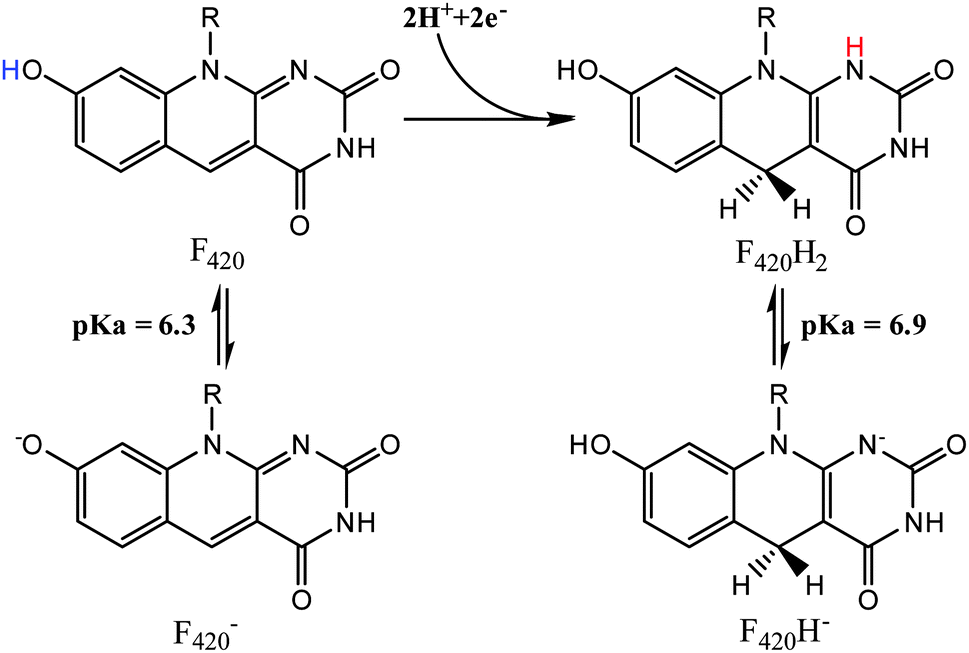 | ||
| Fig. 2 The protonated and deprotonated species of F420 (left) and F420H2 (right).14 | ||
The protonation state of the cofactor will significantly influence the catalytic mechanism owing to the availability or unavailability of labile hydrogens. However, in the current literature this has not been addressed and F420H2 is often presented as its neutral species.6 In this study we have determined the protonation state of F420 and F420H2 when bound to Ddn, and a homolog of Ddn from Mycobacterium smegmatis, MSMEG_2027, using a combination of UV-visible spectroscopy and time-dependent density functional theory (TD-DFT) calculations.4 Initially, absorbance spectra of both F420 and F420H2 were measured in an aqueous environment as a function of pH so as to assign the neutral and deprotonated spectra (Fig. 3; Fig. S1 in ESI†). However, in order to ensure that these assignments do not change in the lower polarity environment within the enzyme active site, we also used TD-DFT to calculate the neutral and deprotonated spectra under both aqueous and low polarity solvent environments (Fig. 4).
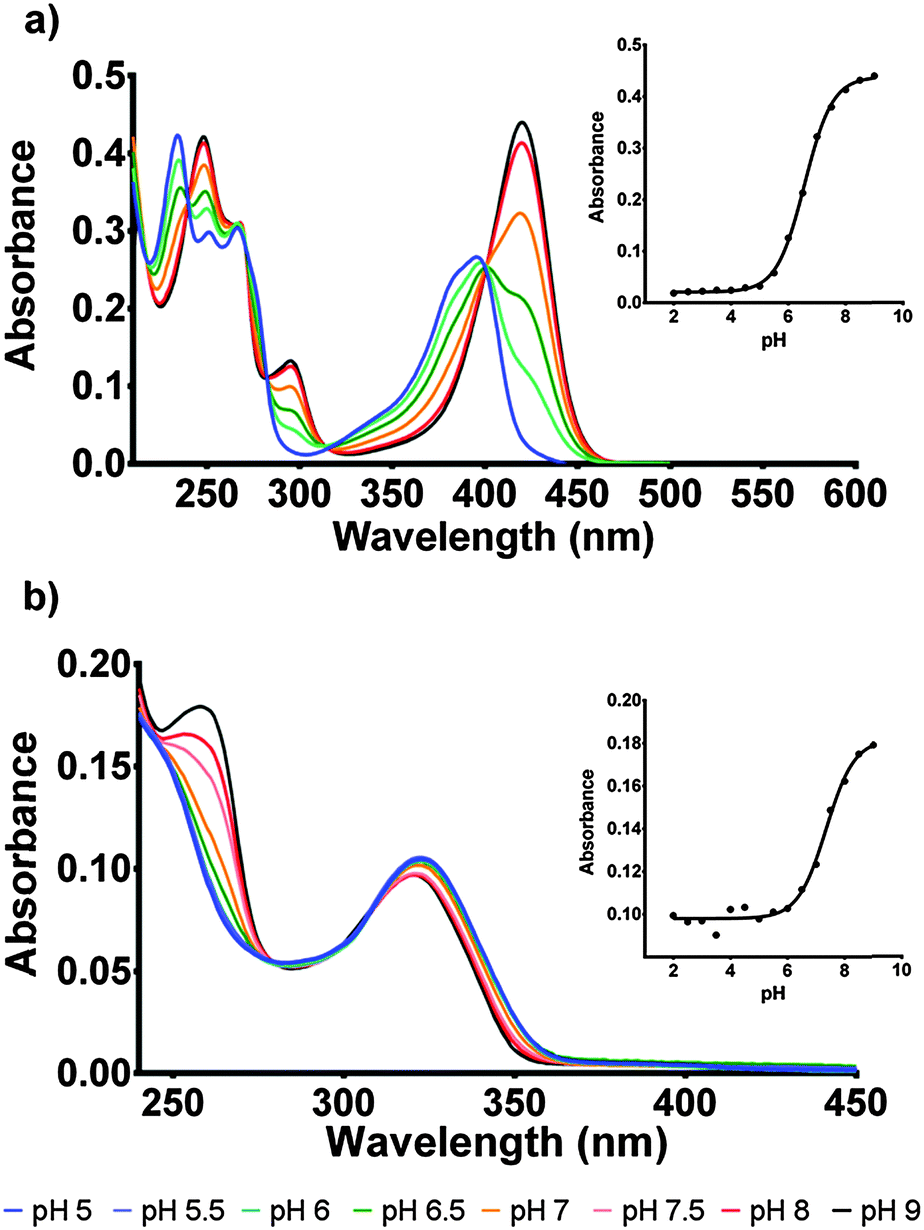 | ||
| Fig. 3 (a) pH curve of F420 for the pH range of 5–9 showing the spectroscopic changes observed with pH change. Inset: The curve of absorbance change at 420 nm across the pH range which determined the pKa for deprotonation was 6.5. (b) pH curve of F420H2 for pH range 5–9 with associated spectroscopic changes. Inset: The curve of absorbance change at 260 nm across the pH range which determined the pKa for deprotonation was 7.1. | ||
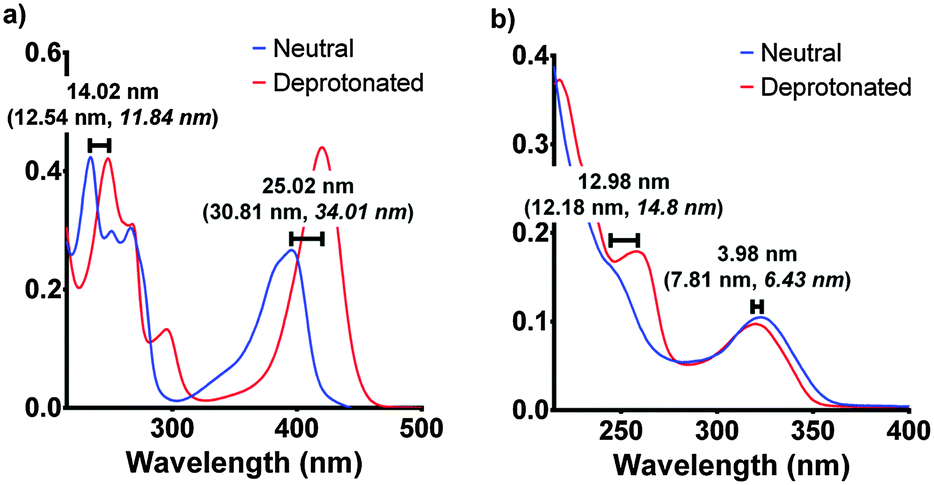 | ||
| Fig. 4 The experimental spectroscopic peak shifts for F420 (a) and F420H2 (b) compared with the calculated values (in parenthesis) for both aqueous and low polarity (in italics). The experimental results are those for pH 5 and pH 9 for neutral and deprotonated respectively. The electronic transitions were calculated using the ωB97XD/6-311+G(2d,p) level of theory in Gaussian 09.17,18 | ||
For F420, the characteristically intense absorbance peak at 420 nm gives way to a weaker peak at 400 nm at lower pH values with the observed pKa of 6.5 (Fig. 3a) agreeing with previously reported values of 6.3.14,15 The spectroscopic red-shift from 400 to 420 nm is also indicative of stabilization of the π–π* transitions stemming from the deprotonation of the 8-OH group. For F420H2, its characteristic peak at 320 nm did not exhibit the strong red-shift upon deprotonation observed in F420, instead it was the neutral species that was slightly red-shifted to 322 nm with the deprotonated species at 319 nm and consistent with literature for both F420 as well as the structural analogue 1,5-dihydrodeazariboflavin.15,16 Rather, a more noticeable spectral change is seen at 260 nm with a peak present in the deprotonated species. Both the 320 nm and 260 nm peaks exhibit a pKa near neutrality at 7.1 (Fig. 3b). These results agree with previously published literature values for F420H2 of 6.9 and 7.2 for F420H2 and 1,5-dihydrodeazariboflavin.15,16
Although the computationally derived electronic transitions for both species deviated from the experimental values, their relative values, and the trends that are observed, agree closely with the experimental spectrum (Fig. 4). For both species it was also noted that the electronic transitions responsible for the absorbance peaks remained in agreement for both the aqueous environment with a dielectric constant ε = 80.1 and the low polarity environment with ε = 12. This uniform behaviour enables us to directly compare experimental UV-visible spectra of the cofactor in aqueous conditions with that of when the cofactor is bound within the active site to complete the holoenzyme. The full comparisons of the simulated electronic transitions with experiment are in Fig. S2 in ESI.†
To determine the protonation state in the enzyme, the bound absorbance spectrum was measured with F420H2 bound to the Ddn and 2027 enzymes (Fig. 5; Fig. S4 in ESI†). The results identified the cofactor as being stabilized more in its deprotonated state even at neutral pH where the conversion usually takes place. Upon binding to the enzyme active site, the electronic transitions that give rise to the absorbance spectrum are stabilized due to electrostatic interactions with the surrounding amino acid residues leading to most spectroscopic features being red-shifted by approximately 5–10 nm. This observation proved to be a convenient indicator for whether the species being measured was actually bound to the active site or only present in the surrounding environment.
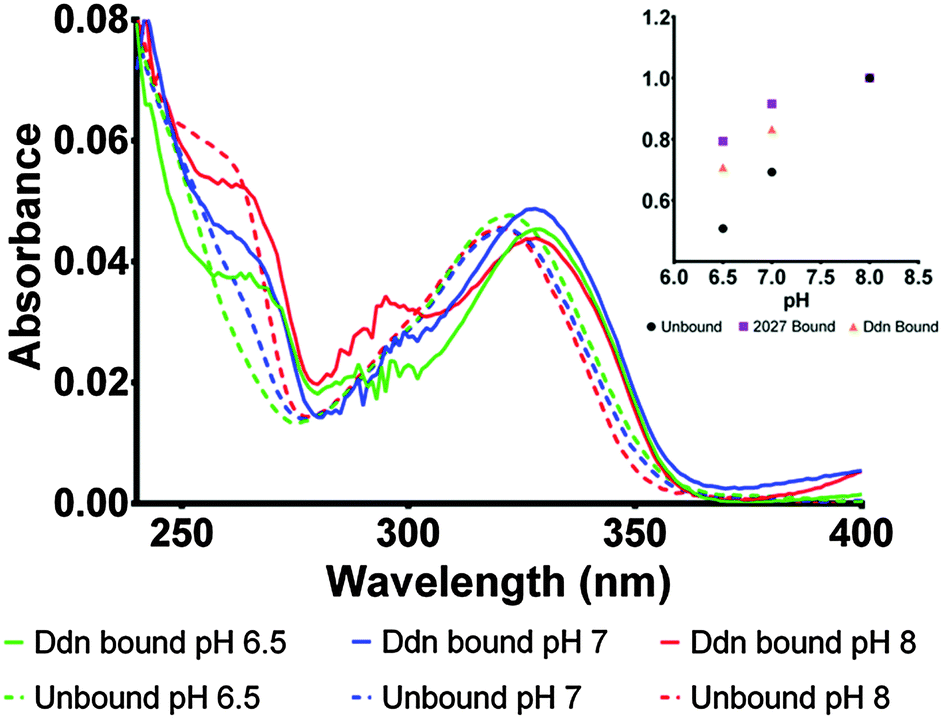 | ||
| Fig. 5 Comparison of the Ddn bound F420H2 absorbance spectra at different pH with the unbound aqueous species. The spectroscopic features are red-shifted by approximately 5–10 nm due to further stabilization from binding. The assignment of the protonation state can be made due to the presence of the 260 nm peak at both pH 7 and pH 6.5 which is absent in the aqueous spectra. The inset demonstrates the increase in absorbance (at 262 nm) at pH 6.5, 7 and 8 for both Ddn and 2027 bound when compared with the aqueous conditions. The values were normalised at pH 8 to account for concentration fluctuations. | ||
Owing to the limited spectroscopic changes at the 320 nm peak associated with the deprotonation, the secondary 260 nm peak, which exhibits greater differences between the two states, was used to determine the species present. At pH 8, where the cofactor is normally deprotonated in aqueous solution, no change was observed as both bound and unbound F420H2 exhibited the deprotonated peak at 260 nm. However, at pH 7 where the 260 nm peak is greatly diminished under aqueous conditions as it is shifted to the neutral state, the bound spectrum still shows a notable contribution of the 260 nm peak indicating the presence of the deprotonated species. Furthermore, the comparison of the bound spectrum at pH 6.5 and unbound spectrum at pH 6.5 corroborate this assessment as the 260 nm deprotonated peak is still observed at pH values below the (aqueous) pKa in the bound species while, as expected, is completely absent under aqueous conditions. On this basis we can conclude that the cofactor F420H2 is bound and stabilized within the active site in its deprotonated form.
This same observation is made using the experimental spectra of F420H2 bound to the Ddn homolog MSMEG_2027 (Fig. S4 in ESI†), which shares a similar active site structure.4Fig. 6 displays a docking simulation of the cofactor and substrate within the active site, which shows how the deprotonated species might be stabilized by the surrounding positively-charged electrostatic environment.
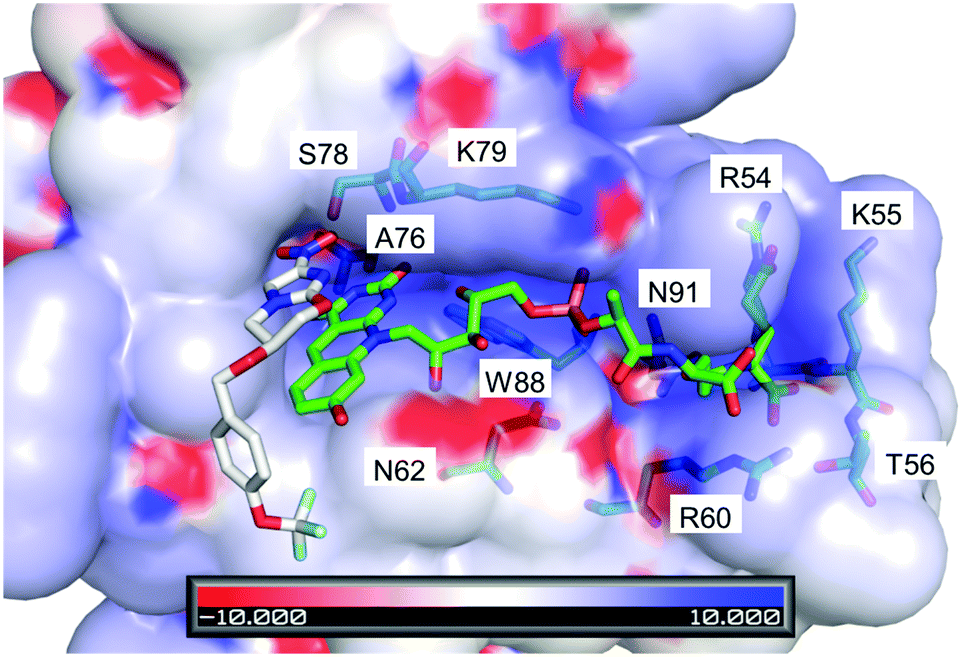 | ||
| Fig. 6 Crystal structure of F420 bound within Ddn (PDB ID: 3R5R), with the substrate pretomanid docked via simulation.4,6 The electrostatics of the surrounding residues reveals the positively charged surroundings which help bind and stabilize the negatively charged deprotonated cofactor. The residues that contribute to the positive environment are shown and labelled. Scale is in kT/e. | ||
Correctly assigning the protonation state of F420/F420H2 is essential for further mechanistic studies involving the cofactor F420H2 and Ddn. This result suggests that the proton transfer step of the reaction, which completes the 2H+/2e− reduction, has to be facilitated via a source other than from the cofactor. Additionally, the deprotonation of the reduced cofactor enables it to readily form the more stable neutral molecule F420 following the hydride transfer instead of a less stable carbocation that would result if the protonated species were involved. There are several ionisable sidechains in the vicinity of the cofactor that could act as general acids in this reaction; their potential involvement is currently under investigation. Conversely, it has also been suggested that the protonation of pretomanid is completed by water molecules from the surrounding environment.1 A proposed mechanism utilizing deprotonated F420H2 is presented in Fig. 7, where initial hydride transfer is followed by a proton transfer event. These steps result in the formation of oxidized F420, and reduced pretomanid-H2, which then undergoes heterolytic bond cleavage and bond formation to produce the anti-mycobacterial nitric oxide species.
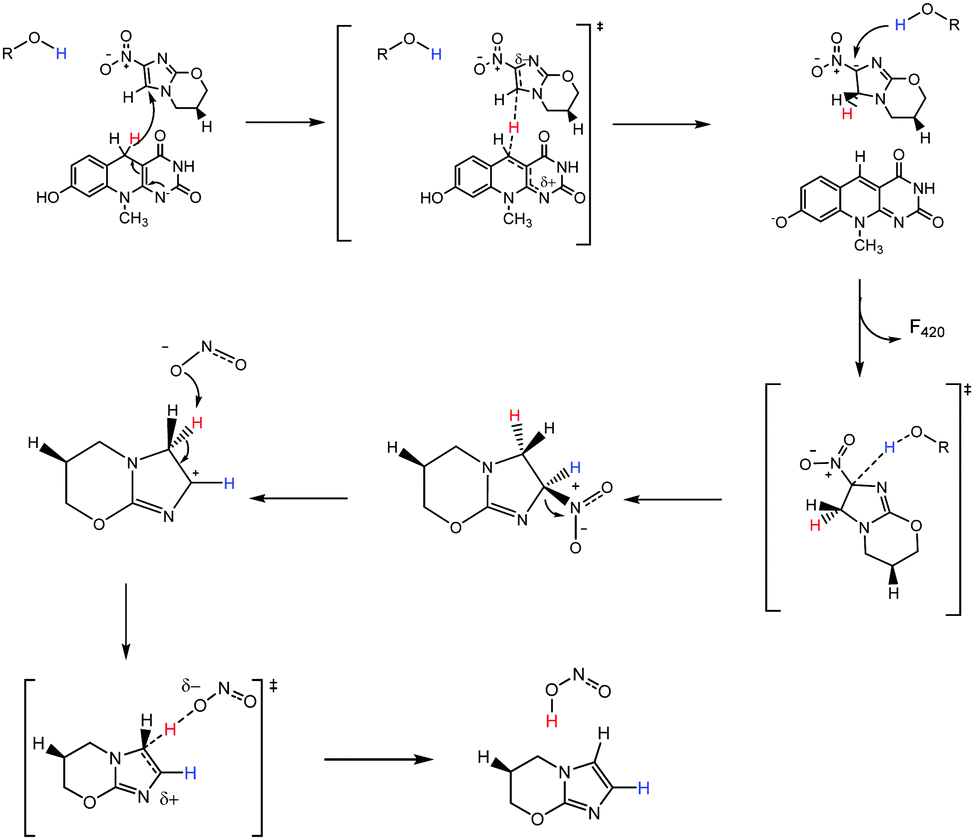 | ||
| Fig. 7 Proposed mechanism for the activation reaction of pretomanid by F420H2. The R group signifies the possibility of the proton source for being a tyrosine residue, serine residue or a water molecule and is currently under investigation. | ||
The holoenzyme structure solved by Cellitti et al. was obtained with the oxidized species F420 at pH 6.5.6 Therefore the cofactor would be deprotonated, as would be true for F420H2 at physiological pH: both molecules would lack the N1 hydrogen, although the hydroxyl group at C8 would most likely be deprotonated in F420. Since the C8–OH group extends into the solvent, this difference is unlikely to significantly affect substrate binding, making the crystal structure a good model for the holoenzyme structure. Our results provide some insight into the substrate specificity of Ddn since the substrate range would be relatively broad if the proton transferred to C2 could be donated by F420H2. Instead, because the proton appears to be donated from a side-chain or protein-stabilized water molecule, there are significant constraints on the substrate range, given that the substrates must be in sufficiently close contact with the general acid for proton transfer to occur. This model is consistent with previous work that showed there is little enantioselectivity with smaller pretomanid analogs, such as CGI-17341 and phenyl oxazole, since the C2 atom will be in the same approximate position in either enantiomer.11
In summary, we determined the protonation state of the cofactor F420H2 when bound to the Ddn active site. This has a significant impact on our understanding of the F420H2 mediated activation mechanism as it implies that maintaining the deprotonated state of F420H2 contributes to the catalytic efficiency of the enzyme and that the proton source to complete the reaction is not F420H2 itself, but a separate residue or nearby molecule involved within the active site.
MLC and CJJ gratefully acknowledge funding from the Australian Research Council in the form of Discovery Project funding (DP130102144) and ARC Future Fellowships. MLC also acknowledges generous allocations of supercomputing time on the National Facility of the Australian National Computational Infrastructure.
Notes and references
- R. Singh, U. Manjunatha, H. I. M. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim, L. Zhang, S. Kang, T. H. Keller, J. Jiricek and C. E. Barry, Science, 2008, 322, 1392–1395 CrossRef PubMed.
- M. C. Taylor, C. J. Jackson, D. B. Tattersall, N. French, T. S. Peat, J. Newman, L. J. Briggs, G. V. Lapalikar, P. M. Campbell, C. Scott, R. J. Russell and J. G. Oakeshott, Mol. Microbiol., 2010, 78, 561–575 CrossRef CAS PubMed.
- L. Daniels, N. Bakhiet and K. Harmon, Syst. Appl. Microbiol., 1985, 6, 12–17 CrossRef CAS.
- F. H. Ahmed, P. D. Carr, B. M. Lee, L. Afriat-Jurnou, A. E. Mohamed, N.-S. Hong, J. Flanagan, M. C. Taylor, C. Greening and C. J. Jackson, J. Mol. Biol., 2015, 427, 3554–3571 CrossRef CAS PubMed.
- J. D. Selengut and D. H. Haft, J. Bacteriol., 2010, 192, 5788–5798 CrossRef CAS PubMed.
- S. E. Cellitti, J. Shaffer, D. H. Jones, T. Mukherjee, M. Gurumurthy, B. Bursulaya, H. I. Boshoff, I. Choi, A. Nayyar, Y. S. Lee, J. Cherian, P. Niyomrattanakit, T. Dick, U. H. Manjunatha, C. E. Barry III, G. Spraggon and B. H. Geierstanger, Structure, 2012, 20, 101–112 CrossRef CAS PubMed.
- M. Gurumurthy, M. Rao, T. Mukherjee, S. P. S. Rao, H. I. Boshoff, T. Dick, C. E. Barry and U. H. Manjunatha, Mol. Microbiol., 2013, 8, 744–755 CrossRef PubMed.
- C. K. Stover, P. Warrener, D. R. VanDevanter, D. R. Sherman, T. M. Arain, M. H. Langhorne, S. W. Anderson, J. A. Towell, Y. Yuan, D. N. McMurray, B. N. Kreiswirth, C. E. Barry and W. R. Baker, Nature, 2000, 405, 962–966 CrossRef CAS PubMed.
- U. Manjunatha, H. I. M. Boshoff and C. E. Barry, Commun. Integr. Biol., 2009, 2, 215–218 CrossRef CAS PubMed.
- R. Dawson, A. H. Diacon, D. Everitt, C. van Niekerk, P. R. Donald, D. A. Burger, R. Schall, M. Spigelman, A. Conradie, K. Eisenach, A. Venter, P. Ive, L. Page-Shipp, E. Variava, K. Reither, N. E. Ntinginya, A. Pym, F. von Groote-Bidlingmaier and C. M. Mendel, Lancet, 2015, 385, 1738–1747 CrossRef CAS.
- M. Gurumurthy, T. Mukherjee, C. S. Dowd, R. Singh, P. Niyomrattanakit, J. A. Tay, A. Nayyar, Y. S. Lee, J. Cherian, H. I. Boshoff, T. Dick, C. E. Barry and U. H. Manjunatha, FEBS J., 2012, 279, 113–125 CrossRef CAS PubMed.
- C. Walsh, Acc. Chem. Res., 1986, 19, 216–221 CrossRef CAS.
- P. Cheeseman, A. Toms-Wood and R. S. Wolfe, J. Bacteriol., 1972, 112, 527–531 CAS.
- L. M. de Poorter, W. J. Geerts and J. T. Keltjens, Microbiology, 2005, 151, 1697–1705 CrossRef CAS PubMed.
- L. D. Eirich, G. D. Vogels and R. S. Wolfe, Biochemistry, 1978, 17, 4583–4593 CrossRef CAS PubMed.
- R. Spencer, J. Fisher and C. Walsh, Biochemistry, 1976, 15, 1043–1053 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, et al., Gaussian 09, Rev D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00033a |
| This journal is © The Royal Society of Chemistry 2016 |