
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of bacterially expressed erythropoietin by single site-specific introduction of short branched PEG chains at naturally occurring glycosylation sites†
E.
Hoffmann
a,
K.
Streichert
b,
N.
Nischan‡
b,
C.
Seitz
c,
T.
Brunner
c,
S.
Schwagerus
b,
C. P. R.
Hackenberger
*b and
M.
Rubini
*a
aDepartment of Organic Chemistry, University of Konstanz, D-78464 Konstanz, Germany. E-mail: marina.rubini@uni-konstanz.de; Fax: +49 7531884150; Tel: +49 7531 882398
bLeibniz Institute of Molecular Pharmacology, D-13125 Berlin and Humboldt Universität zu Berlin, D-12489 Berlin, Germany. E-mail: hackenbe@fmp-berlin.de; Fax: +49 30 94793109; Tel: +49 30 94793181
cDepartment of Biochemical Pharmacology, University of Konstanz, D-78464 Konstanz, Germany
First published on 11th January 2016
Abstract
The covalent attachment of polyethylene glycol (PEG) to therapeutic proteins can improve their physicochemical properties. In this work we utilized the non-natural amino acid p-azidophenylalanine (pAzF) in combination with the chemoselective Staudinger-phosphite reaction to install branched PEG chains to recombinant unglycosylated erythropoietin (EPO) at each single naturally occurring glycosylation site. PEGylation with two short 750 or 2000 Da PEG units at positions 24, 38, or 83 significantly decreased unspecific aggregation and proteolytic degradation while biological activity in vitro was preserved or even increased in comparison to full-glycosylated EPO. This site-specific bioconjugation approach permits to analyse the impact of PEGylation at single positions. These results represent an important step towards the engineering of site-specifically modified EPO variants from bacterial expression with increased therapeutic efficacy.
Introduction
Erythropoietin (EPO) is a single domain glycoprotein that promotes the maturation of erythrocytes.1 EPO consists of 165 amino acids with two structural disulfide bonds, three N-glycosylations (Asn24, Asn38, Asn83) and one O-glycosylation (Ser126).2 In contrast to the O-glycosylation, N-linked glycans are essential for the regulation of the circulatory half-life of EPO, as unglycosylated EPO is rapidly cleared from the bloodstream.3 Moreover, N-glycosylations seem to protect the protein against proteases and enhance its solubility and stability against unspecific aggregation4 in a composition-dependent manner.5Several recombinant EPO derivatives have tremendous relevance for the treatment of patients suffering from different kinds of anemia.6 Major cell systems for expression of glycosylated EPO are eukaryotic organisms like S. cerevisiae, P. pastoris, and Chinese hamster ovary (CHO) cell line. Although relative homogeneous glycosylated recombinant EPO isoforms have been obtained,7 the glycosylation patterns of commonly used hosts for therapeutic production are typically heterogeneous, differ from the one found in humans, and present a high level of microheterogeneity which impedes a proper characterization of the complex sugar chains.8 Moreover, while expression of EPO in yeast leads to hypermannosylated products with increased immunogenicity,9 mammalian hosts present the drawbacks of low yields and high costs of production.9,10 In order to circumvent these problems, several synthetic and semisynthetic methods have been described for the synthesis of homogeneously glycosylated EPO.11–15
For long-term stabilization of protein pharmaceuticals, and in particular EPO, further modifications that increase protein stability and prolong its half-life in blood have been developed. PEGylation has been widely used for improving pharmacokinetics properties, increasing solubility, and protecting against proteolysis.16,17 Despite the fact that recent studies aroused the suspicion that administration of PEGylated therapeutics could be less effective in patients that display anti PEG antibodies,18 PEGylation remains a safe protein modification.19–21 In the last decade, the covalent attachment of PEG chains has been applied several times to both fully glycosylated and unglycosylated EPO.22–29 The latter is usually produced in bacterial cells which presents the advantage of high expression yields of recombinant protein combined with low production costs. On the other hand, unglycosylated EPO is very hydrophobic and aggregation prone.30 Mostly, PEGylation has been achieved by random coupling of N-hydroxysuccinimide-PEG chains to primary amines in proteins;28,29 however, no technique allows for the isolation of site-specifically PEGylated EPO from random PEGylated samples. Random PEGylation does not only affect the homogeneity of the resulting product, thus hindering its characterization, but can also decrease biological activity by undesired modification of residues involved in the binding to the receptor. This is the case for certain lysine residues (K20; K45; K97; K152) on EPO that are situated in a region that is critical for the binding to the receptor.31 In fact, extensive modifications of these residues were found to reduce EPO's bioactivity in vitro on hematopoietic cells by over 100-fold.32 To circumvent these drawbacks, several methods have been developed to perform site-specific PEGylation of proteins.33,34 In the case of recombinant EPO, selective modification was achieved by reductive amination of its N-terminus,25,28 or by modification of additionally introduced cysteine residues for selective coupling with PEG-maleimide.22,24,26 However, the introduction of cysteine residues can not only destabilize the target protein, but can also drop the yields of this approach due to incorrect disulfide bond formation and protein dimerization.16,17 Irrespective of its therapeutic advantages, the impact of PEGylation on protein conformational stability at a molecular level is poorly understood and its thermodynamic origin is still a matter of debate.35
Experimental methods
Bacterial expression and purification of pAzF-containing EPO variants
E. coli strain BL21(DE3) were co-transformed with plasmids pEVOL-pAzF36 and plasmid pet11a carrying the EPO genes of interest with an amber stop codon either at position 24, 38, or 83. Cells were grown at 37 °C in Luria Bertani (LB) Medium until OD600 = 0.7 was reached and centrifuged at 4000 g for 20 min pAzF was dissolved in 80% acetic acid and was added to fresh LB medium to 3 mM final concentration. The pH of the medium was adjusted to 7.0 with NaOH. Cells were resuspended in medium containing pAzF and were shaken at 37 °C for 15 minutes. IPTG was added (1 mM final concentration) to induce protein expression. The gene encoding for the engineered aminoacyl-tRNA-synthetase specific for pAzF was expressed constitutively under the control of the GlnRS promoter. After expression overnight at 37 °C, cells were centrifuged and pellet was stored at −20 °C until further processing.EPO was recovered from inclusion bodies and purified by Nickel-affinity chromatography under denaturing conditions. Cell pellet was resuspended in 6 M guanidinium chloride (GdmCl), 20 mM tris/HCl pH 8.0 and the solution was stirred at RT for 1 h. After centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g, 40 min, 4 °C) the supernatant was applied on a Ni-Nta column. The column was washed with 10 volumes wash buffer (3 M GdmCl, 20 mM tris/HCl, 40 mM Imidazole, pH 8.0). EPO was eluted by increasing imidazole concentration (elution occurs at 100 mM Imidazole).
000 g, 40 min, 4 °C) the supernatant was applied on a Ni-Nta column. The column was washed with 10 volumes wash buffer (3 M GdmCl, 20 mM tris/HCl, 40 mM Imidazole, pH 8.0). EPO was eluted by increasing imidazole concentration (elution occurs at 100 mM Imidazole).
PEGylation of EPO via the Staudinger-phosphite reaction
Staudinger-phosphite reaction was performed in 3 M GdmCl, 20 mM tris-HCl, pH 8 at 40 °C for 3 days.37 A concentration of 40 μM EPOxpAzF with 200 eq. PEG 750 phosphite or PEG 2000 phosphite was used. PEG phosphite was added in 4 portions to the reaction. After 3 days 10 mM β-mercaptoethanol (final concentration) was added to reduce disulfide bonds. The reaction mix was concentrated up to 1 ml via centrifugal concentrators and refolded by 200-fold dilution in optimized refolding buffer (20 mM tris-HCl pH 8.0, 500 mM L-arginine, and 0.3 mM ox. Glutathione, 1 mM red glutathione).29 After refolding overnight at 4 °C, volume was reduced to 0.5–1 ml via centrifugal concentrators. PEGylated EPO was separated from the non-PEGylated form by size exclusion chromatography (Superdex 75, 20 mM tris-HCl, 300 mM NaCl, pH 8.0).Cell proliferation assay
The human erythroleukemia cell line TF-1 was obtained from DSMZ.38Cells were cultivated in RPMI 1640 medium, supplemented with 16% heat inactivated foetal bovine serum (FBS), a mixture of penicillin and streptomycin, and 10% conditioned medium from cell line 5637. The latter contains several cytokines (but not EPO) which are essential for the survival of TF-1 cells.
A defined volume of TF-1 cell suspension was washed twice with cultivation medium without conditioned medium 5637 and the cell number was adjusted to 1 × 105 ml−1. The cell suspension was distributed into the wells of a 96-well plate. The final cell number per well was 1 × 104. After incubating the cells at 37 °C, 5% CO2 for four hours without any cytokines, the dilutions of several EPO variants were added to each well. Final EPO concentrations ranging from 0.001 to 500 ng ml−1 were used. The AlamarBlue® Reagent39 was added after 48 hours of incubation, and after further 48 hours, the fluorescence was read out (excitation wavelength: 560 nm, emission wavelength: 590 nm). All experiments were performed in triplicates.
Colony forming unit assay
Bone marrow from mouse tibiae and femur were isolated flushing bones with PBS.40 Cell solution was filtered through a 70 μm cell strainer and centrifuged for 5 min at 500 g. To lyse existing erythrocytes, cell pellet was resuspended in 5 ml erythrocyte lysis buffer.41 After incubating for 5 min at room temperature, IMDM + 2% FBS were used to dilute to 50 ml. Cell suspension was filtered using a cell strainer and centrifuged subsequently. The pellet was resuspended in 10 ml of IMDM + 2% FBS and again filtered. Cells were counted and a concentration of 5 × 105 cells per ml was adjusted with IMDM + 2% FBS. Next steps were performed according to the protocol of R&D systems® 2015. 4 ml of mouse methylcellulose complete medium without EPO (HSC008) were mixed with 400 μl of prepared cell suspension (total cell number: 4.5 × 104 cells per ml). A final concentration of 50 ng ml−1 EPO was added. As negative control, Tris buffer was used. According to the protocol, three times 1.1 ml of the mix was distributed in 35 mm culture dishes (to generate triplicates). To maintain humidity, two culture dishes were placed together with a culture dish containing sterile water in a 100 mm culture dish. It was incubated seven days at 37 °C, 5% CO2. For a better determination of haemoglobin containing cells, a staining according to Gallicchio and Murphy was performed.42 Briefly, 1 ml of a staining solution (0.2% benzidine dihydrochloride in 0.5 M acetic acid and 0.1% H2O2) was added directly on the cultures. After 30 min, total colony number and BFU-E colonies (deep blue) could be determined.Mass spectrometry
MALDI-TOF analyses were carried out on a Bruker Microflex mass spectrometer (Bruker Daltonics, Bremen, Germany), equipped with a nitrogen UV laser. A saturated solution of α-cyano-4-hydroxy-cinnamic acid in acetonitrile/0.1% trifluoroacetic acid in water (3:2, v/v) was used as a matrix. An aliquot of 1 μl of sample solution was mixed on the target with 1 μl of matrix solution and allowed to dry. Mass spectra were acquired in positive ion mode, at an acceleration voltage of 20 kV.
Results and discussion
In the current study we intended to systematically address whether a site-specific PEGylation with a short branched PEG chain at a single residue has the same stabilizing effect as the natural occurring glycosylations while still maintaining the function of EPO. Therefore, we utilized EPO's conserved N-glycosylation sites as natural candidates for the introduction of PEG chains into bacterially expressed unglycosylated EPO. In addition, we took advantage of a recently developed chemoselective PEGylation strategy using the Staudinger-phosphite reaction, which generates branched PEG-phosphoramidate linkages at pAzF residues preinstalled by amber suppression (Fig. 1).37,43,44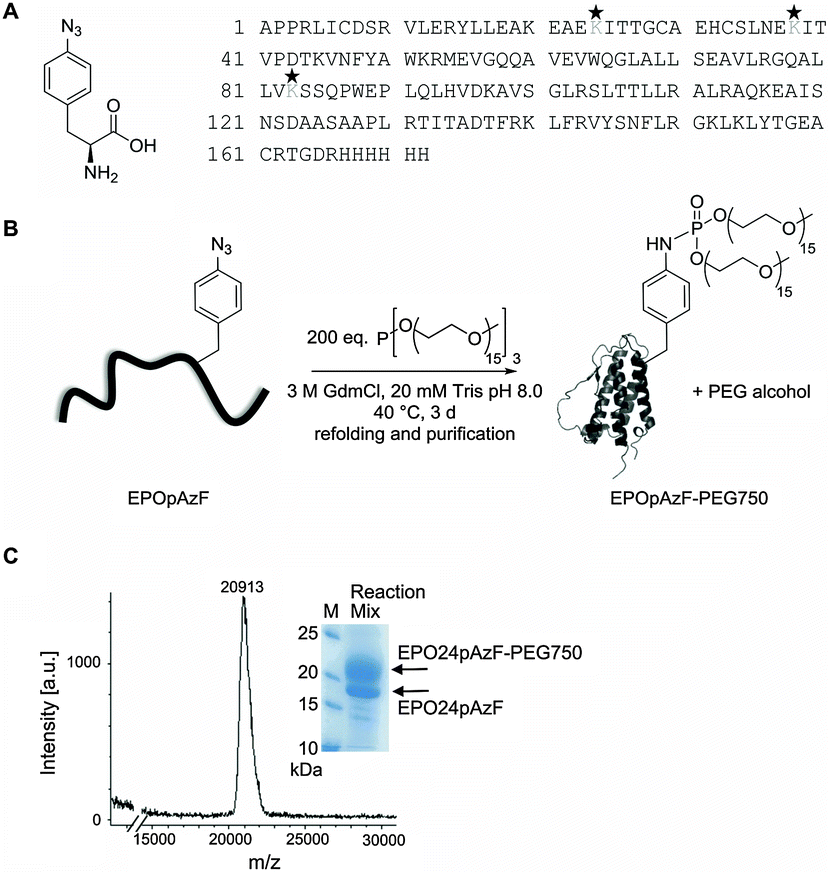 | ||
| Fig. 1 Approach for site-specific PEGylation of EPO. (A) The non-natural amino acid pAzF was incorporated either at position 24, 38, or 83, which are the naturally occurring glycosylation sites. (B) Staudinger-phosphite reaction: site-specific PEGylation of EPO. (C) Representative deconvoluted MALDI spectrum of PEGylated EPO after refolding and purification (variant EPO24pAzF-PEG750). Inset: SDS-PAGE of Staudinger reaction mix after stopping reaction and refolding of EPO. | ||
We envisioned that the simultaneous attachment of two phosphoramidate-linked PEG chains (MW 750 or 2000 Da each chain) at a branching point has the advantage to protect a protein more efficiently from proteolytic degradation even with low molecular weight PEGs due to the so-called “umbrella effect”,45 which we recently utilized for the intracellular stabilization of proapoptotic peptides.37
The non-natural amino acid pAzF was incorporated with high efficiency (ESI,† Fig. S1) through an amber stop codon at position 24, 38, or 83 by exploiting an engineered orthogonal Tyrosyl-tRNA-synthetase/tRNATyr pair from M. jannaschii encoded on a pEVOL plasmid.36 The gene carrying a modified EPO gene sequence (N24K; N38K; N83K)30 with an His6 tag at the C-terminus and with the amber stop codon (UAG) at the desired position was cloned into the vector pET11a. Both vectors were co-transformed into BL21 (DE3) E. coli strain and expression of EPO was induced with IPTG after addition of 3 mM pAzF. EPO was recovered from inclusion bodies and purified by Nickel-affinity chromatography under denaturing conditions by increasing imidazole concentration. The partially purified EPO bearing pAzF was coupled to branched PEG750-phosphite or PEG2000-phosphite that were synthesized as described previously.37 Other than previously reported,37 we performed the Staudinger-phosphite reaction under denaturing conditions. This represents a big advantage when working with proteins with a strong aggregation tendency like unglycosylated EPO, as after PEGylation refolding yields are substantially enhanced. The reaction yields varied with respect to the coupling position and to the molecular mass of the PEG chains.
While PEGylation at positions 24 or 38 with PEG750 gave always conversion yields about 30%, PEGylation at position 83 led only to small amounts of PEGylated protein. PEGylation of EPO24pAzF with PEG2000 led to the best conversion yields with a final amount of over 60% PEGylated protein (ESI,† Fig. S2). After refolding the reaction mixture was concentrated and the PEGylated EPO variants were separated from the unmodified protein by gel filtration (ESI,† Fig. S3), which allows also the separation of incorrect refolded species. Typically, yields of refolded purified PEGylated EPO variants were in the range between 0.2–1 mg. The successful coupling to PEG-phosphite was confirmed by MALDI mass spectrometry (expected: ∼21 kDa, found: 20913 Da; polydisperse PEG was used for the reaction) (Fig. 1, ESI,† Fig. S4).
Unglycosylated EPO tends to aggregate even at room temperature, thus making the protein handling difficult.30 The refolding of unglycosylated EPO after recovery from inclusion bodies is a crucial step which often results in a great loss of protein due to aggregation in the refolding buffer.46 The attachment of the short branched PEG positively influenced the refolding of EPO, resulting in approximately five-fold increase of the refolding yields of the modified protein in comparison to non-PEGylated EPO, probably as a result of the enhanced hydrophilicity. The incorporation of pAzF into EPO at position 83 led to a severe destabilization of the protein as reflected in an increased aggregation propensity; therefore this EPO variant could not be further characterized. However, the attachment of a branched PEG at position 83 by Staudinger-phosphite reaction under denaturing conditions allowed the successful refolding of the modified protein EPO83pAzF-PEG750 and EPO83pAzF-PEG2000 and its partial characterization.
We could confirm by CD spectroscopy and thermal denaturation that PEGylated EPO-derivatives displayed a far UV CD spectrum that is typical for EPO and an intact tertiary structure (ESI,† Fig. S5 and S6). No thermodynamic analysis was performed as the process was not reversible because EPO aggregated at temperatures above 40 °C.
Afterwards, we tested the impact of PEGylation on aggregation of EPO at physiological temperature (Fig. 2A). After 66 h at 37 °C, ∼50% and ∼70% of EPO24pAzF-PEG750 and EPO38pAzF-PEG750, respectively, were still in solution while the non-PEGylated form had almost completely aggregated. The maintenance of higher amounts of PEGylated EPO in solution over 48 h has been reported, but only when high molecular weight linear PEG chains of at least 20 kDa were used.29 Our results confirm that a short branched PEG chain has an impact comparable to high molecular weight PEG, due to the aforementioned “umbrella effect”.
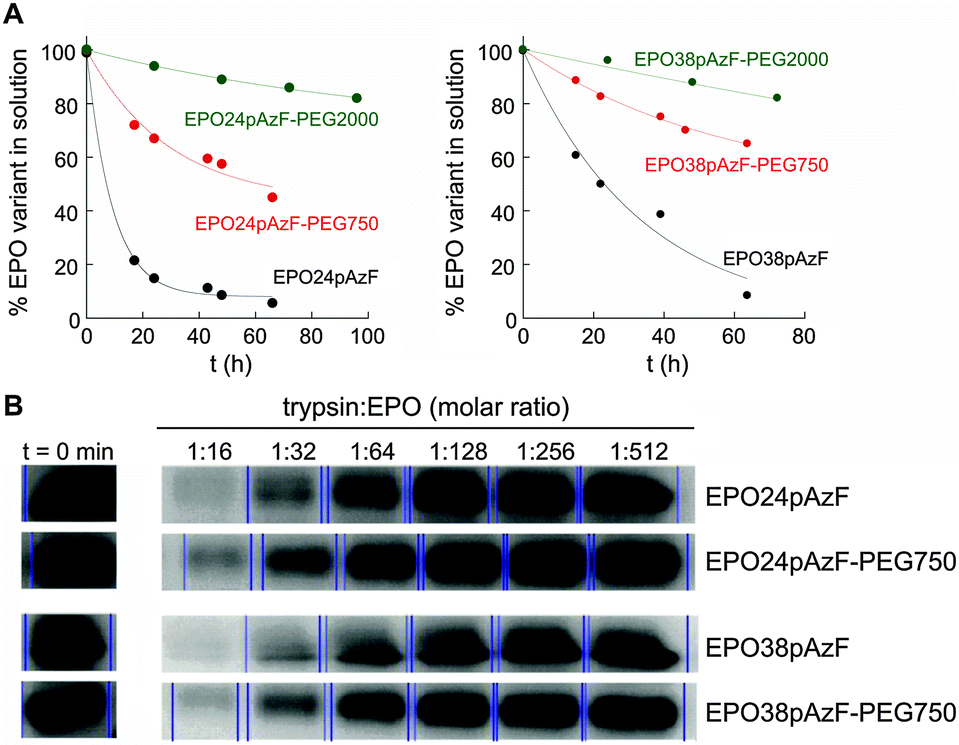 | ||
| Fig. 2 (A) Percentage of soluble EPO variant, as determined by CD spectroscopy at different incubation times at 37 °C. (Starting concentration of each sample: 10 μM.) (B) 15% SDS-PAGE (Coomassie staining) showing the amount of intact EPO variants after 15 min proteolytic digests. | ||
To test the proteolysis resistance of the PEGylated EPO variants in comparison to their non-PEGylated counterparts we incubated the created EPO variants (EPO24pAzF-PEG750 and EPO38pAzF-PEG750) for 15 minutes with trypsin at different concentrations. Briefly, the concentration of EPO was kept constant, while trypsin was two-fold serial diluted in 10 steps, beginning with a 1:1 EPO to trypsin ratio. After 15 minutes the proteolytic digest was stopped by addition of protease inhibitor and the amount of intact protein was analyzed by SDS PAGE. At protease:EPO molar ratios ranging from 1:1 to 1:8, no EPO band was detectable on the gel. At lower protease concentrations we observed a decrease in the protease activity for both PEGylated variants EPO24pAzF-PEG750 and EPO38pAzF-PEG750 (Fig. 2B).
Apparently, the branched-PEG indeed displays a discrete protection against proteolytic digest.
Next, we investigated the biological activity of the PEGylated variants in cell proliferation assays using TF-1 EPO dependent cells.38 Full-glycosylated EPO, produced in Chinese hamster ovary cells (CHO-EPO), was employed as positive control. While unglycosylated EPO24pAzF and EPO38pAzF had similar stimulating effect on cell proliferation, EPO83pAzF was completely inactive (ESI,† Fig. S7). This complete loss of activity could be explained by incorrect folding of this variant and by its pronounced aggregation propensity. All PEGylated variants, except for EPO83pAzF-PEG750 exhibited EC50 values that were in the range of CHO-EPO (Fig. 3 and Table 1).
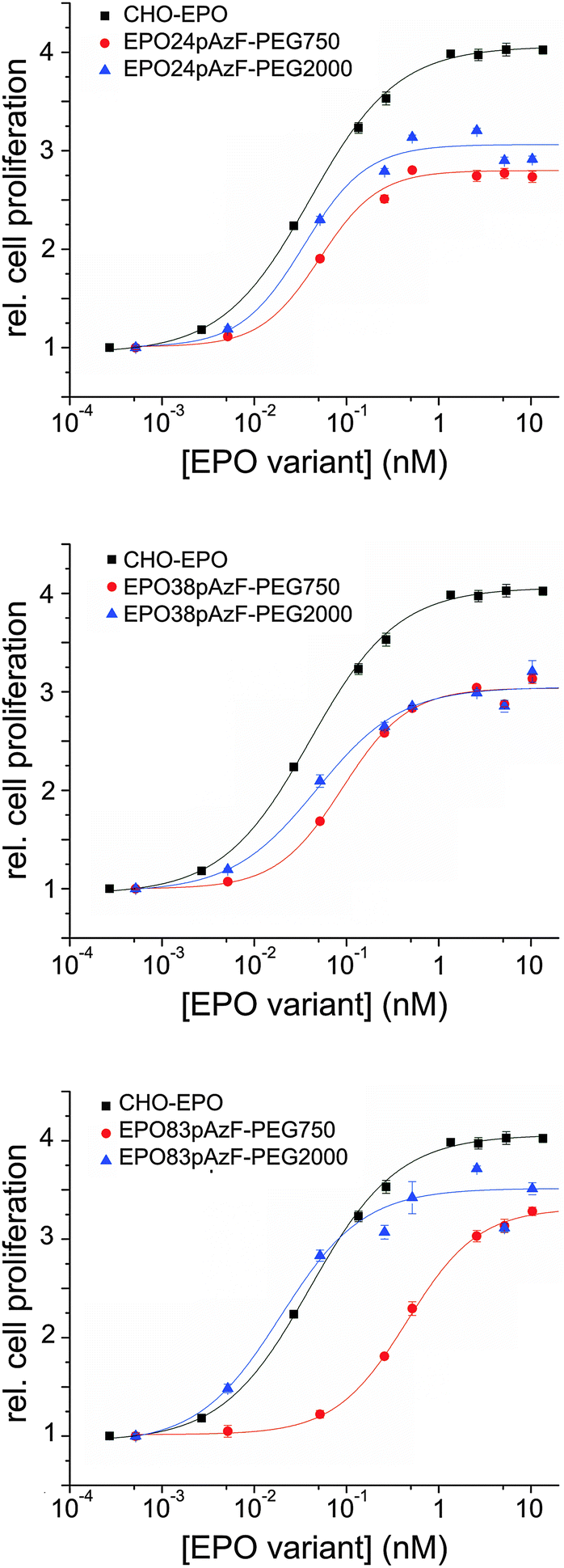 | ||
| Fig. 3 In vitro bioactivity of EPO variants measured by cell proliferation assay with TF-1 cells. The relative increase in cell number was plotted against EPO concentration, and the data were fitted to a non-cooperative binding reaction with a single binding site (Hill coefficient = 1) (solid lines). | ||
| EPO variant | EC50 (nM) | Proliferation activity (%) |
|---|---|---|
| CHO-EPO | 0.042 ± 0.00 | 100 |
| EPO24pAzF-PEG750 | 0.051 ± 0.01 | 69 |
| EPO24pAzF-PEG2000 | 0.034 ± 0.01 | 75 |
| EPO38pAzF-PEG750 | 0.090 ± 0.01 | 75 |
| EPO38pAzF-PEG2000 | 0.047 ± 0.01 | 75 |
| EPO83pAzF-PEG750 | 0.452 ± 0.02 | 82 |
| EPO83pAzF-PEG2000 | 0.019 ± 0.01 | 87 |
Surprisingly, both PEGylated EPO83pAzF variants were active even though EPO83pAzF was not. Moreover, EPO83pAzF-PEG2000 was the most active variant with a twofold better EC50 value in comparison to CHO-EPO (0.019 nM vs. 0.042 nM) and a proliferation activity very close to that of full-glycosylated EPO (∼90%). It is evident that while the attachment of PEG750 favored the refolding of the protein and the partial rescue of its biological activity, the installation of PEG2000 enabled the complete recovery of the engineered protein.
Our results support the positive effect of a short branched PEG chain on the foldability and solubility of the protein which is reflected also on the in vitro bioactivity of the tested variants. The introduction of the non-natural amino acid pAzF is likely to cause a destabilization of the protein scaffold due to steric or stereoelectronic effects, which becomes by far compensated by PEGylation. The fact that also EPO24pAzF-PEG2000 and EPO38pAzF-PEG2000 display EC50 values that are 1.5 to 2-fold lower than EPO24pAzF-PEG750 and EPO38pAzF-PEG750 seems to confirm this reasoning. The in vitro assays include cell incubation for at least 96 hours at 37 °C after addition of EPO into the medium, and at this point most of unmodified EPO has already aggregated (Fig. 2A). Our results corroborate the assumption that site-specific PEGylation can enable the creation of EPO variants with preserved in vitro bioactivity.22,26 Contrarily, for EPO PEGylated with conventional random chemistry, higher EC50 values relative to unmodified non-glycosylated EPO have been reported.28,29 It is likely that a considerable portion of the resulting heterogeneously modified proteins was actually inactive due to steric hindrance of the receptor binding by the PEG-chains and to PEGylation of lysine residues necessary for the receptor binding. This thesis is also supported by results reported in the literature where two distinct defined polymers were site-specific covalently attached to synthetic EPO analogs.34,47
Interestingly, the attachment of PEG2000 to EPO83pAzF led to a variant that was even more active than CHO-EPO. In fact, the decrease in cell proliferation activity that sometimes accompanies in vitro refolded proteins is only very faint for this variant and it is largely counterbalanced by a more than 2-fold lower EC50 value. These results suggest that this position plays a significant role for the overall protein architecture.
We also performed a colony-forming unit assay to compare the erythropoietic activity of EPO83pAzF-PEG2000 to that of CHO-EPO. The engineered variant showed the same activity as full-glycosylated EPO in triggering differentiation into mature burst-forming unit-erythroid (BFU-E) in mouse bone marrow (ESI,† Fig. S8).
Conclusions
In summary, we have successfully created site-specific PEGylated EPO at single native glycosylation sites, combining genetic code expansion with a chemoselective PEGylation protocol. We showed that even a short branched PEG with a total molecular weight of 1500 Da increased solubility, decreased the aggregation propensity of EPO, and protected the protein against proteolysis. The attachment of PEG2000 (4000 Da overall) enabled us to create EPO variants with a similar or even superior in vitro bioactivity compared to CHO-EPO. It has to be underlined that in nature glycosylations account for ∼40% of the molecular mass of CHO-EPO, while PEGylations presented in this study amount only from 7 to 17%. In contrast to previously reported studies using randomly engineered EPO variants with high molecular weight PEGs, our PEGylated variants displayed an increased in vitro bioactivity. Moreover, the here presented methodology will enable to dissect the specific roles played by glycans and PEGs in protein stability, as glycan replacements by hydrophilic polymer chains can be used to distinguish between sugar-specific or mere solubility-based effects. Even if the impact of the non-natural amino acid on protein stability is not always neutral, this study clearly shows the tremendous potential of the Staudinger-phosphite reaction for the installation of site-specific PEG-units, enabling the evolution of novel therapeutic proteins and further studies on the impact of post-translational modifications.Acknowledgements
MR and CPRH acknowledge the DFG (SPP1623, SFB765) for financial support. CPRH acknowledges support from the Boehringer Ingelheim Foundation (Plus 3 award), the Einstein Foundation Berlin and the FCI. MR and EH acknowledge the Graduate School Chemical Biology (University of Konstanz) for financial support and Marilena Manea for support for mass spectroscopy.References
- M. J. Koury and M. C. Bondurant, Science, 1990, 248, 378–381 CAS.
- P. H. Lai, R. Everett, F. F. Wang, T. Arakawa and E. Goldwasser, J. Biol. Chem., 1986, 261, 3116–3121 CAS.
- L. C. Wasley, G. Timony, P. Murtha, J. Stoudemire, A. J. Dorner, J. Caro, M. Krieger and R. J. Kaufman, Blood, 1991, 77, 2624–2632 CAS.
- Z. Kiss, S. Elliott, K. Jedynasty, V. Tesar and J. Szegedi, Eur. J. Clin. Pharmacol., 2010, 66, 331–340 CrossRef CAS PubMed.
- W. Jelkmann, Eur. J. Haematol., 2002, 69, 265–274 CrossRef CAS PubMed.
- J. W. Adamson and J. W. Eschbach, Annu. Rev. Med., 1990, 41, 349–360 CrossRef CAS PubMed.
- S. R. Hamilton, R. C. Davidson, N. Sethuraman, J. H. Nett, Y. Jiang, S. Rios, P. Bobrowicz, T. A. Stadheim, H. Li, B. K. Choi, D. Hopkins, H. Wischnewski, J. Roser, T. Mitchell, R. R. Strawbridge, J. Hoopes, S. Wildt and T. U. Gerngross, Science, 2006, 313, 1441–1443 CrossRef CAS PubMed.
- P. L. Storring, R. J. Tiplady, R. E. Gaines Das, B. Rafferty and Y. G. Mistry, J. Endocrinol., 1996, 150, 401–412 CrossRef CAS PubMed.
- T. U. Gerngross, Nat. Biotechnol., 2004, 22, 1409–1414 CrossRef CAS PubMed.
- T. Dingermann, Biotechnol. J., 2008, 3, 90–97 CrossRef CAS PubMed.
- D. Macmillan, R. M. Bill, K. A. Sage, D. Fern and S. L. Flitsch, Chem. Biol., 2001, 8, 133–145 CrossRef CAS PubMed.
- K. Hirano, D. Macmillan, K. Tezuka, T. Tsuji and Y. Kajihara, Angew. Chem., Int. Ed. Engl., 2009, 48, 9557–9560 CrossRef CAS PubMed.
- M. Murakami, R. Okamoto, M. Izumi and Y. Kajihara, Angew. Chem., Int. Ed. Engl., 2012, 51, 3567–3572 CrossRef CAS PubMed.
- R. M. Wilson, S. Dong, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed. Engl., 2013, 52, 7646–7665 CrossRef CAS PubMed.
- P. Wang, S. Dong, J. H. Shieh, E. Peguero, R. Hendrickson, M. A. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS PubMed.
- V. Gaberc-Porekar, I. Zore, B. Podobnik and V. Menart, Curr. Opin. Drug Discovery Dev., 2008, 11, 242–250 CAS.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729–1734 CrossRef CAS PubMed.
- R. A. Cohan, A. Madadkar-Sobhani, H. Khanahmad, F. Roohvand, M. R. Aghasadeghi, M. H. Hedayati, Z. Barghi, M. S. Ardestani, D. N. Inanlou and D. Norouzian, Int. J. Nanomed., 2011, 6, 1217–1227 CAS.
- K. Jolling, J. J. Ruixo, A. Hemeryck, V. Piotrovskij and T. Greway, J. Pharm. Sci., 2004, 93, 3027–3038 CrossRef CAS PubMed.
- A. Maleki, A. Madadkar-Sobhani, F. Roohvand, A. R. Najafabadi, A. Shafiee, H. Khanahmad, R. A. Cohan, N. Namvar and H. Tajerzadeh, Eur. J. Pharm. Biopharm., 2012, 80, 499–507 CrossRef CAS PubMed.
- A. Maleki, A. R. Najafabadi, F. Roohvand, A. Shafiee, H. Khanahmad, H. Faghihi, M. H. Hedayati and H. Tajerzadeh, Drug Delivery, 2011, 18, 570–577 CrossRef CAS PubMed.
- D. L. Long, D. H. Doherty, S. P. Eisenberg, D. J. Smith, M. S. Rosendahl, K. R. Christensen, D. P. Edwards, E. A. Chlipala and G. N. Cox, Exp. Hematol., 2006, 34, 697–704 CrossRef CAS PubMed.
- I. C. Macdougall, Curr. Hematol. Rep., 2005, 4, 436–440 CAS.
- Y. J. Wang, S. J. Hao, Y. D. Liu, T. Hu, G. F. Zhang, X. Zhang, Q. S. Qi, G. H. Ma and Z. G. Su, J. Controlled Release, 2010, 145, 306–313 CrossRef CAS PubMed.
- Y. J. Wang, Y. D. Liu, J. Chen, S. J. Hao, T. Hu, G. H. Ma and Z. G. Su, Int. J. Pharm., 2010, 386, 156–164 CrossRef CAS PubMed.
- L. O. Narhi, T. Arakawa, K. Aoki, J. Wen, S. Elliott, T. Boone and J. Cheetham, Protein Eng., 2001, 14, 135–140 CrossRef CAS PubMed.
- R. S. Syed, S. W. Reid, C. Li, J. C. Cheetham, K. H. Aoki, B. Liu, H. Zhan, T. D. Osslund, A. J. Chirino, J. Zhang, J. Finer-Moore, S. Elliott, K. Sitney, B. A. Katz, D. J. Matthews, J. J. Wendoloski, J. Egrie and R. M. Stroud, Nature, 1998, 395, 511–516 CrossRef CAS PubMed.
- M. Leist, P. Ghezzi, G. Grasso, R. Bianchi, P. Villa, M. Fratelli, C. Savino, M. Bianchi, J. Nielsen, J. Gerwien, P. Kallunki, A. K. Larsen, L. Helboe, S. Christensen, L. O. Pedersen, M. Nielsen, L. Torup, T. Sager, A. Sfacteria, S. Erbayraktar, Z. Erbayraktar, N. Gokmen, O. Yilmaz, C. Cerami-Hand, Q. W. Xie, T. Coleman, A. Cerami and M. Brines, Science, 2004, 305, 239–242 CrossRef CAS PubMed.
- N. Nischan and C. P. Hackenberger, J. Org. Chem., 2014, 79, 10727–10733 CrossRef CAS PubMed.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS PubMed.
- P. B. Lawrence, Y. Gavrilov, S. S. Matthews, M. I. Langlois, D. Shental-Bechor, H. M. Greenblatt, B. K. Pandey, M. S. Smith, R. Paxman, C. D. Torgerson, J. P. Merrell, C. C. Ritz, M. B. Prigozhin, Y. Levy and J. L. Price, J. Am. Chem. Soc., 2014, 136, 17547–17560 CrossRef CAS PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- N. Nischan, A. Chakrabarti, R. A. Serwa, P. H. Bovee-Geurts, R. Brock and C. P. Hackenberger, Angew. Chem., Int. Ed. Engl., 2013, 52, 11920–11924 CrossRef CAS PubMed.
- T. Kitamura, T. Tange, T. Terasawa, S. Chiba, T. Kuwaki, K. Miyagawa, Y. F. Piao, K. Miyazono, A. Urabe and F. Takaku, J. Cell. Physiol., 1989, 140, 323–334 CrossRef CAS PubMed.
- G. R. Nakayama, M. C. Caton, M. P. Nova and Z. Parandoosh, J. Immunol. Methods, 1997, 204, 205–208 CrossRef CAS PubMed.
- X. Zhang, R. Goncalves, D. M. Mosser, Current Protocols in Immunology, 2008, ch. 14, unit 14 11 Search PubMed.
- A. M. Kruisbeek, Current Protocols in Immunology, 2001, ch. 3, unit 3 1 Search PubMed.
- V. S. Gallicchio and M. J. Murphy, Jr., Exp. Hematol., 1979, 7, 219–224 CAS.
- R. Serwa, I. Wilkening, G. Del Signore, M. Muhlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. Hackenberger, Angew. Chem., Int. Ed. Engl., 2009, 48, 8234–8239 CrossRef CAS PubMed.
- R. Serwa, P. Majkut, B. Horstmann, J. M. Swiecicki, M. Gerrits, E. Krause and C. P. R. Hackenberger, Chem. Sci., 2010, 1, 596–602 RSC.
- C. Monfardini, O. Schiavon, P. Caliceti, M. Morpurgo, J. M. Harris and F. M. Veronese, Bioconjugate Chem., 1995, 6, 62–69 CrossRef CAS PubMed.
- E. D. B. Clark, Curr. Opin. Biotechnol., 1998, 9, 157–163 CrossRef PubMed.
- S. Y. Chen, S. Cressman, F. Mao, H. Shao, D. W. Low, H. S. Beilan, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, L. Savatski, J. W. Adamson, C. E. Bozzini, A. Kung, S. B. Kent, J. A. Bradburne and G. G. Kochendoerfer, Chem. Biol., 2005, 12, 371–383 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and supplementary figures. See DOI: 10.1039/c5mb00857c |
| ‡ Present address: University of Texas Southwestern Medical Center, Dallas, Texas, USA. |
| This journal is © The Royal Society of Chemistry 2016 |