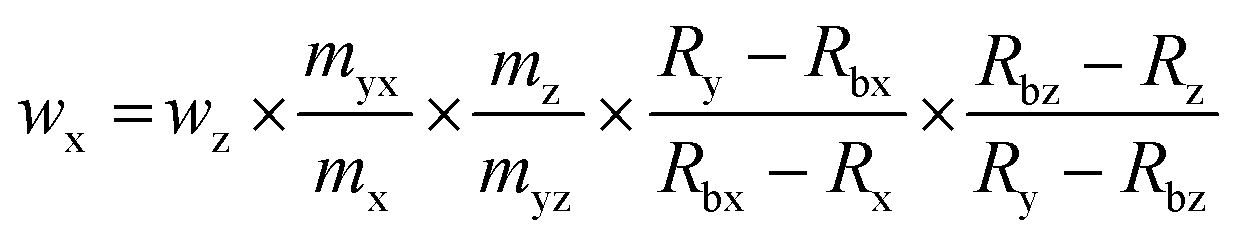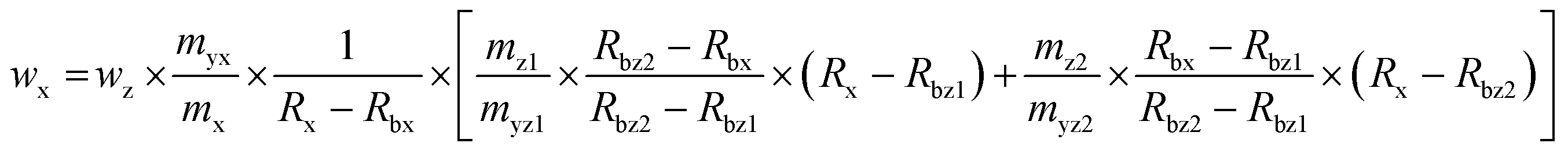DOI:
10.1039/C5JA00459D
(Technical Note)
J. Anal. At. Spectrom., 2016,
31, 1922-1928
Accurate and precise quantification of Cu,Zn-SOD in human red blood cells using species-specific double and triple IDMS†
Received
19th November 2015
, Accepted 17th March 2016
First published on 17th March 2016
Abstract
Cu,Zn-superoxide dismutase (SOD1) is a protein involved in the antioxidant defense system of the body responsible for the dismutation of the superoxide anion. It contains two Cu and two Zn ions per molecule. As this protein is also involved in several diseases it is used in clinical diagnostics as a biomarker, which requires the accurate and reliable determination of SOD1. Therefore, a candidate reference measurement procedure for the quantification of this protein in human erythrocytes was developed using species-specific isotope dilution mass spectrometry (IDMS), a method giving results traceable to the International System of Units (SI). The measurement procedure was validated with regard to a metrological point of view. Commercially available SOD1 was thoroughly characterized to be used as a pure protein calibration standard in IDMS approaches. Furthermore, 65Cu and 67Zn labeled SOD1 was produced to be used as a spike material required for species-specific IDMS. Finally, SOD1 was quantified in human erythrocytes using both double and triple IDMS and a complete uncertainty budget for both approaches was estimated according to the Guide to the Expression of Uncertainty in Measurement (GUM). A calculated mass fraction of SOD1 with its associated expanded uncertainty of (63.94 ± 0.93) μg g−1 (n = 30) for double and (64.02 ± 0.96) μg g−1 (n = 30) for triple IDMS was obtained.
Introduction
Cu,Zn-superoxide dismutase (SOD1)1 is an homodimeric enzyme which contains a closely spaced non-covalently bound Cu and Zn ion pair2,3 and has a molecular mass of about 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1 for the human species. It catalyzes the disproportionation of the superoxide anion (O2−) into oxygen (O2) and hydrogen peroxide (H2O2).4 The Cu ion is responsible for the enzymatic activity as the disproportionation of O2− is driven by the [Cu(II)/Cu(I)] ion redox cycle.3 Besides a single disulfide bond and a complex network of hydrogen bonds, the Zn ion accounts for the great stability of SOD1. The enzymatic activity is linked to the Cu bound to the protein. The involvement of SOD1 in preventing oxidative stress can be used as a bioindicator for environmental impact assessments.5 Additionally, the protein plays a role in medical diagnostics. SOD1 activity is known to decrease in malignant lymphoma,6 chronic alcoholism7 and medullar carcinoma,8 its activity is increased in leukemia.9 It can be used as a prenatal marker for Down syndrome10 and is involved in neurodegenerative diseases such as Alzheimer's disease.11 Furthermore, it is a potential marker for gastric cancer.12 When results from the quantitative determination of SOD1 are used for clinical diagnostics, it is of great importance that the results of the protein quantification are comparable and reliable (inter- and intra-laboratory).13 In routine or clinical laboratories, immunoassays such as enzyme-linked immunosorbent assays (ELISA)14 or spectrophotometric assays15,16 are used to quantify SOD1, but there are no reference methods or materials available yet. Comparison of some of these methods led to large differences in the results, the limits of quantification (LODs) and the reproducibility.17 This shows that reference methods and materials are urgently required. None of these clinical assays is based on metrological principles and results are not traceable to the International System of Units (SI) as demanded by directives such as Directive 98/79/EC.18 Furthermore, no uncertainty budget according to the Guide to the Expression of Uncertainty in Measurement (GUM) was estimated for the quantification of SOD1 for these methods. Within the project HLT05, a reference method traceable to the SI19 was developed for the first time using species-specific double and triple isotope dilution mass spectrometry (IDMS) for the quantification via the Cu content of SOD1. By measuring the Cu containing portion of the SOD1, only the amount of active protein is quantified. Thus, the method can serve as a reference method of higher order for routine measurements. The basic principle of IDMS is the change of the natural isotopic composition of a chosen element in the sample by the addition of a substance enriched in a minor isotope of the chosen element, called spike. For the double IDMS approach,20 a blend of a reference material with the spike, the reference spike blend, is prepared and both sample spike and reference spike blends are measured using inductively coupled plasma mass spectrometry (ICP-MS). In the reference material, the analyte is well characterized and acts as a back spike. A further approach recently developed is triple IDMS,21–23 in which additionally a second reference spike blend is prepared. This is especially recommendable when a spike material is used with a comparable low enrichment leading to a high uncertainty in the measurement of the isotope ratio of the spike material as this isotope ratio is canceled from the equation. A more detailed description of the advantages of triple IDMS is given by Vogl.21 Although the IDMS approach is well established and accepted for elemental analysis,24 the application in elemental speciation, especially for metalloproteins, is still challenging.25 For the use of IDMS in speciation analysis there are two basic approaches: the first one is species-unspecific IDMS also referred to as post-column IDMS,26,27 where the elemental spike is added to the analyte after sample preparation and separation and the second one is species-specific IDMS.28 Here, the species-specific spike material is added to the sample as soon as possible before any sample preparation and separation which has the advantage that any losses or changes of the analyte during the sample preparation process can be compensated for.
800 g mol−1 for the human species. It catalyzes the disproportionation of the superoxide anion (O2−) into oxygen (O2) and hydrogen peroxide (H2O2).4 The Cu ion is responsible for the enzymatic activity as the disproportionation of O2− is driven by the [Cu(II)/Cu(I)] ion redox cycle.3 Besides a single disulfide bond and a complex network of hydrogen bonds, the Zn ion accounts for the great stability of SOD1. The enzymatic activity is linked to the Cu bound to the protein. The involvement of SOD1 in preventing oxidative stress can be used as a bioindicator for environmental impact assessments.5 Additionally, the protein plays a role in medical diagnostics. SOD1 activity is known to decrease in malignant lymphoma,6 chronic alcoholism7 and medullar carcinoma,8 its activity is increased in leukemia.9 It can be used as a prenatal marker for Down syndrome10 and is involved in neurodegenerative diseases such as Alzheimer's disease.11 Furthermore, it is a potential marker for gastric cancer.12 When results from the quantitative determination of SOD1 are used for clinical diagnostics, it is of great importance that the results of the protein quantification are comparable and reliable (inter- and intra-laboratory).13 In routine or clinical laboratories, immunoassays such as enzyme-linked immunosorbent assays (ELISA)14 or spectrophotometric assays15,16 are used to quantify SOD1, but there are no reference methods or materials available yet. Comparison of some of these methods led to large differences in the results, the limits of quantification (LODs) and the reproducibility.17 This shows that reference methods and materials are urgently required. None of these clinical assays is based on metrological principles and results are not traceable to the International System of Units (SI) as demanded by directives such as Directive 98/79/EC.18 Furthermore, no uncertainty budget according to the Guide to the Expression of Uncertainty in Measurement (GUM) was estimated for the quantification of SOD1 for these methods. Within the project HLT05, a reference method traceable to the SI19 was developed for the first time using species-specific double and triple isotope dilution mass spectrometry (IDMS) for the quantification via the Cu content of SOD1. By measuring the Cu containing portion of the SOD1, only the amount of active protein is quantified. Thus, the method can serve as a reference method of higher order for routine measurements. The basic principle of IDMS is the change of the natural isotopic composition of a chosen element in the sample by the addition of a substance enriched in a minor isotope of the chosen element, called spike. For the double IDMS approach,20 a blend of a reference material with the spike, the reference spike blend, is prepared and both sample spike and reference spike blends are measured using inductively coupled plasma mass spectrometry (ICP-MS). In the reference material, the analyte is well characterized and acts as a back spike. A further approach recently developed is triple IDMS,21–23 in which additionally a second reference spike blend is prepared. This is especially recommendable when a spike material is used with a comparable low enrichment leading to a high uncertainty in the measurement of the isotope ratio of the spike material as this isotope ratio is canceled from the equation. A more detailed description of the advantages of triple IDMS is given by Vogl.21 Although the IDMS approach is well established and accepted for elemental analysis,24 the application in elemental speciation, especially for metalloproteins, is still challenging.25 For the use of IDMS in speciation analysis there are two basic approaches: the first one is species-unspecific IDMS also referred to as post-column IDMS,26,27 where the elemental spike is added to the analyte after sample preparation and separation and the second one is species-specific IDMS.28 Here, the species-specific spike material is added to the sample as soon as possible before any sample preparation and separation which has the advantage that any losses or changes of the analyte during the sample preparation process can be compensated for.
Commercially available SOD1 was characterized for use as a reference material and the species-specific spike material was prepared from this. As a proof of concept, SOD1 was quantified in a human erythrocyte sample by species-specific double and triple IDMS and the results were compared.
Experimental
Materials and reagents
All chemicals used were of analytical grade or better unless stated otherwise. High-purity deionized water (18 MΩ cm, Merck Millipore, Darmstadt, Germany) was used for the preparation of buffers and solutions. The chemicals for the preparation of buffers and solvents include ammonia acetate (NH4Ac), tris(hydroxymethyl)methylamine (Tris, both purchased from Sigma-Aldrich, St. Louis, USA), acetic acid, nitric acid, hydrochloric acid (HCl), potassium hydroxide (KOH, all purchased from Merck, Darmstadt, Germany), ethylenediaminetetraacetic acid (EDTA), guanidinium chloride (GndCl, both purchased from Carl Roth, Karlsruhe, Germany), sodium chloride (NaCl, J.T. Baker, Center Valley, USA) and potassium chloride (KCl, Fisher, Loughborough, UK). Furthermore, ethanol (Carl Roth) and chloroform (AppliChem, Darmstadt, Germany) were used. For the activity assay pyrogallol and diethylenetriaminepentaacetic acid (DTPA), purchased from BDH (Dorset, UK), were used. Polyacrylamide gel electrophoresis (PAGE) was performed with hand-cast native gels and with native tris–glycine buffer. Reagents were purchased from Carl Roth. SOD1 from bovine erythrocytes as well as from human erythrocytes was purchased from Sigma-Aldrich. External calibration was performed using Cu and Zn standard solutions (AccuTrace, Bryn Mawr, USA). 65Cu and 67Zn, which were used for isotopic labelling of the bovine SOD1, were from CK Gas Products (Hampshire, UK) with a certified isotopic abundance of 63Cu 0.8% and 65Cu 99.2% and 64Zn 0.38%, 66Zn 0.33% and 68Zn 99.0%. For labelling the human SOD1, 65Cu and 67Zn were obtained from Chemotrade (Leipzig, Germany) with a certified isotopic abundance of 63Cu 0.3% and 65Cu 99.7% as well as 64Zn 0.67%, 66Zn 1.19%, 67Zn 97%, 68Zn 1.11% and 70Zn < 0.02%. Stock solutions of isotopically enriched Cu and Zn were prepared by dissolving the metal in concentrated subboiled nitric acid and diluted gravimetrically to an appropriate working solution. Human erythrocytes were obtained from the German Blood Bank (Braunschweig, Germany) and the bovine erythrocytes were obtained from a local abattoir (Aberdeen, UK).
Instrumentation
The IDMS measurements as well as the characterization of the potential reference material were accomplished with a hyphenated high performance liquid chromatography (HPLC)-ICP-MS system. The chromatographic separation was performed with an Agilent 1200 (Agilent Technologies, Santa Clara, USA) equipped with a degasser, a binary pump, a thermostatted autosampler, a column oven and a diode array detector (DAD). The separation was achieved on a strong anion exchange column (MonoQ™ 5/50 GL, GE Healthcare, Uppsala, Sweden). The coupling of the HPLC to the ICP-MS was realized with a PEEK capillary. The isotopes of interest were detected with a quadrupole ICP-MS system (Agilent 7700cx) in the pulse-counting mode. Typical instrumental working conditions for HPLC and ICP-MS are summarized in ESI Table s1.† For the post-column IDMS measurements the mass flow of the isotopically enriched spike was monitored continuously using a balance Genius ME215S (Sartorius, Göttingen, Germany) applying a method improved by Swart et al.29 The balance sent the mass reading to the data acquisition software SartoConnect provided by the manufacturer of the balance. To determine the molecular mass of the native protein, electrospray ionisation (ESI)-MS was used. The measurements were performed on a quadrupole time-of-flight (TOF) MS micrOTOF-Q (Bruker Daltonics, Billerica, USA). It was used in a positive detection mode with an extended mass range from 300–3000 Da. The obtained spectra were deconvoluted with the Data Analysis software provided by the manufacturer.
Characterization of commercially available SOD1 for use as reference in double IDMS
The identity of the purchased protein was verified using molecular MS (ESI-TOF-MS). The sample was separated on a reversed phase column (Jupiter300-C4, Phenomenex, Torrance, USA) using an Agilent 1200 HPLC system. To determine the purity of the prepared protein solutions concerning the Cu amount, post-column IDMS was used. The isotopic composition of the used spike material was fully characterized with a sector-field ICP-MS in high resolution mode. Furthermore, the protein was determined by UV-vis, which was integrated into the HPLC, at 259 nm and 280 nm.
Preparation and characterization of isotopically enriched SOD1
Three published methods were compared regarding their demetallation efficiency and the possibility to remetallate the apo-protein to produce the isotopically labelled SOD1. In method A, the protein was dialyzed against NH4Ac buffer at pH 2.830 and in method B against NH4Ac buffer containing EDTA at pH 3.8,31 whereas in method C the demetallation was performed using GndCl.32 For remetallation of the produced apo-SOD1, 68Zn was added in surplus, firstly, and 65Cu, secondly. Both the apo- and remetallated SOD1 as well as the native protein were characterized regarding metal content, protein integrity and activity.15 Further information about instrumentation and methods is given in the ESI.†
Determination of SOD1 in human erythrocytes using IDMS
For homogenization all solutions, erythrocytes, reference stock solution and spike stock solution, were vortexed at r.t. for at least one hour before use. The stock solutions of the reference material and spike material were diluted to a final concentration of 0.025 mg g−1 (reference material) and 0.1 mg g−1 (spike material). The reference spike blends as well as the erythrocyte spike blends for the IDMS measurements were prepared gravimetrically and homogenized for about an hour at r.t. before further use. To precipitate the hemoglobin, a mixture of ethanol/chloroform (3:1) was added to the blends in the ratio of 1:1. The blends were shaken for 20 min and then centrifuged at 12000 rpm and 4 °C for 15 min. The supernatant was transferred to another tube and the residue was discarded. To remove the organic solvents, the blends were concentrated using MWCO centrifugal filters and the residues were brought to their final volume with water. Finally, the prepared blends were measured using HPLC-ICP-MS. Peak areas were integrated with MassHunter software from Agilent. The SOD1 amount in the erythrocytes was calculated using the equations for double IDMS and triple IDMS (equations are given in the Results and discussion section). The expanded uncertainty for both approaches was estimated with the GUM workbench (Metrodata GmbH, Weil am Rhein, Germany).
Results and discussion
Characterization of commercially available SOD1 for use as reference in double IDMS
The identity of the purchased human SOD1 was checked by means of ESI-TOF-MS (data given in ESI Fig. s1†). To exclude the occurrence of protein impurities the protein was determined after separation by UV-vis. The column recovery was calculated to be (68.62 ± 0.90)% for SOD1 (n = 10) on the SAX column regarding the Cu content. A purity of > 80% of the protein material by Biuret is stated by the manufacturer. To check this, the purity of the prepared protein solutions was determined by performing post-column IDMS as developed by Heumann et al.26 and optimized by Swart et al.,29 in which the isotopically enriched spike material is added continuously to the effluent from the HPLC with a peristaltic pump via a T-piece. With the SartoConnect software the mass flow of the spike was recorded continuously over the whole time of the chromatogram. The 63Cu isotope serves as the reference isotope. In all measurements, the isotope ratios were corrected for mass bias by the determination of the correction factor K calculated by setting the natural isotope ratio of a measured sample in relationship to the “true” isotope ratio according to the International Union of Pure and Applied Chemistry (IUPAC). Protein solutions and spike solutions, which were added to the sample post-column, were prepared gravimetrically. The deployed masses were corrected for air buoyancy. To take into account the sample loss during the separation procedure, the column recovery is considered in the calculation of the Cu mass fraction of the sample. The used equation is given in the ESI.†29 For the used SOD1 material, a Cu mass fraction of (0.290 ± 0.014) μg g−1 was calculated (n = 20), which corresponds to a SOD1 amount of (0.073 ± 0.004) mg g−1. The purity of the purchased material and its estimated uncertainty was found to be (72.80 ± 4.9)% with regard to the Cu amount. The uncertainty budget was estimated with the GUM workbench as extended uncertainty (k = 2).
Production of enriched SOD1
The first step of the SOD1 spike production is demetallation followed by remetallation with isotopically enriched metals. Due to the high stability of the Cu and Zn bonds in SOD1, relatively harsh conditions are required for demetallation, which may lead to irreversible changes of the secondary structure. Three methods are used predominantly in the literature and these were tested for the preparation of the bovine SOD1 spike material (*SOD1) and compared to each other to get a spike fit for purpose in species-specific IDMS. One of the first methods for the removal of both Cu and Zn was the treatment of the protein with EDTA at low pH (called method B in the present work).33–35 Another method includes the use of even lower pH but omits the use of EDTA (method A).30 One method, which does not expose the enzyme to low pH, uses the denaturing ability of GndCl and EDTA as complexing agents (method C).32,36,37 The demetallation and remetallation efficiency as well as isotope enrichment was determined by SEC-ICP-MS (ESI Table s3 and Fig. s3 and s4†). The correct incorporation of the metals into their native sites was checked by performing native gel electrophoresis and activity assay (ESI Table s3 and Fig. s2†). To ensure that the produced *SOD1 is stable in the biological matrix, the spike was added to bovine erythrocytes and the isotope ratios were measured (ESI Fig. s5 and s6†). Compared to the other two methods *SOD1 produced according to method B has the highest remetallation efficiency and isotopic enrichment. The structure as well as activity of dimeric protein is restored nearly completely. Furthermore, *SOD1 is stable when added to biological samples, which shows that method B is the most suited method for producing species-specific spike materials.38,39
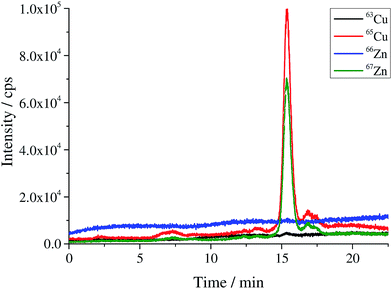 |
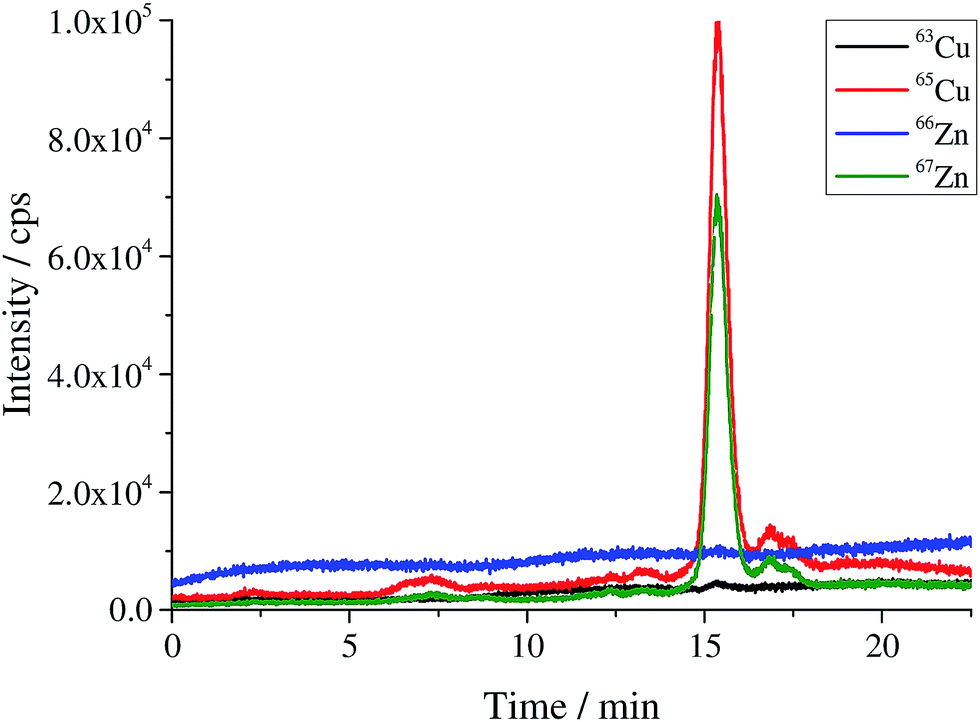
| | Fig. 1 Chromatogram of the produced human SOD1 spike material. | |
Quantification of SOD1 in human erythrocytes using species-specific double and triple IDMS
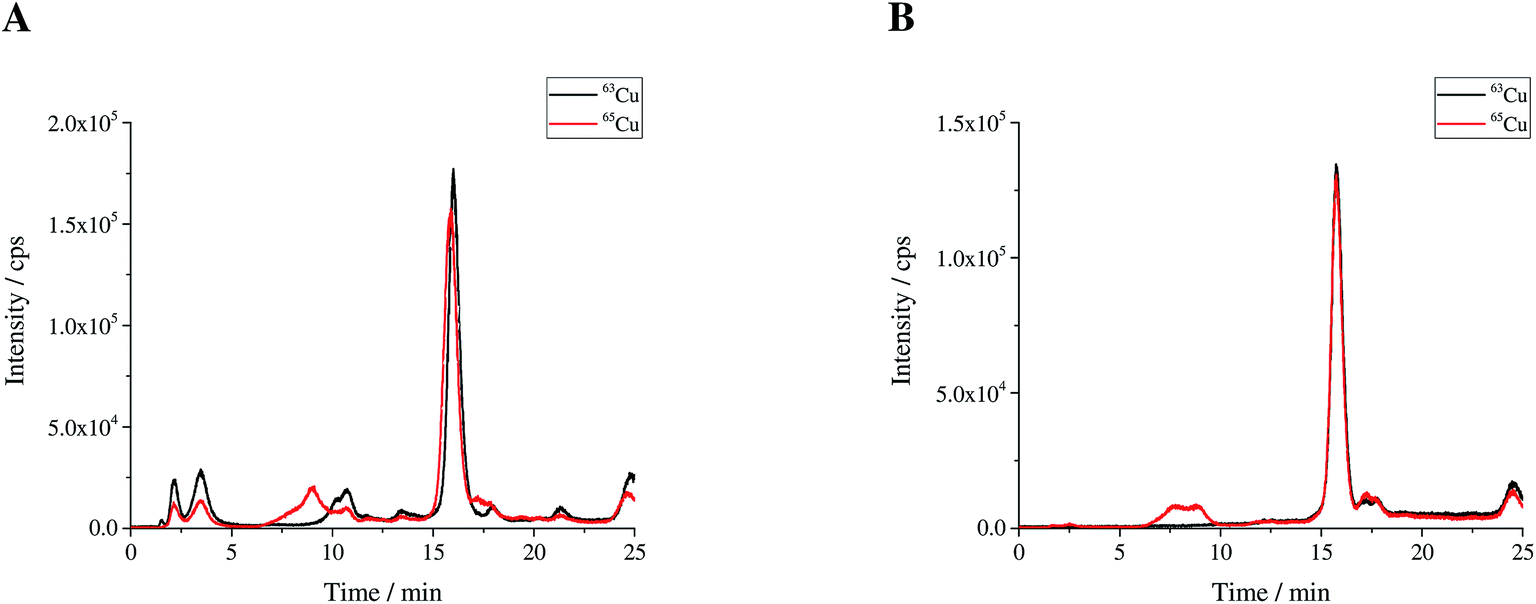
The bovine SOD1 spike showed different stabilities in samples of human origin and a different retention time compared to human SOD and is, therefore, not suitable as the spike for human erythrocyte lysate as shown by Nuevo Ordóñez et al.39 Hence, for the determination of SOD1 in human erythrocyte lysate, a spike was prepared from commercial human SOD1 following method B. A chromatogram of the produced human *SOD1 is shown in Fig. 1. To determine if the human *SOD1 is applicable for use in species-specific IDMS, the stability of the spike in human erythrocytes was tested over a time period of 48 h and found to be satisfactory (ESI Fig. s7†), which makes it fit for use in double and triple IDMS experiments. The advantage in performing double species-specific IDMS is that, compared to single species-specific IDMS,39 the purity of the used species-specific spike need not be known as it is canceled from the equation when using exact matching IDMS. However, a key step in double IDMS measurements is the characterization of the purchased protein to be used as the reference. The sample spike blends (bx) and the reference spike blends (bz) were prepared three times each independently on three different days. Each blend was measured ten times in a bracketing sequence. For the double IDMS experiments, the reference as well as the erythrocyte sample were mixed with the isotopically enriched spike material to result in an isotope ratio of 65Cu/63Cu of approximately 1:1 (Fig. 2). For all measurements, the isotope ratios were corrected for mass bias. Eqn (1)20 was used for the calculation of the SOD1 amount. Meanings of the used symbols are described in Table 1.| | 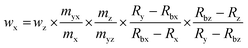 | (1) |
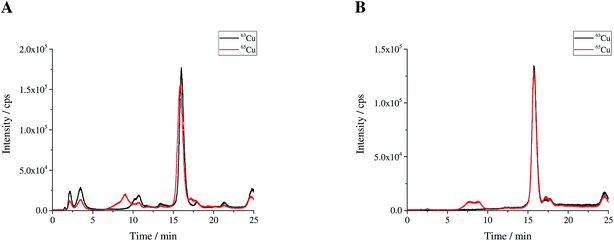 |
| | Fig. 2 (A) Sample spike blend (bx) and (B) reference spike blend (bz) prepared for use in double IDMS measurements. | |
Table 1 Meaning of the symbols used in the eqn (1) and (2)
| Symbol |
Unit |
Definition |
|
w
x, wz |
g kg−1 |
Mass fraction of Cu in sample x and reference z, resp. |
|
R
x, Ry, Rz |
mol mol−1 |
Isotope amount ratio of the spike and reference isotope (65Cu/63Cu) in sample x, spike y and reference z, resp. |
|
R
bx, Rbz, Rbz1, Rbz2 |
mol mol−1 |
Isotope amount ratio of the spike and reference isotope (65Cu/63Cu) in blends bx (sample x + spike y) and blends bz, bz1 and bz2 (reference z, z1 and z2 + spike y), resp. |
|
m
x, mz, mz1, mz2, myx, myz, myz1, myz2 |
g |
Masses of sample x, references z, z1 and z2 and spike y solutions, resp. |
The deployed masses of the reference solution, spike solution and samples were corrected for air buoyancy. The calculated SOD1 mass fraction in the erythrocyte sample as well as the estimated expanded uncertainty is listed in Table 2. As shown in Table 3, the main contributions to the uncertainty were found to be the uncertainty of the purity of the used reference (wpur), the measured isotope ratios in the sample spike blend (Rbx) and the reference spike blend (Rbz). For the double IDMS measurements the variations of the individual results and the average are shown in Fig. 3A. For triple IDMS the same erythrocyte sample was mixed with *SOD1 to result in an approximated 1:1 ratio. Unlike for double IDMS, the triple IDMS approach requires two reference spike blends, both with a slightly different final isotope ratio but both close to 1:1. The final isotope ratio 65Cu/63Cu was chosen to be approximately 0.9 for the first reference spike blend (Rbz1) and approximately 1.1 for the second reference spike blend (Rbz2). In this approach the sample spike blend (bx) and reference spike blends (bz1 and bz2) were prepared three times independently and on three different days. While bx is measured ten times, bz1 and bz2 were measured six and five times, respectively, in a bracketing sequence (means bz1_1, bx_1, bz2_1, bx_2, bz1_2, bx_3, bz2_2, bx_4, bz1_3, bx_5, bz2_3, …). The deployed masses during the preparation of the blends were corrected for air buoyancy. Eqn (2)23 was used to calculate the SOD1 amount in the sample following the triple IDMS approach. Meanings of the used symbols are described in Table 1.
| | 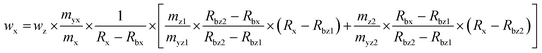 | (2) |
Table 2 Calculated SOD1 amount in a human erythrocyte sample and the appropriate expanded uncertainty for species-specific double and triple IDMS approaches
|
|
SOD1 amount in erythrocytes in μg g−1 |
Expanded uncertainty (k = 2) in μg g−1 |
Expanded uncertainty (k = 2) in % |
| Double IDMS |
63.94 |
0.93 |
1.45 |
| Triple IDMS |
64.06 |
0.96 |
1.50 |
Table 3 Major contributions to the uncertainty of the amount of SOD1 in erythrocytes using species-specific double and triple IDMS, respectively
|
|
Double IDMS in % |
Triple IDMS in % |
|
w
pur
|
71.7 |
70.4 |
|
R
bx
|
15.3 |
15.6 |
|
R
bz
|
10.6 |
— |
|
R
bz2
|
— |
11.4 |
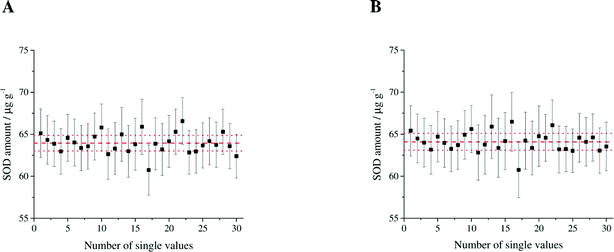 |
| | Fig. 3 Variation of the individual results for (A) double IDMS measurements and (B) triple IDMS measurements. The error bars represent the expanded uncertainty (k = 2) of the single values. The average with its estimated uncertainty is illustrated in red. | |
The calculated SOD1 amount in erythrocytes and the appropriate expanded uncertainty given by the triple IDMS approach are shown in Table 2. Major contributions to the uncertainty were found to be wy followed by the measured isotope ratios Rbx and Rbz2 as shown in Table 3. The variation of the individual results and the average of the triple IDMS measurements are shown in Fig. 3B. In comparison to double IDMS the advantage of triple IDMS is that the isotopic composition of the spike (Ry) need not be determined as it is cancelled from the equation. This is offset by a slightly higher workload due to the preparation of a second reference spike blend and an increased measurement time. The results obtained from the double IDMS and triple IDMS approaches for the determined human erythrocyte sample fit together very well within the estimated uncertainty which is also confirmed by the t-test as the empirical t-value is smaller than the critical t-value (0.502 < t < 2.776, α = 0.05). The high precision of the applied methods is pointed out by the achieved estimated expanded uncertainty of 1.45% and 1.50%. This demonstrates the usefulness of species-specific double and triple IDMS to provide reference values for clinical laboratories as there are no reference methods or materials for SOD1 available yet.
Conclusion
Two methods based on double and triple IDMS were applied for the precise quantification of SOD1 in human erythrocytes. Purchased human SOD1 was characterized using single post-column IDMS, UV-vis and ESI-TOF-MS before use as a reference, which is the key component in IDMS. Furthermore, a species-specific spike material was produced and characterized. A detailed comparison study of three methods published in the literature showed that using low pH and EDTA is the most suitable method for demetallation in the SOD1 spike preparation procedure. The final quantification of SOD1 in a real sample of human erythrocytes via double and triple IDMS shows a good agreement for the results of both methods within the expanded uncertainty. A complete uncertainty budget was estimated using the GUM workbench. The expanded uncertainty (with k = 2) of below 2% for both approaches demonstrates the high precision of the applied methods. The developed methods enable traceability of the results to the SI and can, therefore, be used as reference methods of higher order for routine analysis in clinical laboratories as required by the European in vitro diagnostica directive (Directive 98/79/EC) for the use of in vitro diagnostica in the samples of human origin.18 Furthermore, there is a chance to characterize SOD1 in a biological matrix for the use as reference material for validating quantification methods in medical routine laboratories. Thus, providing reference values to clinical laboratories will help to make the results of different routine measurement procedures more comparable. As SOD1 is an important marker amongst others for neurodegenerative diseases, such as Alzheimer's disease or amyotrophic lateral sclerosis, where the concentration is much lower (0.5 μg g−1 in cerebrospinal fluid40) than in erythrocytes, in the future prospective strategies for the enrichment of SOD1 have to be developed so that quantification by species-specific IDMS can be applied for this purpose.
Acknowledgements
This research was undertaken within the EMRP project HLT05. The EMRP was jointly funded by the EMRP participating countries within EURAMET and the European Union. We gratefully acknowledge support by the Braunschweig International Graduate School of Metrology B-IGSM.
References
- J. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CAS.
- J. A. Tainer, E. D. Getzoff, K. M. Beem, J. S. Richardson and D. C. Richardson, J. Mol. Biol., 1982, 160, 181–217 CrossRef CAS PubMed.
- Y. Yamazaki and T. Takao, Anal. Chem., 2008, 80, 8246–8252 CrossRef CAS PubMed.
- D. Chen, Q. Wang, H. Zhang, S. Mi, J. Wang, Q. Zeng and G. Zhang, Int. J. Quantum Chem., 2010, 110, 1394–1401 CAS.
- C.-T. Lin, T.-L. Lee, K.-J. Duan and J.-C. Su, Zool. Stud., 2001, 40, 84–90 CAS.
- M. Bewick, W. Coutie and G. R. Tudhope, Br. J. Haematol., 1987, 65, 347–350 CrossRef CAS PubMed.
- G. Ucar, B. Demir and B. Ulug, Cell Biochem. Funct., 2005, 23, 29–37 CrossRef CAS PubMed.
- R. Badraoui, S. Blouin, M. F. Moreau, Y. Gallois, T. Rebai, Z. Sahnoun, M. Baslé and D. Chappard, Chem.–Biol. Interact., 2009, 182, 98–105 CrossRef CAS PubMed.
- X. L. Zuo, J. M. Chen, X. Zhou, X. Z. Li and G. Y. Mei, Biol. Trace Elem. Res., 2006, 114, 41–53 CrossRef CAS PubMed.
- B. W. Brooksbank and R. Balazs, Dev. Brain Res., 1984, 16, 37–44 CrossRef CAS.
- J. A. Serra, E. R. Marschoff, R. O. Domínguez, S. de Lustig Eugenia, A. L. Famulari, E. L. Bartolomé and E. M. Guareschi, Clin. Chim. Acta, 2000, 301, 87–102 CrossRef CAS.
- M. Monari, A. Trinchero, C. Calabrese, O. Cattani, G. P. Serrazanetti, J. Foschi, A. Fabbri, D. Zahlane, G. Di Febo, V. Tonini, M. Cervellera, M. R. Tosi and V. Tugnoli, Biomarkers, 2006, 11, 574–584 CrossRef CAS PubMed.
- G. H. White, Ann. Clin. Biochem., 2011, 48, 393–409 CrossRef CAS PubMed.
- S. Oka, K. Ogino, S. Matsuura, S. Yoshimura, K. Yamamoto, Y. Okazaki, T. Takemoto, N. Kato and T. Uda, Clin. Chim. Acta, 1989, 182, 209–220 CrossRef CAS.
- S. Marklund and G. Marklund, Eur. J. Biochem., 1974, 47, 469–474 CrossRef CAS PubMed.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS PubMed.
- F. Attar, E. Keyhani and J. Keyhani, Appl. Biochem. Microbiol., 2006, 42, 101–106 CrossRef CAS.
- Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998.
- P. de Bièvre and H. S. Peiser, Fresenius' J. Anal. Chem., 1997, 359, 523–525 CrossRef.
-
M. Sargent, C. F. Harrington and R. Harte, Guidelines for Achieving High Accuracy in Isotope Dilution Mass Spectrometry, Royal Society of Chemistry, Cambridge, 2002 Search PubMed.
- J. Vogl, J. Anal. At. Spectrom., 2007, 22, 475–492 RSC.
- J. Vogl, Rapid Commun. Mass Spectrom., 2012, 26, 275–281 CrossRef CAS PubMed.
- C. Frank, O. Rienitz, C. Swart and D. Schiel, Anal. Bioanal. Chem., 2013, 405, 1913–1919 CrossRef CAS PubMed.
- O. Rienitz, A. Pramann and D. Schiel, Int. J. Mass Spectrom., 2010, 289, 47–53 CrossRef CAS.
- P. Rodríguez-González, J. M. Marchante-Gayón, García Alonso, J. Ignacio and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef.
- L. Rottmann and K. G. Heumann, Fresenius' J. Anal. Chem., 1994, 350, 221–227 CrossRef CAS.
- K. G. Heumann, S. M. Gallus, G. Rädlinger and J. Vogl, Spectrochim. Acta, Part B, 1998, 53, 273–287 CrossRef.
- C. F. Harrington, D. S. Vidler, M. J. Watts and J. F. Hall, Anal. Chem., 2005, 77, 4034–4041 CrossRef CAS PubMed.
- C. Swart, O. Rienitz and D. Schiel, Talanta, 2011, 83, 1544–1551 CrossRef CAS PubMed.
- C. L. Deitrich, A. Raab, B. Pioselli, J. E. Thomas-Oates and J. Feldmann, Anal. Chem., 2007, 79, 8381–8390 CrossRef CAS PubMed.
- J. A. Fee, J. Biol. Chem., 1973, 248, 4229–4234 CAS.
- M. J. Lindberg, J. Normark, A. Holmgren and M. Oliveberg, PNAS, 2004, 101, 15893–54566 CrossRef CAS PubMed.
- K. M. Beem, W. E. Rich and K. V. Rajagopalan, J. Biol. Chem., 1974, 249, 7298–7305 CAS.
- U. Weser and H. J. Hartmann, FEBS Lett., 1971, 17, 78–80 CrossRef CAS PubMed.
- B. Sutter, P. L. Bounds and W. H. Koppenol, Protein Expression Purif., 2000, 19, 53–56 CrossRef CAS PubMed.
- Y. Furukawa, K. Kaneko, K. Yamanaka, T. V. O'Halloran and N. Nukina, J. Biol. Chem., 2008, 283, 24167–24176 CrossRef CAS PubMed.
- P. A. Doucette, L. J. Whitson, X. Cao, V. Schirf, B. Demeler, J. S. Valentine, J. C. Hansen and P. J. Hart, J. Biol. Chem., 2004, 279, 54558–54566 CrossRef CAS PubMed.
- Y. Nuevo Ordóñez, M. Montes-Bayón, E. Blanco-González and A. Sanz-Medel, Anal. Chem., 2010, 82, 2387–2394 CrossRef PubMed.
- Y. Nuevo Ordóñez, C. L. Deitrich, M. Montes-Bayón, E. Blanco-González, J. Feldmann and A. Sanz-Medel, J. Anal. At. Spectrom., 2011, 26, 150–155 RSC.
- L. Winer, D. Srinivasan, S. Chun, D. Lacomis, M. Jaffa, A. Fagan, D. M. Holtzman, E. Wancewicz, C. F. Bennett, R. Bowser, M. Cudkowicz and T. M. Miller, JAMA Neurol., 2013, 70 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ja00459d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a