
A simple dilute-and-shoot approach for the determination of ultra-trace levels of arsenic in biological fluids via ICP-MS using CH3F/He as a reaction gas
M. R.
Flórez
ab,
E.
García-Ruiz
a,
E.
Bolea-Fernández
b,
F.
Vanhaecke
b and
M.
Resano
 *a
*a
aUniversity of Zaragoza, Department of Analytical Chemistry, Aragón Institute of Engineering Research (I3A), Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: mresano@unizar.es
bGhent University, Department of Analytical Chemistry, Krijgslaan 281-S12, 9000 Ghent, Belgium
First published on 16th October 2015
Abstract
The performance of a mixture of CH3F/He (1/9) as a reaction gas for the determination of As in biological fluids using a quadrupole ICP-MS instrument has been explored. A simple (dilute-and-shoot) interference-free method has been developed to quantify As concentrations at trace and ultra-trace levels in matrices with a high Cl content. As+ reacts with CH3F (through CH3F addition, followed by HF elimination) with high efficiency forming AsCH2+ as the primary reaction product, which can be monitored at a mass-to-charge ratio of 89, free from the Cl-based interferents (e.g., 40Ar35Cl+ and 40Ca35Cl+) that hamper the monitoring of 75As+. Matrix effects are overcome by the use of Te as an internal standard and the addition of 3% v/v ethanol to all samples and calibration standard solutions. The method presented was validated by analysing a set of reference materials (blood, serum and urine) and by assessing As recovery from a set of real blood samples. With this method, the limit of detection was calculated to be 0.8 ng L−1 As, favourably comparable to the vast majority of values reported in the literature, even with those obtained using more sophisticated sector-field instrumentation.
1. Introduction
Arsenicosis or As poisoning is most commonly due to long-term exposure to As-rich drinking water.1,2 The World Health Organization (WHO) sets a guideline value of 10 μg L−1 of As in drinking water, but this value is mainly governed by the traditional limits of detection (LODs).3 There is still a demand for simple, direct and interference-free methodologies that allow better LODs to be obtained and, therefore, provide more reliable results for lower As concentrations, for instance when analysing biological fluids as a proxy for tracking potentially elevated As exposure and the related risks.Several spectroscopy techniques have been employed for As determination, although none of them is free from hindrances.4 Due to its high detection power, inductively coupled plasma-mass spectrometry (ICP-MS) has been widely used to monitor low As contents in a variety of samples. However, accurate determination of As at trace and ultra-trace levels by ICP-MS is strongly jeopardized by the occurrence of spectral overlap of the signals from ArCl+ (40Ar35Cl+, 38Ar37Cl+) and 40Ca35Cl+ ions with that of the mono-isotopic target element at the mass-to-charge ratio (m/z) of 75.5 This interference represents a great challenge when aiming to determine As in matrices such as blood, urine or serum due to the high amount of dissolved chloride salts present and the very low As concentrations expected.
Several analytical methodologies have been developed to overcome the aforementioned spectral interference, but many of them require the separation of the analyte from the matrix prior to the measurement. An obvious choice for a direct and complete resolution of the spectral interference would be using a sector-field ICP-MS (SF-ICP-MS) instrument, capable of working at a higher mass resolution.6 However, at the resolution required, there is a significant drop in sensitivity, which is not desirable when aiming to determine very low concentrations of the analyte.7
The use of quadrupole-based ICP-MS (Q-ICP-MS) instrumentation equipped with a collision/reaction cell is a good alternative, especially as such instrumentation is more affordable for routine labs than SF-ICP-MS and, thus, it is more widespread.8–10 Several gases/mixtures of gases have been explored in the context of As determination in Cl-containing matrices, in an attempt to alleviate the spectral interference. Examples of reactive gases widely used and documented in the literature are H2, O2 or N2O. H2 reacts with ArCl+ helping in reducing the interference at m/z = 75 to some extent.11–15 On the other hand, O2 reacts with As+, forming the oxide ion 75As16O+, which can be monitored at m/z = 91, free from Cl-based polyatomic interference.16,17 However, it has to be kept in mind that Co can form 59Co32O2+, which also shows a m/z = 91.18 It is likely that this fact led Funk et al. to state that “quantification of As in the DRC mode resulted in interfering ions at m/z 91” when aiming at blood analysis.19 N2O reacts with As+ in the same way as O2 does, as an O-atom donor.20,21
NH3 could be another possibility. NH3 reacts with 40Ar35Cl+ successfully removing the interference, but it also reacts with As+ according to what has been described as an “unusually complex condensation reaction” by Baranov and Tanner,22 leading to the formation of various species, of which As(NH3)NH2+ seems to be the most abundant ion. To the best of the authors' knowledge, the formation of such a product has not been used yet for analytical purposes.
He is also typically used as a collision gas in combination with kinetic energy discrimination to reduce the contribution from polyatomic interference, although such an approach tends to significantly reduce the sensitivity, providing a signal-to-noise ratio that is not competitive with those obtained with the reactions described above.23
Despite these possibilities, As determination at the low levels required in some complex clinical samples remains a challenge. A clear example of these difficulties can be found in a very recent publication, that reports on As values obtained using various approaches, based on collision/reaction cell ICP-MS and SF-ICP-MS, for a new blood CRM material. The results obtained showed so much scatter (one order of magnitude range) that the As value could not be certified finally, and only an informative <5 μg L−1 value was provided instead.4 It is worth mentioning that the typical As level in human blood is precisely a few μg L−1, and thus clinical labs should be able to routinely determine such levels reliably.
High efficiency has been reported for the reaction of As+ with CH3F, with the predominant formation of AsCH2+ (m/z = 89), upon CH3F addition and subsequent elimination of HF.24 However, this reactive gas has been tested very seldom in reaction cells.25 Nevertheless, very good results (both in terms of sensitivity and LODs) have been obtained recently for As determination in some biological CRMs (tissues and plants) using a tandem ICP-mass spectrometer (ICP-MS/MS), a new type of ICP-MS instrumentation with a collision/reaction cell situated in-between two quadrupole units.26 Such instrumentation is more expensive than a traditional quadrupole ICP-MS unit, but enables a better control of the reactions taking place within the reaction cell (an octopole) by removing all ions with a m/z different from that defined by the first quadrupole, and subsequently selecting the desired m/z ratio to be monitored using the second quadrupole.
It is the purpose of this work to investigate the potential of CH3F for As determination in a more simple and standard (and thus, more widespread) reaction cell ICP-MS instrument. Plasma, urine and, especially, blood have been selected as target samples owing to their intrinsic interest and the problems mentioned above. The goal of the work was to develop a simple, but accurate dilute-and-shoot approach.
2. Experimental
2.1. Instrumentation
All the measurements were carried out using a quadrupole-based ICP-MS instrument (NexION 300X), which is commercially available from Perkin Elmer (Waltham, USA). This instrument is equipped with a cell (a quadrupole) that can be used both as a collision cell in combination with kinetic energy discrimination (KED) and as a dynamic reaction cell (DRC). The instrument is equipped with a triple cone interface, with an additional hyper skimmer cone, providing a more gradual pressure reduction within the interface, which results in less dispersion of the ion beam. A quadrupole ion deflector reflects the ion beam over a 90 degree angle, focusing it into the cell.The sample introduction system comprises a 0.4 mL min−1 concentric quartz nebulizer and a quartz cyclonic spray chamber, with a 0.38 mm inner diameter PVC flared tubing for the peristaltic pump.
2.2. Samples and standards
HNO3 solutions were prepared from 14 mol L−1 HNO3 SupraPur, obtained from Merck Millipore (Darmstadt, Germany), and used for dilutions.
As and Te solutions were prepared from commercially available 1 g L−1 single-element standards (Merck) by appropriate dilution with 0.14 mol L−1 HNO3 Suprapur. Anhydrous, denatured ethanol of spectrophotometric purity grade (90% alcoholic purity) was purchased from Alfa Aesar (Karlsruhe, Germany). KCl solid salt (pro analysis purity grade) was obtained from Merck.
In addition, whole blood samples from healthy volunteers were obtained from the University Hospital Miguel Servet (Zaragoza, Spain).
2.3. Analytical method for sample analysis
All the samples and reference materials were analysed using the instrumental settings and data acquisition parameters listed in Table 1.| NexION 300X | As determination |
|---|---|
| Mode | DRC |
| Reaction cell gas | CH3F/He (1/9) |
| Reaction cell gas flow rate | 1.6 mL min−1 |
| Nebulizer gas flow rate | 1.02 L min−1 |
| Auxiliary gas flow rate | 1.20 L min−1 |
| Plasma gas flow rate | 18.00 L min−1 |
| RF power | 1600 W |
| RPa | 0.00 |
| RPq | 0.65 |
| Nuclides monitored | 89AsCH2+, 126Te+ |
| Sweeps/reading | 100 |
| Readings/replicate | 1 |
| Replicates | 10 |
| Dwell time | 20 ms |
The samples were not treated prior to analysis, except for dilution. Different dilution factors were evaluated (25-, 50-, 100- and 200-fold), but the final solutions always contained 3% ethanol and 1% HNO3.
A new set of 5 calibration standard solutions of suitable concentrations, adapted to the expected As sample levels and also containing 3% ethanol and 1% HNO3, was prepared for every measurement session.
SupraPur grade HNO3 and TraceSelect purity grade water were used for dilutions, in order to ensure low blank signals.
Te was added as the internal standard to all sample and standard solutions for a final concentration of 20 μg L−1 Te.
3. Results and discussion
3.1. Study of the reaction between As and CH3F/He within the reaction cell
In order to avoid the major interference found at m/z = 75, the possibility to promote a reaction between As and CH3F was investigated.Zhao et al.24 listed AsCH2+ (in a 97% distribution) as the primary product of the reaction between As and CH3F/He, with a minor (3%) formation of the addition product As(CH3F)+, using ICP/selected-ion flow tube-MS (ICP/SIFT-MS). Bolea-Fernandez et al.26 confirmed this behaviour using ICP-MS/MS. This type of reaction with CH3F (CH3F addition and ulterior elimination of HF) is not very common. Fluorination or CH3F addition is much more usual. In fact, As is the only element listed in ref. 18 to undergo this process as the main reaction with CH3F. Recently, it has been shown that the same is true for Se, but in a much less effective way.26 In any case, this reaction between As and CH3F is quite characteristic and has been demonstrated to be very efficient in ICP-MS/MS.26
In this work, CH3F/He is studied as a reaction gas for the determination of As, making use of a more conventional reaction cell-ICP-MS instrumentation. To evaluate the efficiency of the reaction and the products generated under the reaction cell conditions, a 5 μg L−1 As aqueous standard solution was nebulized and measured at different m/z ratios. The reaction cell was pressurized at different CH3F/He flow rates to find the best settings in terms of sensitivity. The results, represented in Fig. 1, show an evident preference of the reaction to evolve towards the formation of AsCH2+, also in the present study.
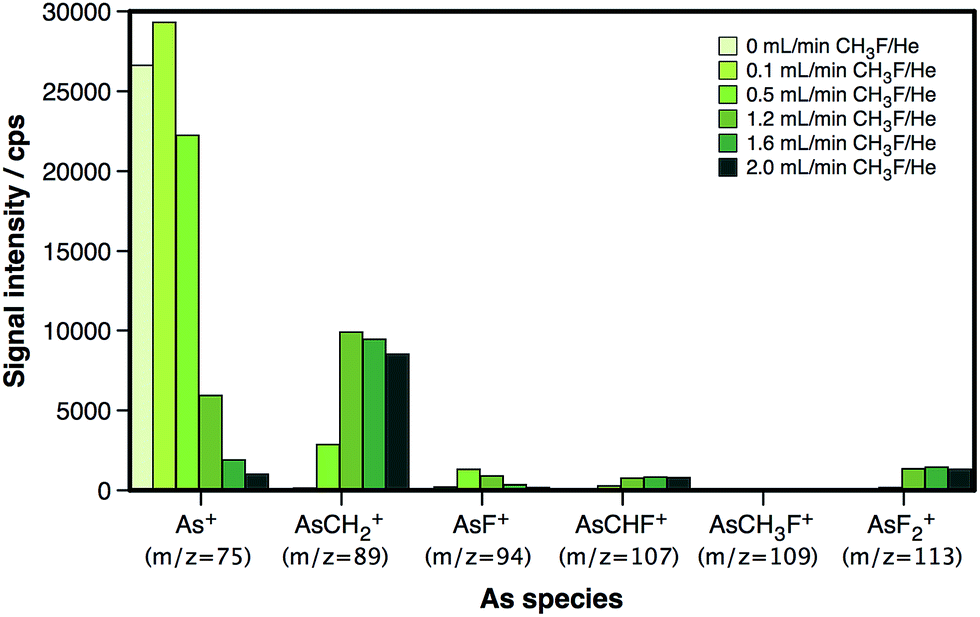 | ||
| Fig. 1 Overview of the As-based reaction products generated within the dynamic reaction cell at different CH3F/He flow rates. | ||
As shown in Fig. 1, the maximum AsCH2+ signal that can be attained is lower than the initial 75As+ signal. The AsCH2+ signal reaches approx. 40% of the initial 75As+ signal at a flow of 1.6 mL min−1 CH3F/He, and even decreases at higher flows. This fact is not due to poor reaction efficiency, as the remaining 75As+ signal and the signals corresponding to other As species are rather low at such CH3F/He flows. Instead, this is most likely due to the presence of He, which, at these high flows, is expected to result in scattering losses. Nevertheless, the addition of He is highly recommended for this type of reaction in order to slow down the ions sufficiently to obtain a good reaction efficiency.24 In any case, this loss of raw sensitivity (roughly a factor of 3) seems like a reasonable price to pay for an interference-free detection of As, as will be demonstrated later on.
3.2. Optimization of the reaction cell parameters
A further optimization of the reaction gas flow rate was carried out by monitoring the signal intensity of AsCH2+ in a 50-fold diluted blood sample at different CH3F/He flow rates. A blank solution containing 4 g L−1 Cl (the concentration typically found in blood), added as KCl, was also measured in the same way, in order to find the conditions providing the best S/N ratio.Fig. 2 shows how the signal intensity at m/z = 89 increases (left y-axis) with the reaction gas flow rate, reaching a maximum sensitivity at 1.6 mL min−1 CH3F/He. The use of higher flow rates leads to signal losses, probably due to scattering. On the other hand, the blank signal (right y-axis) remains rather low (below 40 cps) and stable along all the conditions evaluated. Therefore, an optimum signal-to-noise ratio is obtained at 1.6 mL min−1.
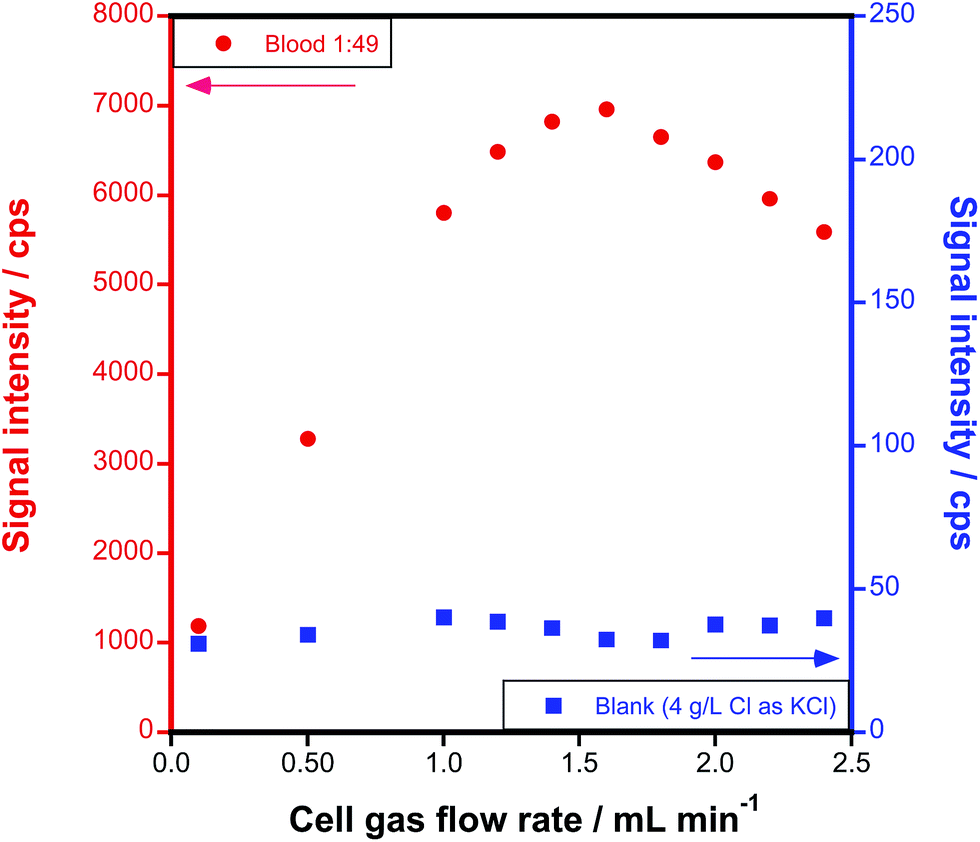 | ||
Fig. 2 Optimization of the CH3F/He reaction gas flow rate for the generation of AsCH2+ (monitored at m/z = 89) from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 diluted blood sample (left y-axis) and from an equivalent Cl-containing blank (right y-axis). :49 diluted blood sample (left y-axis) and from an equivalent Cl-containing blank (right y-axis). | ||
The rejection parameter q of the DRC (RPq), associated with the low-mass cut-off, was also adjusted. An RPq value of 0.55–0.65 was found to bring about the best results in terms of LOD. A summary of the optimized instrument settings is presented in Table 1.
In order to further ensure that no spectral interference is affecting the measurements under these optimized conditions, two sets of 0.5, 1, 2.5 and 5 μg L−1 As aqueous standard solutions were prepared, a first one diluted with 1% HNO3 and a second one diluted with 1% HCl. A calibration curve was plotted based on the results obtained for each set of standard solutions so as to compare the slopes. These results are presented in Fig. 3, where the lack of any significant difference can be appreciated. From this direct comparison, it can be concluded that the presence of a high Cl content in the matrix does not influence the results when monitoring the AsCH2+ species at m/z = 89, thus enabling the determination of As free from spectral interference.
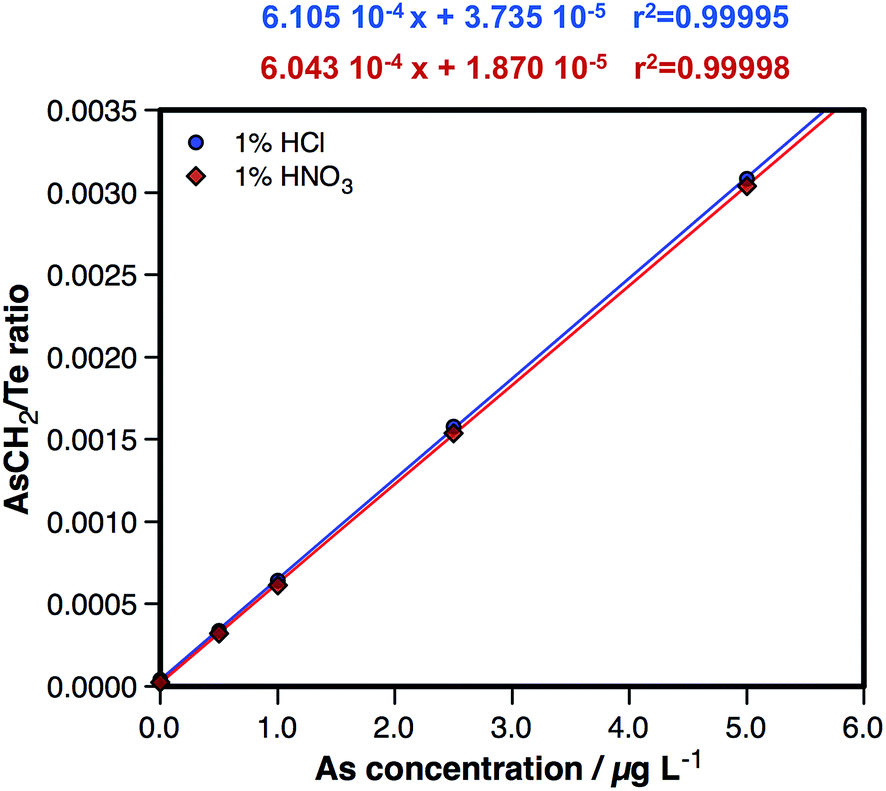 | ||
| Fig. 3 Direct comparison of a calibration curve prepared by dilution with 1% HNO3 and a calibration curve prepared by dilution with 1% HCl, both sets of standard solutions measured under the conditions listed in Table 1. | ||
3.3. Overcoming non-spectral interference
As has a high ionization potential (9.79 eV),27 and displays a very pronounced matrix-induced signal enhancement in some matrices, especially in the presence of organic C (C-effect).5,28–35 Due to this fact, some strategies to correct for matrix effects need to be explored.On the one hand, a suitable internal standard is required, in order to correct for any instrumental instability or signal drift and improve the precision of the measurements. An adequate internal standard has to be absent from the sample matrix, should not lead to or suffer from spectral overlap and must have a mass number close to that of the analyte and a similar ionization potential.36 Additionally, in this case, particular attention has to be paid not to introduce an element that will react with CH3F/He, thus creating a new spectral interference. Ge, for instance, would react with CH3F/He via F atom transfer,24 leading to the formation of 70Ge19F+ at m/z = 89, making the selection of this element, often used for this task when As determination is aimed at, prohibited for our method. Overall, Te (IP = 9.01 eV)27 seems to be the best choice for this work and was used as the internal standard in all following experiments.
On the other hand, differences in the matrix composition may preclude a direct quantification of As in the samples based on external calibration versus aqueous standard solutions. When aiming to determine As at very low concentrations, sample treatment needs to be minimized in order to avoid analyte losses or significant external contamination. Moreover, depending on the concentration, it might not be possible to dilute the sample until no matrix effect is observed. The high organic load of blood and, to a lower extent, serum samples, significantly affects the plasma conditions. Even when applying internal standard correction, such an effect may not be completely compensated for.
Thus, the approach finally developed was based on using a sufficiently high sample dilution factor (at least 1:24) to minimize matrix effects and, also, on adding an amount of ethanol to both the samples and the standard solutions, such that basically its presence would control the plasma conditions, helping in matching the potentially different behaviour expected between standard solutions and samples.
Moreover, it is well-known that the addition of an appropriate amount of an organic compound, such as ethanol, will lead to a more complete ionization of As through the C-effect, thus increasing the sensitivity to this element.5,28–35
An optimization of the amount of ethanol added was carried out, as shown in Fig. 4. As can be seen (Fig. 4A), the presence of ethanol initially increases the sensitivity, until a value of 3–5%. However, the LOD does not follow the same trend. After initially dropping, the LOD tends to increase with higher values, because the blanks are increasing even more than the sensitivity. A value between 1 and 3% ethanol seems optimum from this point of view. Even more important, as Fig. 4B shows, is that overestimated results are obtained if blood is analysed in the absence of ethanol, because the organic matrix tends to increase the As signal. This phenomenon is corrected for when ethanol is added, and good agreement with the reference value is attained at 3% ethanol. The same trend was observed for all the blood samples tested: results biased 15–40% high (depending on the dilution) in the absence of ethanol, and values were in agreement with the expected ones when samples and standards contained 3% ethanol. Thus, this value was used in further work.
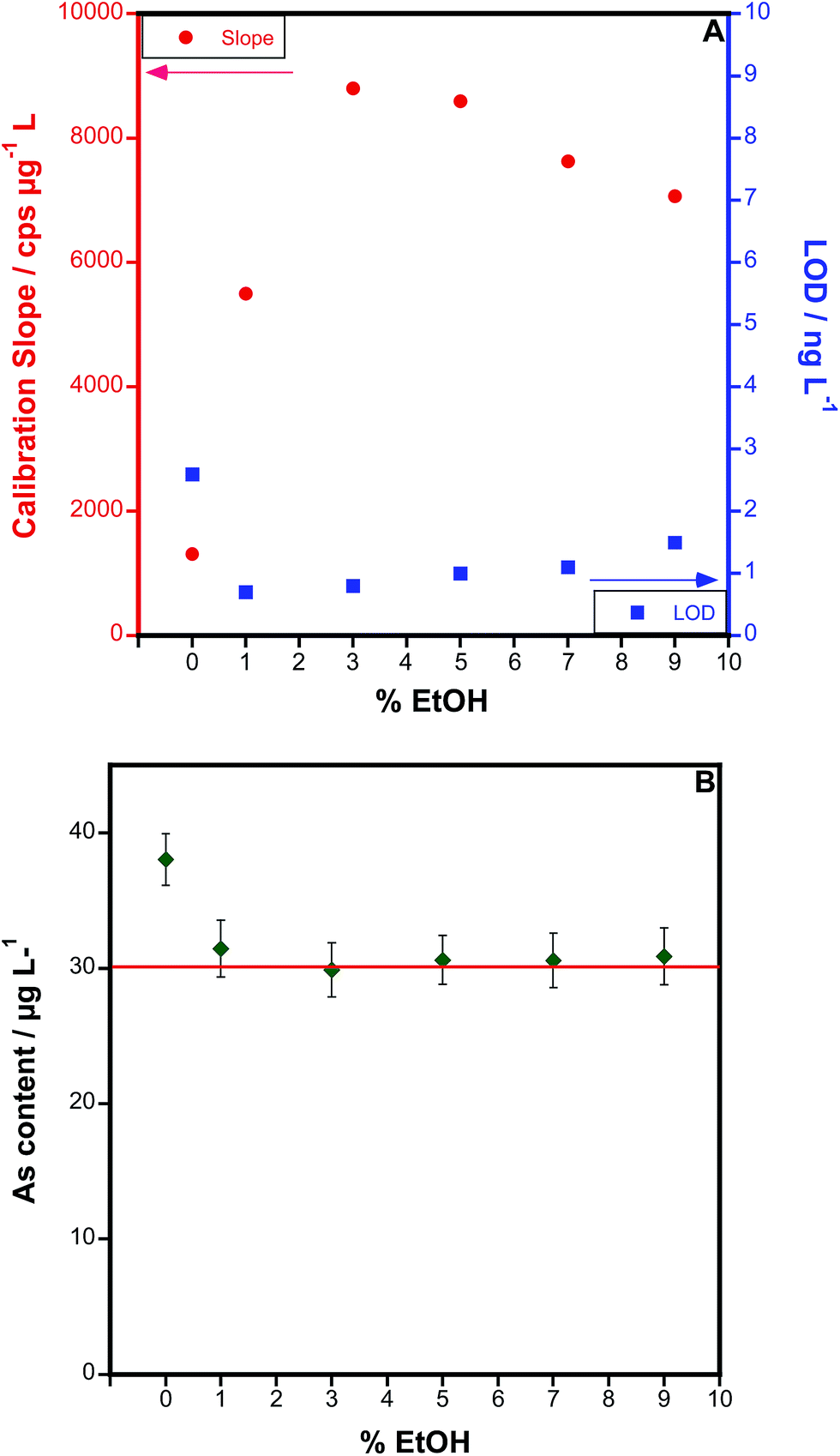 | ||
| Fig. 4 Optimization of the concentration of ethanol in samples and standards. (A) Variation of the sensitivity and the limit of detection with every ethanol concentration tested. (B) Results obtained for the determination of As in a 1:49 diluted reference blood sample (the certified As concentration, 30.4 ± 7.3 μg L−1, is represented by a red line), for all different ethanol concentrations studied. Uncertainty is indicated as the standard deviation of 5 replicates. | ||
Under these conditions, the LOD was calculated to be 0.8 ng L−1. A comparison with the values found in the literature for different interference-free As determination strategies is presented in Table 2. As can be seen, the LOD presented in this work compares favourably with the vast majority of them, even with those obtained using SF-ICP-MS. It seems that only when using CH3F/He as the reaction gas and in the ICP-MS/MS mode a better LOD can be achieved. If CH3F/He is used in a triple quadrupole instrument and the mass window of the first quadrupole is maintained open (no mass filtering), a very similar LOD as the one reported in the current work is obtained.26
| Species monitored | Systemb (gas) | LODs/ng L−1 | Sample | Publication |
|---|---|---|---|---|
| a LODs were calculated as 3 times the standard deviation of the blank signal intensities divided by the slope of the calibration curve. b CC: collision cell; RC: reaction cell; CRI: collision reaction interface; DRC: dynamic reaction cell; SF: sector field; SQ: single quadrupole. | ||||
| As+ | CC (H2/He) | 147 | Biological samples | Ref. 23 |
| As+ | RC (H2) | 25 | Rain water | Ref. 13 |
| As+ | CRI (H2) | 19–95 | Biological samples | Ref. 14 |
| AsO+ | DRC (O2, N2O) | 40 | Sea water | Ref. 21 |
| AsO+ | DRC (O2) | 2.0 | Digested blood | Ref. 17 |
| As+ | SF-ICP-MS | 10 | Solutions | Ref. 26 |
| As+ | SF-ICP-MS | 3.0 | Digested plants | Ref. 37 |
| AsO+ | MS/MS (O2) | 1.0 | Digested plants | Ref. 38 |
| AsO+ | MS/MS (O2) | 1.6 | Drinking water | Ref. 39 |
| AsO+ | MS/MS (O2) | 7.0 | Solutions | Ref. 26 |
| AsCH2+ | MS/MS (CH3F) | 0.2 | Biological samples | Ref. 26 |
| AsCH2+ | MS/MS (CH3F) SQ mode | 1.0 | Biological samples | Ref. 26 |
| AsCH2+ | DRC (CH3F) | 0.8 | Blood, urine, and serum | This work |
It has to be mentioned that the LODs presented in the table are instrumental LODs. Therefore, any dilution required as a consequence of sample pretreatment needs to be factored in as well. Thus, the method proposed in this work ultimately provides a LOD of 20 ng L−1 and a LOQ of 70 ng L−1 (for a dilution factor of 25). These values are sufficiently low to determine As in real blood samples, as will be demonstrated in the next section.
3.4. Method validation and sample results
In order to validate the method developed, all the reference samples listed in Section 2.2.2. were analysed. All of them were diluted in a 3% ethanol solution to four different dilution levels: 25-, 50-, 100- and 200-fold.As determination was carried out according to the optimized instrumental settings listed in Table 1. The results obtained are presented in Table 3. As can be seen, for every sample, the results obtained were found to be in good agreement with the corresponding reference value. The confidence intervals obtained for the different dilutions factors tested always overlapped among them and, also, with the certified range. However, it was also observed that the values obtained with higher dilution factors tended to be a bit lower (see 1:199 values), even if this difference could not be considered as statistically significant. In any case, using such an extreme dilution factor is not really necessary, and it seems preferable to use intermediate dilution factors (1:49 or 1:99) to guarantee the best accuracy, while minimizing residues in the spray chamber.
| Reference material | Reference value | Dil. factor 1:24 |
Dil. factor 1:49 |
Dil. factor 1:99 |
Dil. factor 1:199 |
|---|---|---|---|---|---|
| a Indicative value. | |||||
| Whole blood, level I/μg L−1 | 2.4 ± 0.5 | 3.1 ± 0.2 | 2.9 ± 0.2 | 2.9 ± 0.2 | 2.9 ± 0.2 |
| Whole blood, level II/μg L−1 | 14.3 ± 2.9 | 15.3 ± 1.1 | 14.6 ± 1.2 | 13.4 ± 1.3 | 12.6 ± 1.7 |
| Whole blood, level III/μg L−1 | 30.4 ± 7.3 | 30.0 ± 3.1 | 27.1 ± 3.1 | 26.5 ± 3.7 | 26.1 ± 3.4 |
| Serum, level I/μg L−1 | 0.40a | 0.45 ± 0.07 | 0.52 ± 0.10 | 0.42 ± 0.08 | — |
| Urine, level I/μg L−1 | 43.0 ± 8.6 | 42.9 ± 3.4 | 43.4 ± 3.9 | 44.1 ± 3.5 | 38.8 ± 3.6 |
| Urine, level II/μg L−1 | 83.3 ± 16.6 | 83.2 ± 4.9 | 81.2 ± 5.8 | 78.5 ± 5.1 | 75.9 ± 5.4 |
It is worth mentioning here that, although all the samples and standards were diluted and measured under the same conditions, urine samples do not really require the addition of ethanol, as no significant matrix effect was observed for such samples. It was experimentally confirmed that very similar results were attained for urine when no ethanol was added.
Finally, five real blood samples were also analysed. Since no reference value was available for As, recovery assays were carried out by doping the samples (diluted 1:99) with a 0.1 μg L−1 As standard solution. Samples were measured with and without As spike and the results thus obtained are presented in Table 4. In all cases, recoveries were higher than 90%, with an average recovery of 96.8 ± 3.9%, further proving the validity of the approach proposed for the analysis of real blood samples.
| Sample code | As content/μg L−1 | Recovery assay/% |
|---|---|---|
| MSHBlood01 | 5.10 ± 0.48 | 94.5 |
| MSHBlood02 | 5.18 ± 0.46 | 91.9 |
| MSHBlood03 | 3.13 ± 0.25 | 98.5 |
| MSHBlood04 | 2.84 ± 0.24 | 102.1 |
| MSHBlood05 | 14.6 ± 1.3 | 97.0 |
4. Conclusions
This work presents a simple and straightforward analytical method for the interference-free determination of As at ultra-trace levels in biological fluids with a high Cl content and an organic matrix.A 1/9 gas mixture of CH3F/He was evaluated as the reaction gas in a dynamic reaction cell of a quadrupole ICP-MS instrument. As reacts very efficiently and with a high selectivity with CH3F/He, leading to the formation of AsCH2+ as the primary product.
Despite the strong matrix effects due to the high organic load of the samples, it was possible to develop a simple “dilute and shoot” approach by diluting the samples using 3% ethanol.
This simple method provides very competitive limits of detection (0.8 ng L−1), as well as high accuracy and precision (typically better than 7% RSD).
Acknowledgements
This work has been funded by the Spanish Ministry of Economy and Competitiveness (Project CTQ2012-33494) and the Aragón Government (Fondo Social Europeo). The authors acknowledge Dr Luis Rello from the Miguel Servet Universitary Hospital (Zaragoza, Spain) for providing the real blood samples.References
- A. Gomez-Caminero, P. Howe, M. Hughes, E. Kenyon, D. R. Lewis, M. Moore, J. Ng, A. Aitio and G. Becking, Arsenic and Arsenic Compounds, World Health Organization, 2nd edn, Geneva, 2001 Search PubMed.
- J. C. Ng, J. Wang and A. Shraim, Chemosphere, 2003, 52, 1353–1359 CrossRef CAS PubMed.
- WHO, Guidelines for Drinking-water Quality, World Health Organization, 4 edn, Geneva, 2011 Search PubMed.
- R. L. Paul, W. C. Davis, L. Yu, K. E. Murphy, W. F. Guthrie, D. D. Leber, C. E. Bryan, T. W. Vetter, G. Shakirova, G. Mitchell, D. J. Kyle, J. M. Jarrett, K. L. Caldwell, R. L. Jones, S. Eckdahl, M. Wermers, M. Maras, C. D. Palmer, M. F. Verostek, C. M. Geraghty, A. J. Steuerwald and P. J. Parsons, J. Radioanal. Nucl. Chem., 2014, 299, 1555–1563 CrossRef CAS PubMed.
- J. Goossens, F. Vanhaecke, L. Moens and R. Dams, Anal. Chim. Acta, 1993, 280, 137–143 CrossRef CAS.
- F. Vanhaecke and L. Moens, Anal. Bioanal. Chem., 2004, 378, 232–240 CrossRef CAS PubMed.
- C. Turetta, G. Cozzi, C. Barbante, G. Capodaglio and P. Cescon, Anal. Bioanal. Chem., 2004, 380, 258–268 CrossRef CAS PubMed.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, Part B, 2002, 57, 1361–1452 CrossRef.
- D. R. Bandura, V. I. Baranov, A. E. Litherland and S. D. Tanner, Int. J. Mass Spectrom., 2006, 255–256, 312–327 CrossRef CAS.
- S. D'Ilio, N. Violante, C. Majorani and F. Petrucci, Anal. Chim. Acta, 2011, 698, 6–13 CrossRef PubMed.
- K. Neubauer and U. Völlkopf, At. Spectrosc., 1999, 20, 64–68 CAS.
- M. Resano, E. García Ruiz, V. G. Mihucz, Á. M. Móricz, Gy. Záray and F. Vanhaecke, J. Anal. At. Spectrom., 2007, 22, 1158–1162 RSC.
- J. Darrouzès, M. Bueno, G. Lespès, M. Holeman and M. Potin-Gautier, Talanta, 2007, 71, 2080–2084 CrossRef PubMed.
- C. D. Pereira, E. E. Garcia, F. V. Silva, A. R. A. Nogueira and J. A. Nóbrega, J. Anal. At. Spectrom., 2010, 25, 1763–1768 RSC.
- M. Colon, M. Hidalgo and M. Iglesias, Talanta, 2011, 85, 1941–1947 CrossRef CAS PubMed.
- K. Kawabata, Y. Kishi and R. Thomas, Anal. Chem., 2003, 75, 422A–428A CrossRef CAS.
- S. D'Ilio, N. Violante, M. Di Gregorio, O. Senofonte and F. Petrucci, Anal. Chim. Acta, 2006, 579, 202–208 CrossRef PubMed.
- http://www.chem.yorku.ca/profs/bohme/research/selection_table.html, last accessed, July 2015.
- W. E. Funk, J. K. McGee, A. F. Olshan and A. J. Ghio, Biomarkers, 2013, 18, 174–177 CrossRef CAS PubMed.
- V. Blagojevic, E. Flaim, M. J. Y. Jarvis, G. K. Koyanagi and D. K. Bohme, J. Phys. Chem. A, 2005, 109, 11224–11235 CrossRef CAS PubMed.
- M. Grotti and R. Frache, J. Anal. At. Spectrom., 2007, 22, 1481–1487 RSC.
- V. I. Baranov and S. D. Tanner, J. Anal. At. Spectrom., 1999, 14, 1133–1142 RSC.
- M. Niemelä, P. Perämäki, H. Kola and J. Piispanen, Anal. Chim. Acta, 2003, 493, 3–12 CrossRef.
- X. Zhao, G. K. Koyanagi and D. K. Bohme, J. Phys. Chem. A, 2006, 110, 10607–10618 CrossRef CAS PubMed.
- E. Bolea-Fernández, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Chem., 2014, 86, 7969–7977 CrossRef PubMed.
- E. Bolea-Fernández, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Bioanal. Chem., 2015, 407, 919–929 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press Taylor & Francis group, 89 edn, Boca Ratón, 2008 Search PubMed.
- P. Allain, L. Jaunault, Y. Mauras, J. M. Mermet and T. Delaporte, Anal. Chem., 1991, 63, 1497–1498 CrossRef CAS.
- E. H. Larsen and S. Stürup, J. Anal. At. Spectrom., 1994, 9, 1099–1105 RSC.
- F. Vanhaecke, J. Riondato, L. Moens and R. Dams, Fresenius. J. Anal. Chem., 1996, 355, 397–400 CAS.
- V. L. Dressler, D. Pozebon and A. J. Curtius, Anal. Chim. Acta, 1999, 379, 175–183 CrossRef CAS.
- Z. Hu, S. Hu, S. Gao, Y. Liu and S. Lin, Spectrochim. Acta, Part B, 2004, 59, 1463–1470 CrossRef.
- Z. Hu, S. Gao, S. Hu, H. Yuan, X. Liu and Y. Liu, J. Anal. At. Spectrom., 2005, 20, 1263–1269 RSC.
- W. Guo, S. Hu, X. Li, J. Zhao, S. Jin, W. Liu and H. Zhang, Talanta, 2011, 84, 887–894 CrossRef CAS PubMed.
- G. Grindlay, J. Mora, M. de Loos-Vollebregt and F. Vanhaecke, Spectrochim. Acta, Part B, 2013, 86, 42–49 CrossRef CAS.
- F. Vanhaecke, H. Vanhoe, R. Dams and C. Vandecasteele, Talanta, 1992, 39, 737–742 CrossRef CAS PubMed.
- J. Frank, M. Krachler and W. Shotyk, Anal. Chim. Acta, 2005, 530, 307–316 CrossRef CAS.
- B. P. Jackson, A. Liba and J. Nelson, J. Anal. At. Spectrom., 2015, 30, 1179–1183 RSC.
- C. D. B. Amaral, R. S. Amais, L. L. Fialho, D. Schiavo, T. Amorim, A. R. A. Nogueira, F. R. P. Rocha and J. A. Nóbrega, Anal. Methods, 2015, 7, 1215–1220 RSC.
| This journal is © The Royal Society of Chemistry 2016 |