
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 13 metal complexes containing the bis-(4-methylbenzoxazol-2-yl)-methanide ligand†‡
David-R.
Dauer
,
Melchior
Flügge
,
Regine
Herbst-Irmer
and
Dietmar
Stalke
*
Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 19th November 2015
Abstract
To focus on the high importance of low-valent main group metal complexes, which can be applied to catalytic transformations, this article deals with the promising new ligand system (4-MeNCOC6H3)2CH2 (1). It is patterned on the well known nacnac ligand and further development of the parent bisheterocyclo methanes. In comparison with the results of previous studies based on the bisheterocyclo methanes (NCOC6H4)2CH2 and (NCOC6H4)2CH2 derivative 1 was modified by adding a methyl group to the annulated benzene perimeters to enhance the steric protection of a potentially coordinated main group metal cation. On reaction of 1 with group 13 trimethyl reagents and dialkyl aluminium halides the ligand backbone gets deprotonated and the two endocyclic nitrogen donor atoms coordinate with the remaining organometallic fragment to form a six-membered metalla heterocycle. The synthesis of [Me2Al{(4-MeNCOC6H3)2CH}] (2), [Me2Ga{(4-MeNCOC6H3)2CH}] (3), [Me2In{(4-MeNCOC6H3)2CH}] (4), [ClMeAl{(4-MeNCOC6H3)2CH}] (5), [IMeAl{(4-MeNCOC6H3)2CH}] (6) and [IEtAl{(4-MeNCOC6H3)2CH}] (7) could be accomplished. A structural comparison of those metallated species based on single crystal X-ray analyses identifies them as ideal precursors generating new low-valent main group complexes.
Introduction
In the context of the ongoing increasing demand for the activation of small molecules like H2, N2, NH3 or CO2 the development of new synthetic approaches for generating catalytic active species is of high interest. Even though this research area of catalytic activation is mostly focused on the field of transition metal complexes the transfer to suitable main group metal complexes, which should adopt the same catalytic reactivity compared to the transition metals or even exceed them, plays a key role in current research.1As a figurehead for efficient small molecule activation FLP (frustrated Lewis pairs) chemistry has developed to a promising research area over the last decade.2 In this way the usage of mainly phosphorus/boron, nitrogen/boron or carbon/boron FLP systems offers simple access to the efficient splitting of dihydrogen3 or the ring opening of THF.4 Also the aluminium based FLPs were found to be advantageous for B–H or N–H bond activation,5 polymerisation reaction of methyl methacrylate6 and conversion of CO2.7 Furthermore, the well-known nacnac ligand and the derived main group metal complexes, respectively, facilitate the stabilisation of metal ions in low oxidation states.8 The synthesis of the first Mg(I) compound9 and the corresponding β-diketiminate Ca(II) complexes,10 which were successfully applied for epoxide/CO2 copolymerisation11 or hydrogenation reactions of alkenes with H2,12 should be highlighted as alkaline earth metal complexes performing in catalysis. Switching to group 13 elements also a variety of Al(III) and Ga(III) containing complexes was synthesised and fully characterized.13 Subsequently also the low-valent Al(I)14 and Ga(I)15 species employing the Dipp-substituted (Dipp = 2,6-diisopropylphenyl) nacnac ligand were accessible. More recently the catalytic activity of the related alumoxanes was studied in detail.16 Other common ligand types, which are used for stabilisation of dimeric low-valent group 13 species involving metal–metal multiple bonds, are the substituted terphenyls, which are also prone to interaction with small molecules like H2.17
With this in mind in the field of main group transformation ligand design in particular and the choice of the suitable main group element have significant influence on the stability and the catalytic abilities of the resulting compounds. In this work we tried to mimic the omnipresent nacnac ligand by two methylene bridged substituted benzoxazole moieties. The corresponding group 13 metal complexes were synthesised to get the best from two worlds: the stabilisation abilities to get low-valent metal complexes and the reactivity of group 13 metals. To tie up with earlier studies concerning the related bisheterocyclo methanide18 and amide derivatives19 this work focuses on bis-(4-methylbenzoxazol-2-yl)-methane 1. The previous results of the methanide containing metal complexes have shown that upon deprotonation of the methylene bridge of the parent ligand systems the coordination of the metal cation is preferentially accomplished by the endocyclic nitrogen donor atoms in a chelating fashion.20 Due to this fact and to further shield the coordinated metal the bis-(benzoxazol-2-yl)-methane ligand was additionally methyl substituted in 1 to increase the kinetic stabilisation at the same side of the coordinating nitrogen atoms. Neither in the case of the methanide nor the amide derivatives a coordination via the other feasible O- or S-coordinating sites has been observed in the solid state yet.18,19
The issue of steric shielding of metal centres within the group 13 complexes is an important aspect for further applications and reactions, because in the future we plan to convert these Al(III) containing species into the low-valent Al(I) compounds e.g. [Al{Dipp2nacnac}].14e Because of the high electrophilicity of the Al(I) species steric protection is essential to maintain the monomeric carbene-like structural motif and prevent dimerisation or nucleophilic attacks.21 The challenge to synthesise a low-valent species containing a bisheterocyclo methanide ligand backbone is still the guiding idea of ongoing research and 1 seems to be a promising ligand system for the subsequent reduction steps (vide infra).
Among the nacnac derivatives and the bisheterocyclo methanides efficient metal shielding is comparable within certain limits. In the case of the nacnac ligands the residues at the bulky imine moieties are twisted nearly perpendicularly with respect to the chelating C3N2M-plane, which causes the metal atoms to be most protected. In the case of the complexes derived from 1 the methyl groups shield as well but due to less steric congestion the metal centres are slightly more accessible for allowing some specific substitution reactions. The new metal complexes containing a central six-membered metalla heterocycle are reminiscent of the analogous metal complexes with the omnipresent nacnac ligand. The following similarities between the herein discussed bisheterocyclo methanides and the nacnac ligands can be stated due to the structural comparison of the corresponding metal complexes: Firstly the deprotonation of the bridging moiety leads to the formation of a negative charge in the ligand's backbone, which is completely delocalised to give 6π electrons containing the metalla heterocycle (see Table 1). Secondly, the chelating coordination of the metal cation is achieved exclusively by the two nitrogen donor atoms in the ligand periphery.
| Cipso–C1′ | Cipso–N | C1–C1′–C8 | |
|---|---|---|---|
| (4-MeNCOC6H4)2CH2 (1) | 1.487(2) | 1.291(2) | 110.79(12) |
| [Me2Al{(4-MeNCOC6H4)2CH}] (2) | 1.384(2) | 1.351(2) | 121.13(14) |
| [Me2Ga{(4-MeNCOC6H4)2CH}] (3) | 1.388(3) | 1.341(2) | 121.62(18) |
| [Me2In{(4-MeNCOC6H4)2CH}] (4) | 1.390(13) | 1.337(12) | 123.6(6) |
| [Me2Al{(NCOC6H4)2CH}]18 | 1.380(3) | 1.346(3) | 119.5(2) |
| [Me2Al{(NCSC6H4)2CH}]18 | 1.390(2) | 1.352(2) | 123.54(14) |
| [Me2Al{(NCSC6H4)2N}]19 | — | 1.331(3) | — |
| [Me2Al{(4-MeNCSC6H4)2N}]19 | — | 1.343(2) | — |
Results and discussion
Ligand synthesis
The synthesis of the parent uncharged ligand system is achieved as reported earlier for the corresponding unsubstituted bis-(benzoxazol-2-yl)- and bis-(benzothiazol-2-yl)-methanes.18,22 A cyclocondensation reaction of two equivalents of 2-amino-3-methylphenol and one equivalent of a C3-linker unit derived from malonic acid yields the ligand. Two different synthesis routes were performed and optimised to give a moderate overall yield of 56% (see Scheme 1).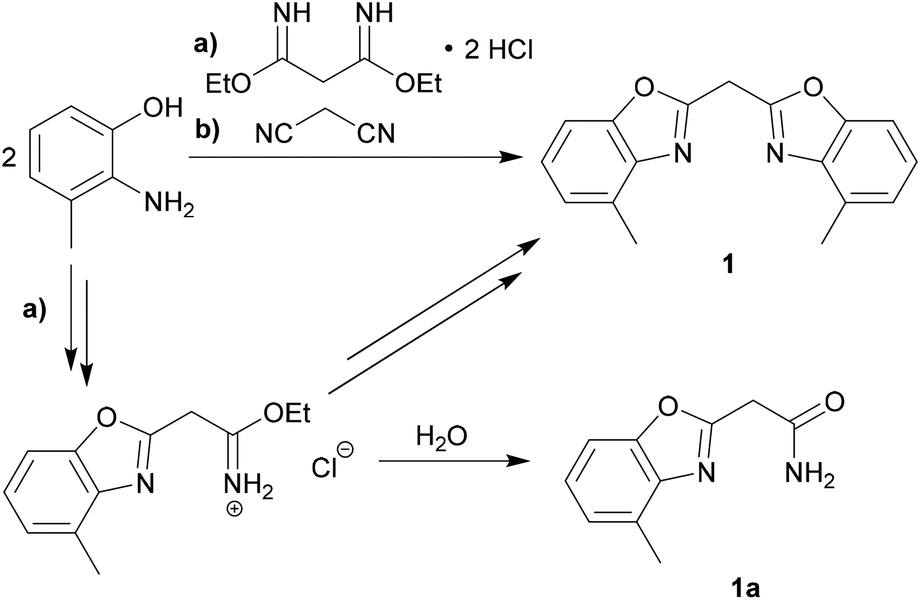 | ||
| Scheme 1 Synthetic routes to the parent ligand system 1 including the side product 1a. | ||
The first route is through the synthesis of the unsubstituted bis-(benzoxazol-2-yl)-methane, in which a bisimidate linker was used for the coupling of the two phenol derivatives. This pathway, however, leads to a smaller yield of the desired product 1 and also promotes the formation of a specific side product 1a. The side product could be identified as 4-methylbenzoxazol-2-yl-carboxamide, which occurs, if just one equivalent of the starting material has cyclised and the remaining imidate unit has been hydrolysed upon further purification. On the second improved route malonic dinitrile was added to the phenolic starting material. In that case polyphosphoric acid (ppa) was added as a solvent and catalytically facilitates the cyclisation reaction to give the parent ligand system 1.
Compound 1 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the asymmetric unit contains one molecule (Fig. 1). Like the other symmetrically substituted bisheterocyclo methanes18 the central carbon atom C1′ is also coordinated in a distorted tetrahedral fashion and the benzoxazole moieties are twisted nearly perpendicularly (81.53(4)°) relative to each other. Due to the higher steric demand of the heterocycles the C1–C1′–C8 angle is widened to 110.8(1)°, whereas all other angles around C1′ are slightly compressed in comparison with an ideal tetrahedral angle. Interestingly in the solid state 1 forms a 3D network of quite remarkable C–H⋯N hydrogen bonds, in which the two acidic methylene hydrogen atoms of the bridge are coordinated by imine nitrogen atoms of two adjacent molecules (see Fig. 2). The experimentally determined values show two hydrogen bonds each, which are energetically more favourable (H1′B⋯N2: 2.39 Å; C1′–H1′B⋯N2: 171.1°), and two which are less pronounced (H1′A⋯N1: 2.62 Å; C1′–H1′A⋯N1: 150.9°).
and the asymmetric unit contains one molecule (Fig. 1). Like the other symmetrically substituted bisheterocyclo methanes18 the central carbon atom C1′ is also coordinated in a distorted tetrahedral fashion and the benzoxazole moieties are twisted nearly perpendicularly (81.53(4)°) relative to each other. Due to the higher steric demand of the heterocycles the C1–C1′–C8 angle is widened to 110.8(1)°, whereas all other angles around C1′ are slightly compressed in comparison with an ideal tetrahedral angle. Interestingly in the solid state 1 forms a 3D network of quite remarkable C–H⋯N hydrogen bonds, in which the two acidic methylene hydrogen atoms of the bridge are coordinated by imine nitrogen atoms of two adjacent molecules (see Fig. 2). The experimentally determined values show two hydrogen bonds each, which are energetically more favourable (H1′B⋯N2: 2.39 Å; C1′–H1′B⋯N2: 171.1°), and two which are less pronounced (H1′A⋯N1: 2.62 Å; C1′–H1′A⋯N1: 150.9°).
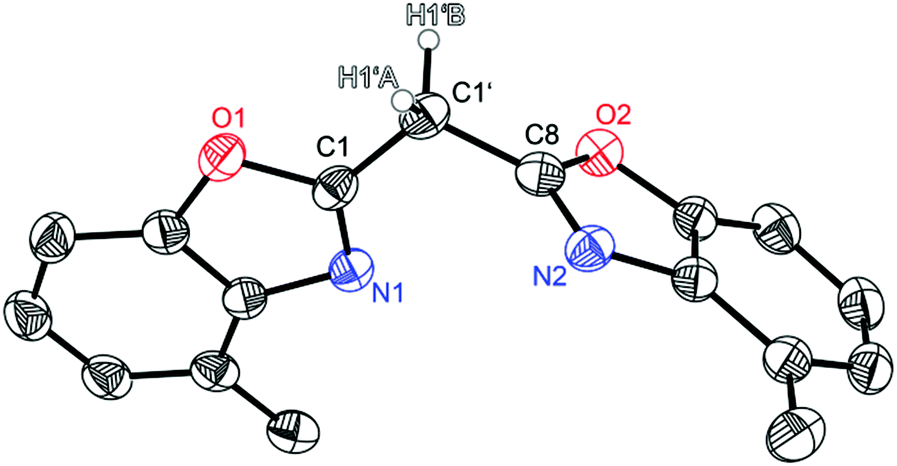 | ||
| Fig. 1 Molecular structure of (4-MeNCOC6H3)2CH2 (1). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the bridging ones. Structural data are given in Tables 1–3. | ||
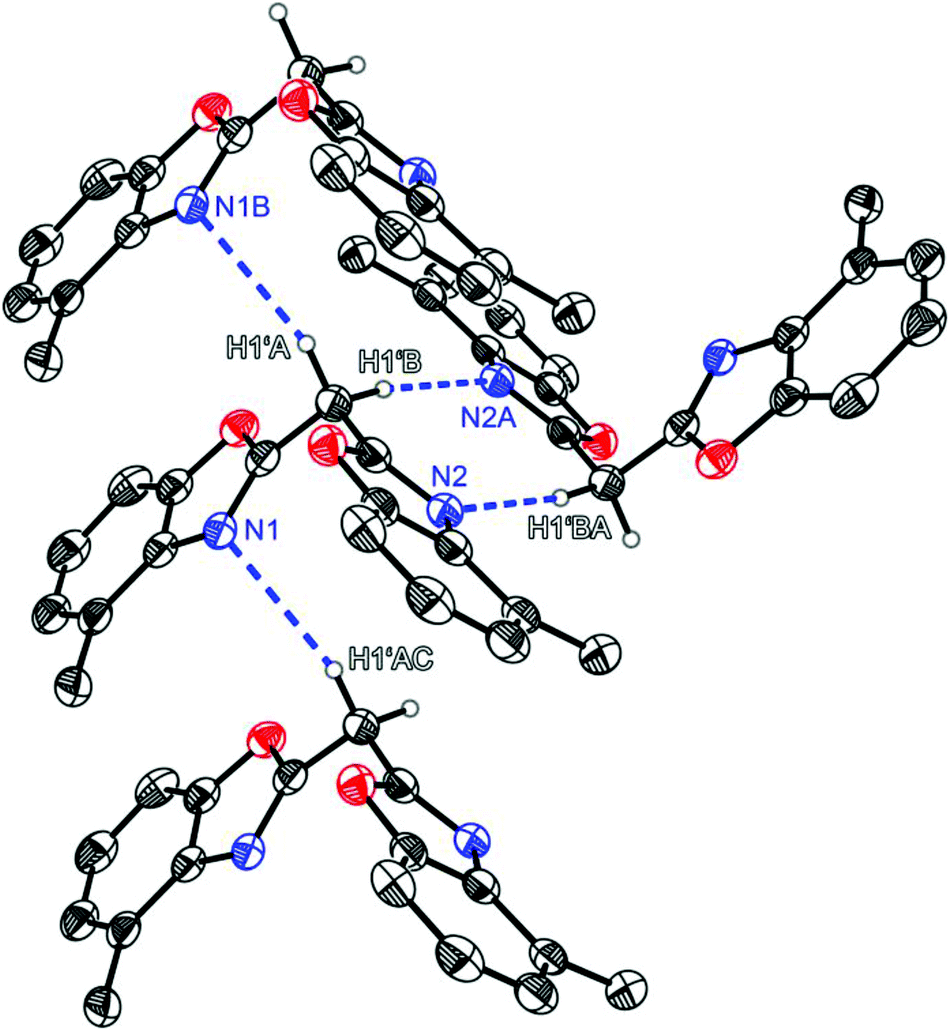 | ||
| Fig. 2 Hydrogen bonding properties of the parent ligand system (4-MeNCOC6H3)2CH2 (1). | ||
Syntheses of the group 13 metal complexes
The reaction of 1 in toluene with pure trimethyl aluminium, gallium or indium leads to the formation of the monoanionic methanides containing the corresponding MMe2 fragment (see Scheme 2). By the addition of these organometallic reagents the acidic methylene bridge gets deprotonated with the evolution of gaseous methane and in a concerted manner the remaining dimethyl group 13 metal cation gets chelated by the two ring nitrogen donor atoms.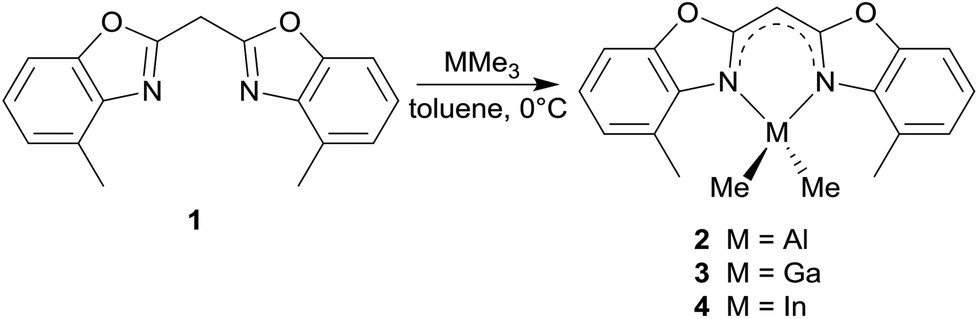 | ||
| Scheme 2 Metallation reactions of 1. | ||
Upon deprotonation the concerned Cipso–Cbridge distances are shortened in comparison with 1 and the Cipso–N distances are slightly elongated (Table 1). This is caused by two facts, which are important for structural features. On the one hand the deprotonation of the central methylene moiety generates a free electron pair. Because of the adjacent conjugated π-systems of the two benzoxazole units this free electron pair tends to be delocalised over the whole ligand framework resulting in different feasible resonance structures: carbanionic, amidic or completely delocalised. On the other hand the hybridisation of the central carbon atom changes from sp3 in the starting material 1 to sp2 in the metallated species 2–4 based on the trigonal planar coordination geometry of C1′.
Structural comparison of 2–4
A closer look at the experimentally determined geometry of 1 shows that the Cipso–Cbridge distance is slightly shorter than what is to be expected for a typical C(sp2)–C(sp3) single bond (1.51 Å). This deviation can be explained by the presence of the four electronegative adjacent heteroatoms, which cause the Cipso–Cbridge bonds to shrink due to their electron withdrawing ability.In comparison with compounds 2–4 the observed Cipso–Cbridge distances (1.384 Å to 1.390 Å) are half way between a typical C(sp2)–C(sp2) single bond (1.47 Å) and a C(sp2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(sp2) double bond (1.34 Å), which is a result of the efficient delocalisation of the double bonds. The same explanation is valid for the Cipso–N bond lengths, which are in a narrow range from 1.337 Å to 1.351 Å. These values for the Cipso–N bond lengths can also be seen as the average of a typical C(sp2)–N(sp2) single bond (1.40 Å) and a C(sp2)N(sp2) double bond (1.29 Å).23 In light of the previous results of the methanides [Me2Al{(NCOC6H4)2CH}] and [Me2Al{(NCSC6H4)2CH}]18 and the analogous amide bridged species [Me2Al{(NCSC6H4)2N}] and [Me2Al{(4-MeNCSC6H4)2N}]19 the discussed structural values match well (Table 1).
C(sp2) double bond (1.34 Å), which is a result of the efficient delocalisation of the double bonds. The same explanation is valid for the Cipso–N bond lengths, which are in a narrow range from 1.337 Å to 1.351 Å. These values for the Cipso–N bond lengths can also be seen as the average of a typical C(sp2)–N(sp2) single bond (1.40 Å) and a C(sp2)N(sp2) double bond (1.29 Å).23 In light of the previous results of the methanides [Me2Al{(NCOC6H4)2CH}] and [Me2Al{(NCSC6H4)2CH}]18 and the analogous amide bridged species [Me2Al{(NCSC6H4)2N}] and [Me2Al{(4-MeNCSC6H4)2N}]19 the discussed structural values match well (Table 1).
Compounds 2 (Fig. 3) and 3 (Fig. 4) are equi-structural and crystallise each in the monoclinic space group P21/c and the asymmetric unit contains one molecule. 4 (Fig. 5) crystallises in the monoclinic space group Pn and contains two metallated molecules in the asymmetric unit (Table 4).
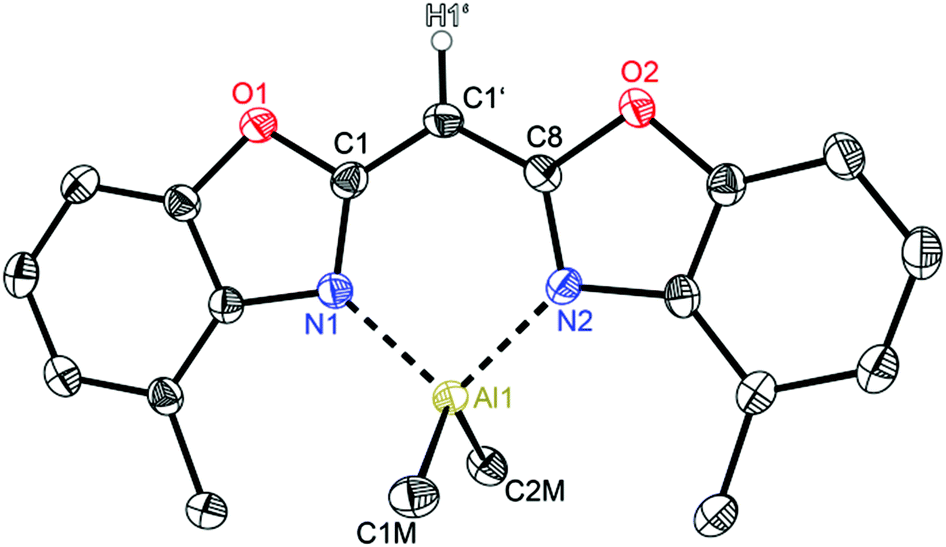 | ||
| Fig. 3 Molecular structure of [Me2Al{(4-MeNCOC6H3)2CH}] (2). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
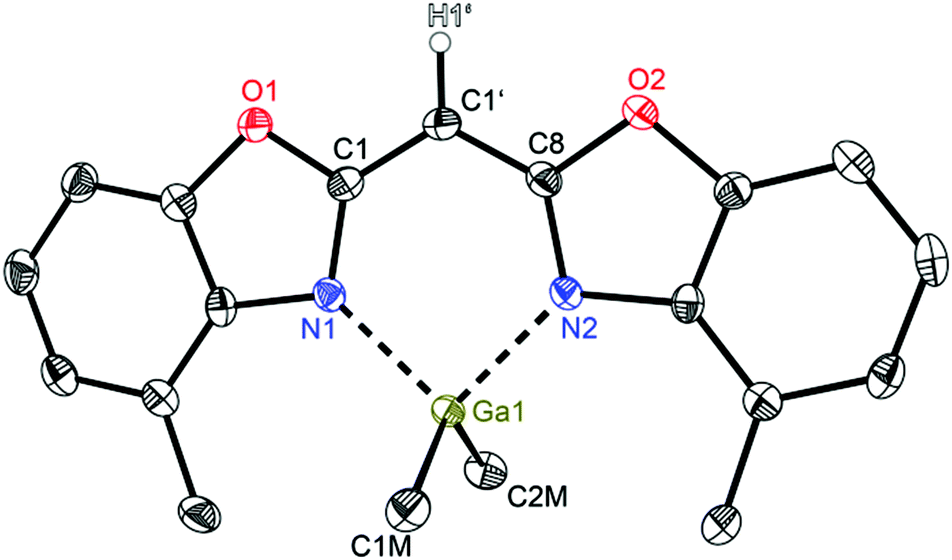 | ||
| Fig. 4 Molecular structure of [Me2Ga{(4-MeNCO C6H3)2CH}] (3). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
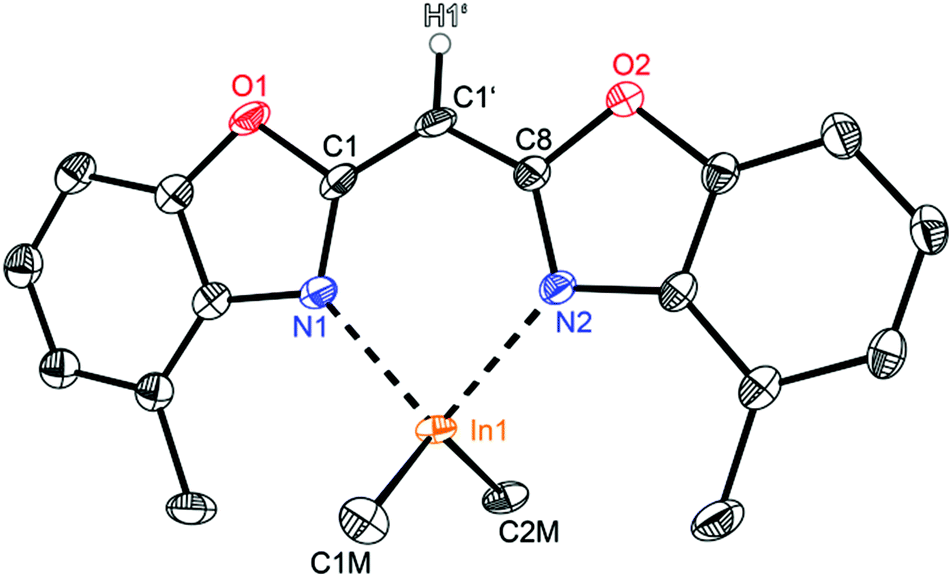 | ||
| Fig. 5 Molecular structure of one molecule of [Me2In{(4-MeNCOC6H3)2CH}] (4). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
The structural comparison of the MMe2 derivatives shows that in each case the metal coordination again is accomplished by the two nitrogen atoms of the benzoxazole moieties and the oxygen atoms are pointing away from the metal (Fig. 3–5). In all three cases this leads to a distorted tetrahedral coordination geometry at the particular metal ion. As expected the ligand framework in 2–4 exhibits nearly no folding of the heterocyclic residues. Additionally the metal ion is located in the C3N2 plane of the central six-membered metalla heterocycle without any significant deviation (see Table 2). Only this arrangement facilitates total conjugation of the whole anionic ligand. Due to the fourfold coordination of the metal ion the methyl residues at that metal are aligned perpendicularly with respect to the N1–M–N2 plane. This V-shaped arrangement of the MMe2 fragments allows the organometallic fragment to slot in between the methyl groups of the ligand. The four methyl groups frame the group 13 metal to stay in plane (see Fig. 6).
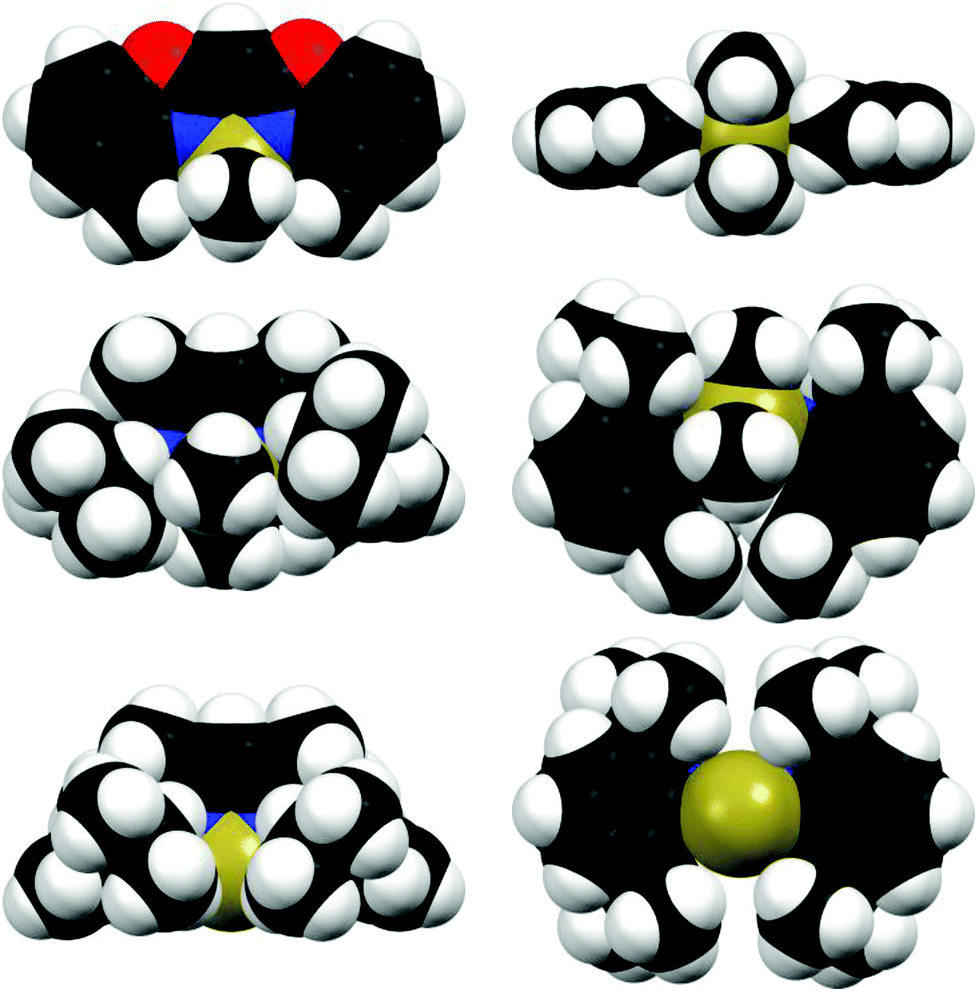 | ||
| Fig. 6 Space filling models from different perspectives (left column: front view, right column: bottom view) of the compared aluminium complexes 2 (top), [Me2Al{Dipp2nacnac}]13i (A) (middle) and [Al{Dipp2nacnac}]14e (B) (bottom). | ||
| Folding angle [°] | M-plane distance [Å] | |
|---|---|---|
| a Two values are given due to the discrepancy of the two molecules within the unit cell. | ||
| 1 | — | — |
| 2 | 3.721(20) | 0.0054(22) |
| 3 | 3.803(20) | 0.0068(23) |
| 4 | 4.925(122) | 0.0572(287) |
| 6.225(263) | 0.1545(269) | |
| 5 | 3.394(20) | 0.0090(19) |
| 6 | 3.042(16) | 0.0226(37) |
| 7 | 4.425(71) | 0.0483(20) |
In the row of the investigated group 13 metal complexes 2–4 of the bis-(4-methylbenzoxazol-2-yl)-methanide ligand some clear trends from the obtained structural data could be deduced (see Table 3):
• The transannular N1⋯N2 distance, which is indicative of the size of the chelated metal atom, increases from 2 over 3 to 4 as expected, because the bigger the coordinated ion, the wider the chelating distance.
• Also the observed N–M distances increase according to the increasing ionic radii of the Al3+, Ga3+ or In3+ ion, respectively.
• As a further consequence of this bond elongation and due to the fact that the metal stays in the ligand plane the resulting N1–M–N2 bite angles decrease from 2 over 3 to 4.
| N1⋯N2 | N1/2–M | C15/16⋯M | N1–M–N2 | |
|---|---|---|---|---|
| 1 | 3.5448(18) | — | — | — |
| 2 | 2.8534(19) | 1.9400(14) | 3.4478(19) | 94.68(6) |
| 3 | 2.9010(23) | 2.0043(15) | 3.4474(22) | 92.73(6) |
| 4 | 3.0485(55) | 2.217(9) | 3.4805(167) | 86.89(14) |
| 5 | 2.8500(20) | 1.9097(14) | 3.4736(19) | 96.52(6) |
| 6 | 2.8775(37) | 1.909(2) | 3.5380(25) | 97.84(12) |
| 7 | 2.8753(21) | 1.9140(16) | 3.5341(22) | 97.38(7) |
| A | 2.870(4) | 1.929(2) | 3.789(3) | 96.18(9) |
| B | 2.764(3) | 1.958(2) | 3.606(3) | 89.86(8) |
In Fig. 6 compound 2 is exemplarily compared to the literature known nacnac derivatives [Me2Al{Dipp2nacnac}]13i (A) and [Al{Dipp2nacnac}]14e (B) containing also the Al(III)Me2 or Al(I) moiety, respectively. From the space filling model the steric demand of each ligand can be visualised and provides a fair estimate of the shielding abilities around the metal atoms.
Starting with the crystal structure of the Al(I) species B it is evident that the metal atom is located almost ideally within the chelating C3N2 plane (Fig. 6, bottom). The N,N′-coordinated metal atom fits well into the pocket made up of the four iPr groups of the Dipp substituents and the carbon atoms in the ortho-position of the associated phenyl rings. Furthermore, these phenyl rings are nearly perpendicularly aligned with respect to the C3N2 plane (89° and 91°). The ortho-carbon atoms provide the closest contact to the aluminium atom (averaged 3.606 Å) and presumably are equally important for the shielding of low-valent Al(I) species (listed in column C15/16⋯M in Table 3), because these distances are located between the nacnac species and the complexes derived from 1. The bottom view also shows that the aluminium atom is not exhaustively coordinated so that coordination of the Lewis acidic site is facilitated. The side view highlights the good shielding abilities of the iPr groups, preventing the molecule from aggregation.
Continuing with the dimethyl aluminium species A the crystal structure clearly deviates from the reduced species B. The central six-membered metalla heterocycle is less planar than in B, because the AlMe2 unit is not located in the chelating C3N2 plane. Furthermore, one of the two phenyl rings of the Dipp-substituents is considerably twisted away from orthogonality with respect to the C3N2 plane (112°). This deviation is caused by the additional methyl groups at the aluminium, which increases the steric demand compared to the naked metal in B. The formed pocket in B is not appropriate in size to accommodate the additional methyl groups. Instead of retaining the perpendicular alignment of the Dipp-substituents one of them is pushed away.
As mentioned earlier the methyl substituents of the chelating ligand in 2 adopt the shielding role of the Dipp residues of the comparable nacnac complexes. These efficient shielding abilities are visualised in the space filling model in the bottom view of 2 in Fig. 6: by chelation of the two nitrogen donor atoms of the ligand the AlMe2 fragment is aligned in the C3N2 plane without any significant deviation. This planarity is also supported by the ligand's methyl groups, which fit nearly perfectly into the pocket made up of the V-shaped coordinated AlMe2 cation. So in comparison with A and B the kinetic shielding considered from the bottom view is even more pronounced. As opposed to this the side view of 2 shows that the planar arrangement of the complex leads to less steric congestion from this point of view. The shielding abilities in 2 are not that encompassing as in the case of A and B, so that beneficial, specific substitution reactions at the metal centre still can take place. This advantageous property is exploited by the synthesis of 5 described in the following paragraph.
Structural comparison of 5–7
In addition to the dimethyl substituted group 13 metal complexes 2–4 three Al(III) derivatives, in which one of the methyl groups is replaced by a halide, were successfully synthesised as well. This substitution is necessary to improve the reactivity of those species compared to the relatively high stability of the dimethyl aluminium complexes. These reaction products should facilitate access to Al(I) or Al(II) species upon reductive dehalogenation reactions.The synthesis of compounds 5 and 7 parallels the procedures of the abovementioned dimethyl derivatives. To a solution of the parent ligand 1 in toluene the pure organometallic reagent AlMe2Cl or AlEt2I, respectively, was added in a slight excess at 0 °C (Scheme 3, pathway a). Differently 6 was prepared starting from the metallated species 2 by reaction with an excess of trimethyl silyl iodide to prompt a methyl iodide exchange at the metal atom (Scheme 3, pathway b).
 | ||
| Scheme 3 Synthesis route for the mono halide substituted aluminium complexes 5–7. | ||
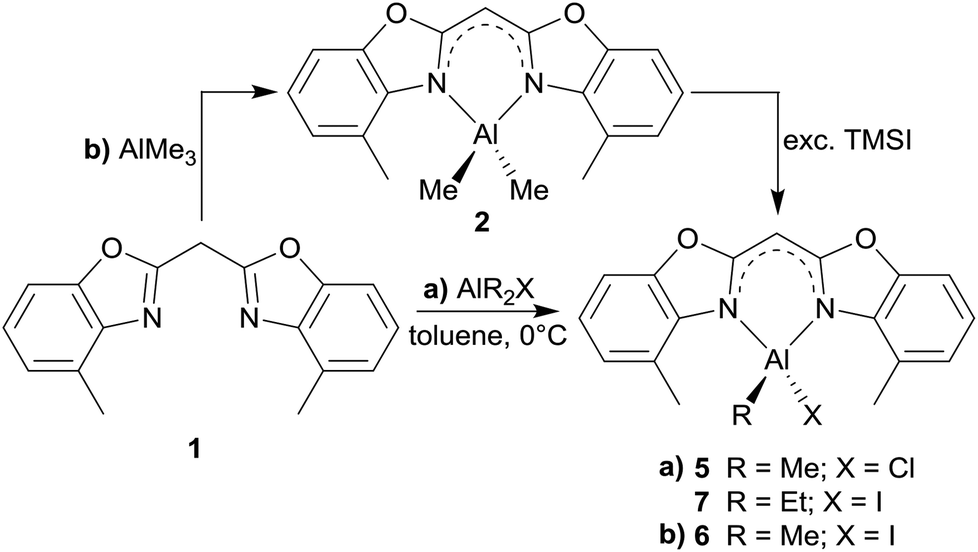
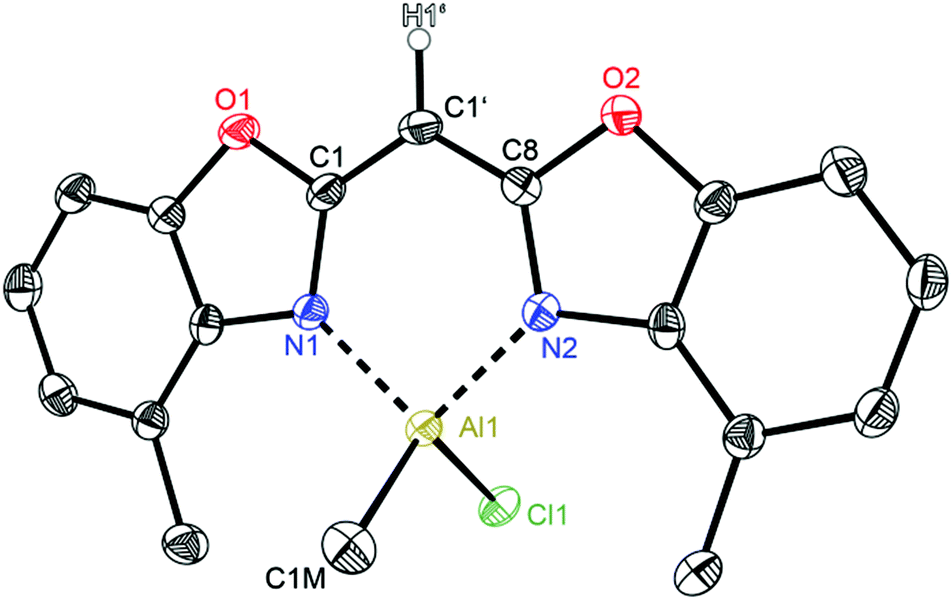
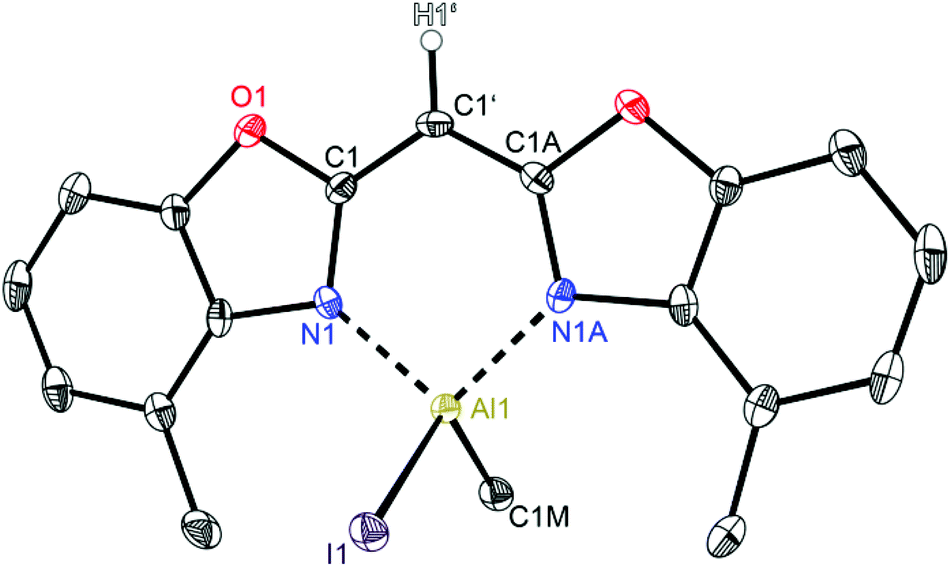
Having a closer look at the geometrical data of those halide substituted species 5–7 the following conclusions can be made: the derivatives 5 (Fig. 7) and 7 (Fig. 9) crystallise in the monoclinic space group P21/c each and the asymmetric unit consists in both cases of one target molecule. Compound 6 (Fig. 8) crystallises in the orthorhombic space group Pbcm and the asymmetric unit contains half a molecule (Table 4).
 | ||
| Fig. 7 Molecular structure of [ClMeAl{(4-MeNCOC6H3)2 CH}] (5). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
 | ||
| Fig. 8 Molecular structure of [IMeAl{(4-MeNCOC6H3)2 CH}] (6). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
 | ||
| Fig. 9 Molecular structure of [IEtAl{(4-MeNCOC6H3)2 CH}] (7). Anisotropic displacement parameters are depicted at the 50% probability level. C–H hydrogen atoms are omitted for clarity except for the one at the bridging CH group. Structural data are given in Tables 1–3. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
a
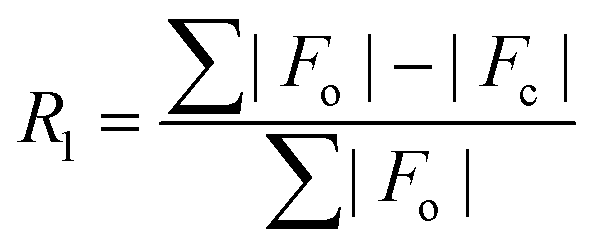 .
b .
b
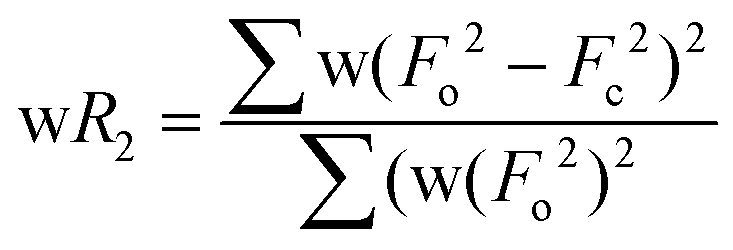 ; ;  ; ;  . .
|
|||||||
| Formula | C17H14N2O2 | C19H19AlN2O2 | C19H19GaN2O2 | C19H19InN2O2 | C18.1H16.3AlCl0.9N2O2 | C18H16AlIN2O2 | C19H18AlIN2O2 |
| Mol. w., g mol−1 | 278.31 | 334.35 | 377.10 | 422.18 | 352.59 | 446.21 | 460.23 |
| CCDC no. | 1429338 | 1429357 | 1429403 | 1429339 | 1429341 | 1429342 | 1429343 |
| Wavelength, Å | 0.71073 | 0.71073 | 0.56086 | 0.56086 | 0.56086 | 0.56086 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group |
P |
P21/c | P21/c | Pn | P21/c | Pbcm | P21/c |
| a, Å | 4.741(2) | 8.029(2) | 7.986(2) | 15.629(3) | 7.982(2) | 8.015(2) | 9.102(3) |
| b, Å | 11.471(3) | 14.773(3) | 14.852(3) | 7.894(2) | 14.727(3) | 13.868(3) | 7.647(2) |
| c, Å | 13.084(3) | 14.072(3) | 14.087(3) | 15.631(3) | 13.890(2) | 15.566(3) | 26.711(4) |
| α, ° | 79.15(2) | 90 | 90 | 90 | 90 | 90 | 90 |
| β, ° | 85.84(2) | 90.03(2) | 90.12(2) | 117.04(2) | 91.73(2) | 90 | 96.59(2) |
| γ, ° | 78.19(2) | 90 | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 683.6(4) | 1669.1(6) | 1670.81(9) | 1717.7(7) | 1632.0(6) | 1730.2(6) | 1846.9(8) |
| Z | 2 | 4 | 4 | 4 | 4 | 4 | 4 |
| Refl. measured | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 615 615 |
57930 |
45271 |
49033 |
20579 |
25400 |
38660 |
| Refl. unique | 2495 | 3060 | 3069 | 8534 | 2904 | 1915 | 4398 |
| R 1 [I > 2σ(I)]a | 0.0364 | 0.0306 | 0.0248 | 0.0248 | 0.0322 | 0.0222 | 0.0210 |
| wR2 (all refl.)b | 0.0938 | 0.0868 | 0.0672 | 0.0546 | 0.0885 | 0.0542 | 0.0534 |
| R int | 0.0369 | 0.0384 | 0.0445 | 0.0481 | 0.0370 | 0.0431 | 0.0231 |
| Abs. stru. para.26 | — | — | — | 0.11(4) | — | — | — |
| Δρfin, e Å−3 | 0.170/−0.162 | 0.222/−0.323 | 0.386/−0.372 | 0.334/−0.463 | 0.206/−0.314 | 0.502/−0.894 | 0.730/−0.406 |
In the halogenated complexes the aluminium atom adopts also the distorted tetrahedral coordination by means of the two nitrogen donors and the remaining substituents at the metal atom. In direct comparison with the AlMe2 derivative 2 the N–Al distances in 5–7 are increased and consequentially the corresponding N–Al–N bite angle is widened (see Table 3). These observed changes are caused by the electron withdrawing halide ligands Cl or I. These substituents increase the partial positive charge at the metal atom due to the negative inductive effect and their reduced electronegativity compared to carbon. The substituted aluminium atom needs less electron density from the donating nitrogen ring atoms and the nitrogen Lewis donors are less attractive.
The influence of the present halide atom on specific binding properties is displayed in Table 3. 5 compared to 6 shows the bigger halide iodide to cause the transannular N1⋯N2 distance to increase by about 0.03 Å (2.8500 Å in 5vs. 2.8775 Å in 6). This widening of the coordination pocket is also displayed in the bite angle, which also gets enlarged from 96.52° to 97.84°. Predominantly the higher steric demand of the iodide compared to the chloride accounts for the structural changes in the ligand. The observed differences among the two iodide derivatives 6 and 7 concerning those values are negligible. Within the triple estimated standard deviations the values for both species are the same, indicating that the slightly enhanced steric demand in 7 due to the ethyl group instead of a methyl group has no significant effect on those structural features.
Conclusions
In this work the successful syntheses and solid state structure determination of six metallated bis-(4-methylbenzoxazol-2-yl)-methanides 2–7 and the parent uncharged methane derivative 1 could be presented. As a consequence of tuning the previously reported bisheterocyclo methanes by introducing methyl groups to the benzannulated heterocycles (in this case benzoxazole) a promising ligand system for metal complexation could be obtained, which exhibits a nearly perfect planar coordination geometry with regard to the ligand side arms in all the discussed solid state structures. This fact is displayed by the small values in the folding angles between both heteroaromatic planes and the just slight deviations of the metal cations from the chelating C3N2 plane in each complex, which is outstanding in the row of the previously investigated methanide and amide complexes. The observed arrangement in the solid state can be explained by the steric strain induced by the additional methyl groups, which fit inside the properly shaped pocket made up the particular V-shaped MR2 fragments, so that a kind of intramolecular interlocking is responsible for the overall planar arrangement.Due to the relatively pronounced inertness of the dimethyl group 13 metal complexes the switch to the mono halide substituted compounds 5–7 offers access to low-valent group 13 complexes, which hopefully can be obtained by reductive salt elimination analogous to the synthesis route of [Al{Dipp2nacnac}] or [Ga{Dipp2nacnac}]. The steric protection around the metal centre resulting from the planar organic ligand is quite efficient for kinetic stabilisation, but in the case of 2 the reaction with an excess of trimethyl silyl iodide showed that specific substitution reactions can still take place, which is beneficial in contrast to the former nacnac derivatives. Nevertheless, it is worth mentioning, that even an excess of TMSI does not lead to a dual halide substituted derivative. Because of the beneficial planar arrangement of the ligand system and the metal cation within the precursors 2, and 5–7 the generation of new Al(I) species is feasible. The planar coordination geometry should enable optimal orbital overlapping between the endocyclic nitrogen donors and the chelated future low-valent metal atom to obtain a more efficient delocalisation. Further substitution and reduction attempts on aluminium containing complexes of this promising ligand system are under investigation.
Experimental section
General procedures
All manipulations were carried out under an atmosphere of dried and purified N2 or Ar by using Schlenk techniques.24 All solvents used within metallation reactions were distilled from Na or K prior to use. The starting materials were purchased commercially and used as received. 1H, 13C, 15N and 27Al NMR spectroscopic data were recorded on Bruker Avance 500 MHz, Bruker Avance 400 MHz and Bruker Avance 300 MHz spectrometers and referenced to the deuterated solvent (thf-d8).25 Elemental analyses (C, H, N and S) were carried out on a Vario EL3 at the Mikroanalytisches Labor, Institut für Anorganische Chemie, University of Göttingen. All EI-MS spectra (70 eV) were recorded on a Finnigan MAT 95.Method 2: A mixture of 2-amino-3-methylphenol (9.85 g, 80.0 mmol, 2.00 eq.) and malonic acid (4.16 g, 40.0 mmol, 1.00 eq.) was suspended in polyphosphoric acid (100 mL) with the use of a sealed precision glass (KPG) stirrer. Under vigorous stirring the reaction mixture was heated to 150 °C and the resulting dark blue viscous solution was kept under stirring at this elevated temperature for 5 h. Then the solution was allowed to cool to about 90 °C and poured over ice. The formed grey solid was filtered off, washed several times with saturated aqueous NaHCO3 solution (9 × 50 mL) and distilled water (10 × 50 mL) to pH neutrality. The remaining solvent was evaporated in vacuo to obtain 1 (6.26 g, 22.5 mmol, 56%) as a pale grey powder.
Anal. Calcd for C17H14N2O2 (278.31 g mol−1): C, 73.37; H, 5.07; N, 10.07. Found: C, 72.90; H, 5.14; N, 9.90; δ1H (300 MHz, thf-d8): 7.33 (d, 3JHH = 8.1 Hz, 2 H, H3), 7.20 (t, 3JHH = 7.8 Hz, 2 H, H4), 7.11 (d, 3JHH = 7.5 Hz, 2 H, H5), 4.68 (s, 2 H, H1′), 2.55 (s, 6 H, H15); δ13C{1H} (75 MHz, thf-d8): 160.62 (s, 2 C, C1), 152.05 (s, 2 C, C2), 141.74 (s, 2 C, C7), 131.32 (s, 2 C, C6), 125.62 (s, 2 C, C4), 125.58 (s, 2 C, C5), 108.58 (s, 2 C, C3), 29.69 (s, 1 C, C1′), 16.41 (s, 2 C, C15); δ15N{1H} (30 MHz, thf-d8): −135.9 (s); EI-MS, m/z (%): 278 (100) [M]+, 146 (10) [M − NCOC7H6]+.
Metallation reactions
To a solution of the corresponding ligand 1 (1.00 eq.) in toluene a slight excess of the pure organometallic reactant AlMe3, GaMe3, InMe3, AlMe2Cl or AlEt2I, respectively (1.10 eq.) was slowly added at 0 °C. The reaction mixture was stirred overnight and allowed to warm to rt. Afterwards the volume of the solution was reduced to a few mL and the resulting concentrated solution stored at −32 °C in a refrigerator. Crystals suitable for X-ray diffraction experiments could be obtained overnight. The crystals thus formed were filtered, washed twice with pre-cooled toluene or hexane (0 °C) and finally dried in vacuum. The given yields below are just based on the received crystals unless stated otherwise. No further improvement of the yields was attempted.X-ray crystallographic studies
Single crystals were selected from a Schlenk flask under an argon or a nitrogen atmosphere and covered with perfluorinated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow using an X-TEMP2 device.27 An appropriate crystal was selected using a polarised microscope, mounted on the tip of a MiTeGen©MicroMount or glass fibre, fixed to a goniometer head and shock cooled by a crystal cooling device. The data for 1–7 were collected from shock-cooled crystals at 100(2) K. The data of 1, 2 and 7 were collected on an Incoatec Mo Microsource28 and compounds 3–6 were collected on an Incoatec Ag Microsource,29 each equipped with mirror optics and an APEX II detector with a D8 goniometer. All diffractometers were equipped with a low-temperature device and used either MoKα radiation of λ = 0.71073 Å or AgKα radiation of λ = 0.56086 Å. The data were integrated with Saint30 and an semi-empirical absorption correction (Sadabs)29 was applied. The structures were solved by direct methods (Shelxt)31 and refined by full-matrix least-squares methods against F2 (Shelxl2014).32 All non-hydrogen-atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to equal 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond length restraints and isotropic displacement parameter restraints. The positions of the amine hydrogen atoms are taken from the difference Fourier map and refined freely.Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. The CCDC numbers, crystal data and experimental details for the X-ray measurements are listed in the ESI.‡
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Thanks to the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for partial support and the Land Niedersachsen for providing a fellowship in the GAUSS PhD program.Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- (a) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 RSC; (b) W. Tochtermann, Angew. Chem., 1966, 7, 355–375 ( Angew. Chem. Int. Ed. Engl. , 1966 , 5 , 351–371 ) CrossRef; (c) G. Wittig and E. Benz, Chem. Ber., 1959, 92, 1999–2013 CrossRef CAS.
- (a) D. W. Stephan and G. Erker, Angew. Chem., 2010, 122, 50–81 ( Angew. Chem. Int. Ed. , 2010 , 49 , 46–76 ) CrossRef; (b) P. Spies, G. Kehr, K. Bergander, B. Wibbeling, R. Fröhlich and G. Erker, Dalton Trans., 2009, 1534–1541 RSC; (c) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. C. Welch, J. D. Masuda and D. W. Stephan, Inorg. Chem., 2006, 45, 478–480 CrossRef CAS PubMed.
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., 2013, 52, 4256–4259 ( Angew. Chem., Int. Ed. , 2013 , 52 , 4256–4259 ) CrossRef CAS PubMed.
- Y. Zhang, G. M. Miyake and E. Y. X. Chen, Angew. Chem., 2010, 122, 10356–10360 ( Angew. Chem. Int. Ed. , 2010 , 49 , 10158–10162 ) CrossRef.
- (a) G. Ménard and D. W. Stephan, Angew. Chem., 2011, 123, 8546–8549 ( Angew. Chem. Int. Ed. , 2011 , 50 , 8396–8399 ) CrossRef; (b) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796–1797 CrossRef PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed.
- (a) J. Overgaard, C. Jones, A. Stasch and B. B. Iversen, J. Am. Chem. Soc., 2009, 131, 4208–4209 CrossRef CAS PubMed; (b) M. Westerhausen, Angew. Chem., 2008, 120, 2215–2217 ( Angew. Chem. Int. Ed. , 2008 , 47 , 2185–2187 ) CrossRef; (c) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- (a) C. Ruspic and S. Harder, Inorg. Chem., 2007, 46, 10426–10433 CrossRef CAS PubMed; (b) S. Harder, Organometallics, 2002, 21, 3782–3787 CrossRef CAS.
- D. F. J. Piesik, S. Range and S. Harder, Organometallics, 2008, 27, 6178–6187 CrossRef CAS.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., 2008, 120, 9576–9580 ( Angew. Chem. Int. Ed. , 2008 , 47 , 9434–9438 ) CrossRef.
- (a) Y. Cheng, D. J. Doyle, P. B. Hitchcock and M. F. Lappert, Dalton Trans., 2006, 4449–4460 RSC; (b) S. Singh, H.-J. Ahn, A. Stasch, V. Jancik, H. W. Roesky, A. Pal, M. Biadene, R. Herbst-Irmer, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 2006, 45, 1853–1860 CrossRef CAS PubMed; (c) Z. Yang, H. Zhu, X. Ma, J. Chai, H. W. Roesky, C. He, J. Magull, H.-G. Schmidt and M. Noltemeyer, Inorg. Chem., 2006, 45, 1823–1827 CrossRef CAS PubMed; (d) N. Kuhn, S. Fuchs and M. Steimann, Z. Anorg. Allg. Chem., 2002, 628, 458–462 CrossRef CAS; (e) N. Kuhn, S. Fuchs and M. Steimann, Eur. J. Inorg. Chem., 2001, 2001, 359–361 CrossRef; (f) M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794–2799 CrossRef CAS PubMed; (g) N. Kuhn, J. Fahl, S. Fuchs, M. Steimann, G. Henkel and A. H. Maulitz, Z. Anorg. Allg. Chem., 1999, 625, 2108–2114 CrossRef CAS; (h) C. E. Radzewich, I. A. Guzei and R. F. Jordan, J. Am. Chem. Soc., 1999, 121, 8673–8674 CrossRef CAS; (i) B. Qian, D. L. Ward and M. R. Smith, Organometallics, 1998, 17, 3070–3076 CrossRef CAS.
- (a) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS; (b) X. Li, X. Cheng, H. Song and C. Cui, Organometallics, 2007, 26, 1039–1043 CrossRef CAS; (c) H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 4027–4038 RSC; (d) M. N. Sudheendra Rao, H. W. Roesky and G. Anantharaman, J. Organomet. Chem., 2002, 646, 4–14 CrossRef CAS; (e) C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., 2000, 112, 4444–4446 ( Angew. Chem. Int. Ed. , 2000 , 39 , 4274–4276 ) CrossRef.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- (a) P. Hao, Z. Yang, W. Li, X. Ma, H. W. Roesky, Y. Yang and J. Li, Organometallics, 2015, 34, 105–108 CrossRef CAS; (b) B. Li, C. Zhang, Y. Yang, H. Zhu and H. W. Roesky, Inorg. Chem., 2015, 54, 6641–6646 CrossRef CAS PubMed; (c) Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., 2015, 127, 10363–10367 ( Angew. Chem. Int. Ed. , 2015 , 54 , 10225–10229 ) CrossRef; (d) Y. Yang, H. Li, C. Wang and H. W. Roesky, Inorg. Chem., 2012, 51, 2204–2211 CrossRef CAS PubMed.
- (a) Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., 2009, 121, 2065–2068 ( Angew. Chem. Int. Ed. , 2009 , 48 , 2031–2034 ) CrossRef; (b) R. J. Wright, M. Brynda and P. P. Power, Angew. Chem., 2006, 118, 6099–6102 ( Angew. Chem. Int. Ed. , 2006 , 45 , 5953–5956 ) CrossRef; (c) R. J. Wright, A. D. Phillips, S. Hino and P. P. Power, J. Am. Chem. Soc., 2005, 127, 4794–4799 CrossRef CAS PubMed; (d) N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, Angew. Chem., 2002, 114, 2966–2968 ( Angew. Chem. Int. Ed. , 2002 , 41 , 2842–2844 ) CrossRef; (e) J. Su, X.-W. Li, R. C. Crittendon and G. H. Robinson, J. Am. Chem. Soc., 1997, 119, 5471–5472 CrossRef CAS.
- D.-R. Dauer and D. Stalke, Dalton Trans., 2014, 43, 14432–14439 RSC.
- D.-R. Dauer, M. Flügge, R. Herbst-Irmer and D. Stalke, Dalton Trans., 2015 10.1039/C5DT03911H.
- (a) F. Baier, Z. Fei, H. Gornitzka, A. Murso, S. Neufeld, M. Pfeiffer, I. Rüdenauer, A. Steiner, T. Stey and D. Stalke, J. Organomet. Chem., 2002, 661, 111–127 CrossRef CAS; (b) H. Gornitzka and D. Stalke, Angew. Chem., 1994, 106, 695–698 ( Angew. Chem. Int. Ed. Engl. , 1994 , 33 , 693–695 ) CrossRef CAS; (c) H. Gornitzka and D. Stalke, Organometallics, 1994, 13, 4398–4405 CrossRef CAS.
- W. W. Schoeller, Inorg. Chem., 2011, 50, 2629–2633 CrossRef CAS PubMed.
- H. Ben Ammar, J. Le Nôtre, M. Salem, M. T. Kaddachi and P. H. Dixneuf, J. Organomet. Chem., 2002, 662, 63–69 CrossRef CAS.
- (a) P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement A Crystallographer's Guide to SHELXL, Oxford University Press, New York, 2006 Search PubMed; (b) P. Rademacher, Strukturen organischer Moleküle, VCH, Weinheim, 1987 Search PubMed.
- Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/advanced/13_de.html.
- Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/nmr/de.html.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., 2013, 69, 249–259 CAS.
- (a) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (d) Georg-August-Universität Göttingen, Virtuelles Labor, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/special/22_de.html.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- Bruker AXS Inc., in Bruker Apex CCD, SAINT v8.30C, ed. Bruker AXS Inst. Inc., WI, USA, Madison, 2013 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- (a) G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2014, 70, C1437 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Herbert W. Roesky on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Tables of data collection parameters, bond lengths and angles of compounds 1–7 and further information for side product 1a. CCDC 1429338, 1429339, 1429341–1429343, 1429356, 1429357, and 1429403. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03913d |
| This journal is © The Royal Society of Chemistry 2016 |