
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remote control over folding by light
Zhilin
Yu†
and
Stefan
Hecht
*
Department of Chemistry and IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: sh@chemie.hu-berlin.de; Fax: +49-30-2093-6940; Tel: +49-30-2093-7365
First published on 29th March 2016
Abstract
Integrating stimulus-responsive components into macromolecular architectures is a versatile strategy to create smart materials that can be controlled by external stimuli and even adapt to their environment. Helical foldamers, which are omnipresent in Nature and display well-defined yet dynamic structures, serve as an ideal platform to integrate photoswitches to modulate their conformations by light. This feature article summarizes the development of photoswitchable foldamers, focussing on various design approaches that incorporate the photoswitches either at the side chains, as tethered loops, or directly in the main chain. Based on the emerging insight into the folding–switching relationship more advanced molecular designs should enable the development of photoresponsive foldamers with high sensitivity to control and power functional macromolecular and supramolecular systems.
 Zhilin Yu | Zhilin Yu received his BS degree from Tianjin University in 2006. Then he worked with Prof. Yu Liu at Nankai University on the complex of functional cyclodextrins and carbon materials, and obtained his MS degree in 2009. Following that, he continued his graduate study under the supervision of Prof. Stefan Hecht at the Humboldt-Universität zu Berlin, where his research involved the investigation of the structure–feature relationship of photoswitchable foldamers composed of azobenzene units. He was awarded a PhD degree in 2013, and currently he is conducting his postdoctoral training with Prof. Samuel I. Stupp at Northwestern University. |
 Stefan Hecht | Stefan Hecht studied chemistry at Humboldt-Universität zu Berlin and obtained his PhD from the University of California at Berkeley in 2001, working under the guidance of Prof. Jean M. J. Fréchet. After establishing his own research group at Freie Universität Berlin and a subsequent position as a group leader at the Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr, in 2006 he returned to his alma mater as the Chair of Organic Chemistry and Functional Materials. His research interests range from synthetic macromolecular and supramolecular chemistry to surface science, with particular focus on utilizing photochromic molecules for remote controlling materials, devices, and processes. More information at: http://www.hechtlab.de. |
1. Introduction
Photoresponsive molecular systems, allowing for an external control of their structures and resulting properties by light, will be one of the key elements for creating “smart” materials with broad applications ranging from nanotechnology to pharmacology.1–3 Over the past two decades, photochromic moieties4–6 have been incorporated into numerous materials that serve as molecular machines,7–14 remote-controlled catalysts,15–18 memory and storage devices,4,19,20 and optomechanical actuators,21,22 among others.23–29 However, the creation of real “smart” materials from these photoswitches requires maximization of the apparent functional responses to light at the macroscopic level. For this purpose – and alternatively to improve the intrinsic performance of the individual photoinduced events30–32 – incorporation of photoswitches into supramolecular33 and macromolecular34 systems allows for the potential amplification of the light signal by exploiting cooperative mechanisms.35–38 Considering the latter, covalent approach, dynamic helical macromolecules offer the unique potential of cooperative folding39 and, inspired by the importance of helices in nature, the past decade has witnessed the development of a variety of photoresponsive helical structures,38,40–46 which can undergo a light-induced (cooperative) folding and/or unfolding transition (Fig. 1, left).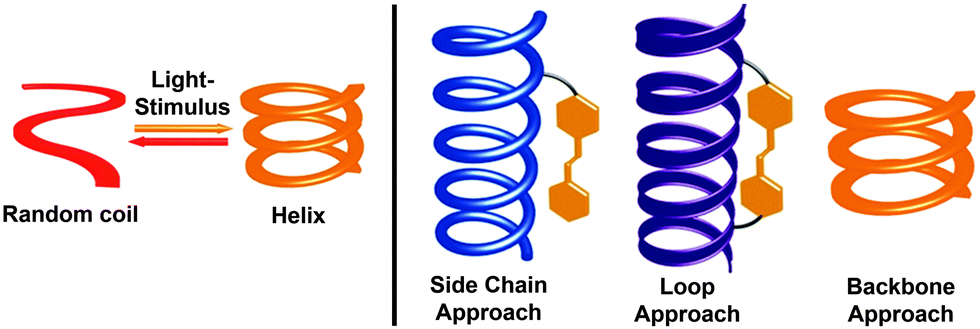 | ||
| Fig. 1 Schematic illustration of a foldamer's (cooperative) helix–coil transition triggered by light (left). Conceptually different approaches to incorporate photoswitches, illustrated here for the most commonly used azobenzene, into the helical structures as side chains, tethered loops, or directly into the backbone (right). | ||
In general, helical structures in foldamers are stabilized by a set of covalent local constraints and non-covalent intramolecular interactions.39,47–50 To successfully render foldamers photoswitchable one has to maximize the geometry change between the two different isomers of the photoswitch that should lead to selective stabilization or destabilization of the helical structure in one and/or the other switching form. Among the many established photoswitches,4–6 azobenzene offers the unique advantage that it features a remarkable change in molecular geometry (and dipole moment) based on the light-induced interconversion between its planar E-isomer and its non-planar Z-isomer.31 These dramatic structural changes occurring in the course of E–Z isomerization allow for facile variation of local constraints in the helical backbone but also influence π,π-stacking interactions and solvation, both of which contribute important enthalpic and entropic effects during helical folding. Alternatively, the light-induced conversion of charge-neutral spiropyrans to their zwitterionic merocyanine form51 has been used to locally enhance polarity and stabilize or destabilize helical structures.52 Thus far, three types of design approaches have been followed by integrating the photoswitches into the helical systems either (i) as side chains linked in a more or less flexible fashion to the main chain, or (ii) as tethered loops giving rise to constrained macrocyclic architectures, or (iii) directly into the helical backbone (Fig. 1, right).
In this feature article, we summarize the development of photoswitchable helical foldamers composed of various oligomeric/polymeric backbones and highlight their resulting light-induced (un)folding behavior in relation to the specific location of the photoswitches within the chain. Throughout we focus on the underlying design principle of the photoresponsive helices, following either side chain, loop, or backbone approaches, and the resulting relationship between local photoswitching and global foldamer conformation.
2. Side chain approach
In a significant body of work photoswitches have been attached to mostly polymeric foldamer backbones as their side chains.40,41,53,54 Therefore, the local geometrical changes induced upon photoswitching do not affect the backbone conformation directly but rather alter the non-covalent interactions between the side chains, thereby indirectly leading to an overall conformational change. This design offers the advantage that minimizes the steric constraint for switching imposed by the backbone, thereby resulting in potentially lower energy barriers and hence more efficient photoswitching. However, the secondary effects of photoswitching on modulating side chain interactions are somewhat harder to predict, rendering a straightforward design more challenging. Note that in particular when using this design approach one should carefully distinguish between photomodulation of folding, i.e. influencing the helix–coil equilibrium by light, and photomodulation of (excess) helicity, i.e. influencing twist sense bias during folding by light. Experimentally, these two scenarios are sometimes not trivial to distinguish in particular when relying entirely on circular dichroism (CD) spectroscopy.55Early work on photoresponsive polymers reported light-induced structural changes of polyelectrolytes caused by non-covalently associated photoswitches.56,57 In these cases, the isomerization events modulate the interactions between photoswitches and polymers and thereby change the ionic repulsion along the hydrophobic polymer backbones, resulting in their extension or shrinkage. This strategy did inspire the development of covalent approaches to attach azobenzene and spiropyran units in the side chains of polymers.58,59 Due to the absence of defined (helical) conformations, researchers initially focused on monitoring viscosity changes of polymer solutions upon exposure to UV-light. The first example able to directly record light-induced conformational transition of photoresponsive polymers by CD spectroscopy reported the attachment of varying amounts of azobenzene units to the carboxyl side chains of poly(L-glutamic acid)s (Fig. 2).60 In organic solvents, such as trifluoroethanol or trimethylphosphate, the modified polymeric backbones adopted an α-helical conformation as detected by CD spectroscopy.61 Exposure to 350 nm UV light led to E → Z photoisomerization of the azobenzenes yet the CD signal did not change indicating that the α-helix structure of the backbone was not disturbed. In contrast, when dissolving the polymer in water at pH = 7.6 assisted by the surfactant dodecylammonium chloride,62 the isomerization of azobenzene moieties gave rise to a reversible transition between random coil and α-helical conformations of the peptide backbone (Fig. 2). The observed solvent dependence of the photoswitchable folding behavior has been attributed to the change in the azobenzene's molecular dipole moment during photoisomerization, which is accompanied by a large change in the hydrophilicity of the side chain. Therefore, in the thermal resting state composed entirely of E-azobenzene side chains, their lipophilicity keeps the amphiphilic poly(L-glutamate) at the interface of the micelles where it adopts a random coil structure. Upon irradiation the metastable Z-isomer enriched peptide can adopt an α-helical conformation, which is able to expose the polar and hence more hydrophilic side chains to the aqueous environment dragging the peptide into the water phase. This behavior results in what we refer to as the photoinduced turn-on helix, i.e. light leads to helix formation while heat returns the system to a random coil conformation due to the thermal instability of the Z-isomer.
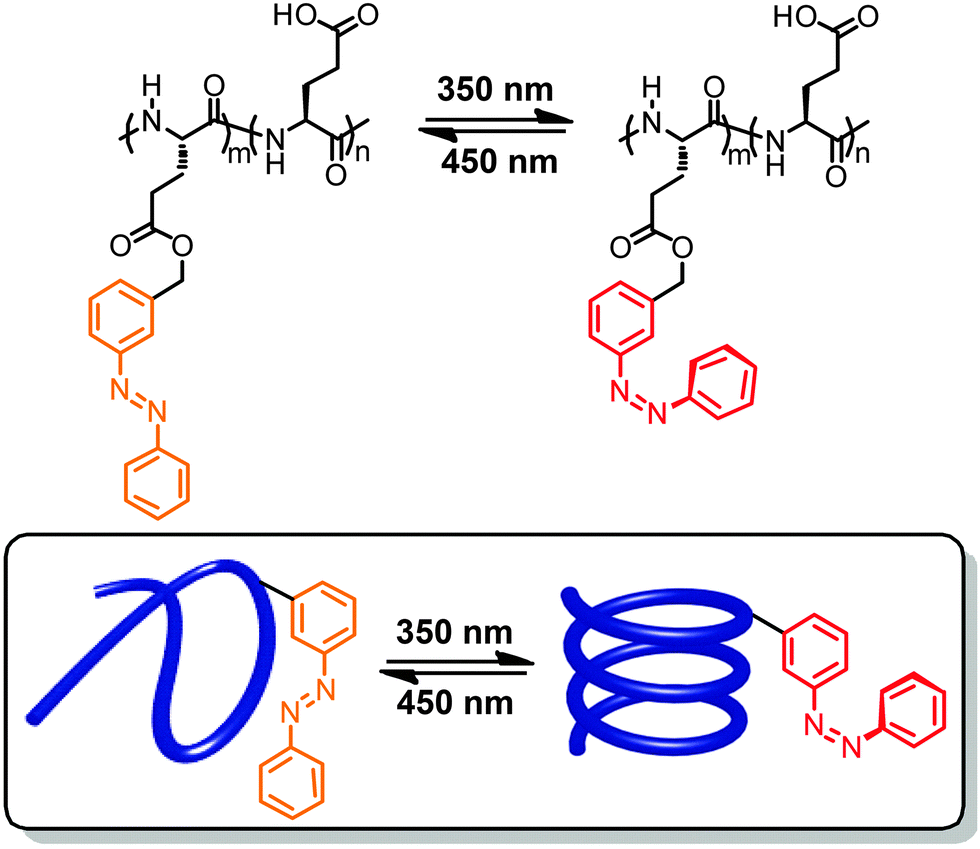 | ||
| Fig. 2 Poly(L-glutamic acid) containing azobenzene units in the side chains undergoes a photoswitchable coil–helix transition in surfactant–water mixtures leading to turn-on helices.62 | ||
At around the same time, Ueno and coworkers reported the modification of poly(benzyl L-aspartate) by varying amounts of phenylazo units that were attached to the benzyl side chains at either their meta- or para-positions, leading to different orientations of the resulting azobenzenes with regard to the peptide backbone.63–65 Interestingly, in this case and using specific organic solvent mixtures the authors were able to show by a thorough analysis of CD spectra that photoisomerization of the azobenzene side chains causes reversible inversion of the helix screw sense.66 When studying poly(benzyl L-aspartate) containing 49% meta-linked azobenzene side chains in a mixture of helix-stabilizing 1,2-dichloroethane and helix-denaturing trimethyl phosphate, 86% of the polymer chains adopt a left-handed helix while after irradiation with UV-light 100% of the polymer chains adopt a right-handed helix (Fig. 3). Even when reducing the azobenzene side chain content to 9.7% a modulation between 91% right-handed helix in the all-E-isomer and 74% in the Z-enriched sample could be observed. Furthermore, the fact that in the latter case only 50% E → Z photoconversion is necessary to cause the effect illustrates that overall only ca. 5% isomerization of the side chains is sufficient to cause a helix inversion of the majority of the sample.
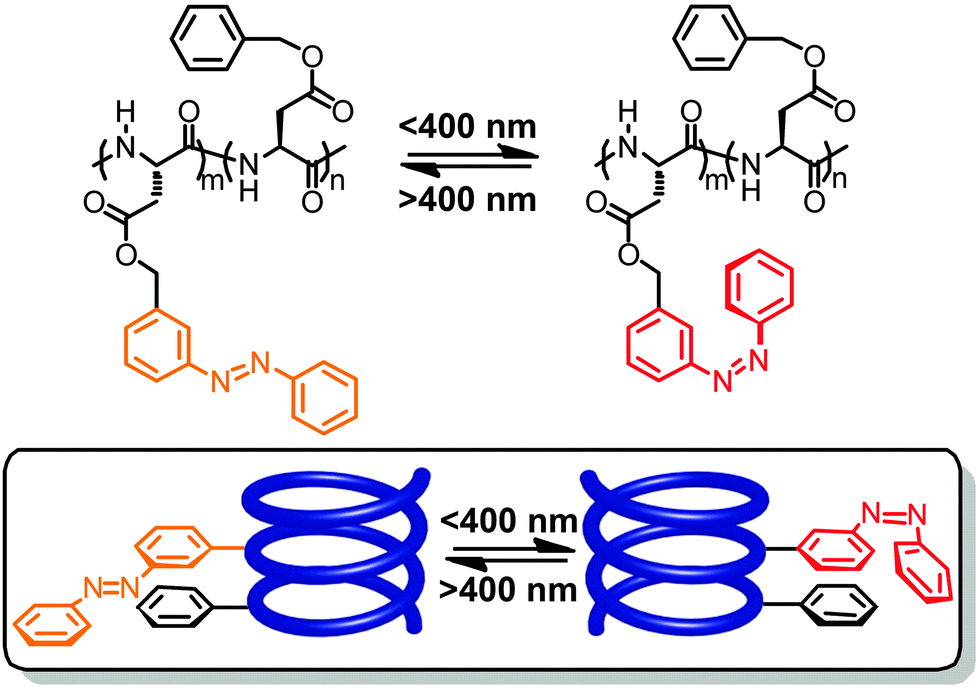 | ||
| Fig. 3 Poly(L-aspartate) containing meta-linked azobenzene units undergoes a photoswitchable helicity inversion (change of helix screw sense) in the mixture of 1,2-dichloroethane and trimethyl phosphate.64 | ||
In addition to azobenzene units, poly(L-glutamic acid) has also been functionalized by spiropyrans in the side chains.66–68 In hexafluoroisopropanol, the merocyanine form is thermodynamically more stable (so-called negative photochromism) and the polypeptide backbone carrying the merocyanine residues adopts a random coil conformation (Fig. 4).66 However, upon irradiation with visible light the colorless spiropyran is formed and the polypeptide backbone adopts a right-handed α-helical conformation. In this particular case and under the specific conditions, the zwitterionic merocyanine has a strong tendency to dimerize69 and therefore forces the polymer chain into a random coil structure. Visible light induces conversion to the spiropyran form and thereby weakens the side chain interactions such that the intrinsic helicogenicity of the backbone drives formation of the α-helix, giving rise to a turn-on helix once again.
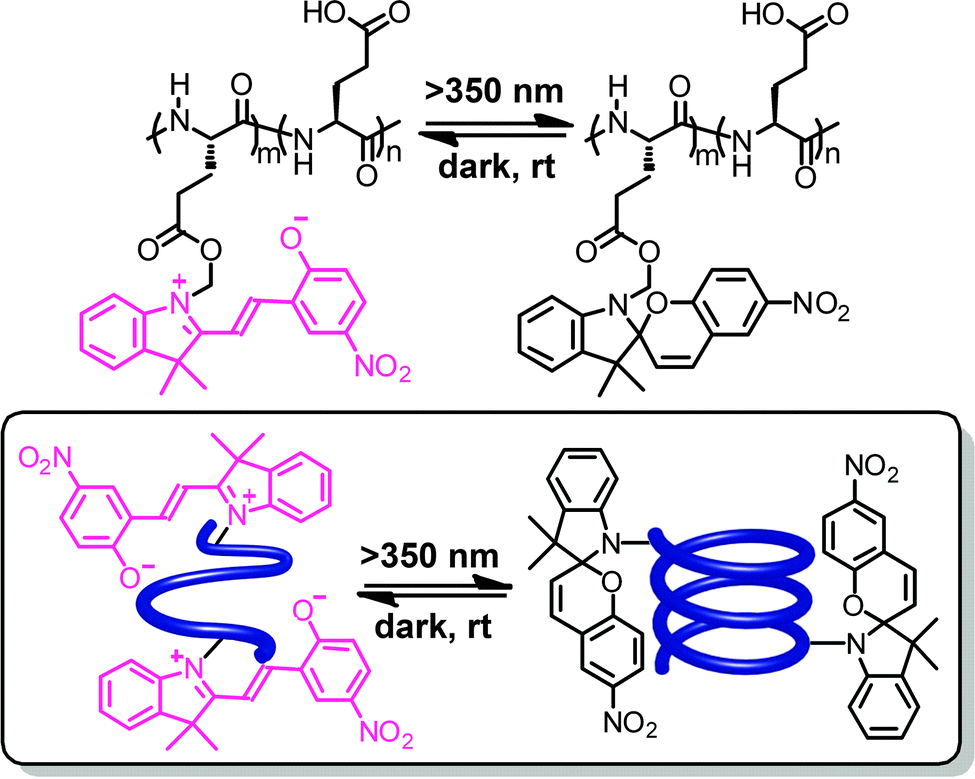 | ||
| Fig. 4 Poly(L-glutamic acid) containing spiropyran units in their side chains undergoes a photoswitchable coil–helix transition in hexafluoroisopropanol leading to turn-on helices.66 | ||
Zentel and coworkers incorporated photoswitches in the side chains of abiotic foldamer backbones, such as polyisocyanates, to photomodulate their conformational behavior (Fig. 5).40 Polyisocyanates composed of achiral monomers typically exist as a racemic mixture of right- and left-handed helices or isolated helical segments within the same polymer chain.70,71 The polyisocyanates decorated with chiral azobenzene side chains carrying one stereogenic center exhibited an intensity change in both ORD and CD spectra upon E → Z photoisomerization, indicating a change in the population of differently biased helical segments (center, Fig. 5).72 However, the E → Z photoisomerization of the azobenzene units with two chiral centers resulted in a more pronounced inversion of the Cotton effect (bottom, Fig. 5),73 suggesting a helicity inversion of the polymers. The chiral amplification of the polyisocyanates was investigated involving a number of copolymers of various chiral azobenzene-containing isocyanates and over the course of their studies the authors observed a fine balance of various effects, including the distance of the stereocenters to the polymeric backbone, the solvent, and the concentration of photoswitches.74–76 By relating the E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio to the molar elipticity in the CD spectra,74 it could be shown that the Z-isomer exhibits a stronger induction power and rules the helix diastereoselection, in accordance with the majority rule.71,77
:Z ratio to the molar elipticity in the CD spectra,74 it could be shown that the Z-isomer exhibits a stronger induction power and rules the helix diastereoselection, in accordance with the majority rule.71,77
 | ||
| Fig. 5 Polyisocyanates containing chiral azobenzene units (top) exhibiting either a photoswitchable shift in their equilibrium between P- and M-helical segments (center)72 or helix inversion (bottom)73 depending on the number of stereogenic centers in the azobenzene side chain. | ||
Complementary to these aliphatic backbones, azobenzene moieties have also been attached to the side chains of aromatic foldamers, in particular poly(meta-phenylene ethynylene)s, prepared via Sonogashira–Hagihara polycondensation of 3,5-bis(diethynyl)azobenzene and 1,3-diiodobenzene derived monomers (Fig. 6).78 Intramolecular H-bonds between the amide and carbamate units allow this backbone to adopt stable helical conformations in a wide range of organic solvents.79 Upon exposure to UV light, only subtle changes in the UV spectra were observed, indicating that just a small (7%) fraction of the azobenzene side chains underwent photoisomerization. This, presumably, is the reason why only limited degree of conformational response could be achieved as evident from the minimal changes in the corresponding CD spectra.
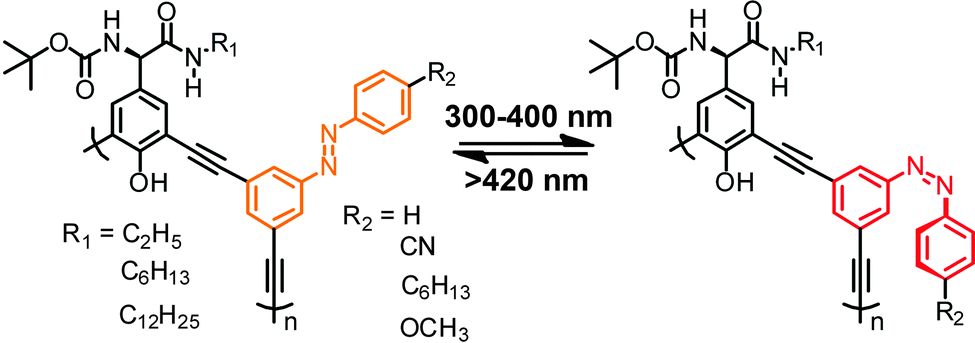 | ||
| Fig. 6 Poly(meta-phenylene ethynylene) with appended phenylazo side chains folds into a helical structure in a variety of organic solvents due to H-bonding yet undergoes only little photoisomerization of the azobenzene moieties.78 | ||
3. Loop approach
Rather than appending the photochromic units to the foldamer main chain on only one of their termini, tethering them at both of their ends to the backbone provides a macrocyclic loop architecture as a structural alternative to create photoswitchable foldamers.46 Thus far peptides have been employed as backbones. Their versatile solid-phase synthesis allows for the positioning of amino acids carrying appropriate reactive groups at specific locations in the backbone, at which photoswitches with suitable geometry and size are subsequently tethered in a loop fashion. In principle, the geometry of one of the two isomers of the incorporated photoswitch – but not the other – is chosen to match the distance between the two reactive side chains in the targeted conformation of the peptide backbone. The photoinduced interconversion between these two isomers, associated with a significant local geometry change as in the case of the most commonly used azobenzenes, therefore reversibly makes and breaks the covalent local constraints favoring the specific backbone conformation, typically a peptide α-helix.This loop approach has been pioneered by Woolley and coworkers, who tethered a variety of symmetrical azobenzene photoswitches via their para-positions to two cysteine residues within a peptide sequence (Fig. 7).80 Molecular modelling suggested that upon attaching the azobenzene to cysteine residues in the i and i + 7 positions the peptide α-helix is only commensurate with the kinked Z-isomer, while the E-isomer should lead to unfolding and population of the random coil structure.81 Consequently, UV-induced E → Z isomerization promoted the formation of the α-helical conformation, while thermal or vis-light induced Z → E isomerization collapsed the helical conformation. Upon comparing the calculated distances between the two cysteine residues in various conformations, the authors could show that the α-helix content was improved from 2% in the E-isomer to 48% in the Z-isomer.82 On the other hand, tethering the peptide using cysteine residues in the more distant i and i + 11 positions renders the α-helix compatible with the extended E-azobenzene, which upon photoconversion to the shorter Z-azobenzene should consequently break the helical conformation. This nicely illustrates how either the E-isomer or the Z-isomer of the azobenzene tether can be utilized to specifically stabilize the peptide's α-helix, resulting in a turn-off and turn-on foldamer, respectively. These two complementary approaches display some additional differences associated with the specific photoswitching behavior of azobenzene. In the turn-off design, the remaining E-isomer due to incomplete photoconversion leads to residual population of the α-helical conformation. In contrast, in the turn-on design helix formation can facilitate E → Z photoisomerization and thermally stabilize the Z-isomer while quantitative (photo)conversion of the Z-isomer back to the E-isomer leads to complete reversal of the process.
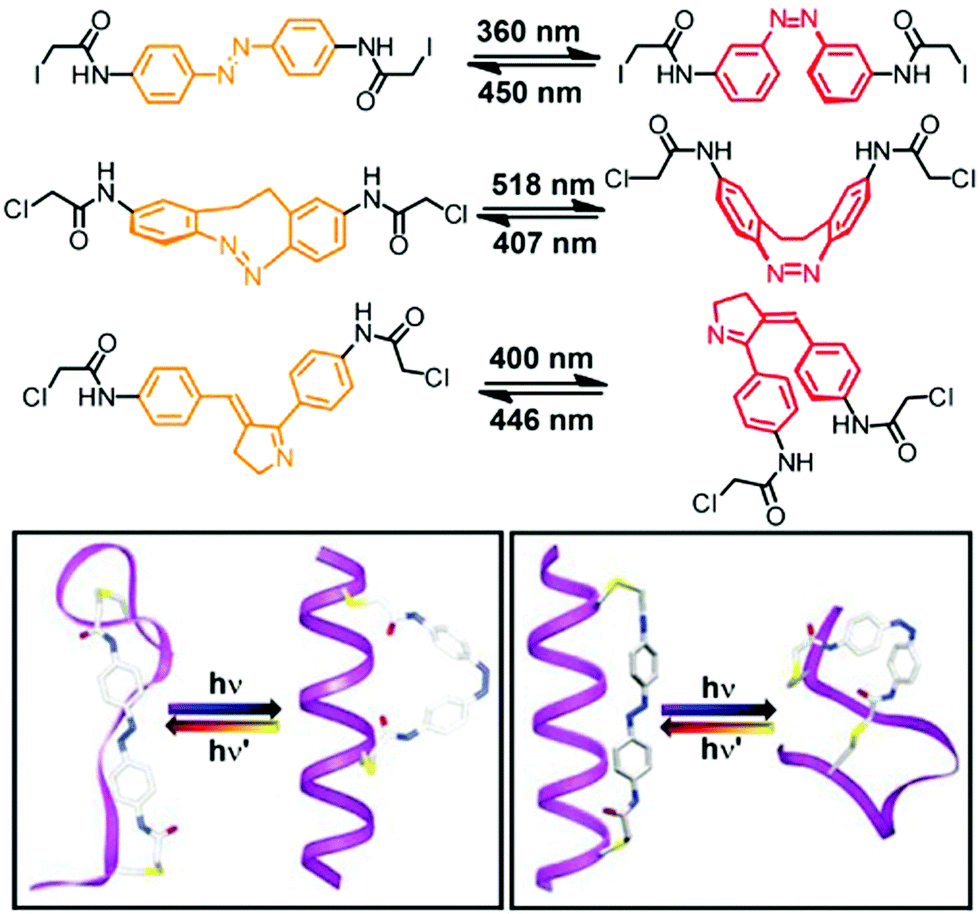 | ||
| Fig. 7 Photoswitches that have been used as tethers to covalently link two side chain residues in peptide based foldamers in the loop approach (top). Turn-on (bottom left) and turn-off (bottom right) helices with the E- and Z-configured tethers connecting the i and i + 7 positions and the i and i + 11 positions of the peptide α-helix, respectively.80 | ||
Rendering the loop approach more applicable, the group of Woolley developed and incorporated photoswitches, which do not require the use of UV-light and can be operated entirely with visible light.25,26 For example, they could trigger a reversible helix–coil transition by visible light and furthermore improve the extent of helical unfolding by using novel photochromic units (Fig. 7).83,84 On the one hand, a cyclic bis(para-acetamido)azobenzene derivative with separated n → π* absorption bands for the two isomers85 was tethered to the i and i + 11 positioned cysteine residues within the backbone.83 As a result, the helix–coil transition could repetitively be switched by an alternating exposure to 407 nm and 518 nm light and thanks to the very efficient photoconversion between the E- and Z-isomers, the change in population of the different conformers was quite significant. On the other hand, to enhance the effect of the local photoswitching event on the conformational transition, i.e. improve the transduction efficiency, a non-symmetrical benzylidene–pyrroline chromophore, mimicking the Schiff base in rhodopsin and giving rise to a larger geometrical change upon photoisomerization, has been linked to the peptide backbone.84 Tethering the photoswitch between the i and i + 11 residues allowed the peptide backbone to adopt an α-helical conformation in the dark, while a random coil conformation was efficiently populated following E → Z photoisomerization of the benzylidene–pyrroline moiety. However, despite the high degree of photoconversion associated with a large structural change in the photochromic tether, the typical α-helix signature in the CD spectrum did not decrease to a large extent, which is probably due to the location of the photoswitch relative to the helix, leaving a larger part of the peptide and hence its conformation unaffected.
4. Backbone approach
The photoswitchable units can also be incorporated into the main chain86,87 of the helical structures to manipulate their folding behavior. This backbone approach is perhaps the most direct way to couple photoswitches with foldamers and both the photoswitching as well as the folding behavior are inextricably linked. Since most photoswitchable moieties consist of a more or less extended π-conjugated chromophore, their integration as monomer units is most readily accomplished using (hetero)aromatic foldamers based on either local constraints50,88 and/or solvophobic interactions.89 Therefore, the geometrical changes during the course of the photoswitching event affect both the local conformational constraints as well as the typically intrastrand π,π-stacking interactions with non-nearest neighbors and of course vice versa. Following this approach thus far mostly azobenzene has been used due to its large geometrical changes, reflected in dramatically different angles and distances, which have also been exploited in the loop approach (see above), and in the inability of the twisted three dimensional Z-isomer to engage in stabilizing π,π-stacking contacts in strong contrast to the flat E-isomer. These azobenzene-containing foldamers have been connected by three different types of bridging units, i.e. carboxamide and triazole linkages, both providing some local conformational preferences,88 as well as ethynylene (and also diethynylene) linkages, yielding three different classes of rigid photoswitchable foldamers, including oligoamides,50 clickamers,90 and oligo(meta-phenylene ethynylenes) (OmPEs)91 as covered below.4.1 Photoresponsive oligoamides
The remarkable properties of the aromatic oligoamide foldamers,49,50 including their strong local conformational preferences, their high chemical stability, and their propensity to crystallize (and thereby enabling accurate structure determination by single crystal X-ray diffraction), inspire the design of their respective photoswitchable counterparts. Parquette and co-workers reported oligomeric backbones composed of alternating sequences of pyridine-2,6-dicarboxamides and meta-(phenylazo)azobenzene moieties (Fig. 8).43 In its all-E configuration the foldamer adopts a helical structure with an approximately 3.4 Å helical pitch as predicted by molecular modelling and confirmed for the shorter oligomer 94 by X-ray crystallographic analysis in the solid state. Helix formation has been attributed to intramolecular H-bonds between the amides' NH-atoms and the pyridine N-atom (and possibly the adjacent azo N-atom) as well as π,π-stacking interactions between the (hetero)aromatic rings. Due to the stability of the helix structure, the E → Z photoisomerization efficiency of the azobenzene units in the folded oligomers 94 and 178 was reduced by approximately a factor of three when compared to unfolded shorter oligomers. By analyzing the composition of the photostationary state (PSS) mixtures the authors found that the terminal azobenzene units isomerize to a larger extent as compared to the internal azobenzene moieties, which are presumably packed more tightly at the inner core of the helix. Therefore, E → Z photoisomerization results primarily in a departure of the terminal azobenzene units from the helical conformation, leading to a shortened and less stable helix, which is characterized by a lower helix inversion barrier. The latter has actually been measured by CD spectroscopy using chiral oligomers 94* and 178*,92 as well as by VT-NMR spectroscopy. Indeed, upon E → Z photoisomerization the helix inversion barrier decreased from 13.8 kcal mol−1 for the initially all-E-configured 178 to 12.8 kcal mol−1 for the PSS mixture. The authors concluded that helix inversion in their system occurs via a stepwise unfolding rather than a global wrap-unwrap process. Somewhat surprisingly and despite successful thermal Z → E isomerization, photochemically induced Z → E isomerization was not possible since presumably photoreduction of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N units occurred upon exposure to visible light.
N units occurred upon exposure to visible light.
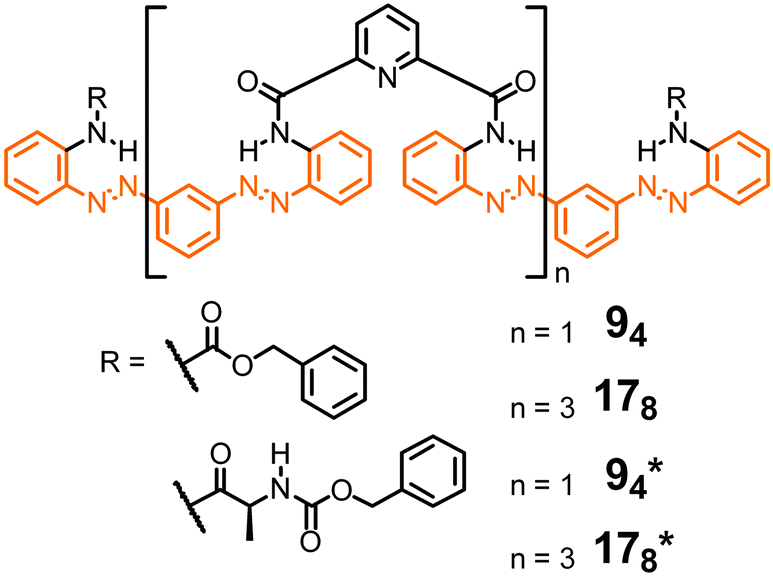 | ||
| Fig. 8 Photoswitchable oligoamide foldamers composed of azobenzene and pyridine moieties terminated with achiral units (94 and 178) and chiral units (94* and 178*), the latter to facilitate analysis by CD spectroscopy.43,92 Throughout, oligomers are named by giving the number of aromatic repeat units (here: 9 or 17) followed by the number of incorporated azo (NN) bonds in subscripts (here: 4 or 8). | ||
Azobenzene has also been incorporated into folded aromatic amide dendrons, adopting a compact, helical conformation to cause a local perturbation that subsequently should lead to large photoinduced structural changes in dendrimers (Fig. 9).93 The 1st and 2nd generation dendrons were composed of a bifunctional pyridine-2,6-dicarboxamide focal point connected via two azobenzene units to either two or four 2-methoxyisophthalamide termini. The helical conformation of the dendrons was stabilized by the syn–syn conformations of both the 2-methoxyisophthalamides as well as the pyridine-2,6-dicarboxamides, owing to the intramolecular H-bonding from the amides' NH-atoms to the methoxy oxygen and the pyridine N-atom, respectively. Similarly to the authors' observations with respect to the folded oligomers detailed above, the E → Z photoisomerization of the azobenzene units was less efficient within the folded dendrons as compared to unfolded model compounds. In the PSS mixtures the hydrodynamic volume of the dendrons (measured by DOSY NMR) was increased by 75% for the 1st generation and by 46% for the 2nd generation dendrons when compared to their initial E,E-isomer, illustrating a substantial structural change upon E → Z photoisomerization in these compact, folded structures.
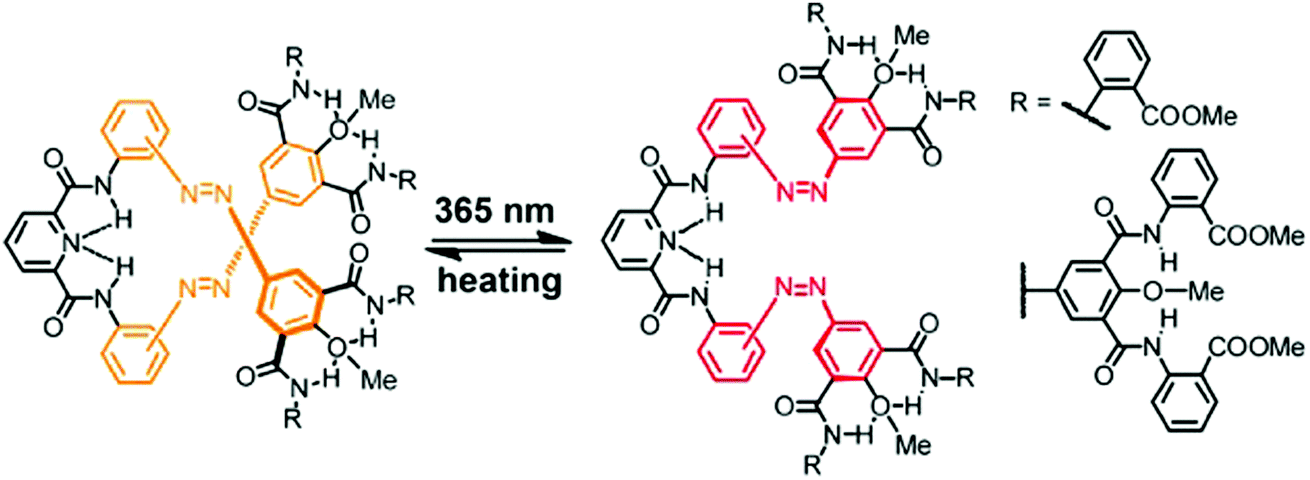 | ||
| Fig. 9 Photoswitchable folded dendrons composed of a pyridine-2,6-dicarboxamide focal point and 2-methoxyisophthalamide termini connected via azobenzene units.93 | ||
4.2 Photoswitchable clickamers
Besides connecting (hetero)aromatic moieties via strategically placed amide residues well-defined helical structures can also result from linking various types of aza-heterocycles, such as pyridine, pyrimidine, and pyridazine, among others.39,94 Due to its strong conformational preferences,95 the 1,4-linked 1,2,3-triazole moiety that can efficiently be generated by the Cu-catalyzed azide–alkyne 1,3-dipolar cycloaddition, the so called “click reaction”, has been utilized to construct heteroaromatic foldamers, referred to as “clickamers”.90,96–99 Photoswitchable versions thereof have been reported by Flood and coworkers, who incorporated azobenzene units at both termini of the amphiphilic clickamer backbone (Fig. 10).44 In this clever design, the E,E-configuration allows both azobenzenes to engage in full π,π-stacking contacts, while in the non-planar Z,Z-configuration the terminal phenylazo moieties are removed from the helical skeleton, thus weakening the π,π-stacking interactions and resulting in a helix to coil transition (turn-off foldamer). In view of the ability of these clickamers to bind anions in their inner cavity,100E → Z photoisomerization of the azobenzene termini should be able to gate the affinity for chloride ions. Indeed, the foldamer's binding constant for chloride ions decreases significantly upon irradiation due to the UV-light-induced shortening of the helical skeleton. In addition, an intermediate binding constant was observed for the mixed E,Z-configured oligomer and therefore the chloride ion affinity follows the order of E,E > E,Z > Z,Z. Consequently, the photoswitchable clickamer can be used to release and recapture chloride ions in solution in response to UV and visible irradiation, respectively.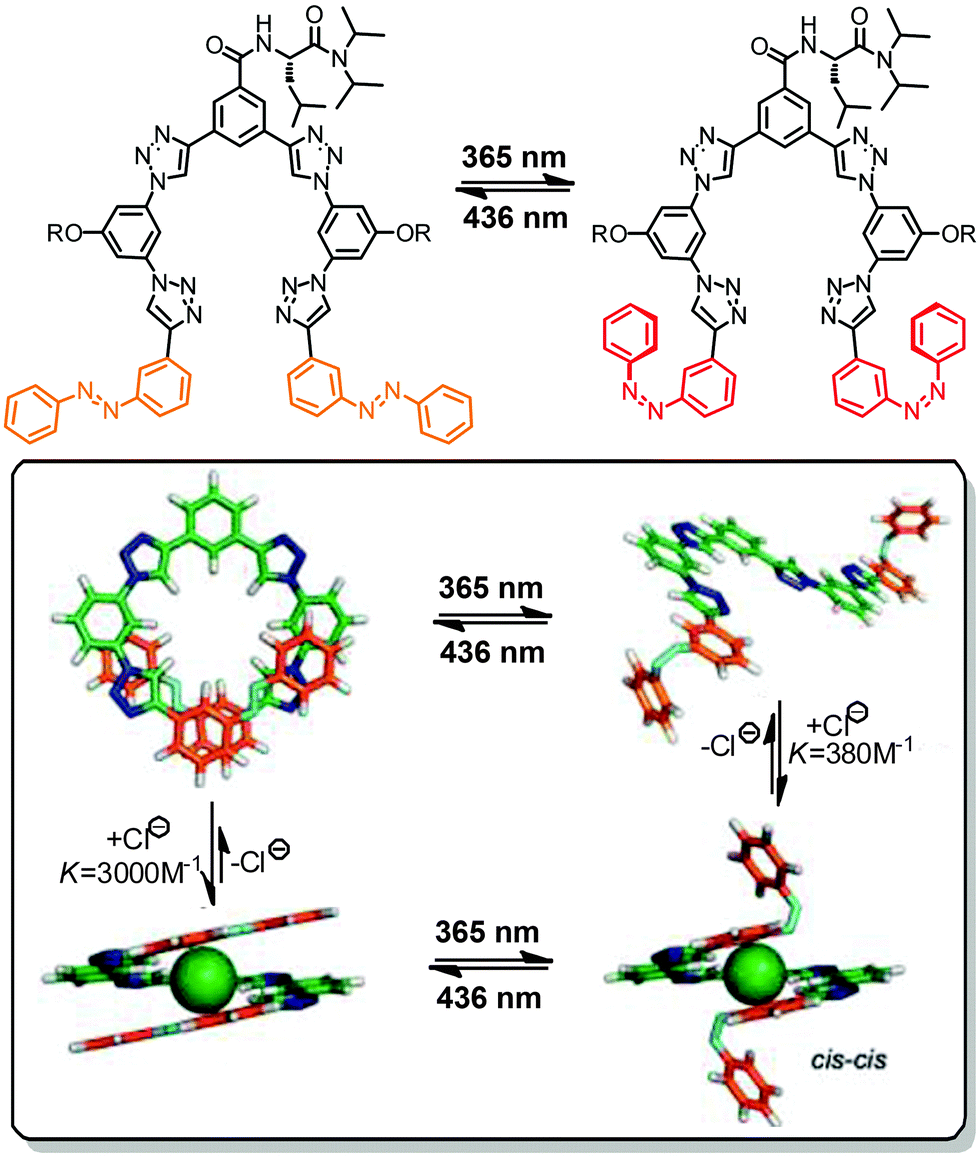 | ||
| Fig. 10 Photoswitchable clickamer with terminal azobenzene units, which gate the helix–coil transition allowing for light-controlled capture and release of chloride ions.44 | ||
To further advance their design, the same group prepared several new clickamers with stronger ion affinity utilizing interlocking and capping approaches.101,102 In the interlocking approach, the L-leucine residues forming β-sheet-type H-bonds were introduced to the central and terminal phenyl groups of the clickamer foldamer to initially stabilize the folded conformation as well as the foldamer–anion complexes by interlocking helical turns (Fig. 11, top).101 The formation of these interlocking H-bonds improved the chloride binding constant by almost one order of magnitude (from 1.26 × 105 M−1 to 9.7 × 105 M−1). Upon exposure to UV-light, the isomerization of the terminal azobenzene units significantly decreased the stability of the foldamer and hence led to unfolding, associated with a 84-fold reduced chloride affinity of the oligomer. In the alternative capping method, two clickamers composed of six triazole and five phenylene units were terminated with two ortho-linked azobenzene units (Fig. 11, bottom).102 These foldamers form single or double helical complexes with chloride ions in aqueous solution. In the latter double helical complex, a much larger percentage of the hydrophobic π-surface is buried (∼80% instead of ∼50% in the single helix) and in addition the terminal E-configured azobenzene units shield the hydrogen-bond donors to chloride, thereby creating a solvent-excluding cavity capable of generating stronger CH⋯Cl− H-bonds. While this work nicely illustrates the importance of creating specific microenvironments to optimize binding, the photoresponse of the clickamers was not discussed.
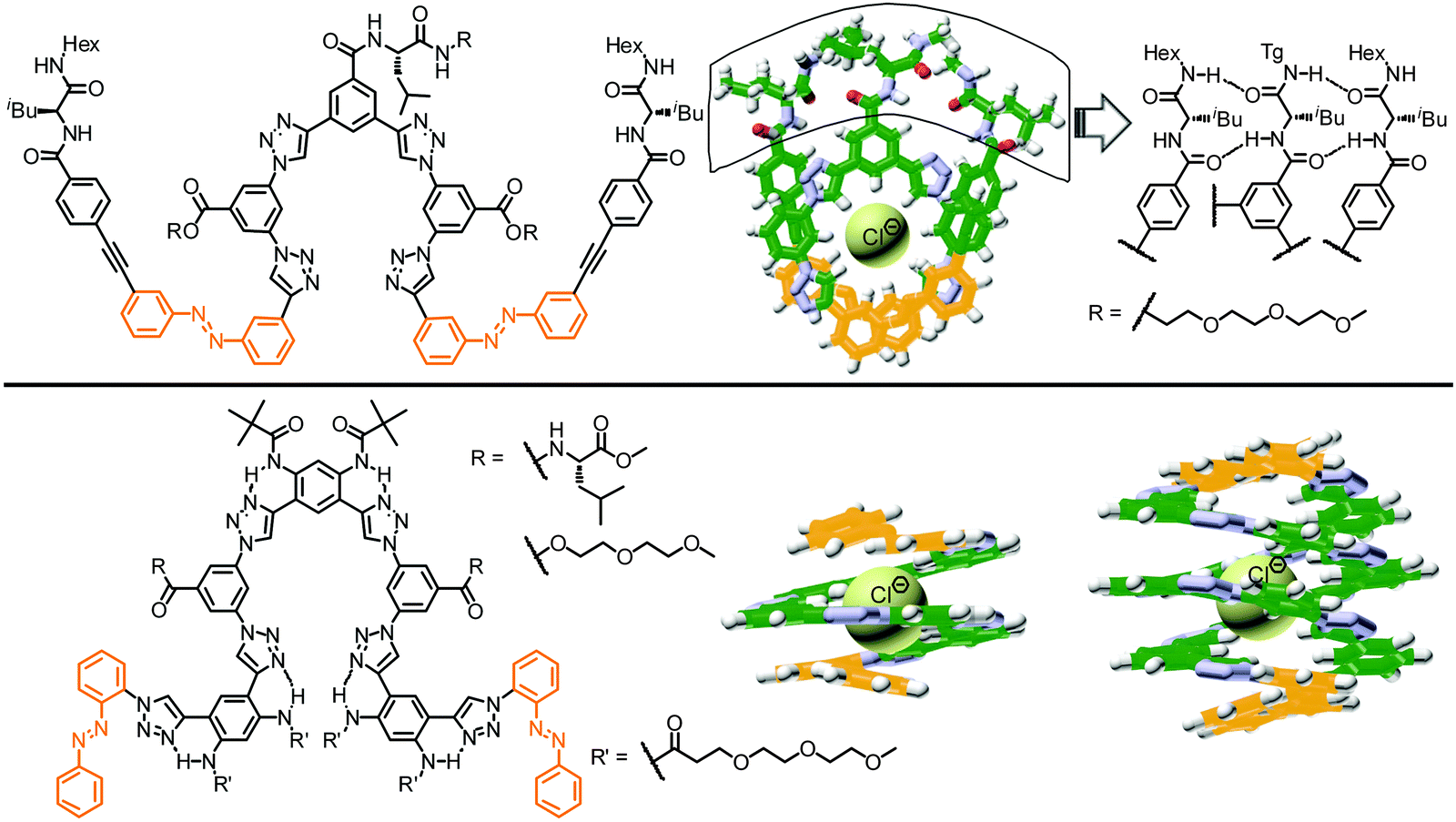 | ||
| Fig. 11 Photoswitchable clickamers with improved chloride affinity by interlocking H-bonds (top) and azobenzene caps (bottom).101,102 | ||
Another example of a photoswitchable clickamer has been reported by Jiang and coworkers, who placed the azobenzene unit at the center of the oligomer (Fig. 12).103 Upon exposure to UV-light, the central azobenzene unit readily underwent E → Z photoisomerization, resulting in 82% Z-azobenzene in the PSS. NMR titration experiments revealed that the foldamer exhibits somewhat increased binding constants (a factor between 1.7 and 4.1) for various anions (halides, nitrate, and bisulfate) in the Z-isomer as compared to the E-isomer. The observed slight increase in binding constant can probably be attributed to the shrinkage of the inner cavity upon switching to the Z-azobenzene. However, the overall relatively weak anion affinity, in particular when compared to the ones by Flood and coworkers,44,101,102 in combination with its only moderate light-induced modulation, limit the applicability of this particular clickamer for light-regulated anion transport and separation.
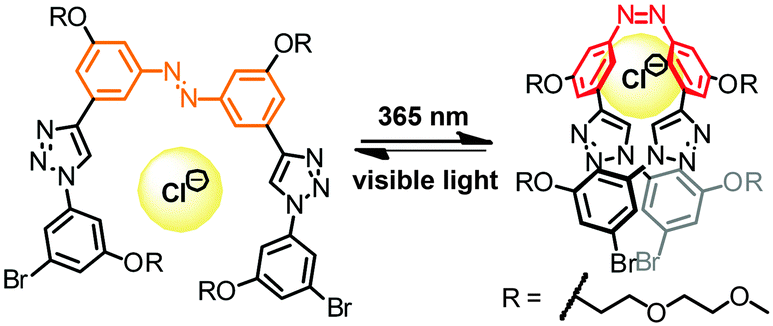 | ||
| Fig. 12 Photoswitchable clickamer with a central azobenzene unit modulates chloride binding by light.103 | ||
4.3 Photoswitchable OmPE foldamers
Oligo(meta-phenylene ethynylene)s (OmPEs) have been demonstrated to fold into a helical conformation in specific environments stabilized by a well-balanced set of supramolecular interactions, such as H-bonding, metal–ligand coordination, π,π-stacking, coulombic, dipolar, and van der Waals interactions.91 Rather than attaching the photoactive groups as the side chains of the OmPE backbones, for example to covalently lock the helical structure containing an inner void,104 we have embedded photochromic azobenzene units into the amphiphilic backbones to replace one (or more) of the diphenylacetylene (tolane) moieties. In the following, we summarize our corresponding research efforts starting from the design of the first prototype with a single azobenzene core42,105 to the investigation of the relationship between the folded structure and the resulting photoresponse in various series of azobenzene-based foldamers.45,106–109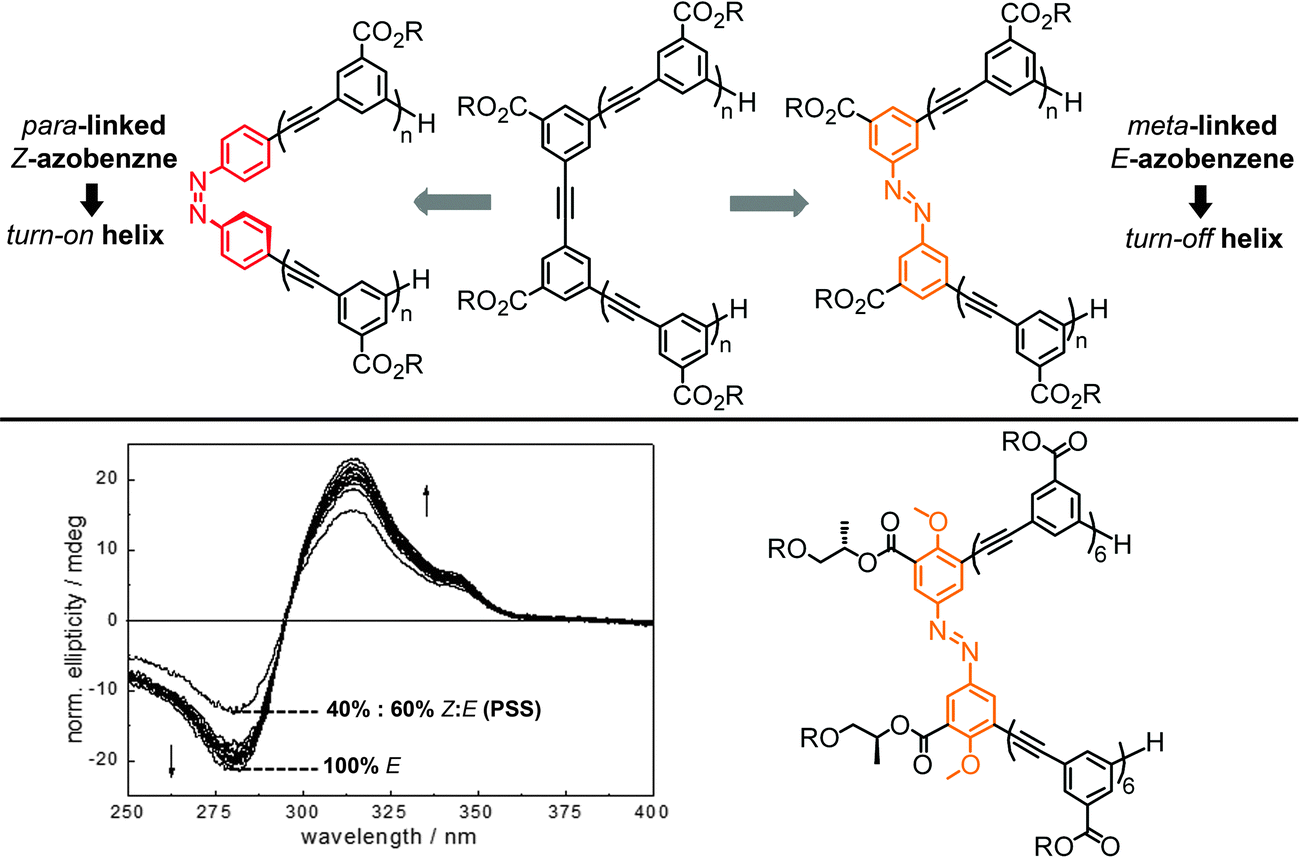 | ||
| Fig. 13 Turn-on and turn-off foldamers containing a single azobenzene core linked in its terminal para- and meta-positions, respectively (top). Photoswitchable tetradecamer with improved methoxy-substituted azobenzene core and the corresponding CD spectra during thermal Z → E isomerization in CH3CN at 25 °C (bottom).42,105 | ||
To ensure selective excitation of the azobenzene core and hence access photoswitchability in the helically folded structure, the π → π* absorption band of the azobenzene core was bathochromically shifted by introducing para-methoxy groups. As a consequence of the slightly weaker π,π-stacking contact of the more electron-rich 4,4′-dimethoxyazobenzene (DMAB) unit, the oligomer had to be elongated from a dodecamer to a tetradecamer (Fig. 13, bottom),42 which adopts a helical structure that is 1.7 kcal mol−1 more stable than the coil structure, i.e. helix stabilization energy = −1.7 kcal mol−1, in acetonitrile.110 The formation of the helix was furthermore supported by the significant Cotton effect in CD spectra in aqueous acetonitrile solution due to the twist sense bias provided by the chiral side chains. As confirmed by both CD and UV/vis spectroscopy, exposure of the foldamer to UV-light induced formation of the Z-azobenzene core, which disrupts π,π-stacking interactions in the helix and consequently led to unfolding of the tetradecamer. This first prototype of a photoswitchable OmPE foldamer displayed approximately 40% unfolding in the PSS and sparked further efforts to design more efficient analogues.
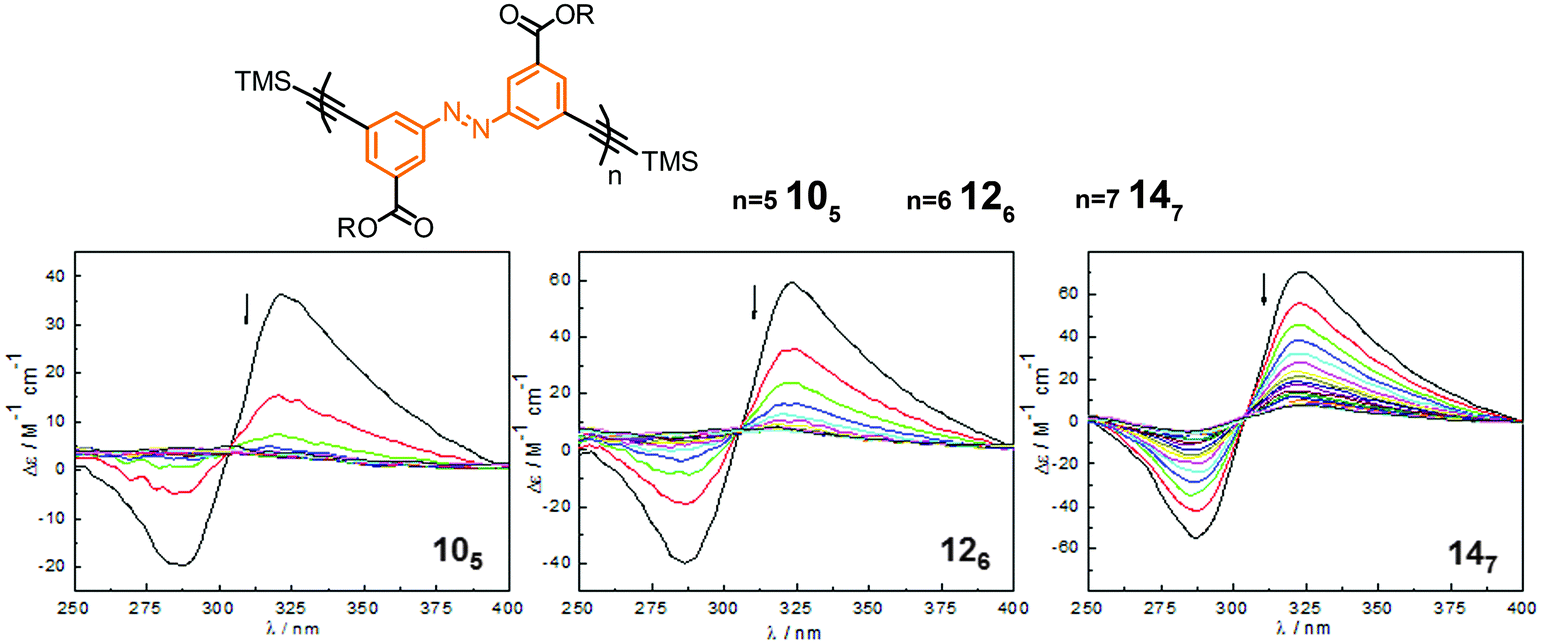 | ||
| Fig. 14 Photoswitchable oligoazobenzene foldamers 105, 126, and 147 (R = –(CH2CH2O)3CH3 or –(CH(S-CH3)CH2O)(CH2CH2O)3CH3) and their corresponding CD spectra during the course of irradiation at λirr = 358 nm in CH3CN at 25 °C.45 | ||
For all three oligomers 105, 126, and 147, the intensity of the Cotton effect in the CD spectra decreased dramatically or even quantitatively upon exposure to UV-light, indicating the occurrence of an efficient photoinduced unfolding transition following the E → Z photoisomerization (Fig. 14). The photoconversion was monitored by UV/vis spectroscopy and the content and distribution of Z-azobenzenes in the PSS was determined by NMR spectroscopy. Similarly to the findings of Parquette and coworkers,43 the efficiency of the E → Z photoisomerization is dependent on the location of the azobenzene unit in the helix and the interior azobenzene units display a lower Z-content as compared to the terminal units. Importantly, the foldamer with a higher content of interior azobenzene units represents a lower rate constant of the photoinduced unfolding, illustrating that the untwisting process of these homogeneous backbones is predominately starting from the terminal positions. In contrast to Parquette's foldamers the helical refolding in this series of foldamers can readily be induced by visible light irradiation and resulting in full recovery of the helical structures over many cycles without any noticeable fatigue.
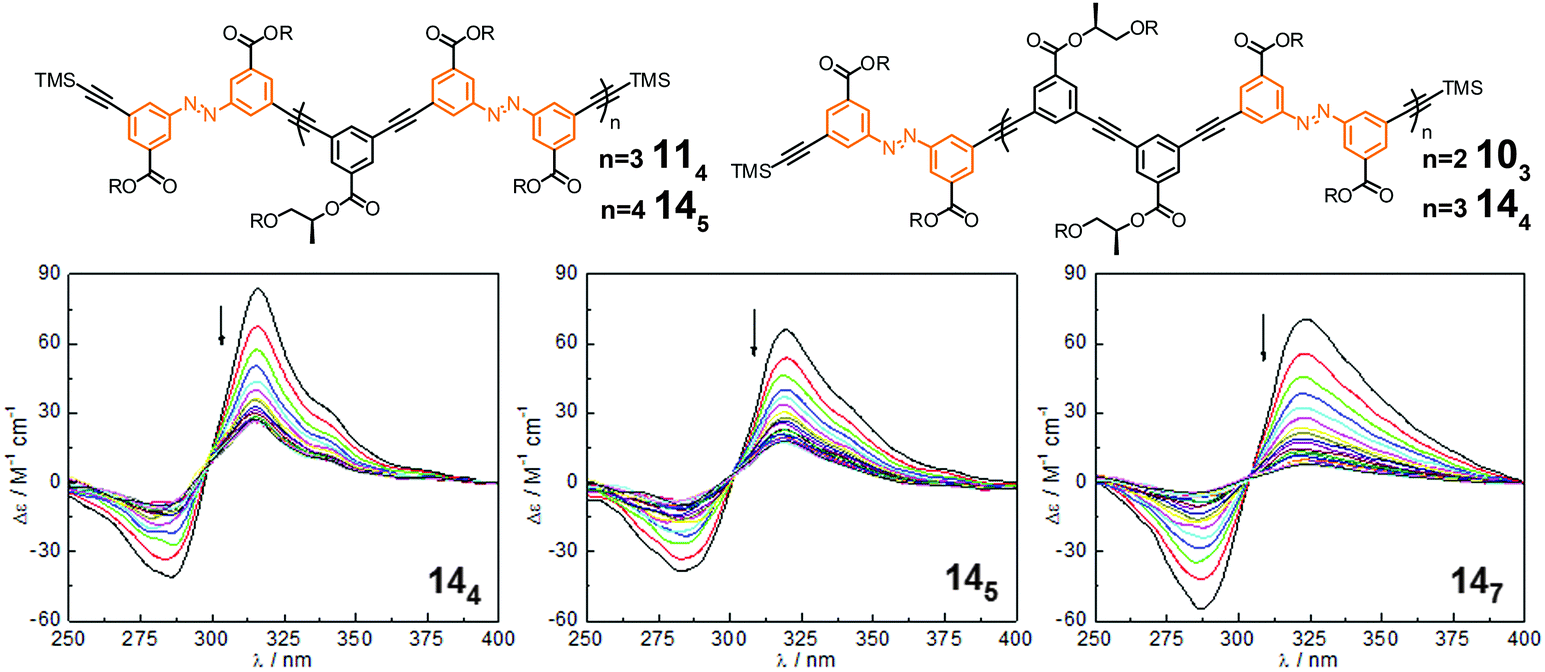 | ||
| Fig. 15 Photoswitchable oligoazobenzene tetradecamers 144, 145, and 147 (R = –(CH2CH2O)3CH3) and their corresponding CD spectra during the course of irradiation at λirr = 358 nm in CH3CN at 25 °C.106 | ||
The variable relative orientations of azobenzenes within the helical structures led to three types of π,π-stacking interactions, i.e. azobenzene–azobenzene (azo–azo), phenylene–phenylene (Ph–Ph), and azobenzene–tolane (azo–tol). Calculated from the stabilization energies ΔG(CH3CN) of all the investigated foldamers determined by solvent titration experiments, the corresponding strengths of the three kinds of π,π-stacking interactions, i.e. azobenzene–azobenzene (azo–azo), phenylene–phenylene (Ph–Ph), and azobenzene–tolane (azo–tol), can be determined as −0.86 kcal mol−1, −0.44 kcal mol−1, and −0.71 kcal mol−1, respectively (Fig. 16). Based on the linear combination of these individual π,π-stacking interactions, the helix stabilization energy for various azobenzene-containing OmPE foldamers in acetonitrile can be estimated by:
| ΔG (CH3CN, kcal mol−1) = (−0.86)a + (−0.44)b + (−0.71)c |
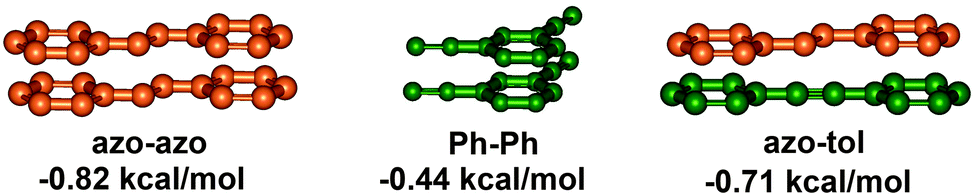 | ||
| Fig. 16 Kinds of π,π-stacking interactions present in the folded backbones and their strengths. | ||
Considering the three tetradecamers 144, 145, and 147, which only vary in the number and relative orientation of azobenzenes within the helix, we found that an increasing azobenzene content leads to both slower photoisomerization and unfolding, yet more complete denaturation according to the CD spectra upon UV-irradiation (Fig. 15). From this finding two competing effects become apparent: on the one hand, enhancing the number of azobenzene units is favorable as it increases the likelyhood of light-induced formation of helix-breaking Z-isomers (statistics); whereas on the other hand, avoiding strong π,π-stacking interactions, in particular between cofacial azobenzene units, enhances the efficiency of E → Z photoisomerization and subsequent unfolding (microenvironment). Therefore, both the chain length and azobenzene distribution throughout the backbone need to be optimized to maximize the photoresponse (efficiency) in these photoswitchable foldamers.
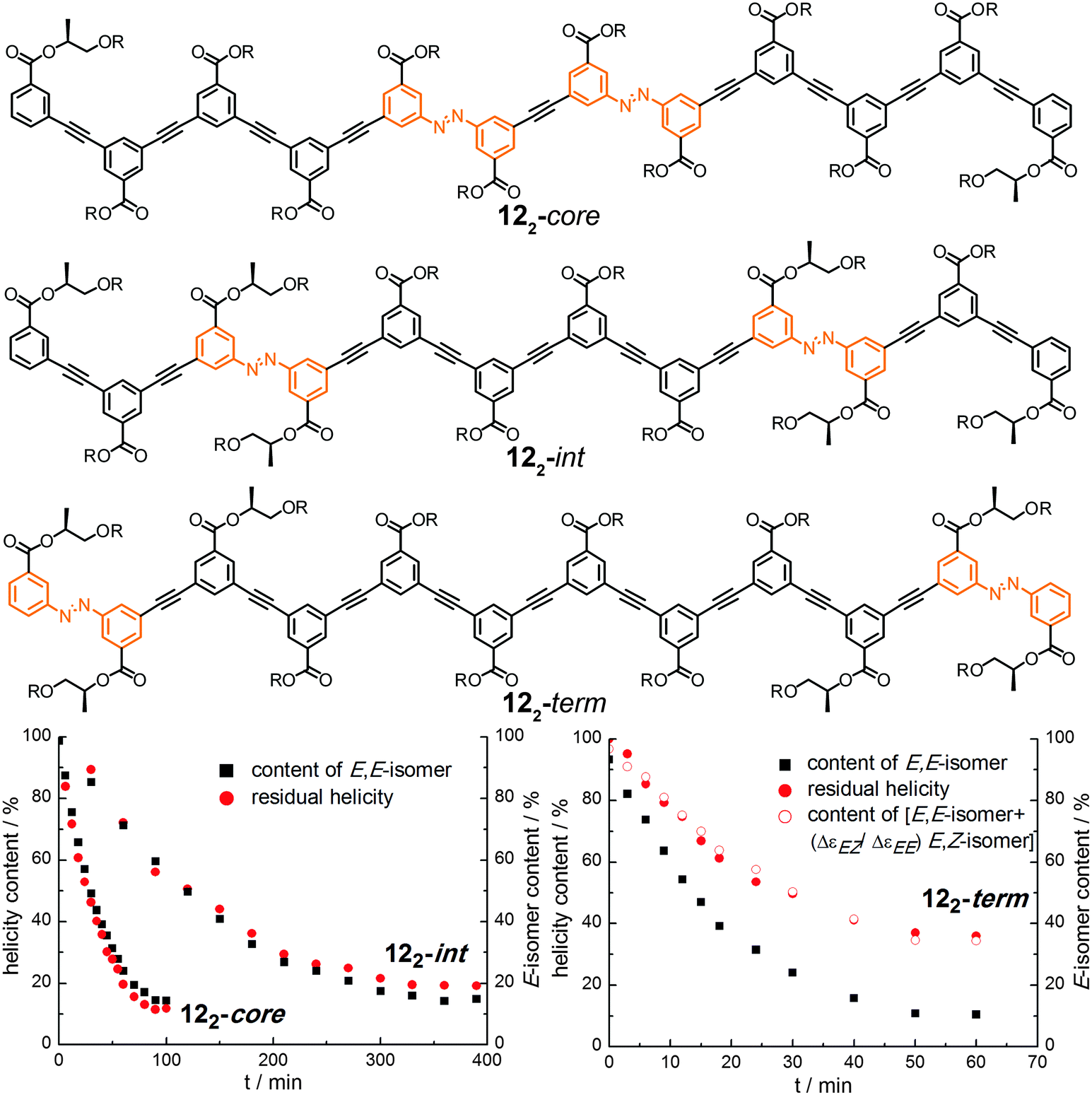 | ||
| Fig. 17 Photoswitchable oligoazobenzene dodecamers 122-core, 122-int, and 122-term (R = –(CH2CH2O)3CH3) and comparison of their CD intensities and isomer ratios during the course of irradiation at λirr = 358 nm in CH3CN at 25 °C.107 | ||
Upon exposure to UV-light, the isomer content as well as the helicity were monitored and both were correlated for the three isomers (Fig. 17). Assuming that the photogenerated Z-isomer acts as a stopper for helix formation, which requires a critical chain length of approximately 10–12 phenylene units,113 the decays of the Cotton effect related to the loss of helicity in all the foldamers could quantitatively be correlated with the isomer contents, i.e. the E,E-isomer for foldamers 122-int and 122-core (Fig. 17, bottom left) or the E,E-isomer as well as the E,Z-isomer for foldamer 122-term (Fig. 17, bottom right). The latter is explained by the fact that terminal isomerization yields a E,Z-configured foldamer, which can still adopt a (partially) helical structure giving rise to a Cotton effect. Importantly, this finding implies that there are no significant changes in the twist sense bias in these foldamers during photoswitching and furthermore no particular preference for untwisting either one of the P- or M-helices. Determination and analysis of the quantum yields for the individual E → Z photoisomerization events shows that photoswitching occurs more efficiently when the respective azobenzene moiety is located either at the helix terminus or in an already (partially) unfolded structure. This implies that once the initial photoswitching event in such structures has occurred, the subsequent photoisomerization event can be substantially facilitated and hence these bisazobenzene systems exhibit (positive) cooperativity36 with regard to its switching ability. Overall, the unfolding transition, which itself is cooperative, serves as a transduction mechanism to confer a cooperative behavior upon the embedded photoswitchable units and thereby creates a photoresponsive macromolecular system with enhanced sensitivity.
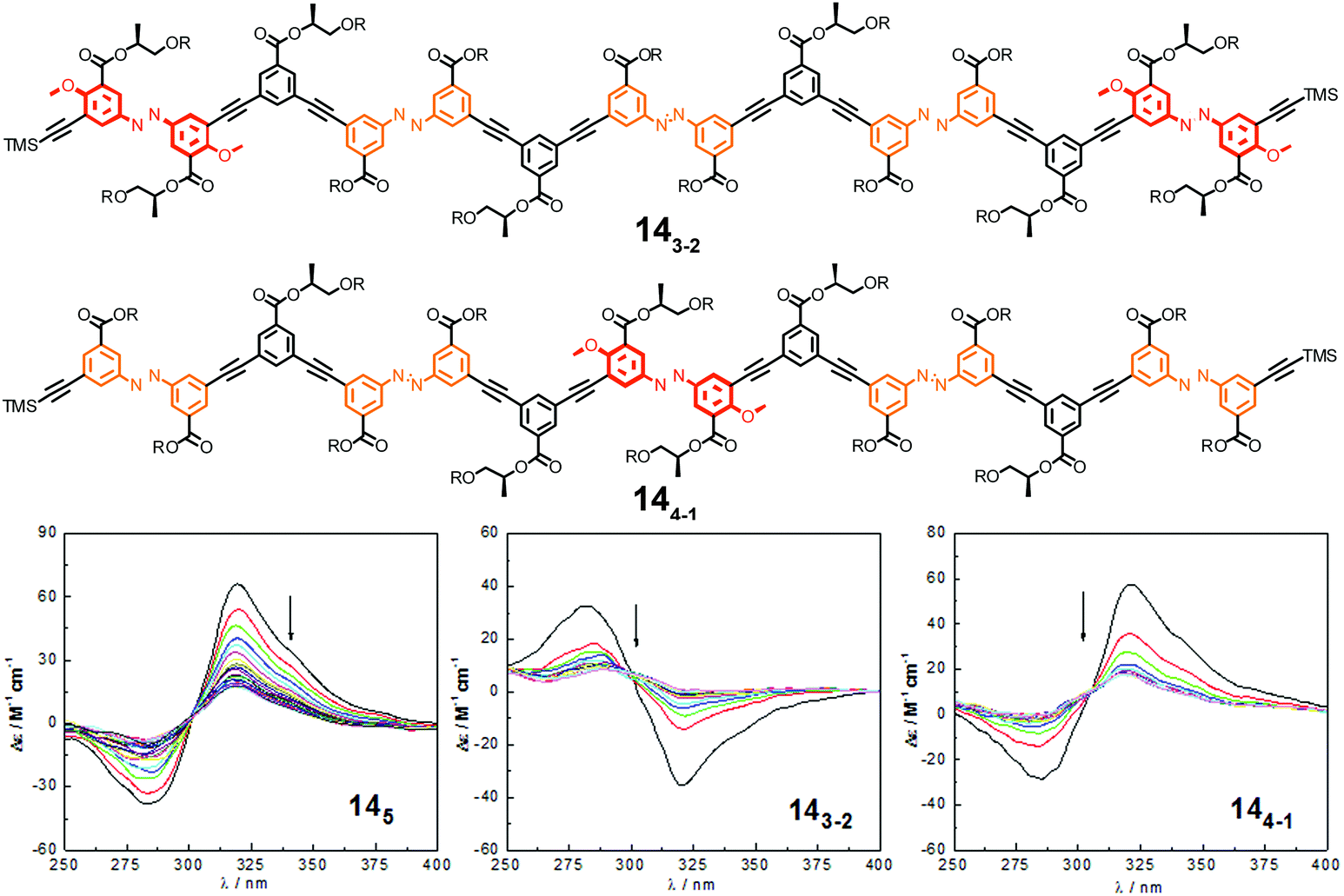 | ||
| Fig. 18 Photoswitchable heterogeneous oligoazobenzene tetradecamers 143-2 and 144-1 (R = –(CH2CH2O)3CH3) and the corresponding CD spectra (145 shown for comparison) during the course of irradiation at λirr = 358 nm in CH3CN at 25 °C.108 | ||
In comparison to the reference foldamer 145 with a homogeneous backbone composed of parent azobenzene units only, the Z-content of photochromic units in the heterogeneous backbones in the PSS is increased specifically in the positions occupied by DMAB units, suggesting that indeed efficient intramolecular excitation energy transfer takes place in our foldamers exhibiting an energy gradient architecture.119–122 In contrast to the homogeneous oligomer 145, in which untwisting the helical structure would predominantly arise from terminal isomerization events as described previously, the priority for isomerizing the DMAB units governs the unfolding process of foldamers 143-2 and 144-1. Specifically, initial terminal isomerization contributes mainly to the outside-in unfolding process of foldamer 143-2, whereas initial interior isomerization dominates in the inside-out unfolding process of foldamer 144-1 (Fig. 19).
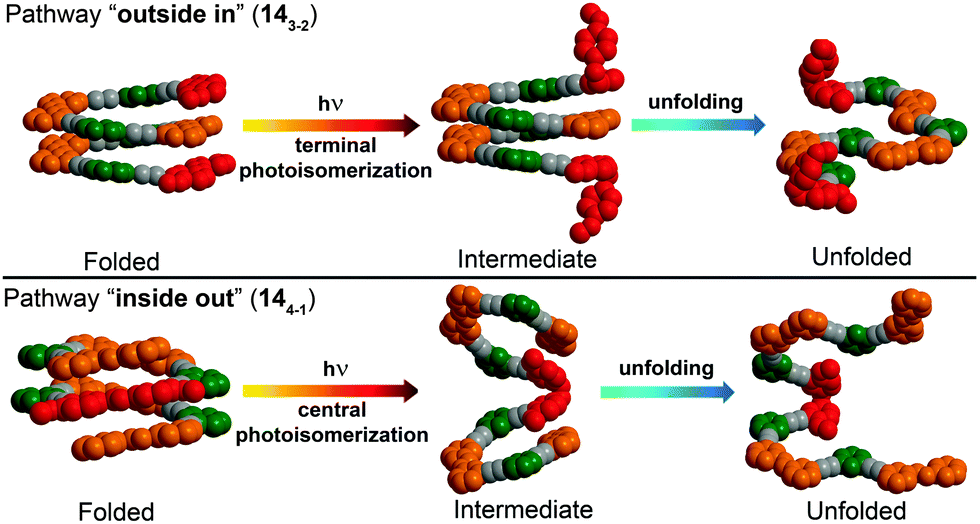 | ||
| Fig. 19 Control over unfolding pathways in photoswitchable heterogeneous oligoazobenzene tetradecamers 143-2 and 144-1 exhibiting an energy gradient architecture leading to localization of the primary photoisomerization events at the DMAB positions (DMAB units are shown in red; parent azobenzene units in orange, meta-phenylene units in green, and ethynylene linkages in grey).108 | ||
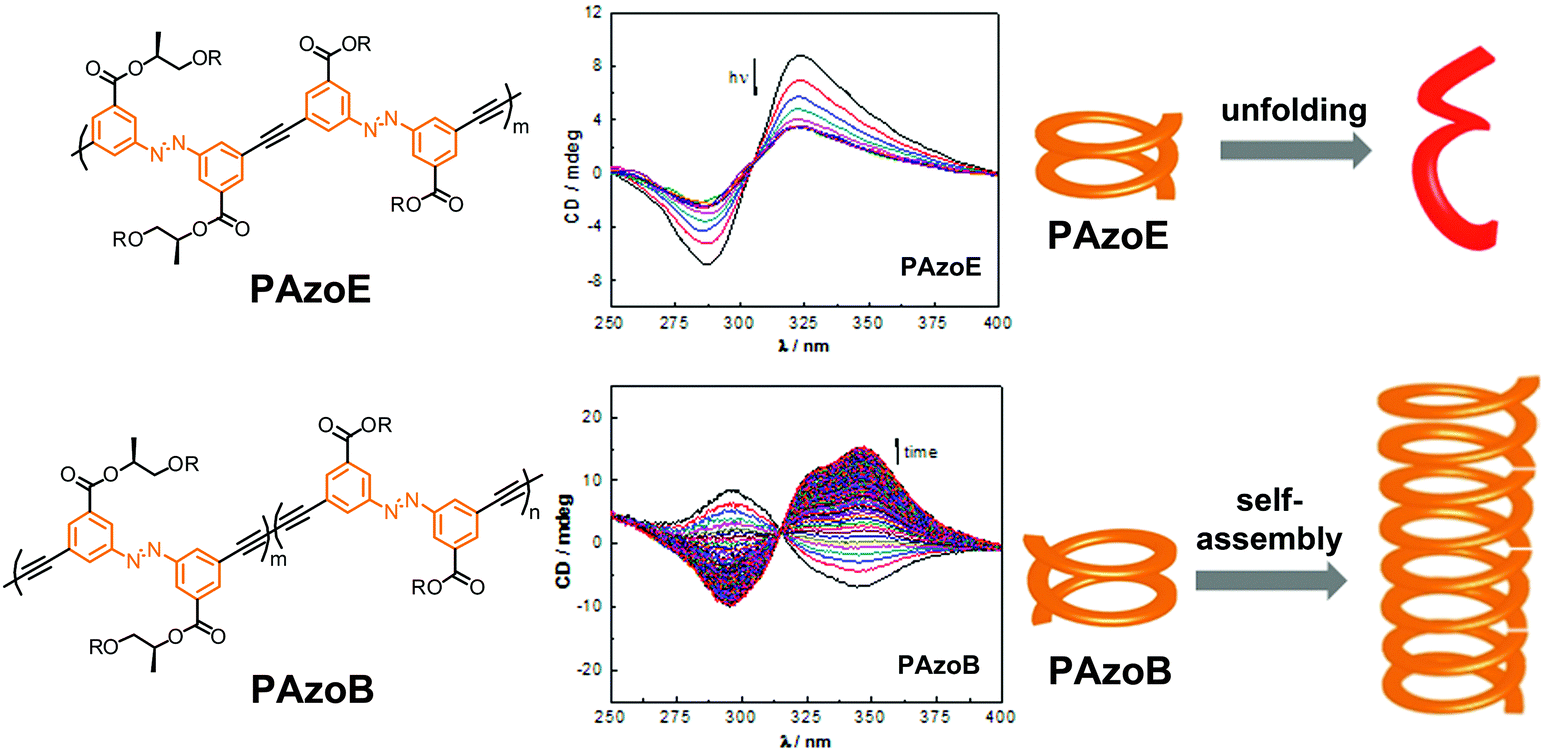 | ||
| Fig. 20 Polyazobenzene foldamers containing ethynylene (PAzoE) and butadiynylene linkages (PAzoB) and CD spectra of PAzoE upon irradiation (λirr = 358 nm) and of PAzoB over time in CH3CN at 25 °C, showing photoinduced unfolding and suppression of photoisomerization due to aggregation, respectively, as illustrated in the cartoon on the right.109 | ||
5. Conclusion and perspectives
In this feature article, we detail the development of the photoswitchable foldamers with particular emphasis on the different design approaches for incorporating the photoswitches into the backbones. Depending on the manner of the attachment and the location of the incorporated photoswitches, the photoinduced interconversion between various (meta)stable isomers affects the local covalent constraints and/or the non-covalent interactions that govern the stabilization of the helix structure, thereby leading to conformational changes, in particular helix inversion and/or helix–coil transition. The coupling of the photoswitching event(s) and the (un)folding behavior is furthermore manifested in the profound effect of the folded architecture on the photoswitching ability and efficiency, leading to an overall “feedback”. Exploiting the strength of the oligomer approach to deduce clear structure-folding–switching relationships several design parameters, including the number, placement, relative orientation, and nature of photoswitches, can be optimized to maximize the conformational consequence of the photoswitching events and hence the sensitivity of photoresponse.Considering the remarkable progress made over the past decade and the detailed understanding gained, we feel that it is time to exploit photoswitchable foldamers as a sophisticated tool to achieve remote control over the properties of various materials and device functions. As shown for photoswitchable clickamers, one of the promising applications of photoresponsive helices is their ability to act as dynamic hosts for the photocontrollable capture and release of guest molecules. Another main advantage is the significant change in the aspect ratio in these systems associated with their cooperative (un)folding that should aid the design of photoresponsive functional macromolecular and supramolecular architectures, including sophisticated light-controlled catalysts15,17 based on photoswitchable foldamer scaffolds as well as molecular walkers10 for optomechanical systems123 based on an extension-shrinkage motion.38 Beyond the molecular level, the collection and amplification of individual molecular photoswitching events still poses a major challenge.1,3,38 Therefore, it is important to organize the helical structures into unidirectional architectures spontaneously124 or assisted by external force fields125 and matrices.126 In addition, non-covalent and covalent assembly of such functional helical architectures127 will greatly enhance their availability and use. Combining all these aspects into new and more advanced molecular designs, we are absolutely confident that photoswitchable foldamers will prove invaluable to remote control and even power functional macromolecular and supramolecular systems operating at the mesoscopic and macroscopic level.
Acknowledgements
Our own contributions to the field have been generously supported by the German Research Foundation (DFG via SFB 765) and the European Research Council (via ERC-2012-STG_308117 “Light4Function”).References
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1683–1890 CrossRef CAS PubMed.
- H. B.-L. Heinz Dürr, Photochromism: Molecules and Systems, Elsevier, Amsterdam, 2003 Search PubMed.
- W. R. Browne and B. L. Feringa, in Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 121–179 Search PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77–92 RSC.
- M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656–3676 RSC.
- V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- Q. Li, G. Fuks, E. Moulin, M. Maaloum, M. Rawiso, I. Kulic, J. T. Foy and N. Giuseppone, Nat. Nanotechnol., 2015, 10, 161–165 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- F. K. Bruder, R. Hagen, T. Rolle, M. S. Weiser and T. Facke, Angew. Chem., Int. Ed., 2011, 50, 4552–4573 CrossRef CAS PubMed.
- T. J. Kucharski, N. Ferralis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441–447 CrossRef CAS PubMed.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149–2180 CrossRef CAS PubMed.
- N. Hosono, T. Kajitani, T. Fukushima, K. Ito, S. Sasaki, M. Takata and T. Aida, Science, 2010, 330, 808–811 CrossRef CAS PubMed.
- S. Hecht, Small, 2005, 1, 26–29 CrossRef CAS PubMed.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS PubMed.
- C. Raimondo, N. Crivillers, F. Reinders, F. Sander, M. Mayor and P. Samorì, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12375–12380 CrossRef CAS PubMed.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and I. I. I. T. H. Epps, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC and entire special issue.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- S. Yagai, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2005, 11, 4054–4063 CrossRef CAS PubMed.
- M. Irie, Adv. Polym. Sci., 1990, 94, 27–67 CrossRef CAS.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 CAS.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- D. Bleger, Z. Yu and S. Hecht, Chem. Commun., 2011, 47, 12260–12266 RSC.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- S. Mayer and R. Zentel, Prog. Polym. Sci., 2001, 26, 1973–2013 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4176 CrossRef CAS PubMed.
- A. Khan, C. Kaiser and S. Hecht, Angew. Chem., Int. Ed., 2006, 45, 1878–1881 CrossRef CAS PubMed.
- C. Tie, J. C. Gallucci and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 1162–1171 CrossRef CAS PubMed.
- Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 1640–1643 CrossRef CAS PubMed.
- G. A. Woolley, Acc. Chem. Res., 2005, 38, 486–493 CrossRef CAS PubMed.
- S. Hecht and I. Huc, Foldamers: Structure, Properties, and Applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- A. Koçer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef PubMed.
- O. Pieroni, A. Fissi, N. Angelini and F. Lenci, Acc. Chem. Res., 2001, 34, 9–17 CrossRef CAS PubMed.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- J. E. G. David and A. Lightner, Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy, Wiley-VCH, Weinheim, 2000 Search PubMed.
- R. Lovrien, Proc. Natl. Acad. Sci. U. S. A., 1967, 57, 236–242 CrossRef CAS.
- G. v. d. Veen, R. Hoguet and W. Prins, Photochem. Photobiol., 1974, 19, 197–204 CrossRef PubMed.
- M. Irie, A. Menju and K. Hayashi, Macromolecules, 1979, 12, 1176–1180 CrossRef CAS.
- L. Matějka and K. Dušek, Makromol. Chem., 1981, 182, 3223–3236 CrossRef.
- O. Pieroni, J. L. Houben, A. Fissi, P. Costantino and F. Ciardelli, J. Am. Chem. Soc., 1980, 102, 5913–5915 CrossRef CAS.
- J. L. Houben, A. Fissi, D. Bacciola, N. Rosato, O. Pieroni and F. Ciardelli, Int. J. Biol. Macromol., 1983, 5, 94–100 CrossRef CAS.
- O. Pieront, D. Fabbri, A. Fissi and F. Ciardelli, Macromol. Rapid Commun., 1988, 9, 637–640 CrossRef.
- A. Ueno, J.-i. Anzai, T. Osa and Y. Kadoma, Bull. Chem. Soc. Jpn., 1979, 52, 549–554 CrossRef CAS.
- A. Ueno, K. Takahashi, J. Anzai and T. Osa, J. Am. Chem. Soc., 1981, 103, 6410–6415 CrossRef CAS.
- A. Ueno, K. Adachi, J. Nakamura and T. Osa, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1161–1170 CrossRef CAS.
- F. Ciardelli, D. Fabbri, O. Pieroni and A. Fissi, J. Am. Chem. Soc., 1989, 111, 3470–3472 CrossRef CAS.
- T. M. Cooper, K. A. Obermeier, L. V. Natarajan and R. L. Crane, Photochem. Photobiol., 1992, 55, 1–7 CrossRef CAS PubMed.
- A. Fissi, O. Pieroni, F. Ciardelli, D. Fabbri, C. Ruggeri and K. Umezawa, Biopolymers, 1993, 33, 1505–1517 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- S. Lifson, M. M. Green, C. Andreola and N. C. Peterson, J. Am. Chem. Soc., 1989, 111, 8850–8858 CrossRef CAS.
- M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef.
- M. Mueller and R. Zentel, Macromolecules, 1994, 27, 4404–4406 CrossRef CAS.
- G. Maxein and R. Zentel, Macromolecules, 1995, 28, 8438–8440 CrossRef CAS.
- M. Müller and R. Zentel, Macromolecules, 1996, 29, 1609–1617 CrossRef.
- S. Mayer, G. Maxein and R. Zentel, Macromolecules, 1998, 31, 8522–8525 CrossRef CAS.
- S. Mayer and R. Zentel, Macromol. Chem. Phys., 1998, 199, 1675–1682 CrossRef CAS.
- M. M. Green, B. A. Garetz, B. Munoz, H. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117, 4181–4182 CrossRef CAS.
- H. Sogawa, M. Shiotsuki, H. Matsuoka and F. Sanda, Macromolecules, 2011, 44, 3338–3345 CrossRef CAS.
- M. Banno, T. Yamaguchi, K. Nagai, C. Kaiser, S. Hecht and E. Yashima, J. Am. Chem. Soc., 2012, 134, 8718–8728 CrossRef CAS PubMed.
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS.
- D. C. Burns, D. G. Flint, J. R. Kumita, H. J. Feldman, L. Serrano, Z. Zhang, O. S. Smart and G. A. Woolley, Biochemistry, 2004, 43, 15329–15338 CrossRef CAS PubMed.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS PubMed.
- S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef CAS PubMed.
- M. Blanco-Lomas, S. Samanta, P. J. Campos, G. A. Woolley and D. Sampedro, J. Am. Chem. Soc., 2012, 134, 6960–6963 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- M. Irie and W. Schnabel, Macromolecules, 1981, 14, 1246–1249 CrossRef CAS.
- M. Irie, Y. Hirano, S. Hashimoto and K. Hayashi, Macromolecules, 1981, 14, 262–267 CrossRef CAS.
- I. Huc and L. Cuccia, Foldamers, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–33 Search PubMed.
- Y. Zhao and J. S. Moore, Foldamers, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 75–108 Search PubMed.
- R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS PubMed.
- J. Moore and C. Ray, Adv. Polym. Sci., 2005, 177, 91–149 CrossRef.
- E. D. King, P. Tao, T. T. Sanan, C. M. Hadad and J. R. Parquette, Org. Lett., 2008, 10, 1671–1674 CrossRef CAS PubMed.
- C. J. Gabriel and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 13708–13709 CrossRef CAS PubMed.
- D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Angew. Chem., Int. Ed., 1997, 36, 1845–1847 CrossRef CAS.
- D. Zornik, R. M. Meudtner, T. El Malah, C. M. Thiele and S. Hecht, Chem. – Eur. J., 2011, 17, 1473–1484 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed.
- Y. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef CAS PubMed.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem. – Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC.
- S. Lee, Y. Hua and A. H. Flood, J. Org. Chem., 2014, 79, 8383–8396 CrossRef CAS PubMed.
- Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630–3633 CrossRef CAS PubMed.
- S. Hecht and A. Khan, Angew. Chem., Int. Ed., 2003, 115, 6203–6206 CrossRef.
- A. Khan and S. Hecht, Chem. – Eur. J., 2006, 12, 4764–4774 CrossRef CAS PubMed.
- Z. Yu, S. Weidner, T. Risse and S. Hecht, Chem. Sci., 2013, 4, 4156–4167 RSC.
- Z. Yu and S. Hecht, Chem. – Eur. J., 2012, 18, 10519–10524 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2013, 52, 13740–13744 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 313–318 CrossRef CAS.
- R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114–3121 CrossRef CAS.
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- P. D. Kiser, M. Golczak and K. Palczewski, Chem. Rev., 2014, 114, 194–232 CrossRef CAS PubMed.
- M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11–20 CrossRef CAS PubMed.
- A. Tanatani, M. J. Mio and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 1792–1793 CrossRef CAS PubMed.
- J.-L. Hou, X.-B. Shao, G.-J. Chen, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386–12394 CrossRef CAS PubMed.
- M. Inouye, M. Waki and H. Abe, J. Am. Chem. Soc., 2004, 126, 2022–2027 CrossRef CAS PubMed.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed.
- Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867–3870 CrossRef CAS PubMed.
- C. Devadoss, P. Bharathi and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 9635–9644 CrossRef CAS.
- T. Weil, E. Reuther and K. Müllen, Angew. Chem., Int. Ed., 2002, 41, 1900–1904 CrossRef CAS.
- W. R. Dichtel, S. Hecht and J. M. J. Fréchet, Org. Lett., 2005, 7, 4451–4454 CrossRef CAS PubMed.
- H. Sogawa, M. Shiotsuki and F. Sanda, Macromolecules, 2013, 46, 4378–4387 CrossRef CAS.
- T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Science, 2002, 296, 1103–1106 CrossRef PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- G. Singh, H. Chan, A. Baskin, E. Gelman, N. Repnin, P. Král and R. Klajn, Science, 2014, 345, 1149–1153 CrossRef CAS PubMed.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef PubMed.
- Z. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS PubMed.
Footnote |
| † Present address: Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3108, USA. |
| This journal is © The Royal Society of Chemistry 2016 |