
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic foldamer chemistry
Bryden A. F.
Le Bailly
and
Jonathan
Clayden
 *
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: j.clayden@bristol.ac.uk
First published on 2nd March 2016
Abstract
Foldamers can be made more than pieces of static, conformationally uniform molecular architecture by designing into their structure the conformational dynamism characteristic of functional molecular machines. We show that these dynamic foldamers display biomimetic properties reminiscent of allosteric proteins and receptor molecules. They can translate chemical signals into conformational changes, and hence into chemical outputs such as control of reactivity and selectivity. Future developments could see dynamic foldamers operating in the membrane phase providing artificial mechanisms for communication and control that integrate synthetic chemistry into synthetic biology.
 Bryden A. F. Le Bailly | Bryden Le Bailly was born in Cambridge in 1987. He finished his Masters degree at the University of Bristol in 2011 where he developed iron-catalysed cross-coupling reactions with Dr Stephen Thomas. In 2015, he completed his PhD with Professor Jonathan Clayden at the University of Manchester, then worked as a post-doctoral researcher in both Manchester and Bristol. His research interests focused on controlling conformation on a nano scale and building synthetic receptor mimics for the relay of information. Bryden now works as an associate editor at Nature Nanotechnology. |
 Jonathan Clayden | Jonathan Clayden completed a PhD at the University of Cambridge in 1992 with Dr Stuart Warren. After postdoctoral work in Paris with Prof. Marc Julia, he moved in 1994 to the University of Manchester, and in 2015 to the University of Bristol. He has research interests in synthesis and stereochemistry, particularly where conformation has a role to play: asymmetric synthesis, atropisomerism, organolithium chemistry, and synthesising biomimetic function using dynamic foldamers. He has published 250 papers, and is a co-author of the widely used undergraduate textbook “Organic Chemistry” (OUP, 2nd edn, 2013), and “Organolithiums: Selectivity for Synthesis” (Pergamon, 2002). |
Synthetic structures and synthetic function
Nature extracts vast diversity of function from a limited collection of molecular structures. The requirement for the structures of functional molecules to be encoded in genetic information in a parsimonious and evolvable way means that the core features of both structure and reactivity in living systems emerge from simple linear sequences of proteins and peptides. The ribosome is the most marvellous molecular structure in the known universe,1 but it can do only one thing: make polymers of a small collection of α-amino acids. The control of biosynthetic pathways by enzymes means that even the wider chemistry of other primary and secondary metabolites is but an ‘extended phenotype’2 of protein function (Fig. 1).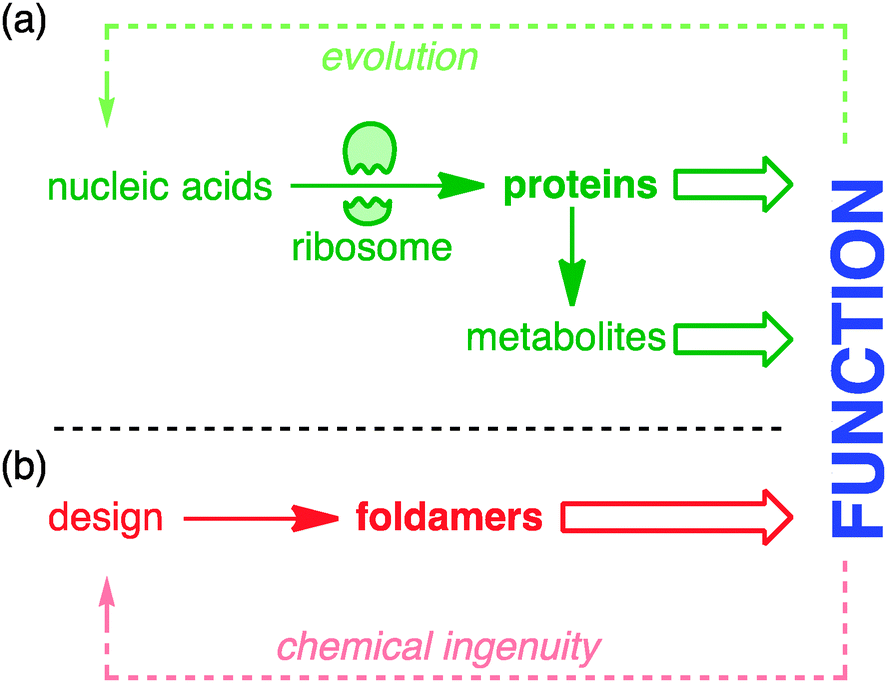 | ||
| Fig. 1 Origin of chemical function in (a) nature and (b) designed systems. | ||
Biological function is honed by Darwinian evolution. Chemical ingenuity is not constrained by evolutionary limitations in such a way.3 In the imagination of a chemist, alternative extended functional molecules can be envisaged built from all kinds of building blocks. But within such huge variety of potential, guiding principles are needed: what molecules to make, and how? For much of the 20th century, chemists took the structures of nature as their lead. The chemical synthesis of proteins and peptides is now routine, and the synthetic construction of the molecules of secondary metabolism without direct recourse to the chemistry of life (the ‘total synthesis of natural products’) reached its zenith in the last decade of the millennium.4 But in parallel a new approach was developing: the direct synthesis of biomimetic function.5 Functional mimicry need not imply structural homology, and molecules were designed and synthesised having some of the key conformational characteristics of their natural counterparts, but built from non-biological precursors. These molecules are foldamers6–10 – extended molecular structures with (in Gellman's definition) ‘a strong tendency to adopt a specific compact conformation’.6 Foldamer chemistry has been immensely successful in mimicking structural motifs such as helices, sheets, columns, and cavities in the solid state, and in many cases also in solution. Well defined foldamer architectures can imitate biomolecular structures by binding guests11–14 or biomolecules,15 or catalysing reactions selectively.16–18
Although a classically ideal foldamer adopts a single conformation, we shall contend in this article that an over-emphasis on conformational uniformity may limit chemists' ability to design and synthesise foldamers that mimic some of the wider functions of biomolecules. Most classes of proteins are not fully rigid,19 and indeed many proteins have segments fully devoid of tertiary structure, or have tertiary structures that become well defined only on binding a ligand.20,21 In several classic examples,22 binding of a ligand induces conformational changes that lend a degree of environmental control to the function of a protein. In allosteric proteins such as haemoglobin, the conformational change induced by binding of one oxygen ligand enhances the protein's affinity for others.22 In membrane-bound receptors of the G protein-coupled receptor (GPCR) class, a change in conformation induced by binding to an appropriate ligand leads to modulation of the affinity of the receptor for its partner G protein.23 In rhodopsin, a switch from E to Z in the geometry of one double bond of a bound ligand (retinal) stimulates switching between alternative alignments of membrane-spanning helices, also inducing binding of the G protein transducin, and leading ultimately to induction of a nerve signal.24
In this review, we describe our response to these aspects of biomolecular function in the design, synthesis and exploitation of foldamers that display dynamic features of conformational switchability and environmental responsiveness. We show how the conceptual range of foldamers may be expanded to include extended molecular structures with a ‘strong tendency’6 to adopt not just one, but multiple favoured conformations, while still retaining structural definition. We shall also review some simple spectroscopic methods that we developed for quantifying that term ‘strong’. Finally we outline some structural features that have been introduced into dynamic foldamer molecules that allow controlled switching between these alternative structures.
Dynamic helical structures
One of the most common structural motifs adopted by foldamers (and indeed by natural biomolecules) is the helix.25–28 This is for good reason: a helix results when a sequence of identical three-dimensional shapes are stacked in such a way that adjacent members of the stack are all related by the same translational vector (Fig. 2).29 Much of the work that has been carried out in the field of foldamer chemistry has been essentially directed towards ensuring the conformational uniformity implied by this repeated translational vector takes on a physical reality. A helix is a chiral structure, and a pair of left and right-handed (M and P) conformations are enantiomeric (and therefore of equal energy) if the helix is built from achiral monomers (Fig. 2a), but diastereoisomeric (and therefore of unequal energy) if the helix is built from chiral monomers (Fig. 2b). Natural biopolymers, and many synthetic foldamers, are built from enantiomerically pure chiral monomers, and hence adopt preferentially one of these two (right-handed, in the case of protein α-helices and a typical DNA double helix; left-handed, in the case of Z-DNA and collagen)30–32 diastereoisomeric conformational alternatives.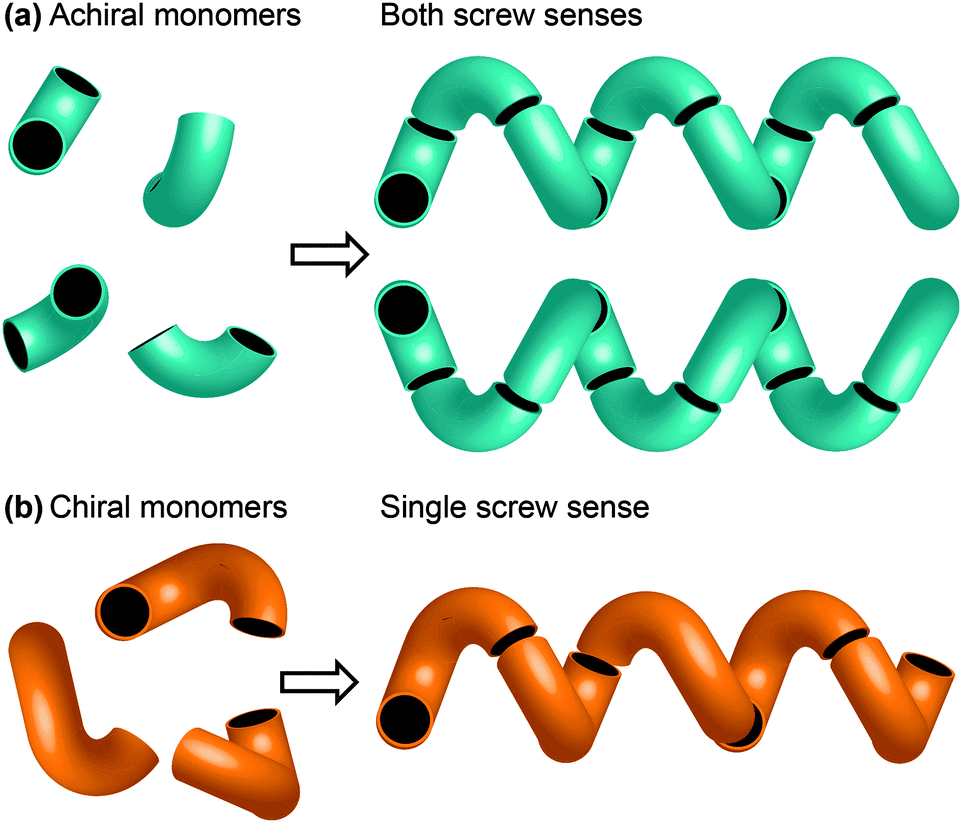 | ||
| Fig. 2 (a) A helix built from achiral monomers can adopt either of two enantiomeric screw senses; (b) a helix built from chiral monomers adopts a single screw sense. | ||
Although they are far less abundant than helical oligomers of chiral monomers (for reasons that we plan to discuss in detail elsewhere), a number of helical polymers and oligomers built from achiral monomers have been described.7,28 Some of these (polyisocyanides,33 and longer polyamides of aromatic amino acids34 for example) exist as stable, separable left- and right-handed screw-sense enantiomers and are chiral structures, even in solution. Others (aromatic polyureas for example35) are helical in the solid state but tend to collapse into random coils in solution.
Importantly, though, a few classes of achiral polymers retain helicity in solution while interconverting rapidly (on a timescale of seconds or less) between their enantiomeric conformers.36 We call these inverting helices ‘configurationally achiral’, because even though their global conformation is chiral, they contain no configurationally stable elements of stereochemistry (Fig. 3). Configurationally achiral helices include polyisocyanates (Fig. 4a),37 shorter polyamides of aromatic amino acids (Fig. 4b),38 polyamides of the achiral aliphatic amino acids 2-aminoisobutyric acid (Aib) (Fig. 4c)39 and related achiral quaternary α-amino acids,40 and polyamides containing alternating Aib and didehydrophenylalanine (ΔPhe) residues (Fig. 4d).41 Oligomers of Aib typically adopt a 310 helical geometry42–44 consisting of repeated β-turns, and Fig. 4e illustrates the three-and-a-half helical turns of the screw-sense labile but reliably helical conformation adopted by an achiral Aib undecamer N3Aib11OCH2CH2SiMe3.45 Related configurationally achiral supramolecular helical structures are also known.27,46–48
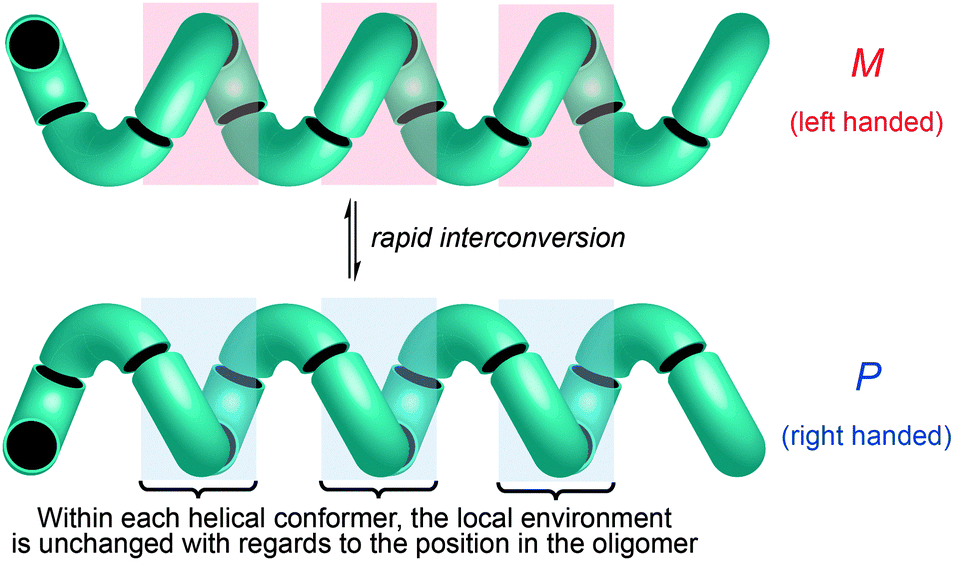 | ||
| Fig. 3 The pair of rapidly interconverting enantiomeric screw sense conformers of a configurationally achiral helix. Local environment within each conformer is unchanged with respect to displacement along the axis of the helix. | ||
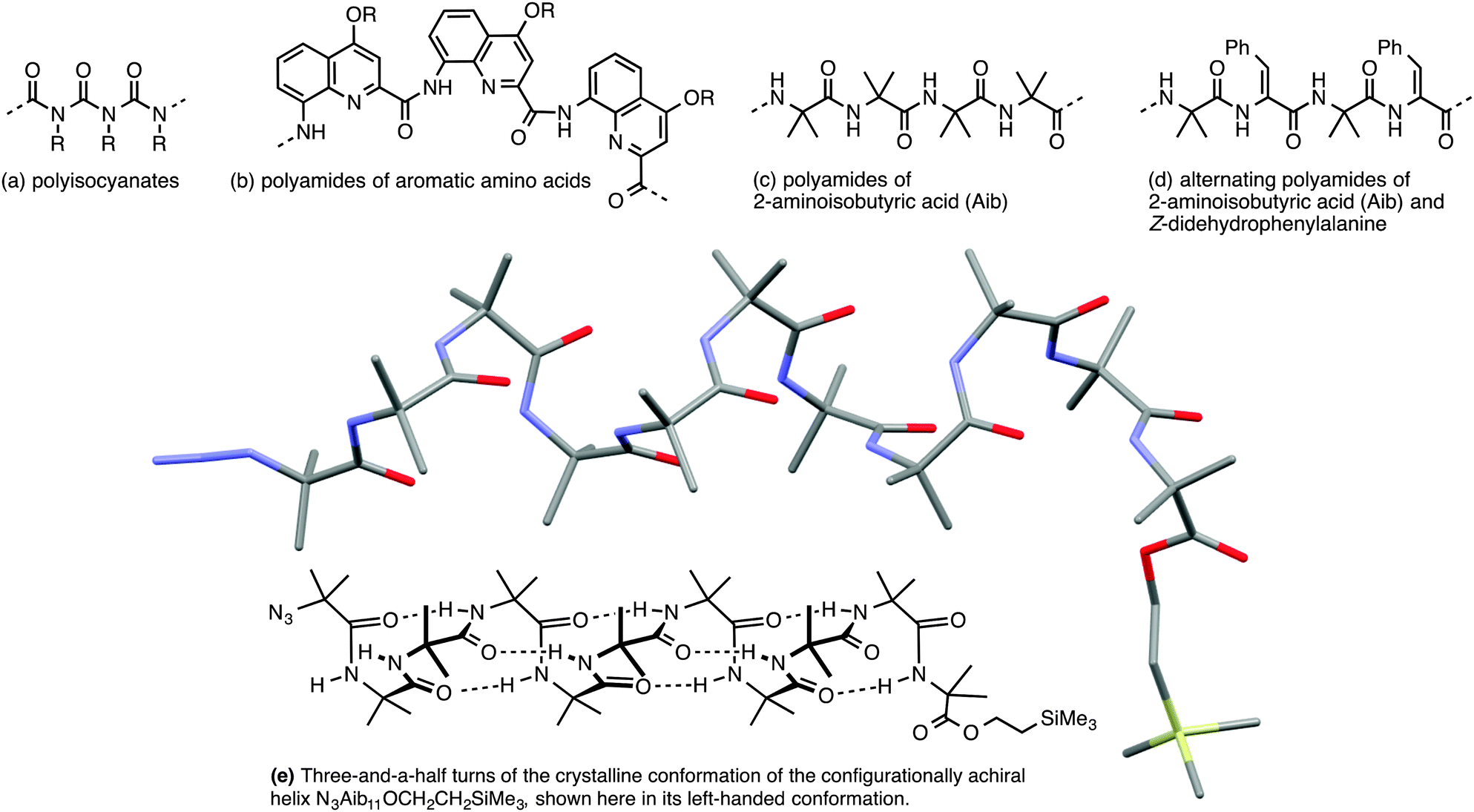 | ||
| Fig. 4 Some oligomers forming configurationally achiral helices: (a) polyisocyanates; (b) oligoamides of aromatic amino acids; (c) oligomers Aibn; (d) oligomers (Aib–ΔPhe)n; (e) a conformational representation and X-ray crystal structure of the 310 helix formed by N3Aib11OCH2CH2SiMe3.45 | ||
These dynamic helical structures have a valuable property that we may loosely term ‘line chirality’ (by analogy with the term ‘point chirality’). This is to say, within each conformer every point in the helix experiences the same local chiral environment, irrespective of its linear coordinate along the axis of the helix (Fig. 3). And, importantly, inversion of helical screw sense in a structure of complete helical fidelity (that is, one in which only uninterrupted helical conformations are populated) likewise inverts the local chiral environment of every point within the helix. A dynamic, screw-sense invertible helix may thus in principle be used as a communication device: conformational uniformity ensures that the adoption of a particular helical screw sense by any point in the helix must be communicated to all other points in the helix.
Circular dichroism (CD) spectroscopy has generally been the technique of choice for the study of helical oligomers in solution: the repeated geometric relationship between chromophores in adjacent monomers gives rise to an identifiable Cotton effect associated with a specific detailed helical geometry.49 However, for configurationally achiral helices, in which left and right-handed screw senses are equally populated, the Cotton effects cancel and no CD spectrum can be observed. This has limited chemists' ability to observe directly the solution-state conformations of helices built purely from achiral monomers. The barriers to inversion of screw sense in achiral polyisocyanates37 (ΔG‡ ≈ 90 kJ mol−1 at 100 °C, Fig. 4a) and oligo-Aib amides50 (ΔG‡ ≈ 46 kJ mol−1 at −8 °C, Fig. 4b) were established by dynamic NMR studies.
These and other helical but achiral oligomers (such as oligomers of aromatic achiral amino acids) trivially fail to conform to Gellman's definition6 of a foldamer: they populate two (enantiomeric) conformations rather than one. This may seem a quibble, but it is a conceptually fecund one, because the equal populations of screw-sense conformers of a configurationally achiral parent oligomer may be usefully perturbed by the introduction of a relatively small chiral conformational influence. In the polyisocyanates, even asymmetric isotopic substitution in a fraction of the monomers (the ‘sergeants and soldiers’ principle)51 or attachment of a (photochemically switchable) chiral terminator unit52 is sufficient to bias the helical conformation towards one screw sense.53,54 In oligo-Aib structures (Fig. 4c),55–57 and their close relatives,58,59 including the didehydrophenylalanine-containing oligomers studied by Inai and coworkers (Fig. 4d),60–63 a single chiral terminal or medial residue is sufficient to induce some degree of preferred screw sense,57 as is a hydrogen-bonded interaction with a chiral partner.41,64–67
Because these dynamic helices can exhibit helicity with or without a preferred screw sense, we find it productive to distinguish clearly between these two terms. A compound displays ‘helicity’ if it adopts a helical secondary structure (rather than a linear, zig-zag, or random coil alternative); we use the term ‘screw sense’ to refer to each enantiomeric conformation of that overall helical secondary structure. Thus ‘helicity preference’ (helical vs. random coil conformations for example) may be distinguished from ‘screw-sense preference’ (M vs. P screw sense): the achiral helices of Fig. 4 have a powerful helicity preference but exhibit no screw-sense preference.
NMR quantifies conformational preference in dynamic foldamers
Detailed NMR studies of the structures of achiral Aib oligomers bearing terminal chiral substituents allowed us to quantify accurately the level of screw-sense control induced in the helix by different chiral terminal residues at either the N39,68 or the C terminus.69 Values of screw-sense preference had previously been derived in other oligomers by comparing CD spectra with compounds supposed to have a single enantiomeric preference,70,71 or by using NMR to measure ratios of diastereoisomeric conformers interconverting slowly on the NMR timescale.38 Our quantitative NMR approach was made possible by a spectroscopic feature displayed by two ‘reporter’ groups, A and B, embedded in a helical but configurationally achiral oligomer (Fig. 5a). In the chiral environment of the helix, A and B become diastereotopic, experiencing different chemical environments and giving distinct signals in an NMR spectrum (13C in the case of A = B = CH3; 1H in the case of A = B = H; 19F in the case of A = B = CH2F or FC6H4). If the screw sense of the helix inverts slowly on the NMR timescale, the two signals will be resolvable, with chemical shift difference Δδslow. If the helix inverts rapidly on the NMR timescale (typically rates of 0.1–1 × 103 Hz) the two signals will coalesce and give rise to a single signal. Fig. 5b shows this effect in the 13C NMR spectrum of an achiral Aib9 helix, in which the middle residue (Aib**) is 13C labelled in both of its methyl groups, on raising the temperature from 233 K through the coalescence temperature to 273 K.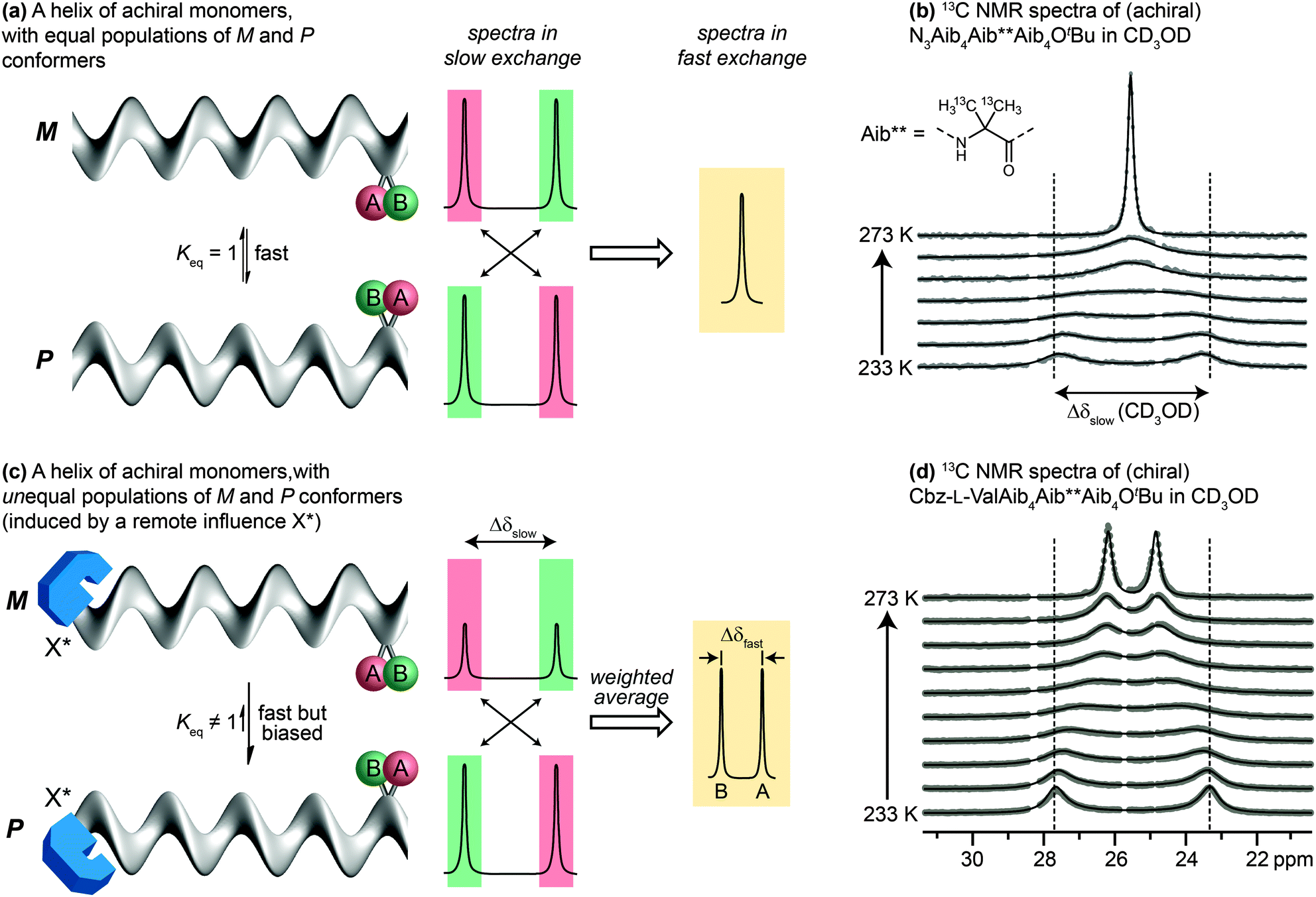 | ||
| Fig. 5 Detection of screw-sense preference by NMR in the fast exchange regime. (a and b) Coalescence between signals in an equally populated pair of enantiomeric conformers; (c and d) coalescence between signals in an unequally populated pair of diastereoisomeric conformers. | ||
The situation is subtly different if the two screw-sense conformers of the helical oligomer are in equilibrium but are not equally populated – for example by virtue of a covalently or non-covalently bound chiral influence that affects the screw-sense conformer distribution but does not directly influence the chemical shift of A and B (represented schematically in Fig. 5c, and with an example in Fig. 5d). In such an event, the slow exchange spectrum will be identical with the equally-populated system, but at fast exchange weighted averaging leads not to a single signal but to two new signals, separated by a new chemical shift difference Δδfast. Fig. 5d illustrates the change in line shape of the 13C NMR signals in oligomer Cbz-L-ValAib9Ot-Bu 13C-labelled in its middle Aib residue, on raising the temperature from 233 K to 273 K.
Because the two screw-sense conformers P and M contributing to the fast exchange spectrum are present in relative populations of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Keq,
:Keq,
| Δδfast/Δδslow = (Keq − 1)/(Keq + 1) |
| Δδfast/Δδslow = ([M] − [P])/([M] + [P]) |
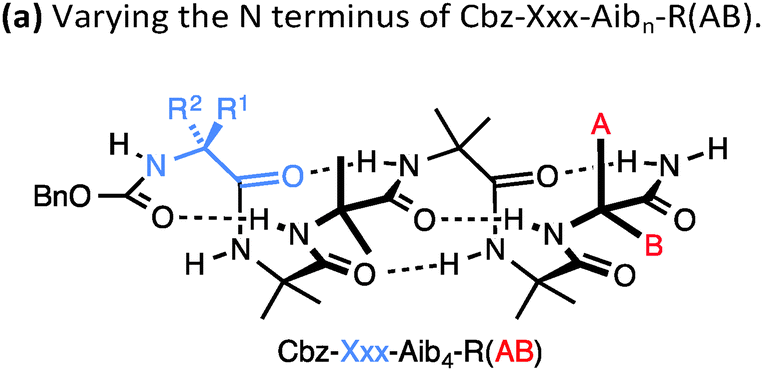
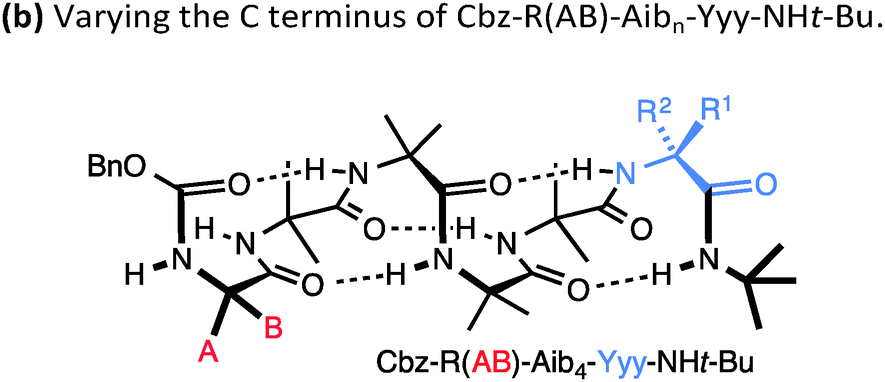
A drawback of this NMR method using a geminal pair of reporter groups in an achiral residue is that it cannot report on whether an M or a P helix is preferred—in other words, the sign of the screw-sense preference remains undetermined. However, CD studies correlated with data from the enantiomerically enriched, isotopically labelled 13C NMR probe discussed below30 have allowed the major screw sense to be assigned with confidence in most cases. Some values for the screw-sense preference induced by covalently linked residues at the N or C terminus are summarised in Table 1.
|
|
|
||
|---|---|---|---|
| Xxx = | h.e. | Yyy = | h.e. |
| Cbz-L-Ser | 4 | L-Ala-NHt-Bu | +98 |
| Cbz-L-Ala | −20 | L-Val-NHt-Bu | +94 |
| Cbz-L-Leu | −33 | L-Phe-NHt-Bu | +92 |
| Cbz-L-Val | −35 | L-tert-Leu-NHt-Bu | +85 |
| Cbz-L-Ser(OTBDPS) | −42 | L-Ser(Ot-Bu)-NHt-Bu | +26 |
| Cbz-L-tert-Leu | −44 | L-Pro-NHt-Bu | +21 |
| Cbz-L-Pro | −45 | L-α-methylvaline-NHt-Bu | +92 |
| Cbz-L-Phe | −52 | (L-α-methylvaline)2-NHt-Bu | +100 |
| Cbz-L-isovaline | +25 | L-Ala-Ot-Bu | −70 |
| Cbz-L-α-methylphenylalanine | +56 | L-Phe-Ot-Bu | −44 |
| Cbz-L-α-methylvaline | +68 | L-Val-Ot-Bu | −60 |
| Cbz-(L-α-methylvaline)2 | +95 | L-tert-Leu-Ot-Bu | −46 |
| L-α-methylvaline-Ot-Bu | −56 | ||
Conformational persistence and signal decay
The values for helical excess (h.e.) obtained from these studies can be considered comparable in meaning to the CD-derived screw-sense excess (s.e.) values reported by Suginome.70,71 However, unlike CD, NMR reports the conformational preference at a single site in the oligomer chain.77 For this reason, it can be used not only to quantify global conformational preferences, but also to assess the variation of conformational preference within a foldamer chain. By synthesising a series of oligomers carrying 13C labels at each position in a chain of Aib residues,78 we were thus able to observe the way that an initial screw-sense preference induced locally by a chiral residue decays exponentially (but remarkably slowly) as the chiral influence becomes more distant (Fig. 6).76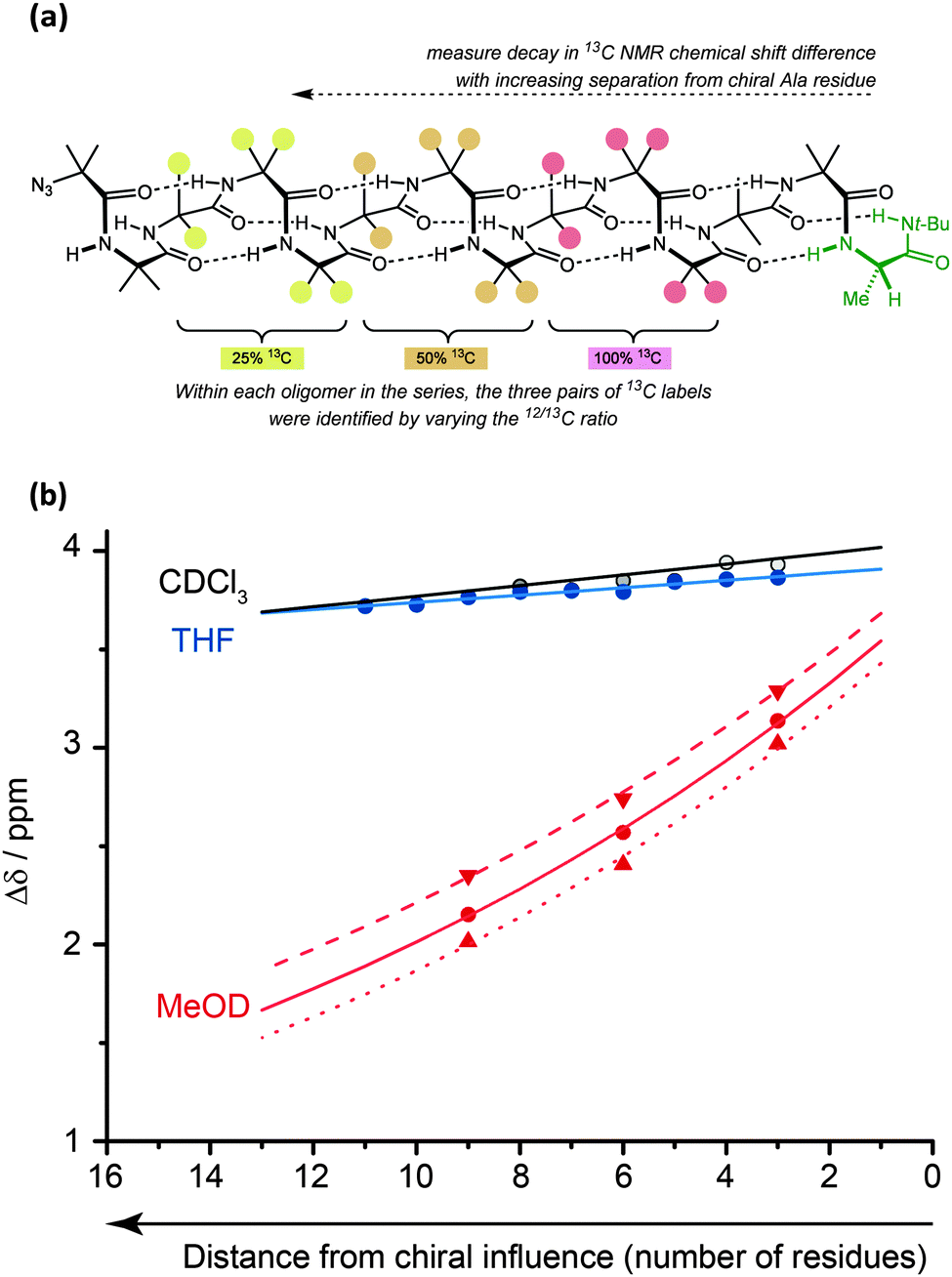 | ||
| Fig. 6 The screw-sense preference exhibited by an Aib oligomer (as reported by chemical shift difference Δδ) decays as a terminal chiral influence becomes more distant. (a) Chemical shift differences were measured in a series of three ‘frame-shifted’ oligomers each containing three pairs of abundance-labelled (100%, 50% or 25%) 13C labels. (b) Decay is more rapid in polar solvents and at higher temperature (dashed lines 0 °C; solid lines 23 °C; dotted lines 40 °C). | ||
The spatial decay constant is at a minimum in non-polar solvents, but is increased by solvents with more powerful dipoles or hydrogen-bonding properties. Quantitative comparisons can be made: in THF, at 23 °C, there is only a 0.5% drop in helical excess at the chain terminus each time the chain is extended by a further Aib residue, while the equivalent value in MeOH is 6.1%. A separate study79 measured the ability of a range of alternative achiral amino acids to ‘communicate’ a screw-sense preference between two helical domains and found that in MeOH quaternary amino acids communicated a conformational preference highly effectively (>90% fidelity), while a single Gly or β-alanine led to loss of about 50% of the conformational signal. More bulky achiral quaternary amino acids (e.g. 1,1-diphenylglycine) disrupted the helical conformation and prevented communication of helical screw sense. Similar NMR measurements should be possible in other achiral helical foldamers containing paired enantiotopic signals, provided NMR spectra can be measured in the fast exchange régime.
This decay in screw-sense preference must arise from the intrusion of occasional non-helical conformational features into the overall helical structure of the molecule – an indication of just how ‘strong’ its ‘strong preference’6 for helicity is. These conformational features are presumably localised, rare helix reversals that occur transiently. Their detailed structure is of interest because they offer an insight into the mechanisms of screw-sense reversal in helical foldamers, and are consequently the subject of further studies currently under way.80
Nature apparently also makes use of the conformational properties of dynamic foldamers which are reliably helical but in which screw sense is poorly controlled. Part of the structural inspiration for our work in this area was the observation that the fungal metabolites known as peptaibiotics81 apparently adopt well defined screw senses in the solid state, despite the fact that parts of their structure contain relatively few chiral residues embedded in a largely achiral chain. The solid state structure of cephaibol C (Fig. 7) and cephaibol A contain at their N terminus a stretch of right-handed 310 helix in which only the N-terminal Phe residue and the Leu at residue 7 are chiral.82 During our work on conformational preferences in structures such as those illustrated in Table 1, we noted that an N-terminal L-amino acid adjacent to a sequence of Aib residues does not typically induce a right-handed screw sense,30,83 and in a sequence Ac-L-Phe-Aib4-Gly-L-Leu⋯ the L-Phe and L-Leu residues are expected to be mismatched in their conformational properties, inducing opposite screw senses and disfavouring the adoption of a uniform conformation. To test this hypothesis, we made four ‘cephaibologues’—diastereoisomers of the N-terminal nonapeptide of the cephaibols (Fig. 7). We found that indeed the diastereoisomers A and D that correspond most closely to the relative configuration of the natural structures are less conformationally uniform than the unnatural diastereoisomers.84 The Trichoderma fungi producing the cephaibols seem to have evolved to produce compounds which have dynamic conformational properties, possibly because these properties enhance the membrane activity required for them to function as antibiotics.45
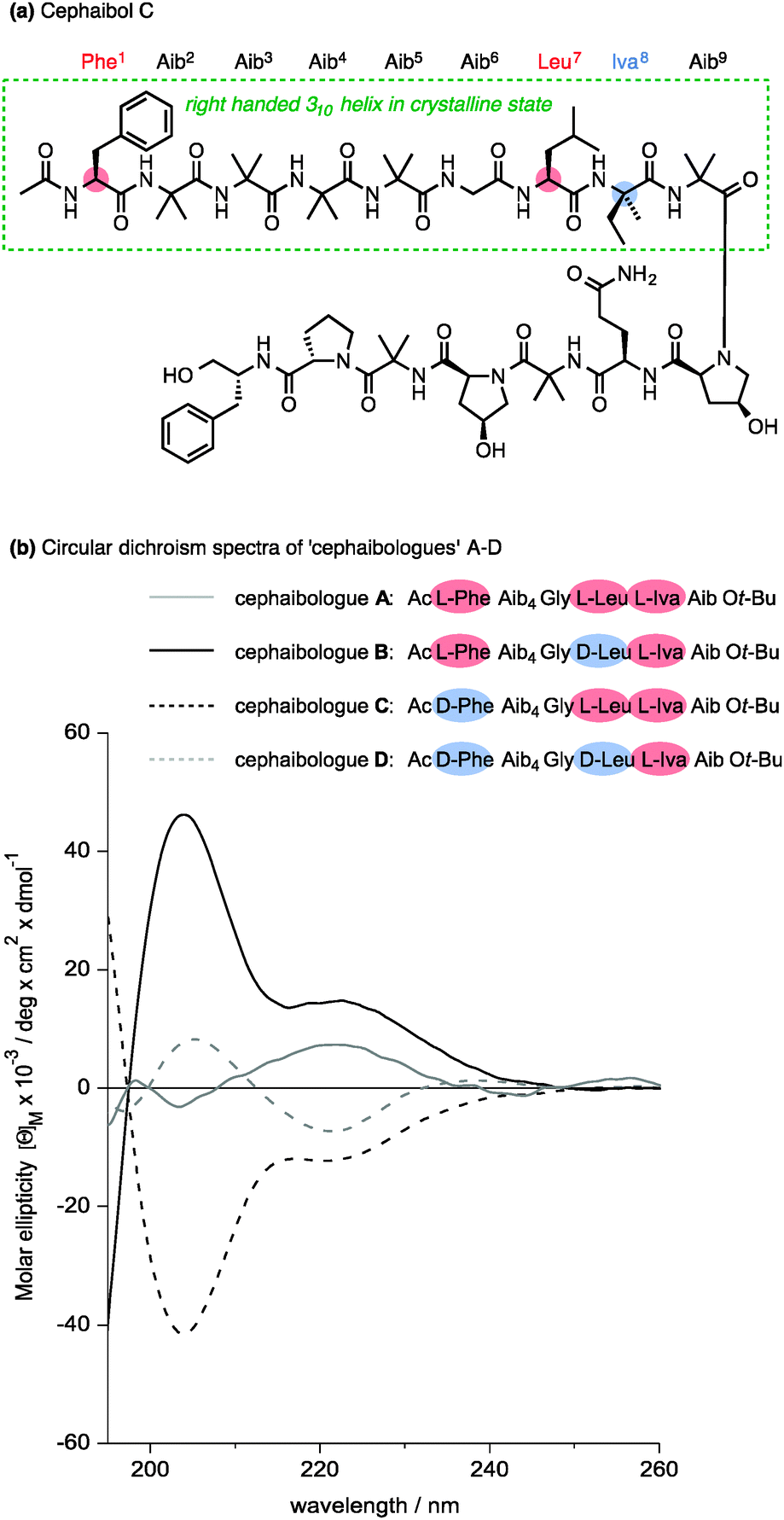 | ||
| Fig. 7 (a) The structure of cephaibol C, and (b) the CD spectra of four diastereoisomeric ‘cephaibologues’ corresponding to its N-terminal nonapeptide sequence. The most conformationally well defined (B and C) are diastereoisomers of the less well defined structures A and D whose structures match most closely the relative configuration of cephaibol C itself. | ||
Dynamic screw sense switching detected by 13C NMR
In the case of 13C labelled Aib foldamers, an additional piece of conformational information may be gained by the incorporation of an isotopic 13C label enantioselectively into one of the two geminal methyl groups of an Aib residue.85 The pro-R methyl group of an Aib residue embedded in a right-handed helix appears reliably at higher field than the pro-S residue86 (though the locations are inverted in an esterified C-terminal Aib residue). Thus, the major 13C signal from a single 13C-labelled residue R-Aib* in a right-handed helix will appear as the upfield member of the diastereotopic pair (so, memorably, R@P = ‘on the right’). While the chemical shift separation of the two signals reports the magnitude of the local screw-sense preference of the oligomer, the location of the major 13C NMR signal in the upfield or downfield signal therefore indicates the left or right-handed chirality of the screw-sense preference at that point in the oligomer.30Thus the starting chiral oligomer shown in Fig. 8, which is preferentially labelled with 13C in the pro-R methyl group of its C-terminal Aib* residue, displays in its 13C NMR spectrum a pair of signals separated by 0.75 ppm. This indicates that it adopts a screw-sense preference of about 33% h.e. In addition the major peak is upfield of the minor, confirming that the helix is an interconverting mixture of which the predominant conformer is left handed (the label being in a C-terminal ester).87 The N-terminal ester-bearing stereogenic centre is however also invertible: methanolysis to the alcohol and treatment under Mitsunobu conditions gives its enantiomer. The 13C NMR spectrum clearly indicates that a local inversion of conformational preference has taken place at the C terminus of the oligomer. Inversion of the configuration of the single controlling centre in the oligomer leads necessarily to inversion of the screw-sense preference of the equilibrating mixture, and information about this stereochemical inversion is transmitted through the induced conformational preference in the helix.
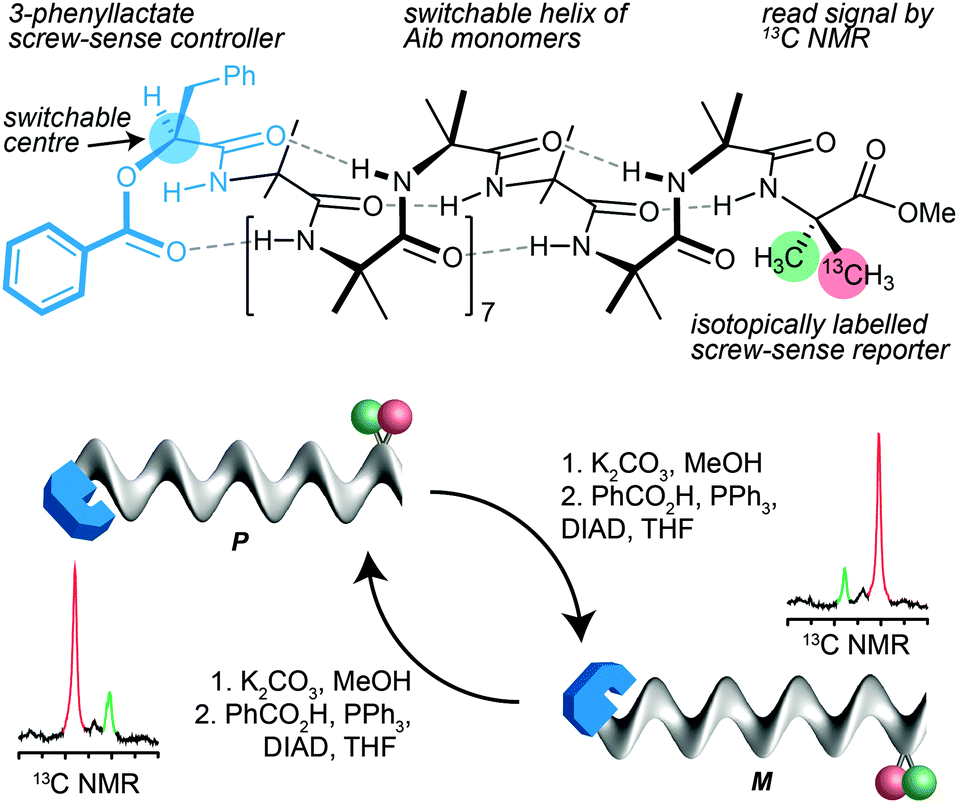 | ||
| Fig. 8 The stereochemical consequence of inversion of configuration at an ester-bearing centre is detected by a remote 13C NMR reporter of conformational preference. The two diastereotopic methyl groups of the C-terminal labelled Aib residue are differentially enriched in 13C (pro-R in red 75% 13C; pro-S in green 25% 13C). Inversion of screw-sense conformation that results from a remote inversion of N-terminal configuration exchanges their positions in the 13C NMR spectrum. Note that in this case, the major signal is (unusually) upfield in an M helix because the R-Aib* residue is a C-terminal ester. | ||
A similar dynamic inversion of screw-sense preference can be achieved by exploiting the fact that the functionality at the C terminus of an Aib oligomer affects local secondary structure in such a way that the presence or absence of a single hydrogen bond can lead to a reversal of screw-sense preference. This feature, first observed in the tendency of C-terminal esters to exhibit reversed screw-sense preference relative to C-terminal secondary amides69 (see Table 1), was exploited in a pH-dependent switch88 in which the removal of a hydrogen bond by base-promoted deprotonation led to inversion of screw-sense preference. The C-terminal NH group in an Aib oligomer was selectively acidified by amidation with a tren ligand and complexation with a Lewis acidic Zn2+ cation (Fig. 9a and b).89 Sequential addition of OH− (Fig. 9c and e) and H+ (Fig. 9d and f) ions caused repeated switching of the foldamer between a predominantly right-handed and a predominantly left-handed conformation.
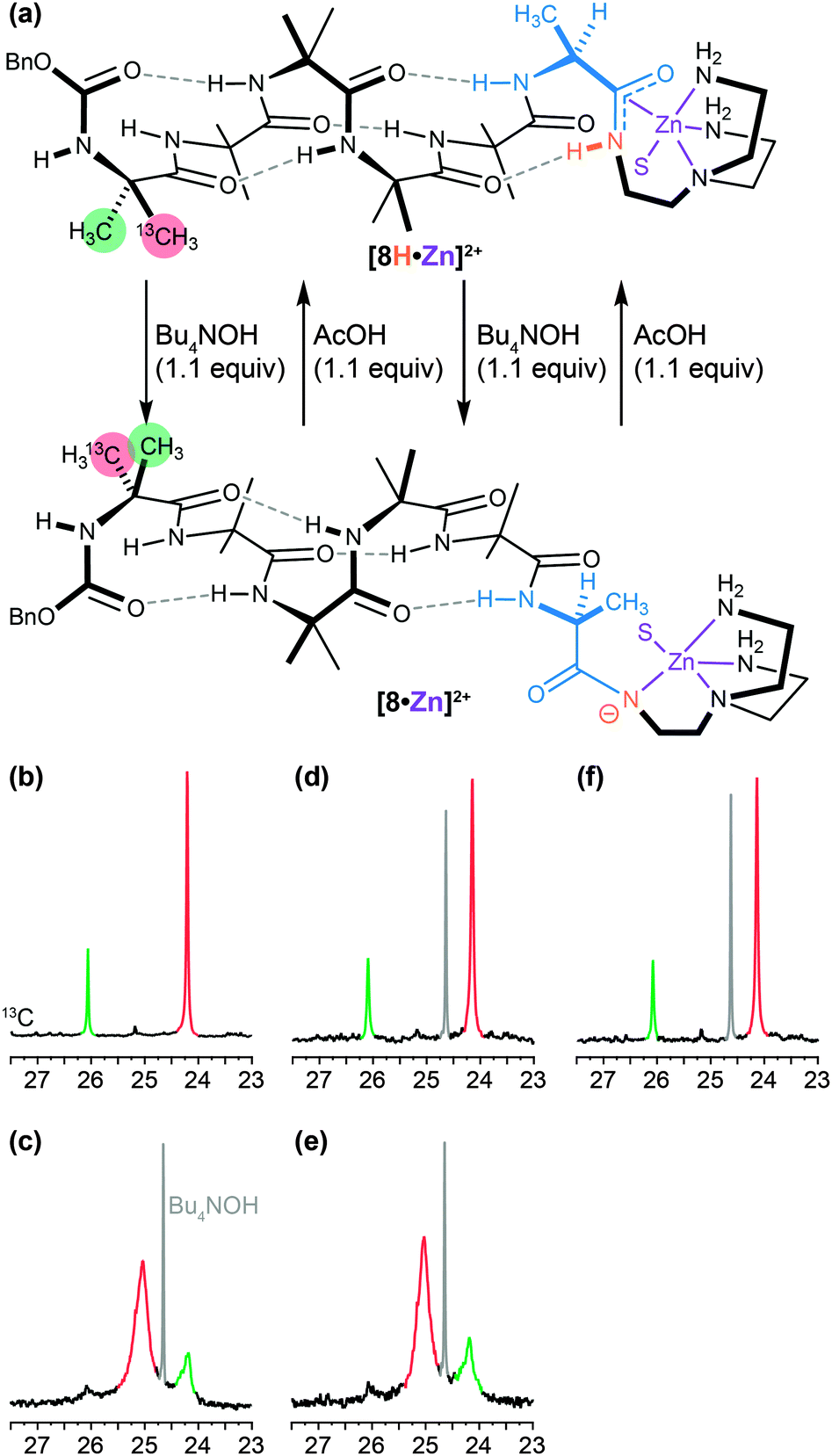 | ||
| Fig. 9 (a) A C terminal tren-Zn moiety acidifies an adjacent NH group (orange) in a dynamic foldamer allowing it to be selectively deprotonated and reprotonated by base and acid. The two diastereotopic methyl groups of the N-terminal labelled Aib residue are differentially enriched in 13C (pro-R in red 70% 13C; pro-S in green 30% 13C). (b–e) 13C NMR spectra illustrating (b) the right handed screw sense of the original foldamer; (c) the left handed screw sense induced on deprotonation by base; (d) the right handed screw sense restored by addition of acid; (e) and (f) further screw sense inversions during a second cycle of base/acid treatment. | ||
A dynamic foldamer as a biomimetic artificial receptor
In a typical biological receptor, a conformational change results from the reversible binding of a ligand with a binding site.90 An artificial mimic of this function would provide an important link between synthetic chemistry and synthetic biology, and offer the possibility of designing complex artificial molecular communication systems. In our early investigations, we chose to ensure an intimate connection between ligand and receptor by using reversible (dynamic) covalent chemistry. Boronic acids are well known to form, reversibly, cyclic boronate esters by condensation with diols.91 A series of Aib*-labelled oligomers were made carrying an amino-boronate function at their N terminus (Fig. 10a).92 In the absence of any diol ligand, 11B NMR indicated that in deuterated methanol the receptor mimic adopted a cyclic azaborolidine structure at the N terminus, and 13C NMR reported, as expected, no screw-sense preference in the achiral foldamer. Adding a chiral diol ligand led to spontaneous formation of the cyclic boronate ester, whose combined steric bulk and Lewis acidity promoted the formation of the methanol-bridged boronate structure. Additionally, with a chiral ligand such as (−)-diisopropyl tartrate (DIPT) a preferred left-handed screw sense was induced in the Aib oligomer that was detectable in the 13C NMR signals of the remote enantioselectively labelled Aib* residue (Fig. 10c). By contrast, an achiral ligand (the meso diastereoisomer of a diol for example) was unable to induce a screw-sense preference.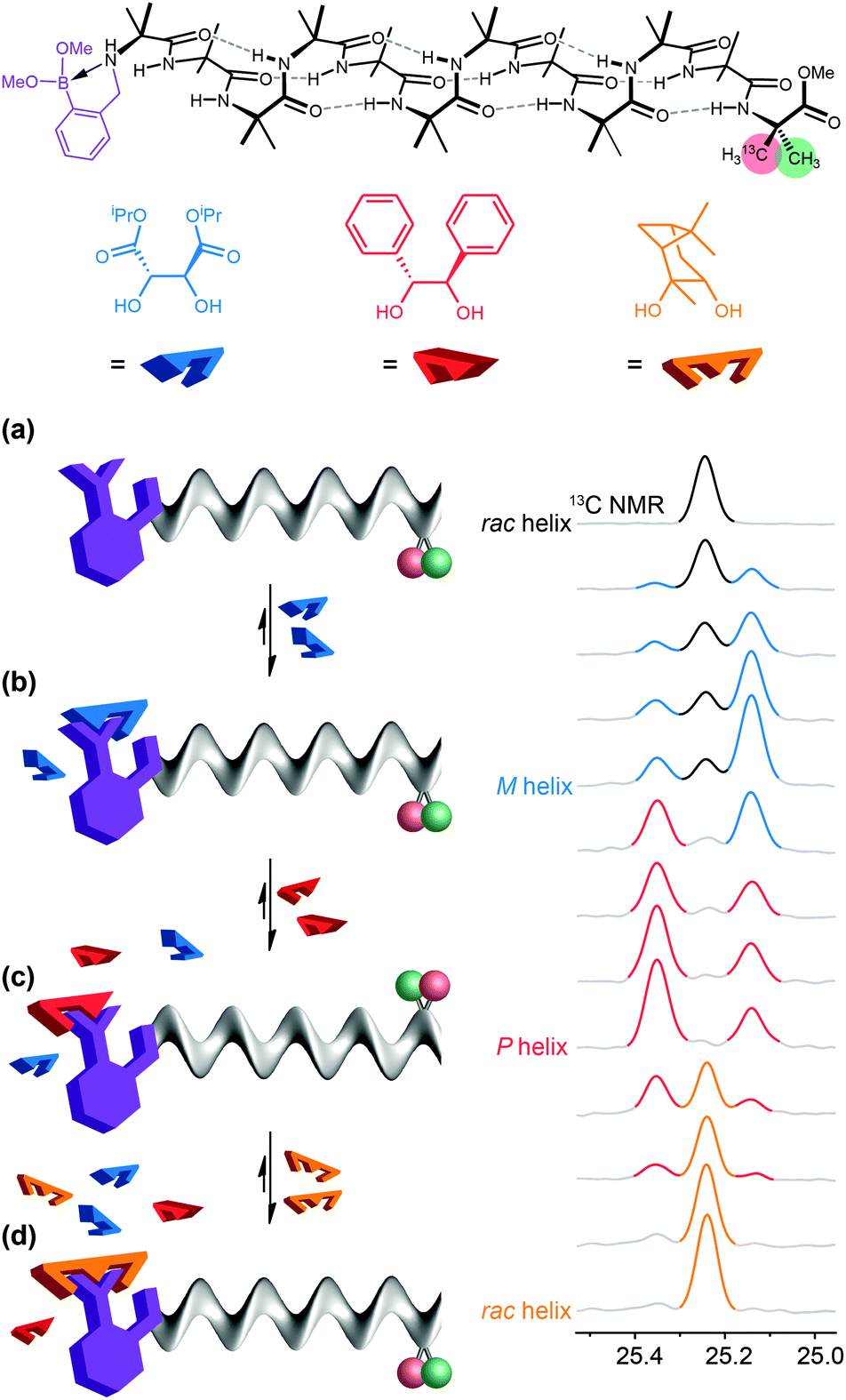 | ||
| Fig. 10 (a) A dynamic foldamer carrying an N-terminal boronate ester bonding site (purple) and a C-terminal labelled Aib residue are differentially enriched in 13C (pro-R in red 75% 13C; pro-S in green 25% 13C). (b) A cartoon of the receptor in its resting state. (c) Reversible esterification with a weakly binding ligand (−)-diisopropyl tartrate (DIPT, blue), K = 300 M−1, induces a left-handed screw-sense preference. (d) A more strongly binding ligand (+)-hydrobenzoin (red), K = 1500 M−1, acts a competitive antagonist, displacing the (blue) DIPT from the binding site and inducing a right-handed screw-sense preference. (e) An even more strongly binding ligand, pinanediol (orange) with K ≫ 2000 M−1 acts as an irreversible inhibitor, displacing all other ligands, and turning the screw-sense preference off. | ||
Just as ligands might vary in their binding strength at a receptor binding site, different diols displayed different equilibrium constants for formation of boronate esters under the conditions of the experiments. The characteristic binding strength of different ligands could hence be exploited in a series of experiments in which sequential ligands compete for the same binding site. After adding 2 equiv. of (−)-DIPT (K = 300 M−1) to the receptor (which induced a predominantly left-handed screw sense) the addition of 2 equiv. of the more strongly binding (+)-hydrobenzoin (K = 1500 M−1) led to the displacement of DIPT from the binding site and the consequent inversion of the screw sense in the receptor to a right-handed preference, indicated by an exchange in position of the major and minor peaks in the 13C NMR spectrum (Fig. 10d). (+)-Hydrobenzoin thus acts as a competitive antagonist of (−)-DIPT. Even stronger binding was exhibited by (−)-pinanediol (K ≫ 2000 M−1). Although chiral, pinanediol induced no screw-sense preference, probably because its greater steric hindrance prevents any interaction of the aminoboronate N atom with B. Pinanediol is an irreversible inhibitor of the artificial receptor. The further biomimetic credentials of the boronate-based receptor were evident in its ability to respond even to biological messengers such as adenosine (K ≈ 625 M−1) and guanosine (K ≈ 600 M−1), which both induced a left-handed screw sense, and uridine and cytidine (K > 2000 M−1) the last of which induced a right-handed screw sense.
Dynamic modulation of foldamer conformation by competitive noncovalent binding in a multicomponent system
Despite the biomimetic character of the reversible and selective ligand binding at this ‘purinergic’ receptor, it nonetheless employs non-biomimetic reversible covalent bonding. Typical biological receptors bind ligands through non-covalent interactions. Inai has shown that achiral oligomers carrying basic binding sites exhibited a conformational response to binding whose strength depended on the nature of the hydrogen bonding between the ligand and the receptor.41,64–66 Advancing this concept, we sought a basic binding site that displayed selective 1:1 binding interactions with acidic ligands, and found that the 2-pyridylacetamide-terminated oligomers illustrated in Fig. 11 showed this property in the presence of either carboxylic or phosphoric acid ligands (Fig. 11a).93 Adding a chiral phosphoric acid to the foldamer induced a switch to a preferred screw-sense preference, evident in the induced 13C NMR chemical shift difference in the C-terminal Aib** labels (Fig. 11b). Adding ammonia to the mixture disrupted the interaction with the chiral acid, turning off the screw-sense preference. Adding hydrochloric acid restored the effect of the chiral ligand (Fig. 11b). Complete transfer of a proton from the acid to the basic binding site to form ion pairs in chloroform is unlikely, so the reversibility of the influence of the chiral acid ligand on the receptor carrying the basic binding site is probably due to competitive hydrogen-bonding interactions of different strengths between the various acidic and basic species in solution.
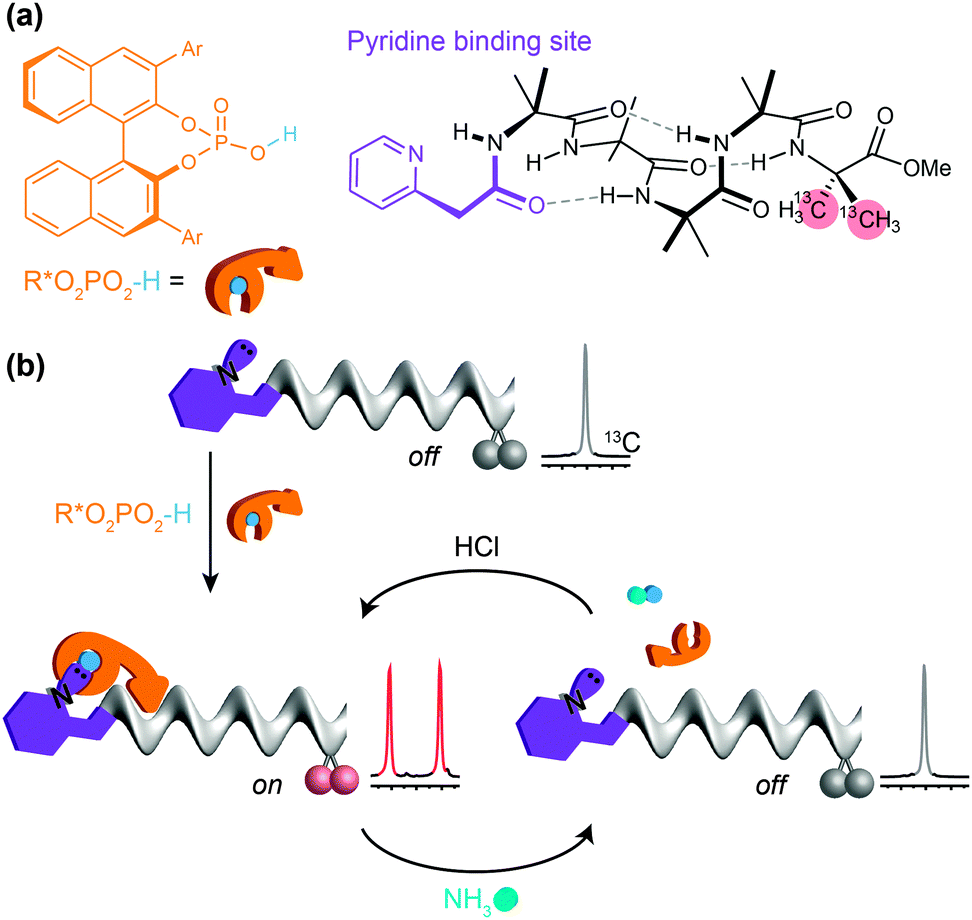 | ||
| Fig. 11 (a) A 13C-labelled dynamic foldamer bearing an N-terminal basic binding site, and a chiral phosphoric acid as a ligand. (b) The hydrogen-bonded complex between the phosphoric acid and the foldamer induces a preferred screw-sense preference that may be turned off and on by addition of ammonia and HCl. | ||
By choosing acids with identifiably characteristic effects on the conformation of the receptor (strongly left-handed, strongly right-handed, or weakly left-handed) we found that we could force a receptor's conformation to cycle between these different preferences either by adding increasingly strong acids (a carboxylic acid, a phosphoric acid, or a triflamide) to the receptor (Fig. 12).93 Even more intriguingly, a mixture of acids of different strengths could also be selectively silenced or activated by addition of increasing amounts of base, with the sequence reversed by acid. Fig. 12 shows how a four-component mixture of receptor and three acids behaves as a chemical system capable of ‘counting’ the number of protons available to it and reporting an output (a left- or right-handed screw-sense preference) as a result.
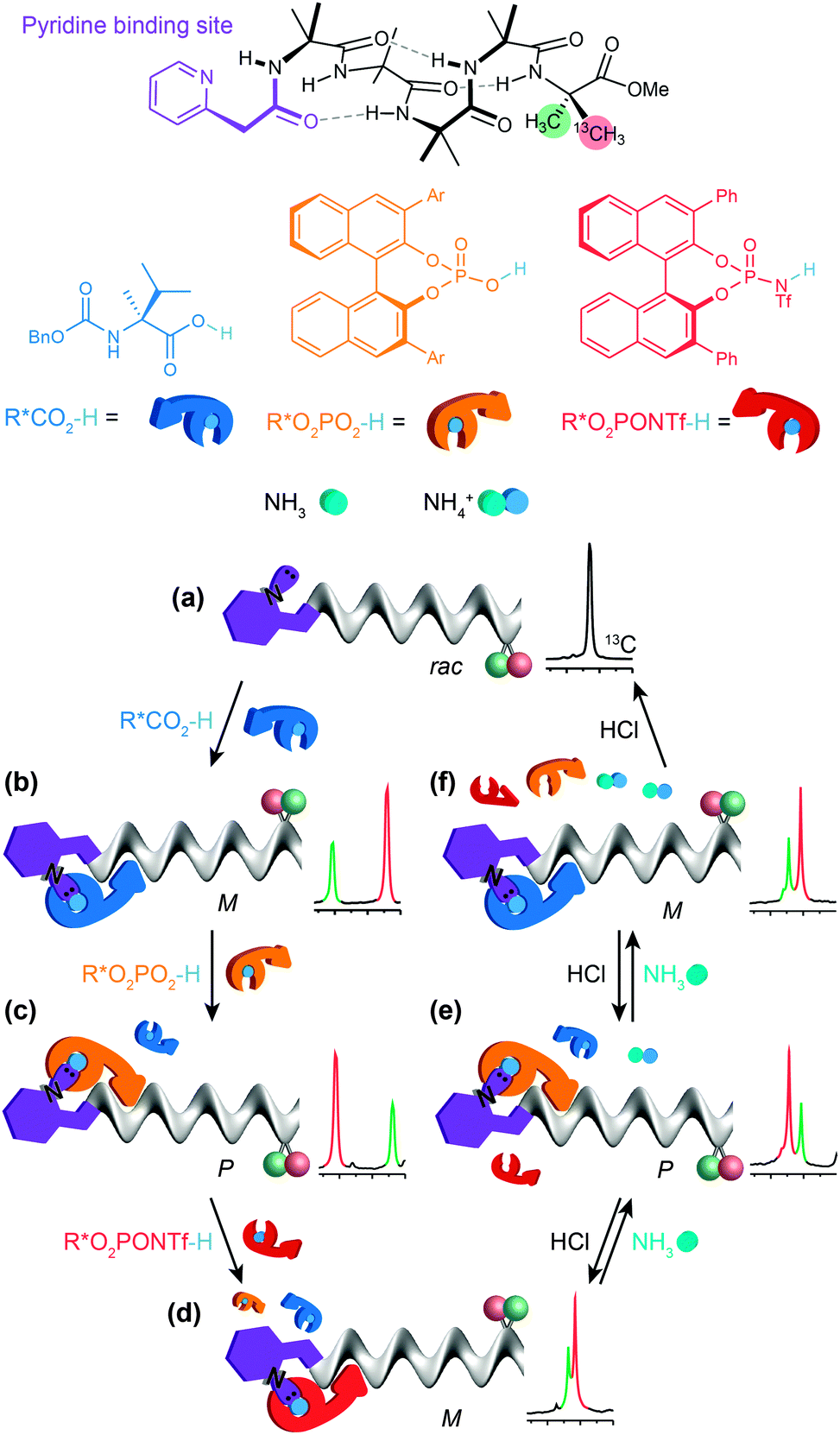 | ||
| Fig. 12 (a) A dynamic foldamer carrying a 13C NMR reporter of conformational preference and functionalised with a basic pyridine binding site. (b) The dynamic foldamer forms a hydrogen-bonded complex in which a chiral carboxylic acid (blue) induces a conformational preference for left-handed screw sense. (c) A chiral phosphoric acid (orange) induces a right-handed screw sense. (d) A chiral triflamide (red) restores a weak preference for left-handed screw sense. (e) Ammonia disrupts the hydrogen-bonded complex firstly of the most acidic triflamide ligand, then (f) of the phosphoric acid ligand and then finally (g) of the carboxylic acid ligand, returning the receptor to its native form. | ||
The uncomplexed receptor (Fig. 12a) responds to the addition of a chiral carboxylic acid (in blue) by adopting a left-handed screw-sense preference (Fig. 12b). Adding a stronger acid, the phosphoric acid shown in orange, displaces the carboxylic acid from the binding site and (for this enantiomer of the acid) induces a switch in screw-sense preference (Fig. 12c). An even stronger acid still, the triflamide shown in red, further displaces the phosphoric acid and results in a second inversion to a weakly left-handed screw-sense (Fig. 12d).
Now, addition of ammonia provides an alternative partner for the most acidic proton available, and thus disrupts the hydrogen bonded complex between the basic foldamer and the most acidic (red) triflamide, leaving the phosphoric acid (orange) free to reassociate with the dynamic foldamer, introducing again a right-handed screw-sense preference (Fig. 12e). A second equivalent of ammonia likewise removes the proton from the phosphoric acid–foldamer complex, and allows the carboxylic acid to restore a preference for left-handed screw sense (Fig. 12f). Finally a third equivalent of ammonia prevents association of any of the three acids with the dynamic foldamer, which thus reverts to its conformationally racemic native form (Fig. 12g). The final three changes can each be reversed by readdition of HCl, which returns a proton to the system and neutralising the effect of the ammonia. The ability of the dynamic foldamer to report on the relative strengths of the different possible interactions between the components of complex mixture has analogies with the network of competitive interactions experienced by proteins in a cell. Future developments could lie in the direction of complex artificial control networks based on a type of molecular computation in which information is encoded in conformation.94
Photoswitching foldamer conformation
Closely related in structure and function to G protein-coupled receptors are the opsins – the proteins of vision. Human vision is mediated by rhodopsin, a modified G protein-coupled receptor that carries a covalently attached ligand in the form of retinal. Photochemical switching of retinal between the E and Z configurations at one particular double bond leads to a conformational change that is communicated through the molecule and ultimately leads to the propagation of the nerve signal. Feringa has shown that a photochemical switch can lead to inversion of helical screw sense of polymeric polyisocyanate,52 although because detection of screw-sense preference used circular dichroism, no data was obtained on how far the information encoded in the conformational switch is communicated. We showed that photochemical stimulation may lead to an inversion of screw sense that propagates through the helical oligomer and consequently inverts the sense of asymmetry induced by a catalytic thiourea unit95 located at the remote terminus (Fig. 13).89 The photochemical inversion made use of the same concept that had been applied to the directed deprotonation of the oligomer shown in Fig. 9. The starting tertiary amide contains no C terminal NH group, and as a result the C-terminal L-Ala residue (in blue) induces the oligomer to adopt a global left-handed helical structure, placing the locally achiral thiourea catalytic site in a chiral environment and allowing it to induce asymmetry in the catalytic addition of malonate to nitrostyrene to give an S product in 37:63 e.r. The transamidation of the (green) tertiary amide to an (orange) secondary amide by photochemical cleavage of the 5-bromo-8-nitroindolinamide96 in the presence of isopropylamine allows a new hydrogen bond to form, reorganizing the terminal portion of the helix into a right-handed screw-sense preference. This resulting screw-sense switched foldamer also catalyses the Michael addition of malonate to nitrostyrene, with the sense of selectivity reversed, giving the R product in 77:23 e.r.
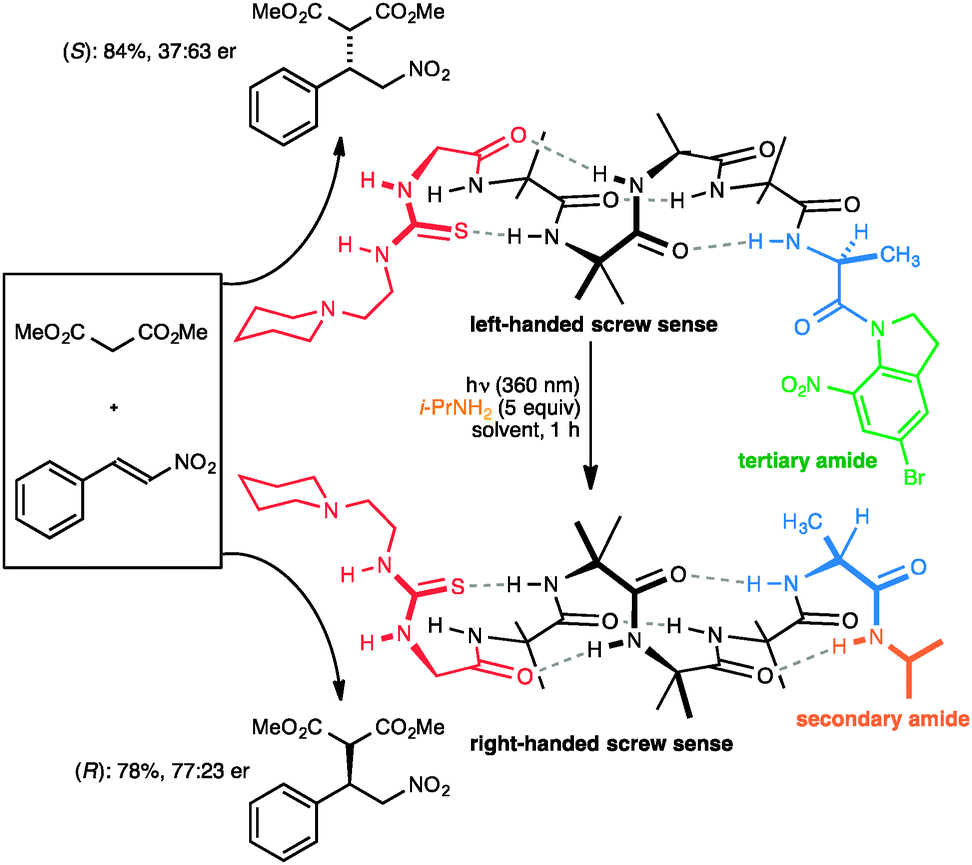 | ||
| Fig. 13 A photolabile tertiary amide function allows global screw-sense inversion of a dynamic foldamer to reverse the enantioselectivity of a remote catalytic site. | ||
Conclusions
Compounds that have a well defined but dynamic conformational structure offer opportunities to use synthetic scaffolds to mimic the features of natural molecules. The principal feature that divides biology from chemistry is information: no artificial chemical system has ever come even close to the informational complexity of even the simplest cell.97 Nonetheless, dynamic foldamers allow spatial communication of information over distances that extend into the multi-nanometre range, in principle allowing communication across cell membranes (whose typical thickness is in the region of 4 nm). Future extensions of this work could allow artificial receptors to be built that function in a membrane environment. Such dynamic foldamers, potentially incorporated into cell membranes, would allow the construction of synthetic, controllable cellular compartments, providing a bridge between synthetic chemistry and synthetic biology. Dynamic foldamers also display the selective responses to competing interactions in a mixture of components, and further developments in this area could see information not only communicated by foldamers but processed by them as well.94Acknowledgements
Our work in this area has been supported by the EPSRC, the BBSRC and the ERC.Notes and references
- A. Yonath, Angew. Chem., Int. Ed., 2010, 49, 4340–4354 CrossRef CAS PubMed.
- R. Dawkins, The Extended Phenotype, OUP, Oxford, 1982 Search PubMed.
- M. Berthelot, La synthèse chimique, Germer Baillière, Paris, 1876 Search PubMed.
- K. C. Nicolaou and S. A. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11929–11936 CrossRef CAS PubMed.
- G. M. Whitesides, Interface Focus, 2015, 5, 20150031 CrossRef PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS.
- S. Hecht and I. Huc, Foldamers: Structure, Properties and Applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC.
- H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC.
- R. B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758–2762 CrossRef CAS.
- D.-W. Zhang, X. Zhao and Z.-T. Li, Acc. Chem. Res., 2014, 47, 1961–1970 CrossRef CAS PubMed.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- M. Reggelin, S. Doerr, M. Klussmann, M. Schultz and M. Holbach, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5461–5466 CrossRef CAS PubMed.
- R. A. Smaldone and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 5444–5450 CrossRef CAS PubMed.
- M. M. Müller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., 2009, 121, 940–943 CrossRef.
- C. A. Smith, D. Ban, S. Pratihar, K. Giller, C. Schwiegk, B. L. de Groot, S. Becker, C. Griesinger and D. Lee, Angew. Chem., Int. Ed., 2015, 54, 207–210 CrossRef CAS PubMed.
- J. Habchi, P. Tompa, S. Longhi and V. N. Uversky, Chem. Rev., 2014, 114, 6561–6588 CrossRef CAS PubMed.
- M. Fuxreiter, Á. Tóth-Petróczy, D. A. Kraut, A. T. Matouschek, R. Y. H. Lim, B. Xue, L. Kurgan and V. N. Uversky, Chem. Rev., 2014, 114, 6806–6843 CrossRef CAS PubMed.
- M. F. Perutz, Q. Rev. Biophys., 1989, 22, 139–236 CrossRef CAS PubMed.
- S. Granier and B. Kobilka, Nat. Chem. Biol., 2012, 8, 670–673 CrossRef CAS PubMed.
- A. Manglik and B. Kobilka, Curr. Opin. Cell Biol., 2014, 27, 136–143 CrossRef CAS PubMed.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed.
- D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349–1372 RSC.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- N. Chouaieb, A. Goriely and J. H. Maddocks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9398–9403 CrossRef CAS PubMed.
- R. A. Brown, T. Marcelli, M. De Poli, J. Solà and J. Clayden, Angew. Chem., Int. Ed., 2012, 51, 1395–1399 CrossRef CAS PubMed.
- J. D. Dunitz, Angew. Chem., Int. Ed., 2001, 40, 4167–4173 CrossRef CAS.
- B. Brodsky and A. V. Persikov, Fibrous Proteins: Coiled-Coils, Collagen and Elastomers, Elsevier, Amsterdam, 2005, vol. 70, pp. 301–339 Search PubMed.
- M. Ishikawa, K. Maeda, Y. Mitsutsuji and E. Yashima, J. Am. Chem. Soc., 2004, 126, 732–733 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC.
- J. Clayden, L. Lemiègre, G. A. Morris, M. Pickworth, T. J. Snape and L. H. Jones, J. Am. Chem. Soc., 2008, 130, 15193–15202 CrossRef CAS PubMed.
- K. Maeda and E. Yashima, Supramolecular Chirality, Springer-Verlag, Berlin/Heidelberg, 2006, vol. 265, pp. 47–88 Search PubMed.
- K. Ute, Y. Fukunishi, S. K. Jha, K. S. Cheon, B. Muñoz, K. Hatada and M. M. Green, Macromolecules, 1999, 32, 1304–1307 CrossRef CAS.
- C. Dolain, H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2005, 127, 12943–12951 CrossRef CAS PubMed.
- J. Solà, G. A. Morris and J. Clayden, J. Am. Chem. Soc., 2011, 133, 3712–3715 CrossRef PubMed.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed.
- Y. Inai, K. Tagawa, A. Takasu, T. Hirabayashi, T. Oshikawa and M. Yamashita, J. Am. Chem. Soc., 2000, 122, 11731–11732 CrossRef CAS.
- B. V. Prasad and P. Balaram, CRC Crit. Rev. Biochem., 1984, 16, 307–348 CrossRef CAS PubMed.
- C. Toniolo, G. M. Bonora, V. Barone, A. Bavoso, E. Benedetti, B. Di Blasio, P. Grimaldi, F. Lelj, V. Pavone and C. Pedone, Macromolecules, 1985, 18, 895–902 CrossRef CAS.
- C. Toniolo and E. Benedetti, Trends Biochem. Sci., 1991, 16, 350–353 CrossRef CAS PubMed.
- J. E. Jones, V. Diemer, C. Adam, J. Raftery, R. E. Ruscoe, J. T. Sengel, M. I. Wallace, A. Bader, S. L. Cockroft, J. Clayden and S. J. Webb, J. Am. Chem. Soc., 2016, 138, 688–695 CrossRef CAS PubMed.
- W. Zhang, W. Jin, T. Fukushima, T. Mori and T. Aida, J. Am. Chem. Soc., 2015, 137, 13792–13795 CrossRef CAS PubMed.
- S. Azeroual, J. Surprenant, T. D. Lazzara, M. Kocun, Y. Tao, L. A. Cuccia and J.-M. Lehn, Chem. Commun., 2012, 48, 2292–2294 RSC.
- F. Dumitru, Y.-M. Legrand, A. Van der Lee and M. Barboiu, Chem. Commun., 2009, 2667–2669 RSC.
- Circular Dichroism: Principles and Applications, ed. K. Nakanishi, N. Berova and R. W. Woody, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed; Circular Dichroism and the Conformational Analysis of Biomolecules, ed. G. D. Fasman, Plenum Press, New York, 1996 Search PubMed.
- R. P. Hummel, C. Toniolo and G. Jung, Angew. Chem., Int. Ed., 1987, 26, 1150–1152 CrossRef.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CAS.
- D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693–3696 CrossRef CAS PubMed.
- E. Yashima, K. Maeda and T. Nishimura, Chem. – Eur. J., 2004, 10, 42–51 CrossRef CAS PubMed.
- E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef CAS.
- B. Pengo, F. Formaggio, M. Crisma, C. Toniolo, G. M. Bonora, Q. B. Broxterman, J. Kamphuis, M. Saviano, R. Iacovino, F. Rossi and E. Benedetti, J. Chem. Soc., Perkin Trans. 2, 1998, 1651–1657 RSC.
- J. Clayden, A. Castellanos, J. Solà and G. A. Morris, Angew. Chem., Int. Ed., 2009, 48, 5962–5965 CrossRef CAS PubMed.
- M. Crisma, M. De Zotti, F. Formaggio, C. Peggion, A. Moretto and C. Toniolo, J. Pept. Sci., 2015, 21, 148–177 CrossRef CAS PubMed.
- N. Ousaka, Y. Takeyama and E. Yashima, Chem. – Eur. J., 2013, 19, 4680–4685 CrossRef CAS PubMed.
- N. Ousaka, Y. Takeyama, H. Iida and E. Yashima, Nat. Chem., 2011, 3, 856–861 CrossRef CAS PubMed.
- Y. Inai, Y. Kurokawa, A. Ida and T. Hirabayashi, Bull. Chem. Soc. Jpn., 1999, 72, 55–61 CrossRef CAS.
- Y. Inai, S. Ashitaka and T. Hirabayashi, Polym. J., 1999, 31, 246–253 CrossRef CAS.
- Y. Inai, Y. Kurokawa and T. Hirabayashi, Biopolymers, 1999, 49, 551–564 CrossRef CAS.
- Y. Inai, Y. Ishida, K. Tagawa, A. Takasu and T. Hirabayashi, J. Am. Chem. Soc., 2002, 124, 2466–2473 CrossRef CAS PubMed.
- Y. Inai, N. Ousaka and T. Okabe, J. Am. Chem. Soc., 2003, 125, 8151–8162 CrossRef CAS PubMed.
- H. Komori and Y. Inai, J. Org. Chem., 2007, 72, 4012–4022 CrossRef CAS PubMed.
- N. Ousaka and Y. Inai, J. Org. Chem., 2009, 74, 1429–1439 CrossRef CAS PubMed.
- For other examples in which small changes of changes of foldamer structure or solvent lead to significant reorganisations of conformational preference, see. F. Mamiya, N. Ousaka and E. Yashima, Angew. Chem., 2015, 54, 14442–14446 CrossRef CAS PubMed; F. Formaggio, G. Ballano, A. Moretto, C. Peggion, M. Crisma, R. Lettieri, E. Gatto, N. Venanzi and C. Toniolo, Biopolymers, 2011, 96, 436 Search PubMed; R. Lettieri, M. Bischetti, E. Gatto, A. Palleschi, E. Ricci, F. Formaggio, M. Crisma, C. Toniolo and M. Venanzi, Biopolymers, 2013, 100, 51–63 CrossRef PubMed.
- M. De Poli, L. Byrne, R. A. Brown, J. Solà, A. Castellanos, T. Boddaert, R. Wechsel, J. D. Beadle and J. Clayden, J. Org. Chem., 2014, 79, 4659–4675 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2014, 50, 7949–7952 RSC.
- Y. Ito, T. Miyake, S. Hatano, R. Shima, T. Ohara and M. Suginome, J. Am. Chem. Soc., 1998, 120, 11880–11893 CrossRef CAS.
- M. Suginome, S. Collet and Y. Ito, Org. Lett., 2002, 4, 351–354 CrossRef CAS PubMed.
- M. De Poli and J. Clayden, Org. Biomol. Chem., 2014, 12, 836–843 CAS.
- L. Byrne, J. Solà, T. Boddaert, T. Marcelli, R. W. Adams, G. A. Morris and J. Clayden, Angew. Chem., Int. Ed., 2014, 53, 151–155 CrossRef CAS PubMed.
- S. J. Pike, M. De Poli, W. Zawodny, J. Raftery, S. J. Webb and J. Clayden, Org. Biomol. Chem., 2013, 11, 3168–3176 CAS.
- B. A. F. Le Bailly, PhD thesis, University of Manchester, 2015.
- B. A. F. Le Bailly, L. Byrne, V. Diemer, M. Foroozandeh, G. A. Morris and J. Clayden, Chem. Sci., 2015, 6, 2313–2322 RSC.
- Inai has nonetheless made use of chromophores with distinctive Cotton effects to establish the preferred screw sense of a localised part of an extended oligomer: see ref. 66.
- In order to limit the number of compounds we needed to make, we incorporated three doubly 13C-labelled Aib residues into each oligomer, and identified the individual labelled residues in each chain by enriching different positions of the chain with different % 13C. The three labels in each oligomer were ‘frame-shifted’ by one residue relative to its neighbours in the oligomer series.
- T. Boddaert, J. Solà, M. Helliwell and J. Clayden, Chem. Commun., 2012, 48, 3397–3399 RSC.
- M. Tomsett, B. A. F. Le Bailly, S. Bijvoets, I. Maffucci, A. Contini and J. Clayden, manuscript in preparation.
- C. Toniolo and H. Brückner, Peptaibiotics, HCA/Wiley-VCH, Zürich, 2009 Search PubMed.
- G. B. Bunkóczi, M. Schiell, L. S. Vértesy and G. M. Sheldrick, J. Pept. Sci., 2003, 9, 745–752 CrossRef PubMed.
- M. De Poli, M. De Zotti, J. Raftery, J. A. Aguilar, G. A. Morris and J. Clayden, J. Org. Chem., 2013, 78, 2248–2255 CrossRef CAS PubMed.
- U. Orcel, M. De Poli, M. De Zotti and J. Clayden, Chem. – Eur. J., 2013, 19, 16357–16365 CrossRef CAS PubMed.
- S. P. Fletcher, J. Solà, D. Holt, R. A. Brown and J. Clayden, Beilstein J. Org. Chem., 2011, 7, 1304–1309 CrossRef CAS PubMed.
- M. De Zotti, B. Biondi, M. Crisma, C. U. Hjørringgaard, A. Berg, H. Brückner and C. Toniolo, Biopolymers, 2012, 98, 36–49 CrossRef CAS PubMed.
- J. Solà, S. P. Fletcher, A. Castellanos and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 6836–6839 CrossRef PubMed.
- E. Kolomiets, V. Berl, I. Odriozola, A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2003, 2868–2869 RSC.
- B. A. F. Le Bailly, L. Byrne and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 2132–2136 CrossRef CAS PubMed.
- J.-H. Ha and S. N. Loh, Chem. – Eur. J., 2012, 18, 7984–7999 CrossRef CAS PubMed.
- T. D. James, M. D. Phillips and S. Shinkai, Boronic Acids in Saccharide Recognition, Royal Society of Chemistry, 2006 Search PubMed.
- R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS PubMed.
- J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680–6691 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. – Eur. J., 2000, 6, 2097–2102 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- B. Amit, D. A. Ben-Efraim and A. Patchornik, J. Am. Chem. Soc., 1976, 98, 843–844 CrossRef CAS.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |