
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen bonding to carbonyl oxygen of nitrogen-pyramidalized amide – detection of pyramidalization direction preference by vibrational circular dichroism spectroscopy†
Siyuan
Wang‡
ab,
Tohru
Taniguchi‡
c,
Kenji
Monde
c,
Masatoshi
Kawahata
d,
Kentaro
Yamaguchi
d,
Yuko
Otani
*a and
Tomohiko
Ohwada
*a
aGraduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: otani@mol.f.u-tokyo.ac.jp; ohwada@mol.f.u-tokyo.ac.jp
bResearch Foundation Itsuu Laboratory, C1232 Kanagawa Science Park R & D Building, 3-2-1 Sakado, Takatsu-ku, Kawasaki, Kanagawa 213-0012, Japan
cFrontier Research Center for Post-Genome Science and Technology, Faculty of Advanced Life Science, Hokkaido University, Kita 21 Nishi 11, Sapporo 001-0021, Japan
dFaculty of Pharmaceutical Sciences at Kagawa Campus, Tokushima Bunri University, 1314-1 Shido, Sanuki, Kagawa 769-2193, Japan
First published on 18th February 2016
Abstract
Nitrogen-pyramidalization of amide increases electron density on nitrogen and decreases that on carbonyl oxygen. We identified hydrogen-bonding to carbonyl of nitrogen-pyramidalized bicyclic β-proline derivatives by crystallography, and by NMR and vibrational circular dichroism (VCD) spectroscopy in solution. Such hydrogen-bonding can switch the preferred nitrogen-pyramidalization direction, as detected by VCD spectroscopy.
The amide bond is a key linkage in proteins, peptides and peptide mimics, serving to connect two neighbouring amino acids or analogues. Most amide bonds are planar, but nonplanar amide structures have been suggested to occur even in proteins and peptides.1 Although the magnitude of nonplanarity found in proteins and peptides is not large, some nonplanar amides with distinct ground states have been reported.2 In such non-planar amides, the nitrogen atom gains a partial sp3-character (i.e., nitrogen-pyramidalization), and at the same time bond-twisting occurs (Fig. 1). This phenomenon results in increased electron density at the nitrogen atom, and decreased electron density at carbonyl oxygen, as compared with the situation in a planar amide.3 While hydrogen-bonding to the pyramidalized electron-rich nitrogen atom has been experimentally and computationally investigated,4 there has been little study on the possibility of hydrogen bonding to the electron-deficient carbonyl oxygen atom of non-planar amides.5
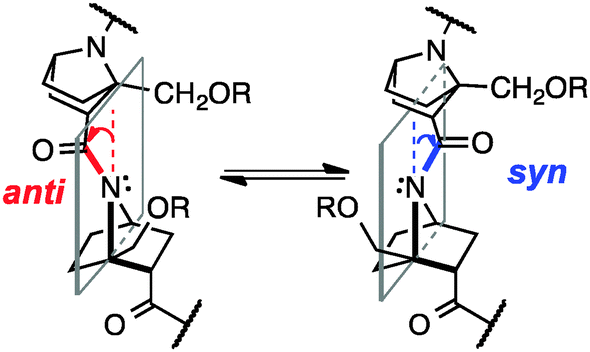 | ||
| Fig. 1 Anti and syn inversion in nitrogen-pyramidal amides of bicyclic β-proline oligomers. | ||
7-Azabicyclo[2.2.1]heptane amides are chemically stable and intrinsically nonplanar (Fig. 1),6 and substitution at the bridgehead position of bicyclic β-proline derivatives (7-azabicyclo[2.2.1]heptane-2-carboxylic acids) can bias amide cis–trans isomerization toward either cis or trans, depending on the position of the bridgehead substituent. Homooligomers of optically active derivatives take helical structures with all-cis or all-trans amide bonds. Non-planarity of amide structures in homooligomers can be detected in crystal structures.6b,c
In solution, there is an equilibrium between two conformers, anti, i.e., the carbonyl group is tilted toward the opposite side of the C-terminal group with respect to the plane of the nitrogen atom and two bridgehead carbons, and syn, in which the carbonyl group is tilted toward the same side of the C-terminal group (Fig. 1). But, because of the small energy difference between the anti and syn conformers and low inversion barrier,6a it has been difficult to detect the direction of pyramidalization in solution.
Herein, we demonstrate the presence of hydrogen-bonding to the carbonyl group of nitrogen-pyramidalized amides by means of crystallographic and NMR analyses and vibrational circular dichroism (VCD) spectroscopy.7 Our results also indicate that such hydrogen-bonding is strong enough to switch the preferred direction of nitrogen-pyramidalization of ground-state amide bonds of bicyclic β-proline derivatives (Fig. 1). Directional preference of nitrogen-pyramidalization of non-planar amides has been little discussed so far, probably because amides with a distinct non-planar ground state are too unstable8 and the magnitude of non-planarity of protein and peptide amides is only marginal. We expected that bicyclic β-proline derivatives would be a good structural platform to observe such effects.
Several homooligomers of the monomeric 7-azabicyclo[2.2.1]heptane unit with the same stereochemical configuration were synthesized (see ESI†) (Fig. 2): the dimer (S)-26c and the trimer (S)-3, which bear a methoxymethyl substituent at the bridgehead 1-position,9 and the dimer (S)-2h and the trimer (S)-3h, which bear a hydroxymethyl substituent at the bridgehead 1-position. X-ray diffraction analysis supported the existence of hydrogen-bonding between the carbonyl oxygen atom and the hydroxymethyl substituent at the bridgehead position of (S)-2h (Fig. 3). The distances between the carbonyl oxygen of carbamate (tButoxycarbonyl, Boc) or amide and hydrogen of the hydroxyl group are 1.897 Å and 1.788 Å, respectively, which are shorter than the sum of the van der Waals radii of hydrogen and oxygen atoms. The crystal-state structures also showed a difference in the direction of nitrogen pyramidalization between (S)-2 and (S)-2h: the nitrogen-pyramidalization directions in (S)-2 are anti for both carbamate and amide, and both the bridgehead –CH2OCH3 groups are oriented in the opposite direction of the nitrogen pyramidalization (see Fig. 1), while those in (S)-2h are syn and both the bridgehead hydroxyl groups are oriented to the same direction to make the hydrogen bonding (Fig. 3 and Table 1). The main-chain dihedral angles ψ are also different (Table S1, ESI†). These results indicate that intramolecular hydrogen bonding to the carbonyl group of nitrogen-pyramidalized amide is possible, and that this inter-residual hydrogen bonding is strong enough to influence the preferred direction of nitrogen-pyramidalization.
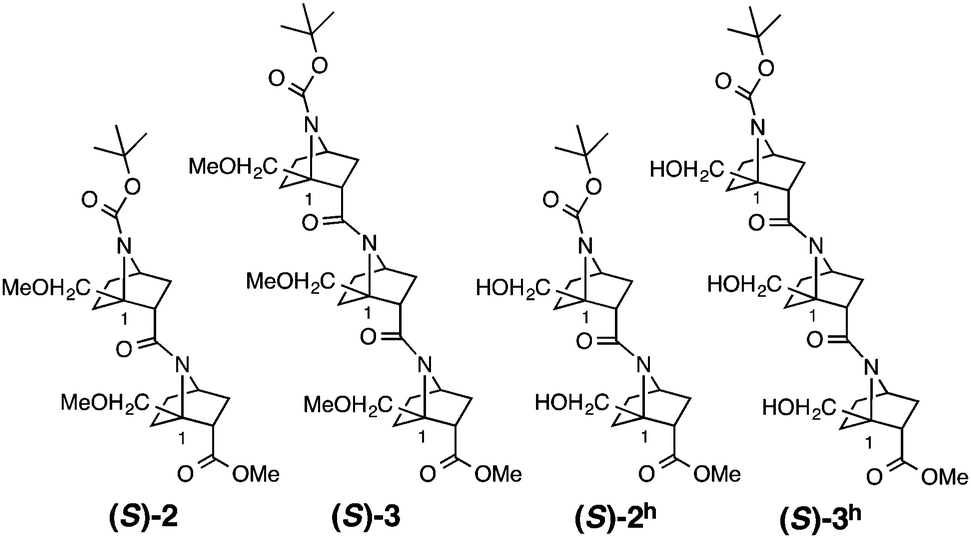 | ||
| Fig. 2 Homooligomers in this study. | ||
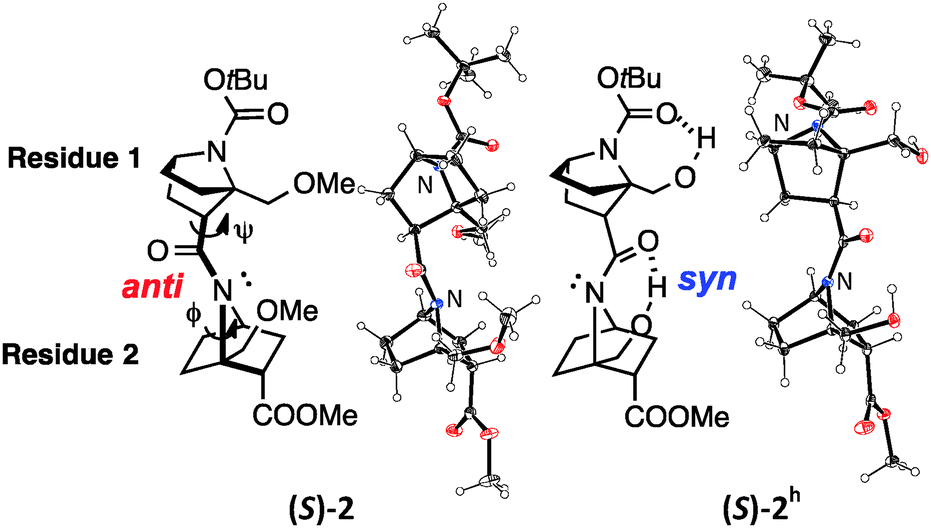 | ||
| Fig. 3 ORTEP drawing (50% probability) of the crystal structures of (S)-2 (CCDC 991065)6c and (S)-2h (CCDC 1445081). Enlarged versions are shown in Fig. S1 (ESI).† | ||
| ΔE (kcal mol−1) (population) | ϕ (N1) (°) (N-Boc) | ϕ (N2) (°) | ϕ (N3) (°) | |
|---|---|---|---|---|
| XRD (S)-2 | — | −166.6 (anti) | −162.9 (anti) | |
| (S)-2 conf 1 | 0.000 (63.2%) | −158.9 (anti) | −154.4 (anti) | |
| (S)-2 conf 2 | 0.567 (24.3%) | −157.1 (anti) | −96.9 (syn) | |
| (S)-2 conf 3 | 1.296 (7.1%) | −159.3 (anti) | −94.5 (syn) | |
| (S)-2 conf 4 | 1.451 (5.4%) | −84.3 (syn) | −97.2 (syn) | |
| (S)-3 conf 1 | 0.000 (73.2%) | −159.2 (anti) | −148.6 (anti) | −153.1 (anti) |
| (S)-3 conf 2 | 0.891 (16.3%) | −159.1 (anti) | −149.1 (anti) | −152.4 (anti) |
| (S)-3 conf 3 | 1.151 (10.5%) | −167.7 (anti) | −149.6 (anti) | −153.3 (anti) |
| XRD (S)-2h | — | −86.5 (syn) | −106.4 (syn) | |
| (S)-2h conf 1 | 0.000 (60.9%) | −96.2 (syn) | −137.9 (anti) | |
| (S)-2h conf 2 | 0.615 (21.6%) | −95.8 (syn) | −120.5 (syn) | |
| (S)-2h conf 3 | 1.123 (9.2%) | −96.2 (syn) | −139.9 (anti) | |
| (S)-2h conf 4 | 1.177 (8.3%) | −95.8 (syn) | −137.6 (anti) | |
| (S)-3h conf 1 | 0.000 (60.2%) | −93.3 (syn) | −105.2 (syn) | −139.0 (anti) |
| (S)-3h conf 2 | 0.666 (19.6%) | −93.6 (syn) | −104.5 (syn) | −121.3 (syn) |
| (S)-3h conf 3 | 1.176 (8.3%) | −148.8 (anti) | −106.1 (syn) | −138.8 (anti) |
| (S)-3h conf 4 | 1.213 (7.8%) | −92.4 (syn) | −105.2 (syn) | −140.5 (anti) |
| (S)-3h conf 5 | 1.594 (4.1%) | −92.8 (syn) | −136.2 (anti) | −109.6 (syn) |
Next, we carried out solution NMR measurements. The 1H-NMR chemical shifts of hydroxyl protons in the bridgehead substituent in (S)-2h were deshielded by 5 ppm (δ), which is consistent with their participation in hydrogen bonding. Temperature coefficient studies and solvent titration studies also supported the existence of strong intramolecular hydrogen bonding between the amide carbonyl and the hydroxyl group in residue 2; i.e., small temperature coefficients10 for the OH resonance in residue 1 (−5.96 ppb K−1) and residue 2 (−3.84 ppb K−1) of (S)-2h were obtained in DMSO-d6 (Fig. 4), and titration of OH with DMSO-d6 in CDCl3 showed a small change in the chemical shift (Δδ (OH)) (Fig. S2, ESI†).
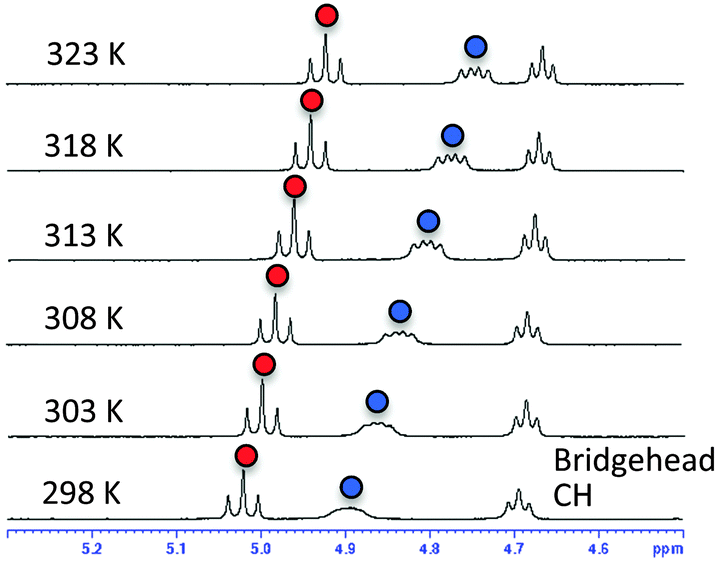 | ||
| Fig. 4 VT 1H-NMR spectra of (S)-2h in DMSO-d6. Signals of OH in residue 1 are marked with blue circles; signals of OH in residue 2 are marked with red circles. | ||
The solution structures of the four homooligomers ((S)-2, (S)-3, (S)-2h, (S)-3h) were further investigated by means of VCD spectroscopy. First, the IR absorption and VCD spectra of (S)-2 were measured in CDCl3 (Fig. 5a). Characteristic vibrational transitions of (S)-2 are as follows: ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1728 cm−1), carbamate CO stretching (1690 cm−1), amide CO stretching (1643 cm−1), amide C–N stretching (∼1350 cm−1), and C–O stretchings/CH bendings (1200–1100 cm−1).11 The conformation of (S)-2 was then assigned by comparison of the observed VCD spectrum with calculated spectra. First, calculations were conducted to find stable conformers of (S)-2, and four conformers were found within a 2 kcal mol−1 energy gap from the most stable one (Fig. 5b). VCD spectra of each conformer were then calculated using DFT (Fig. 5a). Even subtle differences in conformation dramatically changed the entire VCD spectral shape, though the calculated IR absorption spectra of the conformers were all relatively similar (Fig. S3, ESI†). Nevertheless, the averaged VCD spectrum based on the Boltzmann population showed a good agreement with the observed VCD. This finding supports the validity of the calculated relative energies and the structures of the stable conformers of (S)-2.
O stretching (1728 cm−1), carbamate CO stretching (1690 cm−1), amide CO stretching (1643 cm−1), amide C–N stretching (∼1350 cm−1), and C–O stretchings/CH bendings (1200–1100 cm−1).11 The conformation of (S)-2 was then assigned by comparison of the observed VCD spectrum with calculated spectra. First, calculations were conducted to find stable conformers of (S)-2, and four conformers were found within a 2 kcal mol−1 energy gap from the most stable one (Fig. 5b). VCD spectra of each conformer were then calculated using DFT (Fig. 5a). Even subtle differences in conformation dramatically changed the entire VCD spectral shape, though the calculated IR absorption spectra of the conformers were all relatively similar (Fig. S3, ESI†). Nevertheless, the averaged VCD spectrum based on the Boltzmann population showed a good agreement with the observed VCD. This finding supports the validity of the calculated relative energies and the structures of the stable conformers of (S)-2.
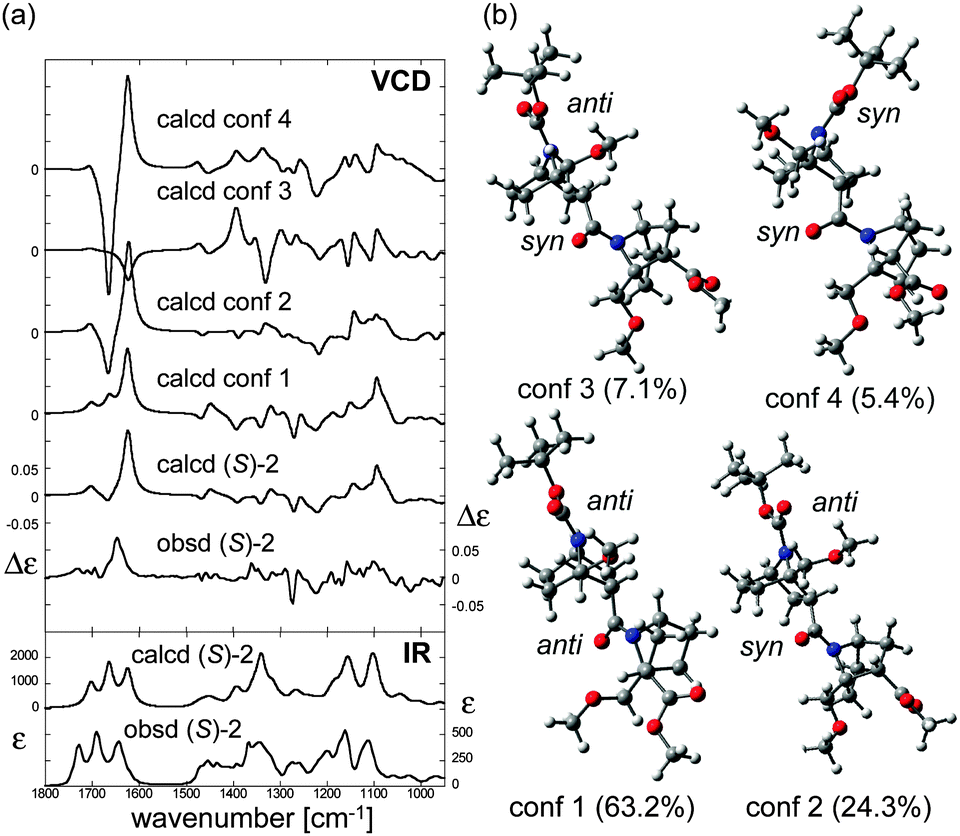 | ||
| Fig. 5 (a) Calculated VCD spectra of each conformer, population-weighed calculated VCD spectrum, and observed VCD spectrum of (S)-2 (top), and population-weighed calculated IR absorption spectrum and observed IR absorption spectrum (bottom). (b) Stable conformers of (S)-2 (for their relative energies, see Table 1). The Boltzmann populations of the conformers simulated at 298 K are shown in parenthesis. Measurement conditions: CDCl3, l = 100 μm, c = 0.18 M, corrected by solvent spectra obtained under the same measurement conditions. Calculations: DFT/B3LYP/6-311+G(d,p) using PCM for chloroform. | ||
All the predicted conformers exhibited nitrogen-pyramidalization in both the carbamate (residue 1) and amide (residue 2) moieties. Importantly, the nitrogen pyramidalization directions in the most stable conformer (conformer 1 in Fig. 5b) were anti for both carbamate and amide, while both the bridgehead –CH2OCH3 groups were oriented to the opposite direction; this corresponds well with the crystal structure. Other minor conformers with higher energies were also predicted by the VCD results, and the structural parameters are summarized in Table 1. Amide nitrogen at residue 2 was prone to adopt syn pyramidalization in the higher-energy conformers, which indicates the flexibility of the terminal residue 2, whereas the carbamate nitrogen atom seems to be restricted to anti.
To confirm the tendency of anti orientation of the amide nitrogen pyramidalization, the trimer (S)-3 was synthesized and studied. For all three predicted stable conformers, the nitrogen-pyramidalization directions were anti for all of residues 1–3 (Table 1 and Fig. S4, ESI†). Therefore, there was a strong preference for the orientation of the amide nitrogen pyramidalization in the case of the bridgehead –CH2OCH3 group.
In fact, the anti nitrogen-pyramidalization was favoured over the syn-pyramidalization by 0.2 kcal mol−1 (DFT/B3LYP/6-311+G(d,p)) even in the case of the simple acetyl amide of 7-azabicyclo[2.2.1]heptane monomer bearing the bridgehead –CH2OCH3 group.
We then applied the VCD technique to dimer (S)-2h and trimer (S)-3h to examine hydrogen bond formation in solution and to investigate whether the presence of hydrogen bonding influences the nitrogen-pyramidalization direction. The IR absorption and VCD spectra of (S)-2h measured in CDCl3 are shown in Fig. 6a. Similar to the case of (S)-2, three CO stretching absorption bands of (S)-2h were observed: the ester CO stretching (observed at 1728 cm−1), carbamate CO stretching (1666 cm−1) and amide CO stretching (1612 cm−1).11 The CO absorption bands of carbamate and amide are significantly shifted to lower wavenumber (long wavelength, bathochromic shift) as compared to those of (S)-2. This is consistent with the presence of hydrogen-bonding interactions to these two CO groups in (S)-2h. In order to elucidate the structure in detail, theoretical calculations of several stable conformers and their IR absorption and VCD spectra were carried out (Fig. 6). The theoretical VCD spectrum of (S)-2h showed moderately good agreement with the experimental spectrum, including a positive band for the amide CO stretching and a negative band for the carbamate CO stretching. Overall, these results support the reliability of the predicted orientations of these carbonyl groups.12
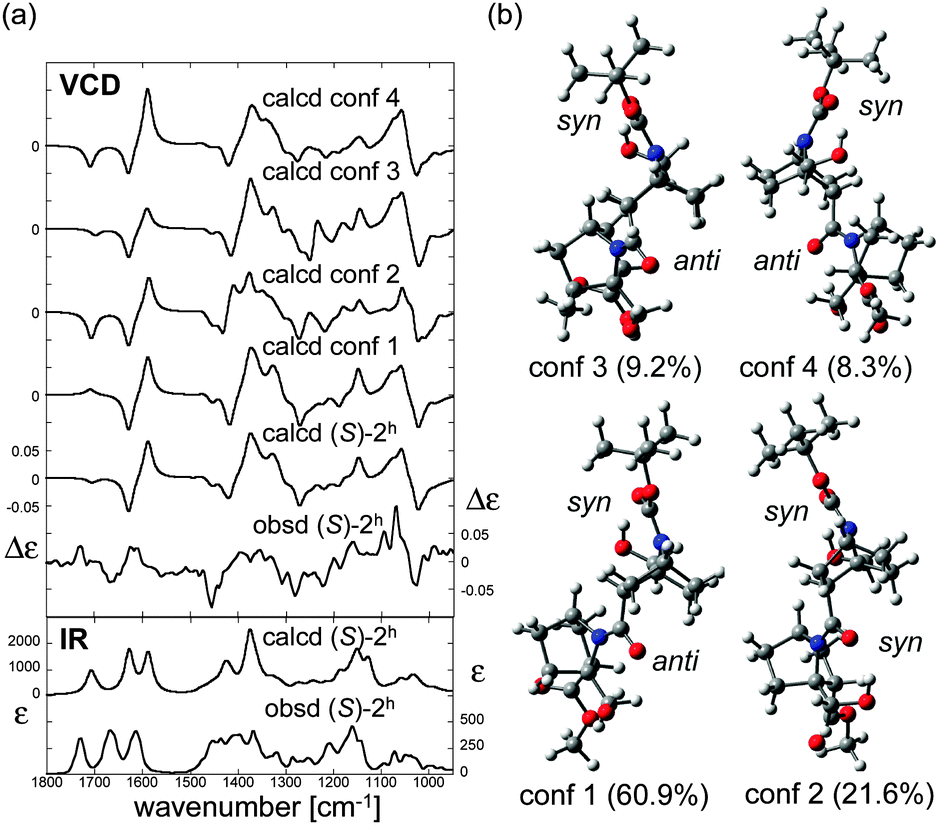 | ||
| Fig. 6 (a) Calculated VCD spectra of each conformer, population-weighed calculated VCD spectrum, and observed VCD spectrum of (S)-2h (top), and population-weighed calculated IR absorption spectrum and observed IR absorption spectrum (bottom). (b) Stable conformers of (S)-2h (for their relative energies, see Table 1). The Boltzmann populations of the conformers simulated at 298 K are shown in parenthesis. Measurement conditions: CDCl3, l = 100 μm, c = 0.06 M, corrected by solvent spectra obtained under the same measurement conditions. Calculations: DFT/B3LYP/6-311+G(d,p) using PCM for chloroform. | ||
The four stable conformers predicted by DFT optimizations are shown in Fig. 6b. In accord with the X-ray diffraction, NMR and IR absorption results, all the conformers showed hydrogen-bonding interactions of the hydroxyl groups and the carbamate or amide of (S)-2h. On the other hand, the nitrogen-pyramidalization direction of residue 2 of (S)-2h was anti for the most stable CO group. The nitrogen-pyramidalization of residue 1 is syn for all conformers, which is consistent with the X-ray diffraction structure conformer, and was syn for the second most stable conformer. The fact that the crystal structure and VCD results of hydrogen-bonding oligomer (S)-2h do not match as well as those of the non-hydrogen bonding oligomer (S)-2 may be at least partially due to the flexibility of the amide conformation in terminal residue 2 in solution, as also seen in the case of (S)-2. Interestingly, the second most stable conformer, whose conformation is the most close to the crystal structure, showed a similar VCD feature to that of the observed spectrum especially in 1100–1500 cm−1 region. Considering the flexibility of the amide conformation of residue 2, it is plausible that its orientation could be easily perturbed by possible intermolecular interactions by the OH group, which were not taken into account in theoretical calculations.11,12
The presence of intramolecular hydrogen bonds in (S)-3h was also examined (Fig. S5, ESI†). The shift of the IR absorption bands for carbamate CO stretching (1666 cm−1) and amide CO stretching (1612 cm−1) was also observed for (S)-3h. Theoretical calculations predicted a high preference for syn orientation of the pyramidized amide nitrogen in the adjoining residue 2 in the most stable conformer, which is in sharp contrast to the high preference for anti at the same position of (S)-3 (Fig. S4, ESI†).
Thus, these results demonstrated that the nitrogen-pyramidalization direction can be regulated by simple substitution of an auxiliary functional group.
In summary, our results show that intramolecular hydrogen bonding to the putative “electron-deficient” carbonyl oxygen atom of non-planar amides is possible, and can influence the preferred direction of nitrogen pyramidalization. Specifically, interresidual hydrogen bonding can change the nitrogen-pyramidalization direction in non-planar amide-bearing 7-azabicyclo[2.2.1]heptane derivatives. This local structural variation may influence the overall ordered structure, and thus may influence the folding of oligopeptides and proteins. We have also demonstrated that VCD spectroscopy is a powerful method for detecting small structural differences of non-planar amide conformation in solution.
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science. Y. O. also thanks a Grant-in-Aid for Scientific Research on Innovative Areas, Studying the Function of Soft Molecular System. A part of computations was performed at the Research Center for Computational Science, Okazaki, Japan. We thank the computational facility for generous allotments of computer time.
Notes and references
- (a) G. N. Ramachandran, Biopolymers, 1968, 6, 1494–1496 CrossRef CAS PubMed; (b) A. Romanelli, A. Shekhtman, D. Cowburn and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6397–6402 CrossRef CAS PubMed; (c) F. K. Winkler and J. D. Dunitz, J. Mol. Biol., 1971, 59, 169–182 CrossRef CAS PubMed; (d) H. M. Sulzbach, P. v R. Schleyer and H. F. Schaefer III, J. Am. Chem. Soc., 1995, 117, 2632–2637 CrossRef CAS; (e) L. Esposito, A. De Simone, A. Zagari and L. Vitagliano, J. Mol. Biol., 2005, 347, 483–487 CrossRef CAS PubMed.
- (a) V. Somayaji and R. S. Brown, J. Org. Chem., 1986, 51, 2676–2686 CrossRef CAS; (b) G. V. Shustov, G. K. Kadorkina, S. V. Varlamov, A. V. Kachanov, R. G. Kostyanovsky and A. Rauk, J. Am. Chem. Soc., 1992, 114, 1616–1623 CrossRef CAS; (c) S. Yamada, Angew. Chem., Int. Ed. Engl., 1993, 32, 1083–1085 CrossRef; (d) H. Shao, X. Jiang, P. Gantzel and M. Goodman, Chem. Biol., 1994, 1, 231–234 CrossRef CAS PubMed; (e) A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785–786 CrossRef CAS; (f) R. Łysek, K. Borsuk, M. Chmielewski, Z. Kałuza, Z. Urbańczyk-Lipkowska, A. Klimek and J. Frelek, J. Org. Chem., 2002, 67, 1472–1479 CrossRef; (g) M. Szostak and J. Aubé, Org. Biomol. Chem., 2011, 9, 27–35 RSC.
- The Amide Linkage, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley-Interscience, Toronto, 2000, and references cited therein Search PubMed.
- (a) N. H. Werstiuk, R. S. Brown and Q. Wang, Can. J. Chem., 1996, 74, 524–532 CrossRef CAS; (b) M. Szostak, L. Yao, V. W. Day, D. R. Powell and J. Aubé, J. Am. Chem. Soc., 2010, 132, 8836–8837 CrossRef CAS PubMed; (c) S. J. Cho, C. Cui, J. Y. Lee, J. K. Park, S. B. Suh, J. Park, B. H. Kim and K. S. Kim, J. Org. Chem., 1997, 62, 4068–4071 CrossRef CAS.
- R. Szostak, J. Aubé and M. Szostak, J. Org. Chem., 2015, 80, 7905–7927 CrossRef CAS PubMed.
- (a) Y. Otani, O. Nagae, Y. Naruse, S. Inagaki, M. Ohno, K. Yamaguchi, G. Yamamoto, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 15191–15199 CrossRef CAS PubMed; (b) M. Hosoya, Y. Otani, M. Kawahata, K. Yamaguchi and T. Ohwada, J. Am. Chem. Soc., 2010, 132, 14780–14789 CrossRef CAS PubMed; (c) S. Wang, Y. Otani, X. Liu, M. Kawahata, K. Yamaguchi and T. Ohwada, J. Org. Chem., 2014, 79, 5287–5300 CrossRef CAS PubMed.
- (a) T. Taniguchi, D. Manai, M. Shibata, Y. Itabashi and K. Monde, J. Am. Chem. Soc., 2015, 137, 12191–12194 CrossRef CAS PubMed; (b) T. Buffeteau, L. Ducasse, L. Poniman, N. Delsuc and I. Huc, Chem. Commun., 2006, 2714–2716 RSC; (c) C. Merten, V. Smyrniotopoulos and D. Tasdemir, Chem. Commun., 2015, 51, 16217–16220 RSC; (d) C. Merten, F. Lee, K. Bravo-Rodriguez, E. Sanchez-Garcia, Y. Xu and W. Sander, Phys. Chem. Chem. Phys., 2014, 16, 5627–5633 RSC; (e) K. Monde, T. Taniguchi, N. Miura and S. I. Nishimura, J. Am. Chem. Soc., 2004, 126, 9496–9497 CrossRef CAS PubMed; (f) N. Miura, T. Taniguchi, K. Monde and S. I. Nishimura, Chem. Phys. Lett., 2006, 419, 326–332 CrossRef CAS; (g) T. Taniguchi and K. Monde, Org. Biomol. Chem., 2007, 5, 1104–1110 RSC.
- (a) M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701–5765 CrossRef CAS PubMed; (b) H. Slebocka-Tilk and R. S. Brown, J. Org. Chem., 1987, 52, 805–808 CrossRef CAS; (c) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS PubMed; (d) I. V. Komarov, S. Yanik, A. Y. Ishchenko, J. E. Davies, J. M. Goodman and A. J. Kirby, J. Am. Chem. Soc., 2015, 137, 926–930 CrossRef CAS PubMed.
- The X-ray diffraction structures of the dimer (S)-2 and the enantiomer of a differently protected trimer have been already reported and discussed in ref. 6c.
- (a) B. Bernet and A. Vasella, Helv. Chim. Acta, 2000, 83, 995–1021 CrossRef CAS; (b) T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249–261 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, Chichester, 3rd edn, 2001 Search PubMed.
- We consider that the poor agreement of simulated spectra at around 1450 cm−1 with the experimental spectrum in this region (Fig. 6) was due to perturbations of the OH bending vibrations by intermolecular interactions and proton exchanges, which could not be taken into account in the theoretical calculations. This observation should not diminish the overall validity of the theoretical predictions.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and supplemental spectra. CCDC 1445081. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00284f |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |