
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Forensic electrochemistry: simultaneous voltammetric detection of MDMA and its fatal counterpart “Dr Death” (PMA)†
Loanda R.
Cumba
ab,
Jamie P.
Smith
b,
Khaled Y.
Zuway
b,
Oliver B.
Sutcliffe
b,
Devaney R.
do Carmo
a and
Craig E.
Banks
*b
aFaculdade de Engenharia de Ilha Solteira UNESP – Universidade Estadual Paulista, Departamento de Física e Química. Av. Brasil Centro, 56 CEP 15385-000, Ilha Solteira, SP, Brazil
bFaculty of Science and Engineering, School of Science and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)161 247 6831; Tel: +44 (0)161 247 1196
First published on 13th November 2015
Abstract
The simultaneous detection of substances present in drugs of abuse is increasingly important since some materials are known for their high mortality rate. One drug that received considerable attention is para-methoxyamphetamine (PMA), commonly known as ‘Dr Death’ – this substance is linked with several deaths internationally and can often be found together with 3,4-methylenedioxymethamphetamine (MDMA) in drugs sold under the alias “ecstasy”, a very popular drug of abuse. This work reports for the first time the detection and quantification of MDMA and PMA simultaneously through an electrochemical technique using screen-printed graphite electrodes (SPEs). The electroanalytical sensing of MDMA/PMA, MDMA and PMA are explored directly at bare unmodified SPEs yielding a detection limit (3σ) corresponding to 0.25 μg mL−1/0.14 μg mL−1 for MDMA/PMA, 0.04 μg mL−1 MDMA and 0.03 μg mL−1 PMA. Raman spectroscopy and presumptive colour tests were also performed on MDMA/PMA, MDMA and PMA using the Marquis, Mandelin, Simon's and Robadope tests but were found to not be able discriminate when PMA and MDMA are both present in the same samples. We report a novel electrochemical protocol for the sensing of PMA and MDMA which is independently validated in a synthetic (MDMA/PMA) sample with HPLC.
Introduction
3,4-Methylenedioxymethamphetamine (MDMA, Scheme 1) is a synthetic entactogen which shares a structural similarity to methamphetamine and acts as a central nervous system (CNS) stimulant producing mood enhancement, increased energy and other empathetic effects by increasing the intra-synaptic concentrations of the key neurotransmitters serotonin, dopamine and norephinephrine.1–5 MDMA was first synthesised by Merck in 1912 as a potential appetite suppressant, and over the next seventy years, a number of researchers explored the psychedelic properties of MDMA with little success.6 During the 1970s and 80s MDMA surfaced on the recreational drugs market, its widespread abuse and potential long-term health effects led many countries to prohibit its possession, supply and manufacture. Currently in the UK, MDMA (or “ecstasy”) is controlled as a Class A, Schedule 1 substance due to its illicit use as a recreational drug and its implication in a number of highly publicized fatalities.7–13 It is usually found in tableted form (containing 60–70 mg MDMA) with each batch being stamped with a particular motif, e.g. a Mitsubishi™ logo, smiley faces or letters.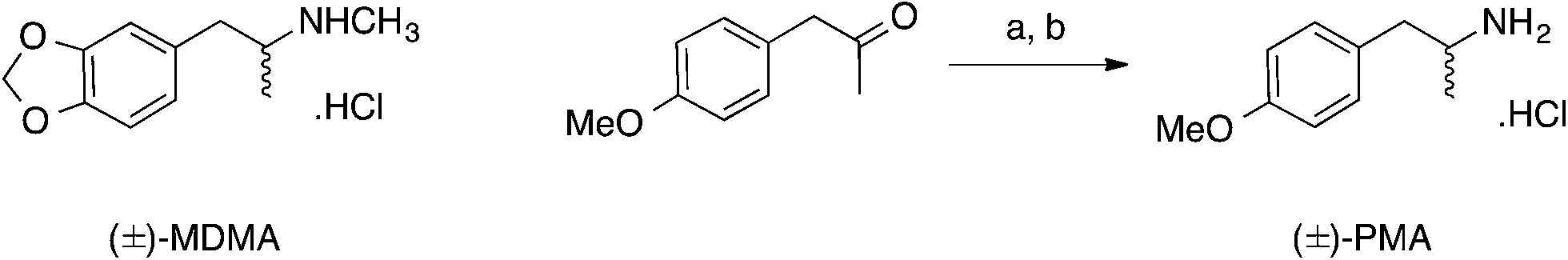 | ||
| Scheme 1 The structure of (±)-3,4-methylenedioxymethamphetamine hydrochloride (MDMA) and the synthesis of (±)-4-methoxyamphetamine hydrochloride (PMA). | ||
Since the global prohibition of MDMA and its precursors (i.e. 3,4-methylenedioxyphenyl-2-propanone, piperonal, safrole and isosafrole) a wide range of structurally-related phenethylamines have appeared on the recreational drugs market, including the designer drug, 4-methoxyamphetamine (PMA, Scheme 1). As a synthetic entactogen, PMA acts on the CNS producing mood enhancement, heightened sexual arousal and increased energy by increasing the release of intra-synaptic concentration serotonin and also inhibiting its reuptake.14 Animal models suggest that PMA is more toxic, than MDMA, resulting in hyperthermia (i.e. serotonin syndrome) due to the enhancement of serotonin release and the delayed onset of action.15,16 It is believed that a synergistic effect of taking PMA in combination with MDMA and alcohol may be particularly hazardous.17,18 PMA (or “Dr Death”) is not a recreational drug, which is sought after, rather it is usually encountered as a misrepresentation of MDMA, leading to serious adverse effects and fatalities.2,17 Due to the high demand of MDMA, illicit drug suppliers have been known to distribute PMA as “ecstasy” leading to a number of hospitalisations and fatalities. PMA is, similar to MDMA, usually found in tableted form (containing 40–70 mg PMA) with each batch being stamped with either a Mitsubishi™ or Superman logo.2 Consequently PMA is linked with several deaths internationally and can often be found together with 3,4-methylenedioxymethamphetamine (MDMA) in drugs sold under the alias “ecstasy”.
A number of groups have reported the simultaneous detection and quantification of MDMA and PMA using HPLC,19,20 GC-MS21,22 and LC-MS,23–25 however, these methods are not suitable for rapid and routine on-site testing. DanceSafe (Oakland, CA) have advocated the use of the Marquis test (1% formaldehyde in concentrated sulphuric acid) for the purposes of pill testing and discrimination between PMA and MDMA,2 however, with no published data (with either a secondary colour test and/or a confirmatory chromatographic assay) verifying this approach in real samples, the method is flawed and the potential for false positives is highly probable.
Electrochemistry is an advantageous analytical tool that is adaptable to an in-the-field device, in light of its portability, and can exhibit sensitivity and selectivity toward many target analytes.26–34 Our previous work on the development of robust electrochemical methods for the sensing of Novel (or New) Psychoactive substances (NPSs), in their pure form33 or in the presence of common adulterants,34 has the potential to be rapid, simple and cost-effective on-the-spot analytical screening tools with screen-printed graphite electrodes (SPEs). In this paper the simultaneous detection of para-methoxyamphetamine (PMA), and 3,4-methylenedioxymethamphetamine (MDMA) is shown to be viable for the first time using screen-printed electrochemical sensing platforms. Prior electrochemical work has reported the electrochemical oxidation of MA, MDA and MDMA at (only) glassy carbon electrodes and was extended to the sensing of MDMA in seized samples and in human serum.35 The electroanalytical sensing of MDMA/PMA, MDMA and PMA are explored in model (buffer) solutions for the first time using a range of commercially available electrode substrates (glassy carbon, boron-doped diamond and screen-printed electrodes). The detection of PMA and MDMA is also explored with presumptive colour tests and Raman analysis. Last, the electrochemical protocol is applied to the sensing of PMA and MDMA in a synthetic street samples and compared to high performance liquid chromatography.
Experimental
The chemicals were of commercial quality (obtained from Sigma-Aldrich, Gillingham, UK) and used without further purification. (±)-3,4-Methylenedioxymethamphetamine hydrochloride (MDMA) was obtained, under Home Office licence, from Sigma-Aldrich (Gillingham, UK) and used without further purification. (±)-4-Methoxyamphetamine hydrochloride (PMA) was prepared as described below. The simulated “ecstasy” sample, containing PMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MDMA (30:70% w/w) was prepared by mixing PMA (17.25 mg) and MDMA (37.4 mg) and homogenising the sample prior to analysis. 1H- and 13C-NMR spectra were acquired on a JEOL AS-400 (JEOL, Tokyo, Japan) NMR spectrometer operating at a proton resonance frequency of 400 MHz. Samples of PMA (10 mg/0.60 mL) were dissolved in DMSO-d6 and filtered prior to analysis. Infrared spectra were obtained in the range 4000–400 cm−1 using a ThermoScientific Nicolet iS10ATR-FTIR instrument (ThermoScientific, Rochester, USA). GC-MS spectra were recorded on an Agilent 6890 gas chromatograph with split-splitless injection (sample volume: 1 μL) and a HP-5MS column (30 m × 0.25 mm, 0.25 μm film thickness). Helium (He) was used as the carrier gas at a flow rate of 1.0 mL min−1. The GC was coupled to an Agilent 5973 MSD (EI, 70 eV, TIC mode scanning m/z 50–500) and injector port was set at 275 °C, the transfer line at 280 °C. The following temperature program was used: 60 °C for 3 min, 20 °C min−1 to 280 °C, 280 °C for 5 min. High-resolution mass spectra were recorded on an Agilent 1260 infinity LC coupled to a 6540 UHV accurate mass Q-TOF mass spectrometer by looped injection using electrospray ionisation (ESI, collision energy: 15 eV). Ultraviolet spectra were obtained using a Unicam 300 UV spectrophotometer (ThermoScientific, Rochester, USA). Thin-Layer Chromatography (TLC) was carried out on aluminium-backed SiO2 plates (Merck, Darmstadt, Germany) and spots were visualised using ultra-violet light (254 nm). Melting points were determined using Gallenkamp 5A 6797 apparatus (Gallenkamp, Germany) and are uncorrected. Optical rotation values [α]D22 (10−1 deg cm2 g−1) were performed on a Bellingham & Stanley ADP-220 polarimeter (Bellingham & Stanley, Tunbridge Wells, UK). All solutions were prepared using deionised water of resistivity no less than 18.2 MΩ cm and were vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen. Solutions containing 1000 μg mL−1 MDMA/PBS (pH 7) and 1000 μg mL−1 PMA/PBS (pH 7) were used on the day of preparation. Working solutions of lower concentrations were prepared by appropriate dilution of the stock solution as mentioned above.
:MDMA (30:70% w/w) was prepared by mixing PMA (17.25 mg) and MDMA (37.4 mg) and homogenising the sample prior to analysis. 1H- and 13C-NMR spectra were acquired on a JEOL AS-400 (JEOL, Tokyo, Japan) NMR spectrometer operating at a proton resonance frequency of 400 MHz. Samples of PMA (10 mg/0.60 mL) were dissolved in DMSO-d6 and filtered prior to analysis. Infrared spectra were obtained in the range 4000–400 cm−1 using a ThermoScientific Nicolet iS10ATR-FTIR instrument (ThermoScientific, Rochester, USA). GC-MS spectra were recorded on an Agilent 6890 gas chromatograph with split-splitless injection (sample volume: 1 μL) and a HP-5MS column (30 m × 0.25 mm, 0.25 μm film thickness). Helium (He) was used as the carrier gas at a flow rate of 1.0 mL min−1. The GC was coupled to an Agilent 5973 MSD (EI, 70 eV, TIC mode scanning m/z 50–500) and injector port was set at 275 °C, the transfer line at 280 °C. The following temperature program was used: 60 °C for 3 min, 20 °C min−1 to 280 °C, 280 °C for 5 min. High-resolution mass spectra were recorded on an Agilent 1260 infinity LC coupled to a 6540 UHV accurate mass Q-TOF mass spectrometer by looped injection using electrospray ionisation (ESI, collision energy: 15 eV). Ultraviolet spectra were obtained using a Unicam 300 UV spectrophotometer (ThermoScientific, Rochester, USA). Thin-Layer Chromatography (TLC) was carried out on aluminium-backed SiO2 plates (Merck, Darmstadt, Germany) and spots were visualised using ultra-violet light (254 nm). Melting points were determined using Gallenkamp 5A 6797 apparatus (Gallenkamp, Germany) and are uncorrected. Optical rotation values [α]D22 (10−1 deg cm2 g−1) were performed on a Bellingham & Stanley ADP-220 polarimeter (Bellingham & Stanley, Tunbridge Wells, UK). All solutions were prepared using deionised water of resistivity no less than 18.2 MΩ cm and were vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen. Solutions containing 1000 μg mL−1 MDMA/PBS (pH 7) and 1000 μg mL−1 PMA/PBS (pH 7) were used on the day of preparation. Working solutions of lower concentrations were prepared by appropriate dilution of the stock solution as mentioned above.
Synthesis of (±)-4-methoxyamphetamine hydrochloride (PMA)
The title compound was prepared, at the University of Strathclyde, using an adaptation of the synthesis reported by Liu et al.36 as an off-white crystalline powder (1.12 g, 56%) after recrystallization from ethanol–diethyl ether: mp (ethanol–diethyl ether) 211–213 °C (Lit. 210–211 °C37); Rf [SiO2, EtOAc–n-hexane (1:3)] = 0.1; [α]D22 = 0 (c = 0.5 g/100 mL, MeOH); IR (ATR-FTIR): 2917.3 (NH3+), 1515.0 (Ar, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); 1037.2 (Ar, O–CH3), 857.5 cm−1 (Ar, 1,4-disub.); 1H-NMR (400 MHz, DMSO-d6) δ1H (ppm) = 8.18 (3H, bs, CH2CH(CH3)NH3+), 7.16 (2H, d, J = 5.0 Hz, AA′BB′), 6.90 (2H, d, J = 5.0 Hz, AA′BB′), 3.73 (3H, s, CH3O–Ar), 3.33 (1H, m), 2.98 (1H, m, J = 13.3, 5.0 Hz), 2.58 (1H, m, J = 13.3, 9.2 Hz) and 1.10 ppm (3H, d, J = 8.0 Hz, CH2CH(CH3)NH3+); 13C-NMR (100 MHz, DMSO-d6) δ13C (ppm) = 17.4 (CH2CH(CH3)NH2), 39.1 (CH2CH(CH3)NH2), 48.0 (CH2CH(CH3)NH2), 54.9 (OCH3), 113.6 (Ar-CH), 128.4 (Ar-qC), 130.2 (Ar-CH) and 158.0 ppm (Ar-qC); LRMS (EI+, 70 eV): m/z = 166 (0.5%, [M + H]), 122 (100), 121 (34), 91 (17), 77 (22) and 44 (45); HRMS (ESI+, 15 eV) calculated for [M + H] C10H16NO: 166.1226, found: 166.1229.
C); 1037.2 (Ar, O–CH3), 857.5 cm−1 (Ar, 1,4-disub.); 1H-NMR (400 MHz, DMSO-d6) δ1H (ppm) = 8.18 (3H, bs, CH2CH(CH3)NH3+), 7.16 (2H, d, J = 5.0 Hz, AA′BB′), 6.90 (2H, d, J = 5.0 Hz, AA′BB′), 3.73 (3H, s, CH3O–Ar), 3.33 (1H, m), 2.98 (1H, m, J = 13.3, 5.0 Hz), 2.58 (1H, m, J = 13.3, 9.2 Hz) and 1.10 ppm (3H, d, J = 8.0 Hz, CH2CH(CH3)NH3+); 13C-NMR (100 MHz, DMSO-d6) δ13C (ppm) = 17.4 (CH2CH(CH3)NH2), 39.1 (CH2CH(CH3)NH2), 48.0 (CH2CH(CH3)NH2), 54.9 (OCH3), 113.6 (Ar-CH), 128.4 (Ar-qC), 130.2 (Ar-CH) and 158.0 ppm (Ar-qC); LRMS (EI+, 70 eV): m/z = 166 (0.5%, [M + H]), 122 (100), 121 (34), 91 (17), 77 (22) and 44 (45); HRMS (ESI+, 15 eV) calculated for [M + H] C10H16NO: 166.1226, found: 166.1229.
High performance liquid chromatography (HPLC)
Reverse phase high-performance liquid chromatography was performed with an integrated Agilent HP Series 1100 Liquid Chromatograph (Agilent Technologies, Wokingham, UK) fitted with an in-line degasser, 100-place auto-injector and diode array UV absorbance detector monitoring at 210 nm. Data analysis was carried out using ChemStation for LC (Ver. 10.02) software (Agilent Technologies, Wokingham, UK). The mobile phase was aqueous potassium dihydrogen phosphate buffer (0.05 M, pH 3.2 ± 0.02):acetonitrile (90:10% v/v); the flow rate was 1.2 mL min−1 with an injection volume of 10 μL. Six replicate injections of each calibration standard were performed. The stationary phase (ACE 3 C18, 150 mm × 4.6 mm i.d., particle size: 3 μm) used in the study was obtained from HiChrom Limited (Reading, UK). The column was fitted with a guard cartridge (ACE 3 C18) and maintained at an isothermal temperature of 22 °C with an Agilent HP Series 1100 column oven with a programmable controller (Agilent Technologies, Wokingham, UK).
Calibration standards
2.0 mg of (±)-3,4-methylenedioxymethamphetamine hydrochloride (MDMA) and 2.0 mg of (±)-4-methoxyamphetamine hydrochloride (PMA) were weighed accurately into a 10.0 mL clear glass volumetric flask and diluted to volume with mobile phase to give a solution containing the two components at 200.0 μg mL−1. This solution was then further diluted with mobile phase to give calibration standards containing 40.0 μg mL−1, 20.0 μg mL−1, 10.0 μg mL−1, 2.5 μg mL−1 and 1.25 μg mL−1 of each analyte respectively.Specificity standards
5.0 mg sucrose, mannitol and lactose were weighed accurately into separate 100.0 mL clear glass volumetric flask and diluted to volume with mobile phase to give solutions containing the components at 50 μg mL−1 of each analyte. This solution was then further diluted (5×) with mobile phase to give specificity standards containing 10.0 μg mL−1 of each analyte respectively.HPLC method validation
The HPLC method was validated in accordance with the ICH guidelines38 using the following parameters: linearity, accuracy, precision, specificity, limit of detection (LOD), limit of quantification (LOQ) and system suitability [resolution (Rs), column efficiency (N), peak asymmetry (As)]. Linearity, precision and system suitability tests: six replicate injections of the calibration standards (vide supra) were performed and the data analysed under the same conditions. The %RSD was calculated for each replicate sample. Accuracy: determined from three replicate concentrations near the test concentration (80%, 100%, and 120%, two replicate injections of each concentration). The percentage recovery and %RSD were calculated for each of the replicate samples. Specificity: six replicate injections of the specificity standards (vide supra) were performed and the data analysed under the same conditions. Limits of detection and quantification: six replicate injections of the calibration standards (vide supra) were performed and the data analysed under the same conditions. The limits of detection and quantification were calculated based on the standard deviation of the response and the slope.Presumptive tests
Presumptive tests were carried out according to the United Nations recommended guidelines.39 The following standard presumptive tests applied in this study: (i) Marquis; (ii) Mandelin; (iii) Simon's and (iv) Robadope test(s). The preparation of the reagents and test procedure is detailed in the ESI.† Six repetitive tests of each compound were conducted and negative control samples were used in all tests. The ESI† contains images of the spotting tiles (after 5 minutes). Test solutions containing 25:75% v/v; 50:50% v/v and 75:25% v/v (PMA:MDMA) were prepared by mixing appropriate volumes of solutions (10 mg mL−1) of the reference standards in methanol.
Electrochemistry
Voltammetric measurements were carried out using a Palmsens 3 (Palm Instruments BV, The Netherlands) potentiostat/galvanostat and controlled by PSTrace software version 4.4 for Windows 7. All electrochemical measurements were performed at room temperature (cyclic voltammetry and differential pulse voltammetry). Differential pulse voltammetry parameters (E step, E pulse, t pulse and scan rate) were optimised prior to experimentation. A conventional three-electrode system was used. For each voltammogram acquired, a new/separate screen-printed graphite electrode was used, with the exception of the scan rate study. This highlights the reproducibility of the SPEs. Experiments were performed using screen-printed graphite electrode (SPE), boron-doped diamond electrode (BDD) and glassy carbon electrode (GC). All the working electrodes used in the analyses had 3 mm diameter working area. Screen-printed graphite electrodes were fabricated in-house with appropriate stencil designs using a DEK 248 screen-printing machine (DEK, Weymouth, U.K.). The screen-printed graphite electrodes (SPEs) utilized consist of a graphite working electrode, a graphite counter electrode and a silver/silver chloride reference electrode. The screen-printed graphite electrodes, which have a 3 mm diameter working electrode were fabricated in-house with appropriate stencil designs using a microDEK 1760RS screen-printing machine (DEK, Weymouth, UK). This screen-printed electrode design has been previously reported.26,28,29,33,40–44 For the case of each fabricated electrode, first a carbon ink formulation (Product Code: C2000802P2; Gwent Electronic Materials Ltd, UK), which is utilized for the efficient connection of all three electrodes and as the electrode material for both the working and counter electrodes, was screen-printed onto a polyester (Autostat, 250 micron thickness) flexible film. After curing the screen-printed carbon layer in a fan oven at 60 degrees Celsius for 30 minutes, next a silver/silver chloride reference electrode was included by screen-printing Ag/AgCl paste (Product Code: C2040308D2; Gwent Electronic Materials Ltd, UK) onto the polyester substrates, which was subsequently cured once more in a fan oven at 60 degrees for 30 minutes. Finally, a dielectric paste (Product Code: D2070423D5; Gwent Electronic Materials Ltd, UK) was then printed onto the polyester substrate to cover the connections and define the active electrode areas, including that of the working electrode (3 mm diameter). After curing at 60 degrees for 30 minutes the SPEs are ready to be used. The reproducibility and repeatability of the fabricated batches of electrodes were explored through comparison of cyclic voltammetric responses using Ru(NH3)2+/3+ redox probe in 1 M KCl. Analysis of the voltammetric data revealed the % relative standard deviation (%RSD) to correspond to no greater than 0.82% (n = 20) and 0.76% (n = 3) for the reproducibility and repeatability of the fabricated GSPEs (for use in electroanalysis). For each electrochemical experiment/scan, a new screen-printed graphite electrode was used. All differential pulse voltammograms were baseline corrected.Raman
Raman Spectroscopy was performed using a ‘Renishaw InVia’ spectrometer with a confocal microscope ×50 objective, spectrometer with an argon laser of 514.3 nm excitation. Spectra were recorded using a 10 s exposure time for 1 accumulation. Three spectra were recorded and an average representation is presented within the manuscript.Analysis of synthetic sample: “ecstasy synthetic”
For the electrochemical determination of MDMA/PMA solutions were prepared using carefully weighed samples of “synthetic ecstasy” in PBS (pH 7). Subsequently, the solution was sonicated for 3 minutes for homogenization and filtered to remove remaining undesirable insoluble materials. A total of five seized ecstasy tablets were analysed via HPLC and the presence of PMA was not detected in either tablet, so a sample containing the two substances was created. PMA is a lethal substance when administered in substantial quantities, but produces similar effects to MDMA. As it is a cheaper substance, it is occasionally added to MDMA in ecstasy tablets to lower the cost of the drug.Results and discussion
Electrochemical detection of MDMA and PMA
First, cyclic voltammetric measurements were performed using screen-printed graphite electrodes (SPEs) in solutions containing PMA, MDMA and MDMA/PMA in phosphate buffer (pH 7). Fig. 1A shows the cyclic voltammograms of SPE in the presence (500 μg mL−1) and absence of PMA. In the case of PMA a well-defined oxidation peak is observed at +1.162 V. Fig. 1B shows the cyclic voltammograms in the presence (250 μg mL−1) and absence of MDMA using the SPEs. The cyclic voltammogram in the presence of MDMA shows the oxidation peaked in +0.924 V. Fig. 1C shows the cyclic voltammograms in the presence of (250 μg mL−1)/(500 μg mL−1) and absence of MDMA/PMA where two oxidation peaks can be observed; peak I at +0.92 V due to oxidation of MDMA and peak II at +1.20 V is due to oxidation of PMA. The sole presence of an oxidation peak for all the analytes suggests an electrochemically irreversible reaction.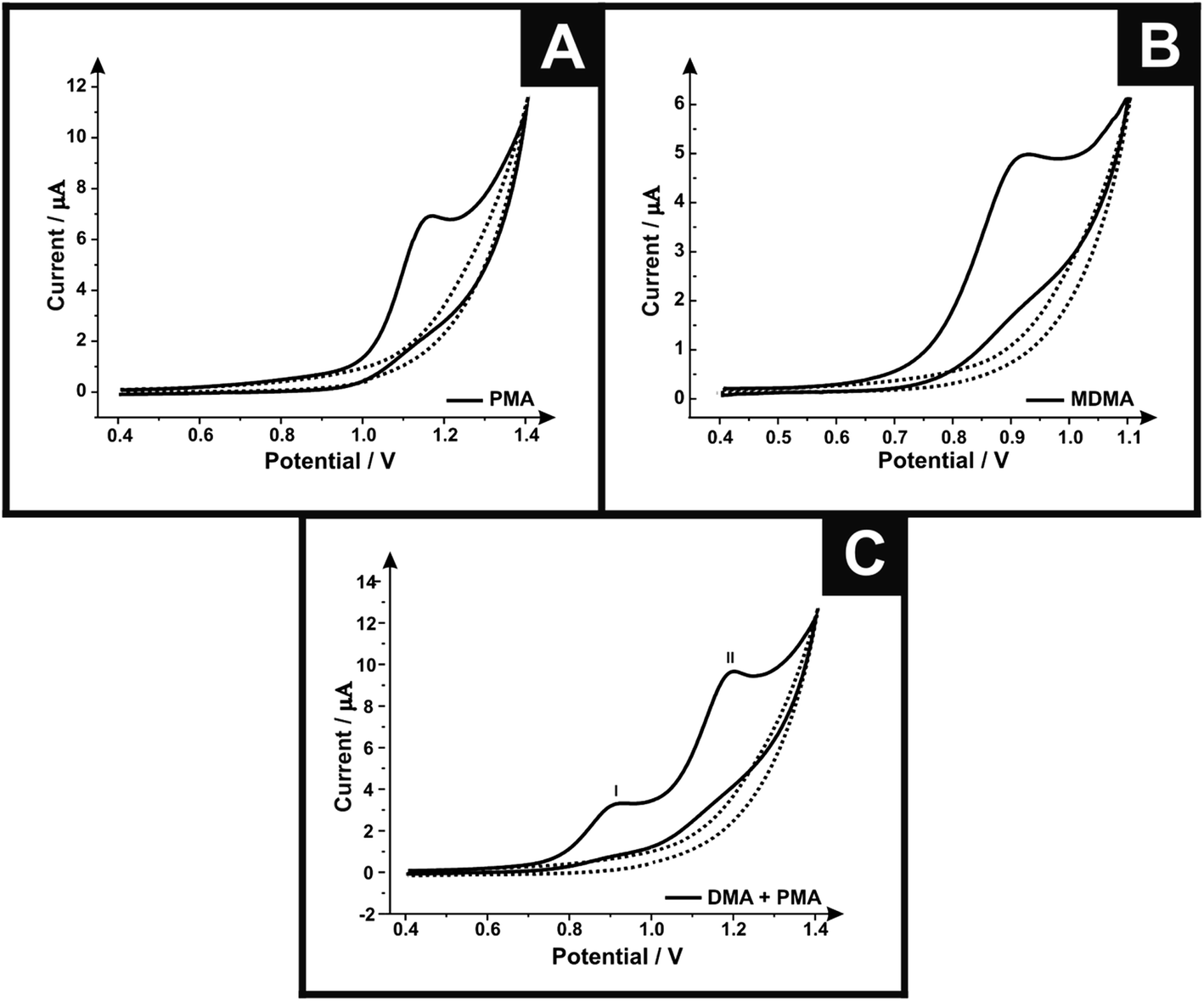 | ||
| Fig. 1 Cyclic voltammograms of SPE (A) in presence (500 μg mL−1) and absence of PMA, (B) in presence (250 μg mL−1) and absence of MDMA and (C) in presence (250 μg mL−1)/(500 μg mL−1) and absence of MDMA/PMA in PBS (pH 7) (scan rate: 100 mV s−1). | ||
Different electrodes were explored towards the electrochemical oxidation of the target analytes to provide a direct comparison with the response obtained using the screen-printed graphite electrode. Fig. SI-1† shows the electrochemical response of the SPE, boron doped diamond (BDD) and glassy carbon (GC). For the target analyte MDMA, the SPE shows an optimal response with the electrochemical oxidation occurring at the lowest overpotential (+0.99 V) with the highest current density (145.42 μA cm−2) when compared with the BDD (+1.19 V and 127.44 μA cm−2) and GC (+1.15 V and 137.77 μA cm−2). The current densities were calculated using the geometric area of the electrode surfaces. For PMA, the GC (+1.28 V) and SPE (+1.29 V) show the potential response similar, but the GC show the highest current density (376.22 μA cm−2) when compared with the BDD (229.70 μA cm−2) and SPE (252.37 μA cm−2). The difference of current density of the GC and SPE does not make the GC advantageous because this electrode needs to go through a pre-treatment step each time it is used, which is not necessary for the SPE because after each use the electrode is disposed thereby gaining time. In the case of the MDMA/PMA, the SPE exhibits the optimal voltammetric response (88.99 μA cm−2 MDMA/89.97 μA cm−2 PMA) and lowest overpotential (+0.97 V MDMA/+1.26 V PMA) when compared with the BDD (+1.15 V and 71.65 μA cm−2 MDMA/+1.45 V and 82.80 μA cm−2 PMA) and GC (+1.18 V and 77.75 μA cm−2 MDMA/+1.51 V and 83.34 μA cm−2 PMA). The SPE was chosen since it exhibits the best electrochemical response, being an electrode for easy handling, low cost and high reproducibility.
Next, the effect of scan rate upon the electrochemical oxidation were explored for the target analytes in PBS (pH 7). A plot of peak height against the square-root of scan rate was constructed for PMA (Ip/A = 7.10 × 10−5 A (V s−1)−0.5 + 1.21 × 10−6 A R2 = 0.997), MDMA (Ip/A = 3.05 × 10−5 A (V s−1)−0.5 + 1.17 × 10−6 A, R2 = 0.993) and MDMA/PMA (Ip/A = 2.87 × 10−5 A (V s−1)−0.5 + 6.61 × 10−8 A, R2 = 0.996/Ip/A = 3.49 × 10−5 A (V s−1)−0.5 − 7.10 × 10−7 A, R2 = 0.990) and found to be linear in all cases indicating a diffusional electrochemical process. The effect of the pH on the voltammetric response (differential pulse voltammetry) were also explored for the target analytes PMA, MDMA and MDMA/PMA in various buffers. Fig. SI-2† shows the voltammograms for the SPE in (A) 500 μg mL−1 PMA/PBS, (B) 500 μg mL−1 MDMA/PBS and (C) 500 μg mL−1 PMA + 250 μg mL−1 MDMA/PBS (pH 7) over a range of pH (2–12). It can be seen that the oxidative peak exhibits a potential at more negative offset to regions with increasing pH (acid to basic) and a fall in current values of the oxidative peak at basic pH with a linear relationship for PMA of (Ep/V = −0.03 V per pH + 1.27 V; R2 = 0.945) with a gradient of 28.30 mV per pH that value is close to that expected for a 1 proton and 2 electron process (30 mV per pH unit at 25 °C), for MDMA to (Ep/V = −0.02 V per pH + 1.00 V; R2 = 0.988) with a gradient of 20.50 mV per pH which is close to that expected for a 1 proton and 3 electron process (20 mV per pH unit at 25 °C) and MDMA/PMA (Ep/V = −0.02 V per pH + 1.01 V; R2 = 0.978)/(Ep/V = −0.03 V per pH + 1.34 V; R2 = 0.993) sustaining the response of individually analysed analytes. The appearance of one new peak (between +0.6 and +0.7 V) above pH 9 is related with the acid–base properties of the MDMA and is probably due to the existence of an electron lone-pair.35 The pH of 7 was chosen due to the good electrochemical responses compared with other pHs.
Differential pulse voltammetry was next utilised to explore the electroanalytical efficiency of the proposed protocol through utilising the SPEs by adding aliquots of MDMA (pH 7.0) at concentrations over the range of 0.50–4.98 μg mL−1 into a pH 7 phosphate buffer; Fig. 2 shows the voltammetry. Through the additions of MDMA there is an increase in the current intensity of the oxidation peak and a small displacement of potential into positive regions. The insert graph shows the linear calibration curve obtained from the differential pulse voltammograms (Ip/μA = 0.11 A mL g−1 [μg mL−1] + 0.01 μA; R2 = 0.999 and N = 3) with a limit of detection (3σ) found to correspond to 0.04 μg mL−1 and a relative standard deviation (%RSD) of 2.14%. Again note that each additions is made with a new SPE. Various methods have been used before in the detection and quantification of MDMA45–48 and Table 1 summarises these with the results of this work also presented and compared demonstrating our approach is competitive.
 | ||
| Fig. 2 Differential pulse voltammograms obtained utilizing the screen-printed graphite electrodes by adding aliquots of MDMA (pH 7.0) at concentrations in the range of 0.50–4.98 μg mL−1 (in phosphate buffer supporting electrolyte); the dotted line represents a blank. (inset) An analytical curve corresponding to the anodic peak for the oxidation of MDMA over the concentration range (E-step: 0.002 V; E-pulse: 0.1 V; t-pulse: 0.05 s; scan rate: 0.005 V s−1; t-eq. 1 s). | ||
| Analyte | Analytical method | Matrix | Analytical linear range | Limit of detection | Reference |
|---|---|---|---|---|---|
| PMA | Gas chromatography equipped with a nitrogen phosphorus detector | Peripheral and heart blood | 0.125 mg L−1 to 1.0 mg L−1 | N/A | 51 |
| MDMA | Square wave voltammetry | Supporting electrolyte | 8–45 μM | 1.2 μM | 35 |
| Human sérum | 12–45 μM | 2.4 μM | |||
| PMA | Capillary electrophoresis with diode array | Plasma | 50–5000 ng mL−1 | 20.92 ng mL−1 | 52 |
| Urine | 24.26 ng mL−1 | ||||
| MDMA | Turn-on fluorogenic probe | Water | N/A – not disclosed | 0.13 μM | 53 |
| MDMA | Thin layer chromatography/fluorescence | Urine | N/A – not disclosed | 50 ng | 54 |
| MDMA | HPLC–chemiluminescence | Plasma | 0.01–1.0 ng mL−1 | 3 ng mL−1 | 55 |
| Hair root | 0.10–10 ng mL−1 | 17 ng mL−1 | |||
| Hair shaft | 0.10–10 ng mL−1 | 14 ng mL−1 | |||
| MDMA | Cyclic voltammetry – dip coating/spin coating | KCl (0.1 mol L−1) | 4.2–48 μmol L−1 | 3.5/2.7 μmol L−1 | 56 |
| MDMA | This work | Phosphate buffer | 0.50–4.98 μg mL −1 | 0.04 μg mL −1 | N/A |
| PMA | 0.50–4.98 μg mL −1 | 0.03 μg mL −1 | |||
| MDMA/PMA (simultaneous) | 2.00–19.60 μg mL −1 (for both) | 0.25 μM/0.14 μg mL −1 |
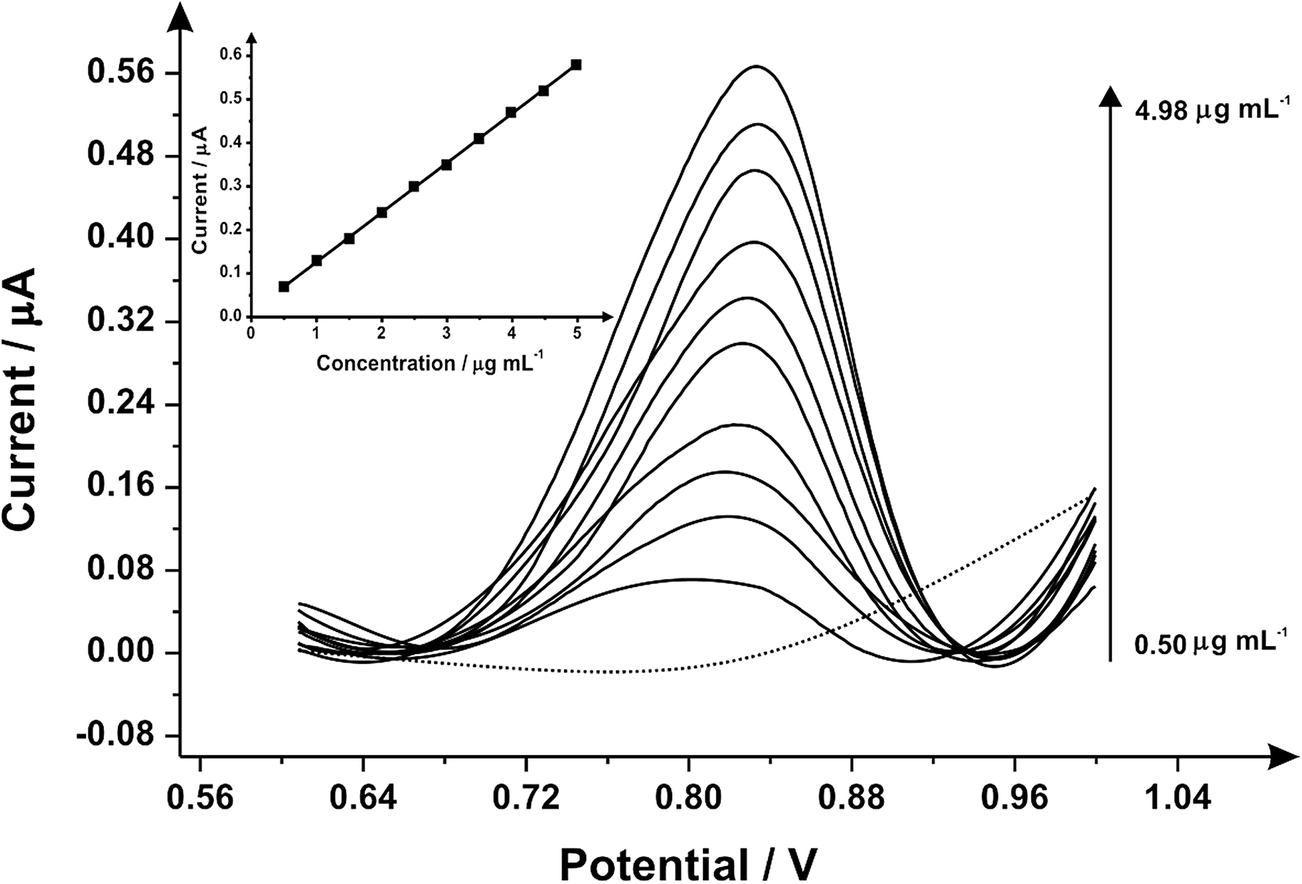
Fig. 3 illustrates the voltammetric behaviour of the SPEs through the addition of aliquots of PMA (in phosphate buffer pH 7.0) at concentrations over the range of 0.50–4.98 μg mL−1. The linear calibration curve of the anodic current of oxidation peak as a function of PMA concentration is presented in the insert of Fig. 3 (Ip/μA = 0.21 A mL g−1 [μg mL−1] + 0.07 μA, R2 = 0.999 and N = 3) with a limit of detection (3σ) found to correspond to 0.03 μg mL−1 and relative standard deviation (%RSD) of 4.3. Last, the analytical performance of MDMA/PMA using SPEs were explored (see Fig. 4) with additions made over the range of 2.00–19.60 μg mL−1 for MDMA (Ip/μA = 0.09 A mL g−1 [μg mL−1] + 0.08 μA; R2 = 0.999; N = 3) and 2.00–19.60 μg mL−1 for PMA (Ip/μA = 0.18 A mL g−1 [μg mL−1] + 0.03 μA; R2 = 0.999; N = 3). The limit of detection (3σ) was found to correspond to 0.14 μg mL−1 for PMA (Fig. 4 inset) and 0.25 μg mL−1 for MDMA (Fig. 4 inset) and relative standard deviations (%RSD) of 3.2% MDMA and 3.8% for PMA. Table 1 summarises these electroanalytical results and benchmarks these against the current literature demonstrating our work is highly competitive.
 | ||
| Fig. 3 Differential pulse voltammograms of the screen-printed graphite electrode by adding aliquots of PMA (in phosphate buffer pH 7.0) at concentrations in the range of 0.50–4.98 μg mL−1 (in phosphate buffer supporting electrolyte); the dotted line represents a blank. (inset) An analytical curve corresponding to the anodic peak for the oxidation of PMA over the concentration range (E-step: 0.002 V; E-pulse: 0.1 V; t-pulse: 0.05 s; scan rate: 0.005 V s−1; t-eq. 1 s). | ||
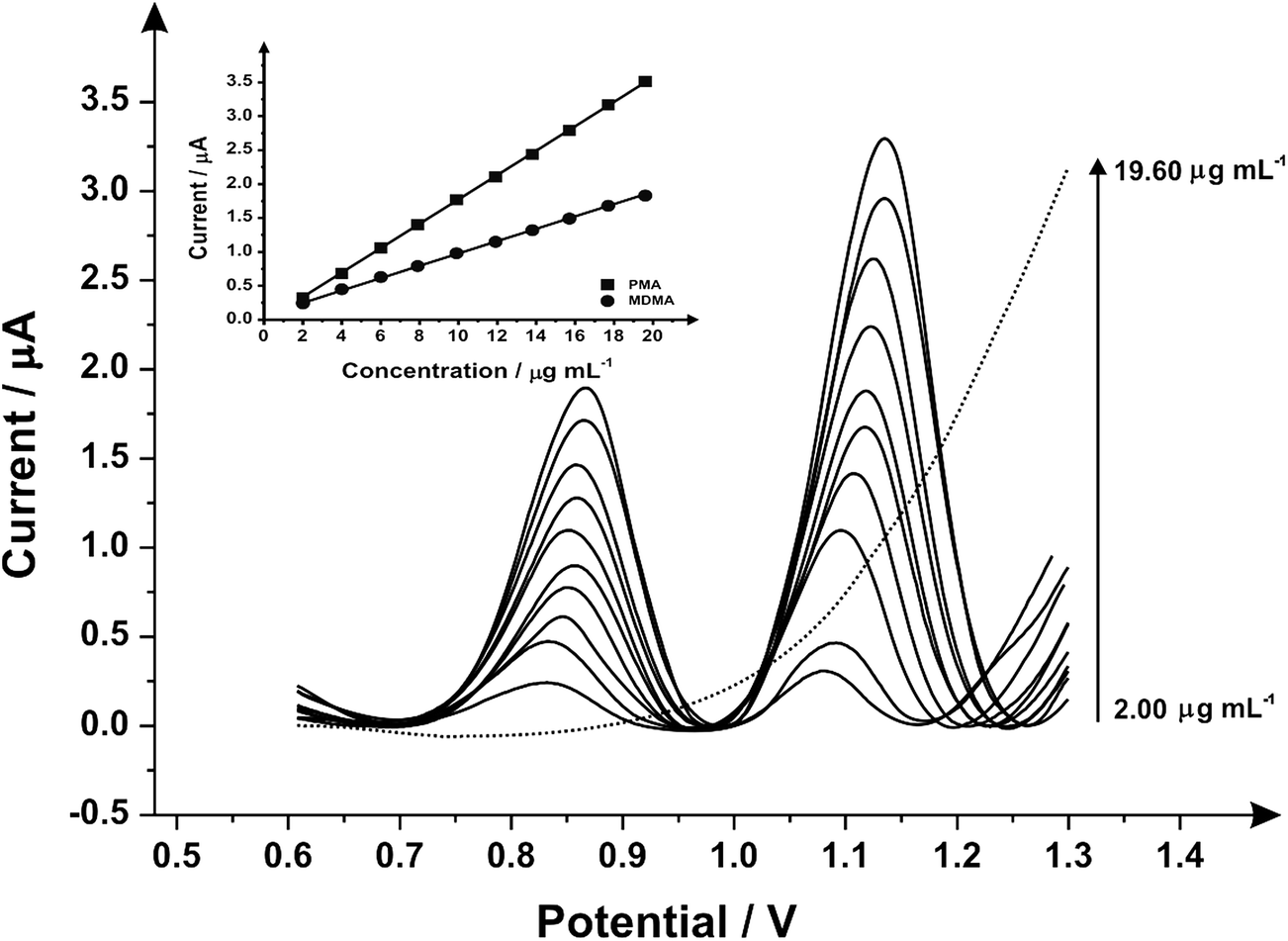 | ||
| Fig. 4 Differential pulse voltammograms obtained utilizing the screen-printed graphite electrodes by adding aliquots of MDMA/PMA (in phosphate buffer pH 7.0) at concentrations in the range of 2.00–19.60 μg mL−1 (in phosphate buffer supporting electrolyte); the dotted line represents a blank. (inset) An analytical curve corresponding to the anodic peak for the oxidation of PMA and MDMA over the concentration range (E-step: 0.002 V; E-pulse: 0.1 V; t-pulse: 0.05 s; scan rate: 0.005 V s−1; t-eq. 1 s). | ||
Alternative approaches for the sensing of MDMA/PMA
:75% v/v; 50:50% v/v and 75:25% v/v (PMA:MDMA) were prepared by mixing appropriate volumes of solutions (10 mg mL−1) of the reference standards in methanol and screened against the standard tests. The data (see SI-5†) indicated that in all cases the test reagents (when compared to the pure standards) confirmed the presence of MDMA (even at concentrations circa. 25% v/v), however, the presence of PMA (indicated by a positive reaction with Robadope reagent) was not as easy to discriminate using this method even when present at high concentrations (circa. 75% v/v), demonstrating that the utilisation of this presumptive colour test in particular for products containing these two compounds is potentially problematic and cannot be relied upon in these cases. The simulated sample of MDMA adulterated with PMA (30% w/w), was screened against the standard tests and observed to give analogous results to the mixture containing 25:75% w/w (PMA:MDMA). The sample gave a positive reaction with Marquis, Mandelin and Simon's reagents indicating that the sample does indeed contain MDMA, however, the colour observed with Robadope reagent was a less intense rose colour than that observed with the pure PMA reference standard, indicating that there may indeed be interference between the mixture of components and test reagents which may be encountered by law enforcement. In summary, Raman spectroscopy and presumptive colour tests fail to distinguish between PMA and MDMA and only electrochemistry provides a useful sensing approach.
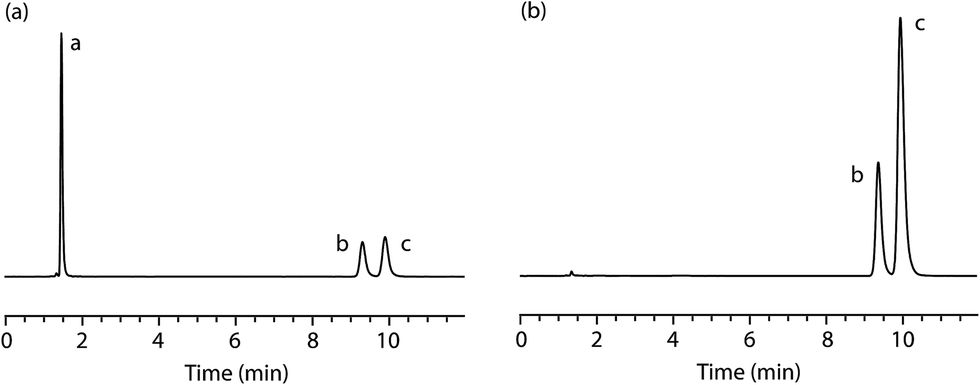 | ||
| Fig. 5 (a) Representative chromatogram of a solution containing PMA (10 μg mL−1, peak b) and MDMA (10 μg mL−1, peak c) obtained using an ACE 3 C18 column (150 mm × 4.6 mm i.d., particle size: 3 μm); flow-rate: 1.2 mL min−1; mobile phase: aqueous potassium dihydrogen phosphate buffer (0.05 M, pH 3.2 ± 0.02):acetonitrile (90:10% v/v); detector wavelength (UV): 210 nm; (b) representative chromatogram of test mixture (10 μg mL−1) containing PMA and MDMA (30:70% w/w) obtained using an ACE 3 C18 column (150 mm × 4.6 mm i.d., particle size: 3 μm); flow-rate: 1.2 mL min−1; mobile phase: aqueous potassium dihydrogen phosphate buffer (0.05 M, pH 3.2 ± 0.02):acetonitrile (90:10% v/v); detector wavelength (UV): 210 nm. The t0 was determined from the tR of a solution of uracil (10 μg mL−1, peak a). | ||
To demonstrate if our proposed method could selectively detect and quantify the components in a potential street sample of MDMA adulterated with PMA a simulated sample was prepared by mixing the two components in a ratio of 70:30% w/w. The simulated sample was homogenised and analysed, in triplicate, using the validated HPLC method at a concentration of 10 μg mL−1 (Fig. 5b). The results confirm that the sample contains the two alleged components (tR = 9.4 min [minor, PMA, 31.51% w/w, %RSD = 0.14%, n = 3] and tR = 9.9 min [major, MDMA, 68.49% w/w, %RSD = 0.07%, n = 3]) in the expected proportions.
The electrochemical protocol was explored towards the ecstasy (synthetic) sample. Fig. 6 shows differential pulse voltammograms for electrochemical oxidation of ecstasy (a mixture of MPA and MDMA) using SPEs at concentrations in the range of 0.00–14.80 μg mL−1 and 0.00–16.70 μg mL−1 for PMA and MDMA respectively (in phosphate buffer supporting electrolyte) it is possible to see two electrochemical oxidation peaks for MDMA in +0.86 V and PMA in +1.14 V. Calibration plots for these two peaks were constructed (see Fig. 6 inset) showing a linear response over the entire concentration range studied. Table 2 lists the parameters obtained of the linear response for PMA/MDMA and the comparison of the electrochemical and high performance liquid chromatography (HPLC) results which agree with each other validating our electroanalytical protocol.
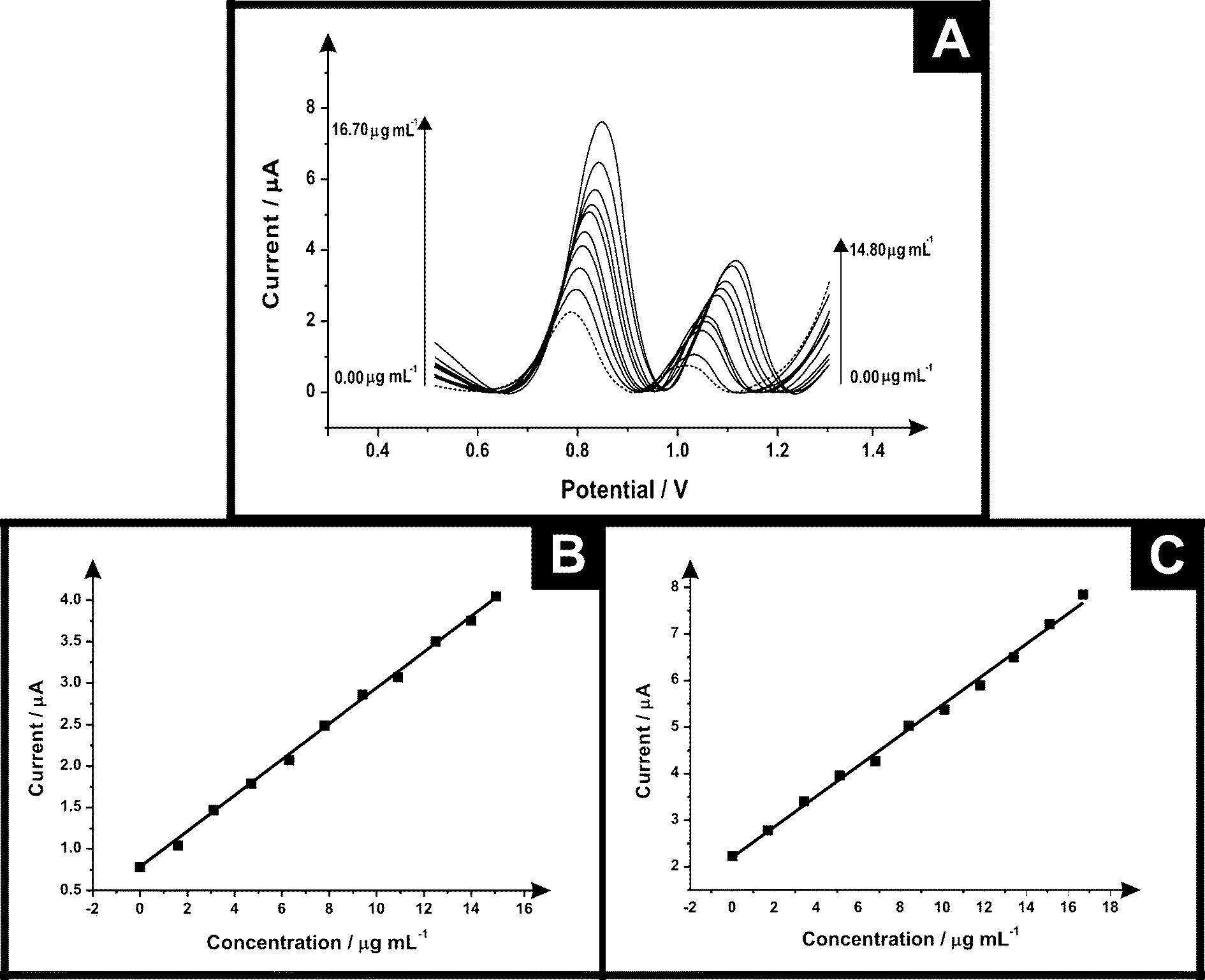 | ||
| Fig. 6 Differential pulse voltammograms following a series of standard additions of “ecstasy” over the range 0.00–14.80 μg mL−1 and 0.00–16.70 μg mL−1 for PMA and MDMA respectively (in phosphate buffer supporting electrolyte); the dotted line represents a blank. (inset) An analytical curve corresponding to the anodic peak for the oxidation of (B) MDMA and (C) PMA over the concentration range (E-step: 0.002 V; E-pulse: 0.1 V; t-pulse: 0.05 s; scan rate: 0.005 V s−1; t-eq. 1 s). | ||
| Ecstasy (synthetic) | PMA | MDMA |
|---|---|---|
| Linear concentration range (μg mL−1) | 0.00–14.80 | 0.00–16.70 |
| Slope of calibration graph (A mL g−1) | 0.24 | 0.33 |
| Intercept (μA) | 0.76 | 2.21 |
| Correlation coefficient | 0.998 | 0.996 |
| Relative standard deviation (%RSD) | 4.38 | 2.33 |
| Purity | Total | PMA | MDMA |
|---|---|---|---|
| Electrochemical w/w% | 99.37 | 31.53 | 67.84 |
| HPLC w/w% | 100.02 | 31.51 | 68.49 |
Conclusions
For the first time, the simultaneous detection of MDMA and PMA using screen-printed graphite electrodes has been reported. This a novel sensing protocol is shown to be both viable and superior to other analytical techniques (HPLC, Raman) in terms of speed and cost. Presumptive testing is also shown to be unable to differentiate between MDMA and PMA which further eludes towards the use of the proposed electroanalytical protocol as an in-the-field sensor. The electroanalytical protocol has been validated in a synthetic street sample with HPLC. Future work is underway to extend this study to seized street samples with independent validation with HPLC/LS-MS.Acknowledgements
We would like to thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) – Process no. 99999.001285/2014-09 and a British Council Institutional Link grant (No. 172726574) for the support in this research.References
- A. R. Pentney, J. Psychoact. Drugs, 2001, 33, 213–221 CrossRef CAS PubMed.
- J. C. Kraner, D. J. McCoy, M. A. Evans, L. E. Evans and B. J. Sweeney, J. Anal. Toxicol., 2001, 25, 645–648 CrossRef CAS PubMed.
- N. A. Buckley and D. G. Barceloux, in Medical Toxicology of Drug Abuse, John Wiley & Sons, Inc., 2012, pp. 126–155, DOI:10.1002/9781118105955.ch9.
- E. B. de Sousa Fernandes Perna, E. L. Theunissen, K. P. C. Kuypers, P. Heckman, R. de la Torre, M. Farre and J. G. Ramaekers, Neuropharmacology, 2014, 87, 198–205 CrossRef CAS PubMed.
- V. Ferraz-de-Paula, A. Ribeiro, J. Souza-Queiroz, M. L. Pinheiro, J. F. Vecina, D. P. Souza, W. M. Quinteiro-Filho, R. L. Moreau, M. L. Queiroz and J. Palermo-Neto, J. NeuroImmun. Pharmacol., 2014, 9, 690–702 CrossRef CAS PubMed.
- M. Isabel Colado, E. O'Shea and A. Richard Green, in Handbook of Contemporary Neuropharmacology, John Wiley & Sons, Inc., 2007, DOI:10.1002/9780470101001.hcn039.
- K. Soar, J. J. D. Turner and A. C. Parrott, Hum. Psychopharmacol., 2001, 16, 641–645 CrossRef PubMed.
- M. E. Liechti and F. X. Vollenweider, Hum. Psychopharmacol., 2001, 16, 589–598 CrossRef CAS PubMed.
- A. E. Fleckenstein, T. J. Volz, E. L. Riddle, J. W. Gibb and G. R. Hanson, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 681–698 CrossRef CAS PubMed.
- M. Verschraagen, A. Maes, B. Ruiter, I. J. Bosman, B. E. Smink and K. J. Lusthof, Forensic Sci. Int., 2007, 170, 163–170 CrossRef CAS PubMed.
- E. A. de Letter, C. P. Stove, W. E. Lambert and M. H. A. Piette, Curr. Pharm. Biotechnol., 2010, 11, 453–459 CAS.
- E. Turillazzi, I. Riezzo, M. Neri, S. Bello and V. Fineschi, Curr. Pharm. Biotechnol., 2010, 11, 500–509 CAS.
- C. White, M. Edwards, J. Brown and J. Bell, J. Psychopharmacol., 2014, 28, 1018–1029 CrossRef PubMed.
- D. G. Caldicott, N. A. Edwards, A. Kruys, K. P. Kirkbride, D. N. Sims, R. W. Byard, M. Prior and R. J. Irvine, J. Toxicol., Clin. Toxicol., 2003, 41, 143–154 CrossRef CAS PubMed.
- D. M. Paton, J. I. Bell, R. Yee and D. A. Cook, Proc. West. Pharmacol. Soc., 1975, 18, 229–231 CAS.
- T. L. Martin, J. Anal. Toxicol., 2001, 25, 649–651 CrossRef CAS PubMed.
- R. Dams, E. A. de Letter, K. A. Mortier, J. A. Cordonnier, W. E. Lambert, M. H. Piette, S. van Calenbergh and A. P. de Leenheer, J. Anal. Toxicol., 2003, 27, 318–322 CrossRef CAS PubMed.
- C. Lora-Tamayo, T. Tena, A. Rodriguez, D. Moreno, J. R. Sancho, P. Ensenat and F. Muela, Forensic Sci. Int., 2004, 140, 195–206 CrossRef CAS PubMed.
- I. B. Müller and C. N. Windberg, J. Chromatogr. Sci., 2005, 43, 434–437 Search PubMed.
- D. R. Stoll, C. Paek and P. W. Carr, J. Chromatogr. A, 2006, 1137, 153–162 CrossRef CAS PubMed.
- A. M. Camilleri and D. Caldicott, Forensic Sci. Int., 2005, 151, 53–58 CrossRef PubMed.
- K. Kudo, T. Ishida, K. Hara, S. Kashimura, A. Tsuji and N. Ikeda, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 855, 115–120 CrossRef CAS PubMed.
- K. A. Mortier, R. Dams, W. E. Lambert, E. A. D. Letter, S. V. Calenbergh and A. P. D. Leenheer, Rapid Commun. Mass Spectrom., 2002, 16, 865–870 CrossRef CAS PubMed.
- R. Stanaszek and W. Piekoszewski, J. Anal. Toxicol., 2004, 28, 77–85 CrossRef CAS PubMed.
- M. D. M. R. Fernández, V. Di Fazio, S. M. R. Wille, N. Kummer and N. Samyn, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 965, 7–18 CrossRef PubMed.
- J. P. Smith, J. P. Metters, C. Irving, O. B. Sutcliffe and C. E. Banks, Analyst, 2014, 139, 389–400 RSC.
- J. P. Smith, O. B. Sutcliffe and C. E. Banks, Analyst, 2015, 140, 4932–4948 RSC.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136, 1067–1076 RSC.
- J. Metters, F. Tan and C. Banks, J. Solid State Electrochem., 2013, 17, 1553–1562 CrossRef CAS.
- J. P. Metters, M. Gomez-Mingot, J. Iniesta, R. O. Kadara and C. E. Banks, Sens. Actuators, B, 2013, 177, 1043–1052 CrossRef CAS.
- A. V. Kolliopoulos, J. P. Metters and C. E. Banks, Anal. Methods, 2013, 5, 851–856 RSC.
- O. Ramdani, J. P. Metters, L. C. S. Figueiredo-Filho, O. Fatibello-Filho and C. E. Banks, Analyst, 2013, 138, 1053–1059 RSC.
- J. P. Smith, J. P. Metters, D. K. Kampouris, C. Lledo-Fernandez, O. B. Sutcliffe and C. E. Banks, Analyst, 2013, 138, 6185–6191 RSC.
- J. P. Smith, J. P. Metters, O. I. G. Khreit, O. B. Sutcliffe and C. E. Banks, Anal. Chem., 2014, 86, 9985–9992 CrossRef CAS PubMed.
- E. M. P. J. Garrido, J. M. P. J. Garrido, N. Milhazes, F. Borges and A. M. Oliveira-Brett, Bioelectrochemistry, 2010, 79, 77–83 CrossRef CAS PubMed.
- J.-T. Liu, M.-P. Sun and Y.-S. Tsai, Forensic Sci. J., 2003, 2, 59–68 Search PubMed.
- A. Y. Savitskii, Zh. Obshch. Khim., 1940, 10, 1819–1826 CAS.
- I. H. T. Guideline, Validation of Analytical Procedures: Text and, Text and Methodology q2(r1) Search PubMed.
- G. Nagy, I. Szollosi and K. Szendrei, Colour Tests for Precursor Chemicals of Amphetamine-Type Substances The Use of Colour Tests for Distinguishing between Ephedrine-Derivatives, http://www.unodc.org/pdf/scientific/SCITEC20-fin.pdf, accessed, August-15-2015 Search PubMed.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2013, 138, 2516–2521 RSC.
- J. P. Metters, F. Tan, R. O. Kadara and C. E. Banks, Anal. Methods, 2012, 4, 3140–3149 RSC.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- E. P. Randviir, D. K. Kampouris and C. E. Banks, Analyst, 2013, 138, 6565–6572 RSC.
- A. V. Kolliopoulos, J. P. Metters and C. E. Banks, Anal. Methods, 2013, 5, 3490–3496 RSC.
- S. Gura, P. Guerra-Diaz, H. Lai and J. R. Almirall, Drug Test. Anal., 2009, 1, 355–362 CrossRef CAS PubMed.
- S. Pichini, M. Navarro, R. Pacifici, P. Zuccaro, J. Ortuno, M. Farre, P. N. Roset, J. Segura and R. de la Torre, J. Anal. Toxicol., 2003, 27, 294–303 CrossRef CAS PubMed.
- I. Riezzo, D. Cerretani, C. Fiore, S. Bello, F. Centini, S. D'Errico, A. I. Fiaschi, G. Giorgi, M. Neri, C. Pomara, E. Turillazzi and V. Fineschi, J. Neurosci. Res., 2010, 88, 905–916 CAS.
- M. Cheze, M. Deveaux, C. Martin, M. Lhermitte and G. Pepin, Forensic Sci. Int., 2007, 170, 100–104 CrossRef CAS PubMed.
- S. E. J. Bell, D. Thorburn Burns, A. C. Dennis, L. J. Matchett and J. S. Speers, Analyst, 2000, 125, 1811–1815 RSC.
- N. Milhazes, P. Martins, E. Uriarte, J. Garrido, R. Calheiros, M. P. M. Marques and F. Borges, Anal. Chim. Acta, 2007, 596, 231–241 CrossRef CAS PubMed.
- T. L. Martin, J. Anal. Toxicol., 2001, 25, 649–651 CrossRef CAS PubMed.
- M. Nieddu, G. Boatto, L. Sini and G. Dessì, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 431–438 CrossRef CAS.
- D. Moreno, B. D. de Grenu, B. Garcia, S. Ibeas and T. Torroba, Chem. Commun., 2012, 48, 2994–2996 RSC.
- N. Kato, S. Fujita, H. Ohta, M. Fukuba, A. Toriba and K. Hayakawa, J. Forensic Sci., 2008, 53, 1367–1371 CAS.
- M. Wada, Y. Ochi, K. Nogami, R. Ikeda, N. Kuroda and K. Nakashima, Anal. Bioanal. Chem., 2012, 403, 2569–2576 CrossRef CAS PubMed.
- M. C. Tadini, M. A. Balbino, I. C. Eleoterio, L. S. de Oliveira, L. G. Dias, G. Jean-François Demets and M. F. de Oliveira, Electrochim. Acta, 2014, 121, 188–193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay02924d |
| This journal is © The Royal Society of Chemistry 2016 |