
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Next generation of labeling reagents for quantitative and multiplexing immunoassays by the use of LA-ICP-MS
S.
Kanje
 a,
A. J.
Herrmann
bc,
S.
Hober
a and
L.
Mueller
*b
a,
A. J.
Herrmann
bc,
S.
Hober
a and
L.
Mueller
*b
aAlba Nova University Center, KTH-Royal Institute of Technology, Division of Protein Technology, Stockholm, Sweden
bBundesanstalt für Materialforschung und-prüfung (BAM), 1.1 Division Inorganic Trace Analysis, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: larissa.mueller@thermofisher.com
cHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Straße 2, 12489 Berlin, Germany
First published on 17th October 2016
Abstract
Immuno imaging by the use of Laser Ablation Inductively Coupled Mass Spectrometry (LA-ICP-MS) is a growing research field in life sciences such as biology and biomedicine. Various element labeling strategies for antibodies have been developed for the application of multiplex immunoassays analyzed by the use of LA-ICP-MS. High multiplexing capabilities, a wide linear dynamic range and the possibility of absolute quantification are the main advantages of ICP-MS. But in the context of immuno imaging by the use of LA-ICP-MS, quantification of analytes is limited due to non-controllable antibody labeling chemistry. In the presented proof-of-principle a novel antibody labeling technique has been investigated which results in a controlled labeling degree. A small affinity protein based on the C2 domain of protein G was modified with conventional metal coded tags (MeCAT) after introducing a cysteine into the C-terminus of the protein. The modified C2 domain photo-crosslinks to the Fc or Fab region of the IgG and allows specific and covalent labeling of antibodies for multiplex immunoassay analysis by the use of LA-ICP-MS. In combination with a house-made calibration membrane the amount of labeled antibody–antigen complexes in a multiplex western blot immunoassay was determined by LA-ICP-MS.
1 Introduction
In recent years Laser Ablation Inductively Coupled Plasma Mass Spectrometry (LA-ICP-MS) has been investigated successfully for immuno imaging of biological, biochemical and clinical samples.1–4 The idea to develop multiplexing immunoassays for ICP-MS detection is based on the simplicity of the “mass fingerprints” in comparison to the data evaluation of multiple fluorescence immunoassays, which are limited by spectral overlap and saturation effects. The combination of immunoassays with ICP-MS is based on the modification of antibodies with artificial element labels. Similar to fluorescence colors, these antibodies are labeled by metals such as lanthanides and act as an indicator for the target protein (antigen) in the biological sample. In addition to the multiplexing capability and the high linear dynamic range of nine orders of magnitude, the possibility of quantification is also a main advantage of LA-ICP-MS compared to other imaging techniques using home-made matrix matched standards.5–10 Because of actual instrumental improvements11 and fast data acquisition,12 LA-ICP-MS stands now on the center stage for novel research in bio medicine and life sciences. Giesen et al. showed for the first time the simultaneous imaging of 32 proteins and protein modifications at subcellular resolution by the use of LA-ICP-TOF-MS (CyTOF).13Common labeling reagents for LA-ICP-MS immuno imaging are bifunctional ligands. They offer a reactive group, which binds to amino groups in the antibody present on lysines and the N-terminus or to thiols present on cysteine residues, and a metal chelate complex allowing the detection via ICP-MS. Often lanthanides are used as the central metal, because of their low natural background and their similar physical properties. The labeling with bifunctional ligands is efficient, but suffers from non-specificity, since all free and available amine/cysteine groups have the possibility to react. This drawback may lead to uncontrolled levels and locations of the labels. Hence, the modified molecules might behave differently and, possibly, the binding site of the antibody can also be affected.14 Additionally, in order to label cysteines, antibodies need to be partially reduced to create free thiol groups that are able to react (Fig. 1a). The complexity of the metal labeled antibody prevents the development of a quantification concept, because a calculation of an exact labeling degree, which is a prerequisite for the determination of the amount of antibody molecules in the sample, is not possible.15 In summary only the artificially introduced amount of metal/element of the selected label could be determined by LA-ICP-MS.6,8 With regard to novel biomedical and bioanalytical questions it is of high interest to know how many antibody molecules bind to their antigens. This would allow estimation about the reactivity and efficiency of the antibody to its antigen and – due to the multiplexing capabilities of LA-ICP-MS – the comparison of the expression profiles of one target protein compared to another protein in the same sample in absolute numbers.
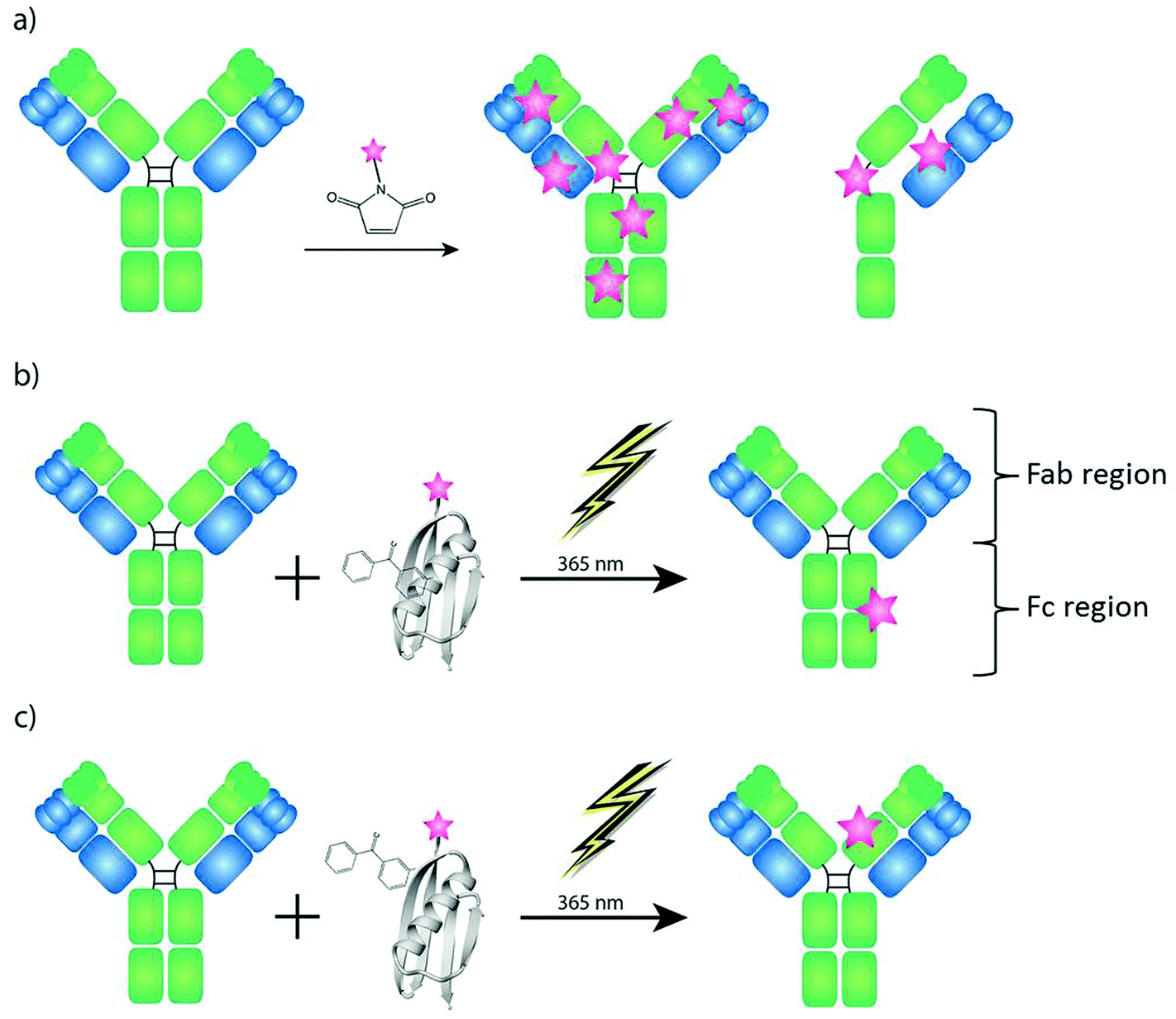 | ||
| Fig. 1 Strategies for labeling of antibodies with MeCAT (pink star). The antibody structure based on two heavy chains (green) and two light chains (blue) connected via disulfide bridges. (a) Labeling of antibodies using maleimide chemistry requires reduction of the antibody and leads to uncontrolled labeling amount and location. (b, c) The labeling strategy presented in this paper is based on a modified C2 domain containing a cysteine that can be conjugated to MeCAT and the unnatural amino acid BPA that can photo-crosslink to nearby amino acids upon UV irradiation. Two different domain label antibodies site specifically either in the region between the CH2 and CH3 domains on the Fc fragment (C2Fc) (b), or on the CH1 domain of the Fab fragment (C2Fab) (c). | ||
In the presented investigation we have used a novel labeling strategy for quantitative immuno imaging by the use of LA-ICP-MS which results in one label per antibody molecule as shown in Fig. 1. Recently small antibody binding domains derived from proteins A and G have been modified with benzophenone groups in order to be able to covalently and site specifically label antibodies.14,16,17 The C2 domain from streptococcal protein G has affinity for both the IgG Fc fragment in the region between the CH2 and CH3 domains18 as well as the constant part (CH1) of the heavy chain on the Fab domain.19 By introducing the unnatural amino acid BPA (p-benzoylphenylalanine) to the C2 domain close to its binding site to IgG, one domain that can label antibodies covalently at the Fc fragment (C2Fc)17 and one domain that can label IgG at the Fab fragment (C2Fab)20 have been developed. As the C2 domain doesn't contain any natural cysteines, a cysteine can easily be introduced into the protein domain and be used for peptide modification via maleimide chemistry. Taking advantage of such a cysteine, the C2 domain, and consequently any antibody, can be labeled with e.g. a MeCAT (metal coded tag) containing a chelated lanthanide metal. Any antibody can then, regardless of stabilizers in its buffer and without any pre-treatment, be labeled using the C2 domain by simply mixing the domains and subjecting them to far-UV light of 365 nm (Fig. 1b and c). The unconjugated C2 is washed away and the labeled antibody is ready to be used for detection in an experiment.
The applicability of the C2 peptide for quantitative LA-ICP-MS based immune imaging is demonstrated on a multiplex western blot assay using monoclonal antibodies derived from the Human Protein Atlas project.
2 Methods
2.1 Design of the C2 peptide
2.2 Antibody labeling
Antibodies were labeled with the MeCAT-conjugated C2 domains and washed as described by Kanje et al.17
To determine the concentration and degree of labeling the washed antibodies were run on an SDS-PAGE as described by Kanje et al.17 next to antibodies of known concentration. The concentration of the antibodies was determined using the ImageJ software, comparing the intensity of the light chain of the labeled antibody to the light chain of antibodies with known concentrations.
For the initial experiments Anti-Actin (A14 Sigma Aldrich, Taufkirchen, Germany), Anti-CYP1A1 (A3), Anti-CYP3A1 (A15), Anti-CYP2B1/2B2 (A16) and Anti-CYP2E1 (A17), (all CYP antibodies were purchased from Abcam, Cambridge, United Kingdom) were labeled on the Fc fragment.
For the multiplex western blot experiments Anti-ARG1 (AMAb90545), Anti-CS (AMAb91005), Anti-SLC27A5 (AMAb90575), Anti-STAT3 (AMAb90776), Anti-SDHB (AMAb90708) and Anti-NAPRT1 (AMAb90725) (all from Atlas Antibodies) were labeled on the constant part of the Fab fragment.
2.2.2 Cysteine labeling by the use of MeCAT
Direct antibody labeling with MeCAT was performed in accordance with the publication of Waentig et al.212.3 Western blot assay
Liver tissue lysate was prepared as described by Älgenäs et al. 2014.22 Pre-stained marker (PageRuler™ Plus (Pierce)) and a total amount of 15 µg protein from the liver lysate suspended in reducing buffer (20 mM Tris, 1 mM EDTA, 88 mM SDS, 720 mM β-mercaptoethanol, 17% glycerol) and heated at 95 °C for 5 min, were loaded on an Any kD™ Mini-PROTEAN®TGX™ gel (Bio-Rad) and run in 1× TGS buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3, Bio-Rad) at 4 °C, 200 V, 30 min. The SDS-PAGE was transferred to an Immune-Blot PVDF membrane (0.45 µm, Bio-Rad) that had been activated with methanol, at 40 V, 90 min, 4 °C in 1× TG (25 mM Tris, 192 mM glycine, pH 8.3, Bio-Rad). The membrane was blocked for 1 h at room temperature (RT) or overnight at 4 °C with blocking buffer (5% milk powder w/v, 0.5% Tween20 v/v). The blocked membrane was incubated for 1 hour at room temperature with the labeled antibody (2–5 µg ml−1) diluted in blocking buffer. The membrane was subsequently washed 4 × 5 min with 1× PBST (1× PBS, 0.1% Tween 20 v/v) and dried before analysis with LA-ICP-MS. For secondary antibody detection the membrane was incubated with the goat-anti-mouse-HRP antibody (Dako) diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8000 in blocking buffer for 1 h at RT. The membrane was washed as described above. The blot was developed using the Immobilon™ western chemiluminescent HRP substrate (Millipore) in a ChemiDoc™ XRS+ imaging system (Bio-Rad).
:8000 in blocking buffer for 1 h at RT. The membrane was washed as described above. The blot was developed using the Immobilon™ western chemiluminescent HRP substrate (Millipore) in a ChemiDoc™ XRS+ imaging system (Bio-Rad).
2.4 Calibration membrane
A matrix matched calibration series was prepared by spotting a mixture of ICP-MS lanthanide standard solutions (CertiPUR, Merck Millipore, Darmstadt, Germany) onto a blot membrane (Whatman® GmbH, 0.45 μm,) by the use of a conventional microarray system (Bio Odyssey Calligrapher Mini Arrayer, Bio-Rad) and a solid pin (946NS3 solid microarray printing pin, Arrayit Corporation). First a validation of the arrayer system was performed. The reproducibility of the spotting process was checked on different days and the delivered volume of the pin was determined (0.61 nL ± 0.14 nL). To determine the delivered volume an empty blot membrane was printed with 10 grids (a grid consist of 4 × 4 spots). These generated grids were cut out; the membrane pieces thus obtained were dissolved in concentrated HNO3, diluted with dest. H2O and measured against a standard calibration series with ICP-MS. Furthermore the relative standard deviation for the printed spots was analyzed by LA-ICP-MS and is less than 10%.For calculation of the amount of lanthanides in the sample, both the printed membrane with the calibration series of lanthanides as well as the western blot membrane were continuously ablated line by line and measured with exactly the same settings.
2.5 LA-ICP-MS
For detection a commercial LA system (NWR213, ESI, Portland, USA) was coupled to an ICP sector field mass spectrometer (Element XR, Thermo Fisher Scientific, Germany). The ICP MS was synchronized with the LA unit in an external triggering mode. Membranes were fixed on a sample holder and inserted into the two volume cell (New Wave 213). The aerosol was transported by helium at a flow rate of 1 L min−1, and argon was added at a typical flow rate of 0.5 L min−1 before the ICP torch. The ICP was tuned daily for maximum ion intensity, keeping the oxide ratio (ThO/Th) below 5% using a microscopic glass slide as the control standard for ablation. The western blot membranes were ablated continuously in line scans (laser spot, 250 µm; scan speed, 250 µm s−1; repetition frequency, 20 Hz; energy, 1 J cm−1). The LA-ICP-MS data files were imported to the program Origin 7 (Originlab Corporations, Northampton, USA) where 2D intensity profiles can be generated by transforming the scan time into a micrometer scale. The measured peak areas of each intensity profile were then processed with Excel (Microsoft, Redmond, USA). Finally, by the use of the calibration data the peak area of the lanthanide in the sample can be converted into the amount of lanthanide [pg] in the sample. For antibodies with a tagging degree of one tag per antibody the determined amount of lanthanide [fmol] is similar to the amount of antibody [fmol].3 Results and discussion
3.1 Optimization of antibody labeling with C2 peptide
To investigate the feasibility of the labeling technique based on C2 in LA-ICP-MS analyses, labeling of monoclonal mouse antibodies was performed on their Fc domain by using C2Fc.17 While labeling with this particular domain works very well on human IgG (almost two labels per antibody), the labeling efficiency for mouse antibodies was not as satisfactory (1 out of 5 antibody molecules were labeled). Still the western blots analyzed by LA-ICP-MS showed adequate performance characteristics as described in section 3.2. Protein G – on which the C2 peptide was based – has different affinities to IgG from different species. The affinity to the Fc-part of mouse IgG1 is very low, the Fab binding is rather good and can therefore be efficiently used for crosslinking.17,20 To further optimize the C2-binder for labeling, a novel C2-domain was developed. This domain is an obligate Fab-binder that shows a higher preference for mouse IgG1, C2Fab.20 The C2Fab domain yields on average one peptide domain, i.e. label, per antibody for mouse IgG1 (Fig. 2) and is therefore more suitable to use when using mouse antibodies for detection in an assay.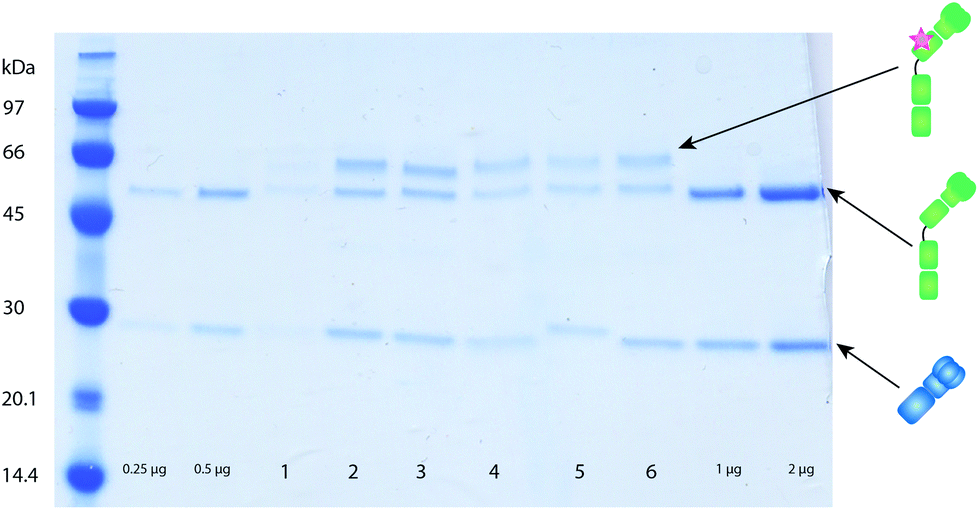 | ||
| Fig. 2 Example of labeling of mouse monoclonal antibodies on the Fab fragment using C2Fab coupled to different lanthanides. To determine concentration and degree of labeling the washed antibodies were run on an SDS-PAGE as described in section 2.2.1. Unlabeled antibodies (0.25, 0.5, 1 and 2 µg) were loaded as a concentration control. Samples 1–6 show different monoclonal antibodies labeled with different lanthanides site specifically on the heavy chain via the C2Fab labeling domain. Successful labeling results in an additional band above the heavy chain from the added weight from the C2Fab-MeCAT. The concentration of the antibodies can be determined using the ImageJ software, comparing the intensity of the light chain of the labeled antibody to the light chain of antibodies with known concentrations. Crosslinking is shown to take place around 50% of the heavy chains, meaning on average one label per antibody. | ||
3.2 Comparison of labeling techniques
After optimization of the antibody labeling on the Fc domain by C2Fc its performance characteristics were investigated in a western blot immunoassay. The results were compared to the performance characteristics of the same antibody labeled with MeCAT on free cysteine residues.21All target analytes could be identified in a liver lysate run on a SDS-PAGE and transferred to a membrane. Monoclonal anti-CYP1A1 (mouse IgG) was selected as the model antibody due to consolidated knowledge from other projects.6,21 The MeCAT and C2 peptide modified antibody samples were applied in two identical western blot immunoassays analyzed by LA-ICP-MS and the results are summarized in Table 1. On the one hand the signal intensities for the MeCAT labeled IgG were higher than for the C2 modified one by a factor of 50. But on the other hand the discrepancy between the signal-to-noise ratios of both methods is not that large (factor of 1.3) which is the result of a higher background signal in the case of the MeCAT labeled IgG. The lowest occupied detection limits (LOD) for both labeling methods lie in the lower pmol range and represent the minimum amount of CYP1A1 antigen detectable in a western blot immunoassay based on LA-ICP-MS analysis. The MeCAT labeling results in a LOD of 0.05 pmol which is only a factor of two lower than for the C2 labeled antibody (0.12 pmol). The reason for the discrepancy is due to the different labeling reactions. MeCAT labeling of cysteine residues after partial reduction of the antibody leads to a mixture of labeling degrees between zero and eight and to a non-controlled fragmentation of antibody molecules. Nevertheless intact antibodies reacting in the immunoassay might have a higher labeling degree or antibody fragments still show reactivity to its antigen, but might also produce background noise by unspecific binding. Hence, labeling chemistry is quite complex and hindered an adequate quantification and understanding of the immune reaction.15 On the other side the C2 peptide leads to a slightly higher detection limit but allows a controlled and robust labeling of antibodies for quantitative immunoassay. The antibody used in this experiment was labeled with the C2Fc domain, which is suboptimal for labeling of mouse IgG, resulting in the labeling of on average one out of five antibody molecules.
| Tag | Peak area | S/N ratio | LOD [pmol] |
|---|---|---|---|
| MeCAT-Tm | 2.08 × 106 | 245.99 | 0.05 |
| C2Fc-MeCAT-Tm | 4.17 × 104 | 187.23 | 0.12 |
In a second step additional CYP antibodies and anti-Actin were labeled with different lanthanides via the C2Fc peptide. Multiplex and single western blots were prepared under the same conditions and analyzed by LA-ICP-MS. The results were comparable with other studies already published in the literature achieving LODs in the lower pmol range.6 As expected the same protein bands were detected in single and multiplex experiments and no cross reactions could be observed (results not shown).
3.3 Quantitative and multiplexing western blot assay analyzed by LA-ICP-MS
For quantitative imaging a multiplex western blot immunoassay with six different monoclonal mouse antibodies (Anti-ARG1, Anti-CS, Anti-SLC27A5, Anti-STAT3, Anti-SDHB, and Anti-NAPRT1) labeled with the MeCAT modified C2Fab domain on the constant part of the Fab fragment was performed as described in section 2.2. All antibodies detect proteins present in a liver lysate run on a SDS-PAGE and transferred to a membrane before detection with the lanthanide labeled antibodies. A dilution series of a mixture of ICP-MS lanthanide standard solutions of known concentration was spotted with a microarray spotter onto a blot membrane. For quantification the calibration series as well as the sample membrane were ablated and analyzed with exactly the same settings. Fig. 3 shows the resulting 2D intensity profiles of one multiplex western blot after quasi simultaneous detection of six selected lanthanide isotopes.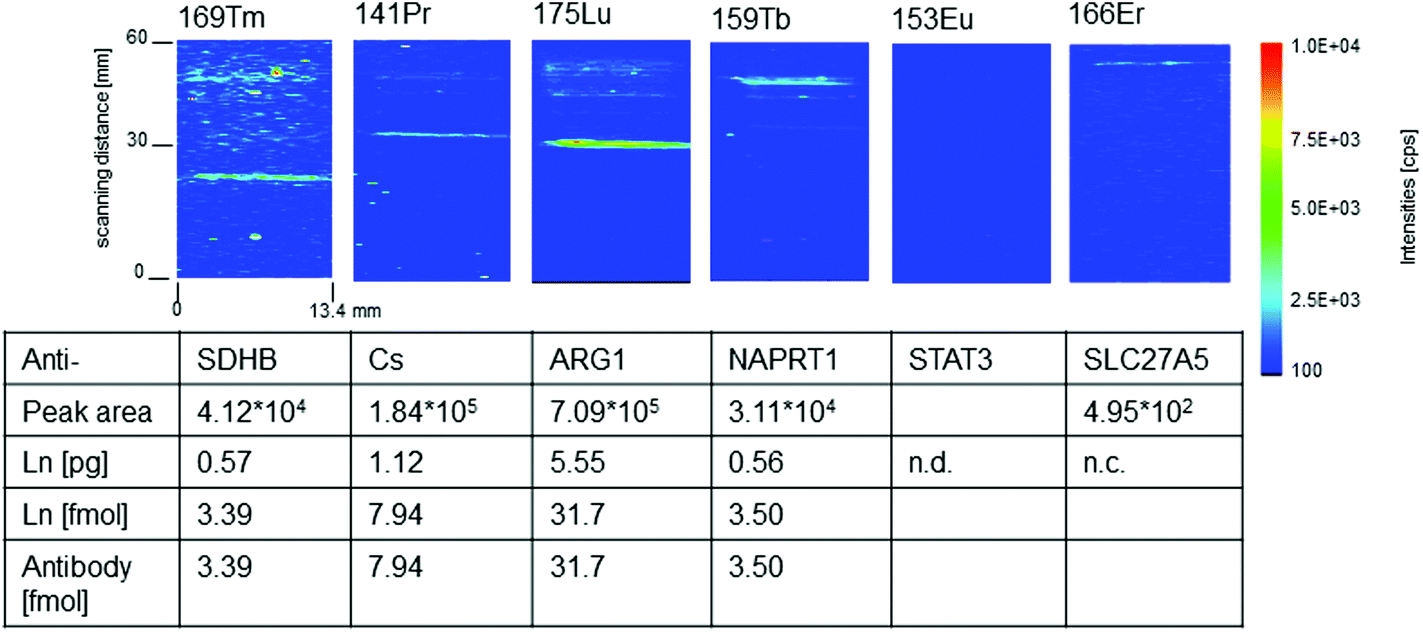 | ||
| Fig. 3 2D intensity profiles of one multiplex western blot membrane analyzed simultaneously with LA-ICP-MS after incubation with a mix of six different antibodies (Anti-ARG1(Lu), Anti-CS(Pr), Anti-SLC27A5(Er), Anti-STAT3(Eu), Anti-SDHB(Tm) and Anti-NAPRT1(Tb)) labeled with MeCAT modified C2Fab domains on the constant part of the Fab fragment. The distribution of the molar amount of the artificial introduced isotopes is given in cps/pixel. The table summarized the quantitative data determined peak areas belonging to the band of the antibody–antigen complex. The determined amount of lanthanide (Ln) is identical to the number of labeled antibody–antigen complexes if the labeling degree of the antibody to C2-peptide is 1:1; n.d., not detected; n.c. not calculated, as the peak area is lower than limit of quantification (LOQ = 9σ). | ||
Four of the six intensity profiles in Fig. 3 show distinct protein bands belonging to anti-SDHB(Tm), anti-NAPRT1(Tb), anti-ARG1(Lu) and anti-CS(Pr). For these antibodies the amount of labeled antibody connected to its antigen could be quantified via the calibration membrane. Linear regression factors (R2) better than 0.99 were calculated for the membrane calibration containing the lanthanides selected as antibody labels. The limit of quantification (LOQ) was determined and range between 0.004 pg (169Tm – lowest) and 0.0022 pg (153Eu – highest). Using the calibration data the peak area of the lanthanide signal according to the protein band can be converted into femtomole of lanthanide. Due to the fact that the labeling degree is one for C2Fab modified antibodies the molar amount of antibody reacted to the target antigen is the same as the molar amount of the detected lanthanide. The amounts of labeled antibody–antigen complexes in the analyzed multiplex western blot assay lie between 31.7 fmol (Lu) and 3.39 fmol (Tm). We identified ten times more anti-ARG1(Lu) than anti-NAPRT1 (Tb) and anti-SDHB(Tm). Anti-CS (Pr) lies in between with 7.94 fmol. In the case of anti-SLC27A5(Er), quantification was not possible due to a peak area (495 equates to 0.0015 pg Er) which was lower than the LOQ of 0.0017 pg.
Subsequently we want to comment on the empty profile in Fig. 3. All labeled antibodies were initially tested against the liver lysate one by one using secondary antibody detection (goat anti-mouse-HRP) in order to verify that they were still functional after labeling. All six antibodies used provided a single band of the expected size, however the developing time in order to get a sufficient signal differed for the different antibodies (from ten seconds to several minutes).
When not all antibodies provided signal in the LA-ICP-MS experiment a multiplex western blot with secondary antibody detection was set up. With all antibodies added to one membrane at the same concentration and detected with a secondary antibody, only four antibody signals that were detected in LA-ICP-MS could be seen (see Fig. 4). The two antibodies that could not be detected (Anti-CS(Pr), Anti-STAT3(Eu)) in a multiplex western blot with the secondary antibody were run in a new triplex western blot together with the antibody that gave a very weak signal in the sixplex western blot (Anti-SLC27A5(Er)). These three antibodies could then all be detected, where the antibody that gave a weak signal in the sixplex blot showed the strongest signal in the triplex blot compared to the other two that were not detectable when running all six antibodies at once. Antibodies will have different affinities for their targets, and in a crude liver lysate the different target proteins will be present at different concentrations. Therefore in the case of HRP detection an antibody that works in a singleplex set up might not work in a multiplex set up together with an antibody that may have a stronger affinity for its target, or a target present in a much higher concentration, which may cause the signal from the weaker antibody to drown out. Detection with a HRP coupled secondary antibody gives an increased signal over time. However if one signal is very strong it may quench the signal from a weaker antibody. In the case of the metal-labeled antibodies no signal amplification can be made. As the multiplex experiment using the secondary anti-IgG antibody confirmed the pattern of detectability from the LA-ICP-MS it is believed that this is caused by the reasons mentioned above. The reason for the singleplex blots giving a signal using secondary antibody detection but not in LA-ICP-MS is believed to be a combination of weaker antibody/lower target concentration and the fact that there is no signal amplification in LA-ICP-MS. Further the label selection might also play a role. The lanthanide erbium contains nine isotopes and we selected 166Er with an abundance of 33% for LA-ICP-MS analysis. This is probably the reason why the intensities are lower for Er compared to the monoisotopic lanthanides thulium, terbium, praseodymium and also 175Lu (97.4%). The same reason is valid for 153Eu with an abundance of 52%. All C2 labeled antibodies were also tested in single immunoassays analyzed by LA-ICP-MS and the results were in accordance with the multiplex experiments discussed here. The experimental data presented in Fig. 3 are a quite good example where the high dynamic range associated with ICP-MS was cut by the limits of the sample preparation. A better detection by LA-ICP-MS could be achieved by labeling the C2 domain with multiple metals.
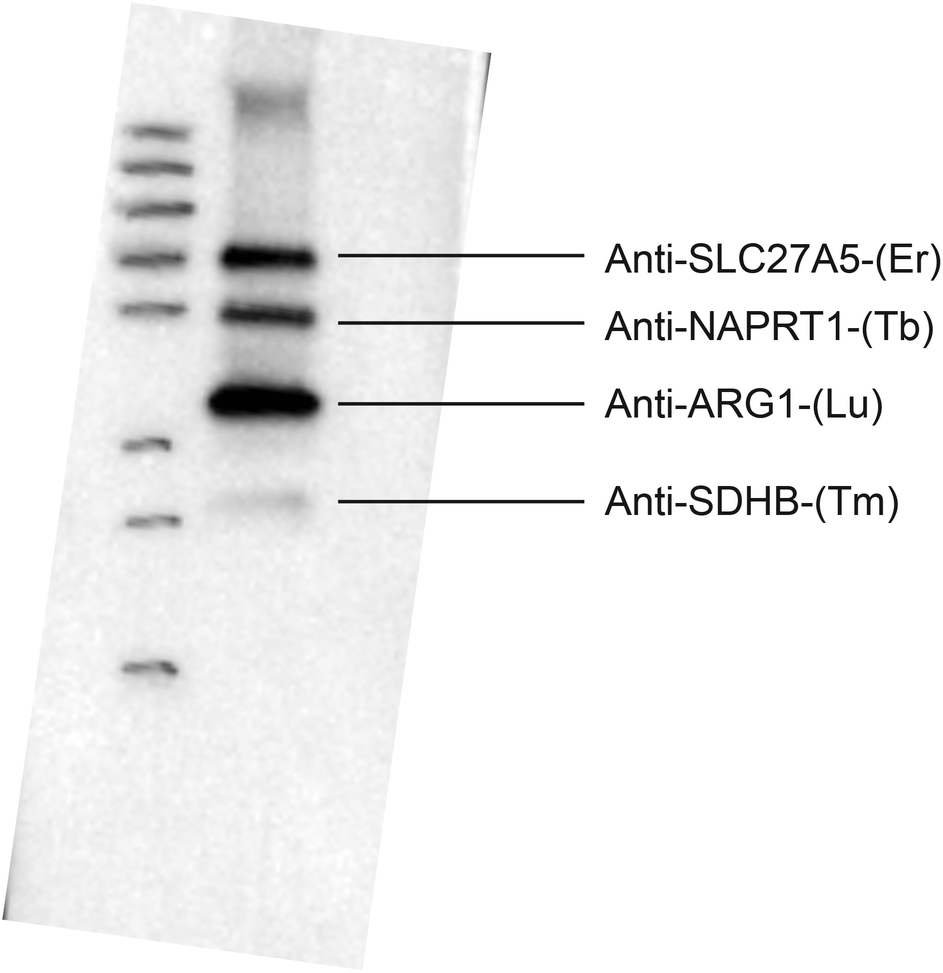 | ||
| Fig. 4 Sixplex western blot of a liver lysate using the lanthanide labeled antibodies and a secondary anti-mouse-HRP detection. The signals with secondary detection correlate well with the antibodies detected in LA-ICP-MS for the sixplex western blot experiment. | ||
4 Conclusion
A novel quantitative labeling concept for immuno imaging with LA-ICP-MS was developed which results in one label per antibody. This allows not only the quantification of the amount of introduced lanthanide ions into the sample but also enables the quantification of the molar amount of the antibody which is bound to the target protein. This is achieved with a labeling technique based on the use of small antibody binding domains, C2Fc and C2Fab, which are modified with metal coded tags (MeCAT). Antibodies can then be labeled with these modified C2 domains by mixing and subjecting them to far-UV light of 365 nm. A further highlight of this elegant labeling methodology is that any antibody can be specifically labeled, regardless of stabilizers in its buffer and without any pre-treatment. The applicability of six C2 tagged antibodies was demonstrated in a LA-ICP-MS based multiplex western blot immunoassay and in a chemiluminescence experiment. Quantification of the labeled antibody–antigen complexes was possible by the use of a calibration membrane and amounts between 30 and 3.0 fmol per protein band were determined. This labeling technique was compared to established cysteine labeling of antibodies and leads to similar performance characteristics e.g. S/N ratio and LODs in the femtomole range. In future the performance characteristics have to be improved to also address the quantitative analysis of low abundant proteins and immuno imaging where a high lateral resolution of the laser equipment is needed e.g. analysis of tissue microarrays. One idea is the use of non-dispersive label complexing many lanthanide ions for signal enhancement.23Acknowledgements
The authors would like to kindly thank Atlas Antibodies for providing us with monoclonal antibodies for this project and Proteomefactory Berlin for providing us with MeCAT reagents. The work of Larissa Mueller was supported by Deutsche Forschungsgemeinschaft DFG (WA 3459/1-1).References
- C. Giesen, T. Mairinger, L. Khoury, L. Waentig, N. Jakubowski and U. Panne, Anal. Chem., 2011, 83, 8177–8183 CrossRef CAS PubMed.
- R. Liu, P. Wu, L. Yang, X. Hou and Y. Lv, Mass Spectrom. Rev., 2014, 33, 373–393 CrossRef CAS PubMed.
- C. Giesen, L. Waentig, U. Panne and N. Jakubowski, Spectrochim. Acta, Part B, 2012, 76, 27–39 CrossRef CAS.
- D. A. Frick, C. Giesen, T. Hemmerle, B. Bodenmiller and D. Günther, J. Anal. At. Spectrom., 2015, 30, 254–259 RSC.
- O. Reifschneider, K. S. Wentker, K. Strobel, R. Schmidt, M. Masthoff, M. Sperling, C. Faber and U. Karst, Anal. Chem., 2015, 87, 4225–4230 CrossRef CAS PubMed.
- L. Waentig, S. Techritz, N. Jakubowski and P. H. Roos, Analyst, 2013, 138, 6309–6315 RSC.
- S. Hoesl, B. Neumann, S. Techritz, M. Linscheid, F. Theuring, C. Scheler, N. Jakubowski and L. Mueller, J. Anal. At. Spectrom., 2014, 29, 1282–1291 RSC.
- T. C. de Bang, P. Pedas, J. K. Schjoerring, P. E. Jensen and S. Husted, Anal. Chem., 2013, 85, 5047–5054 CrossRef CAS PubMed.
- S. Hoesl, B. Neumann, S. Techritz, G. Sauter, R. Simon, H. Schluter, M. W. Linscheid, F. Theuring, N. Jakubowski and L. Mueller, J. Anal. At. Spectrom., 2016, 31, 801–808 RSC.
- D. J. Hare, P. Lei, S. Ayton, B. R. Roberts, R. Grimm, J. L. George, D. P. Bishop, A. D. Beavis, S. J. Donovan, G. McColl, I. Volitakis, C. L. Masters, P. A. Adlard, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. A. Doble, Chem. Sci., 2014, 5, 2160–2169 RSC.
- H. A. O. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. H. Shaw-Stewart, B. Bodenmiller and D. Gunther, Anal. Chem., 2013, 85, 10107–10116 CrossRef CAS PubMed.
- P. J. Schuffler, D. Schapiro, C. Giesen, H. A. O. Wang, B. Bodenmiller and J. M. Buhmann, Cytometry, Part A, 2015, 87A, 936–942 CrossRef PubMed.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schuffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Gunther and B. Bodenmillerthat, Nat. Methods, 2014, 11, 417–425 CrossRef CAS PubMed.
- A. Konrad, A. E. Karlstrom and S. Hober, Bioconjugate Chem., 2011, 22, 2395–2403 CrossRef CAS PubMed.
- L. Mueller, T. Mairinger, G. Hermann, G. Koellensperger and S. Hann, Anal. Bioanal. Chem., 2014, 406, 163–169 CrossRef CAS PubMed.
- Y. W. Jung, J. M. Lee, J. W. Kim, J. W. Yoon, H. M. Cho and B. H. Chung, Anal. Chem., 2009, 81, 936–942 CrossRef CAS PubMed.
- S. Kanje and S. Hober, Biotechnol. J., 2015, 10, 564–U272 CrossRef CAS PubMed.
- A. E. Sauereriksson, G. J. Kleywegt, M. Uhl and T. A. Jones, Structure, 1995, 3, 265–278 CrossRef CAS.
- J. P. Derrick and D. B. Wigley, Nature, 1992, 359, 752–754 CrossRef CAS PubMed.
- S. Kanje, E. von Witting, S. C. C. Chiang, Y. T. Bryceson and S. Hober, Bioconjugate Chem., 2016, 27(9), 2095–2102 CrossRef CAS PubMed.
- L. Waentig, N. Jakubowski, S. Hardt, C. Scheler, P. H. Roos and M. W. Linscheid, J. Anal. At. Spectrom., 2012, 27, 1311–1320 RSC.
- C. Älgenäs, C. Agaton, L. Fagerberg, A. Asplund, L. Bjorling, E. Bjorling, C. Kampf, E. Lundberg, P. Nilsson, A. Persson, K. Wester, F. Ponten, H. Wernerus, M. Uhlen, J. O. Takanen and S. Hober, Biotechnol. J., 2014, 9, 435–445 CrossRef PubMed.
- N. Jakubowski, L. Wäntig and P. Roos, Patent polypeptide markerDE 102012104504A1, 2013 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |