
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticles as sensitive optical probes
Zhiqin
Yuan
*a,
Cho-Chun
Hu
b,
Huan-Tsung
Chang
*c and
Chao
Lu
a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yuanzq@mail.buct.edu.cn; Tel: +86-10-64411957
bDepartment of Applied Science, National Taitung University, Taitung 95002, Taiwan
cDepartment of Chemistry, National Taiwan University, 1, Section 4, Roosevelt Road, Taipei 106, Taiwan. E-mail: changht@ntu.edu.tw; Fax: +1-886-2-33661171; Tel: +1-886-2-33661171
First published on 26th January 2016
Abstract
Gold nanoparticles (Au NPs) have become one of the most popular materials for sensing of analytes of interest in the last decade, mainly because of their ease in preparation and conjugation, stability, biocompatibility, and size-dependent optical properties. We have witnessed many sensitive and selective Au NP based optical systems for the quantitation of metal ions, anions, proteins, and DNA, based on analyte induced changes in their absorption, fluorescence, and scattering. In this tutorial review, we briefly discuss wet chemical approaches for the preparation of Au NPs. Sensing mechanisms and strategies of Au NP based optical systems are provided to show basic concepts in designing sensitive and selective sensing systems. Strategies for signal amplification applied in Au NP based systems are emphasized for the analysis of trace amounts of analytes in real samples. Many excellent Au NP based optical sensing systems are discussed to highlight their practicality for the analysis of complicated biological and environmental samples. The tutorial review ends with the discussion of the challenges and future trends of Au NP based optical sensing systems.
 Zhiqin Yuan | Zhiqin Yuan is currently a school specially appointed Associate Professor of State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology. He obtained his PhD from College of Chemistry and Chemical Engineering, Hunan University, in 2013 with Dr Yan He and Dr Edward S. Yeung. His research interests are focused on the synthesis and analytical applications of metal nanoparticles and nanoclusters. |
 Cho-Chun Hu | Cho-Chun Hu is currently an Associate Professor of the Department of Applied Science, National Taitung University. He received his PhD from the Department of Chemistry, National Taiwan University, in 1995 with Dr Chuen-Ying Liu. His major interest is to understand important chemical processes in analytical chemistry, synthesis of nanomaterials and polymer chemistry. |
 Huan-Tsung Chang | Huan-Tsung Chang is currently a Professor of the Department of Chemistry, National Taiwan University. He is a fellow of the Royal Society of Chemistry. He obtained his PhD from the Department of Chemistry, Iowa State University, in 1994 with Dr Edward S. Yeung. His current research interests include nanotechnology, green chemistry, biosensors, and mass spectrometry. |
 Chao Lua | Chao Lu received his PhD degree in Materials Science from Chinese Academy of Sciences with Dr Jin-Ming Lin in 2004. He is currently a full professor of State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology. In 2011, he was selected to participate in the ‘New Century Outstanding Talent’ scheme of the Ministry of Education. His research is focused on the synthesis and characterization of nanostructured materials, the fabrication of luminescent films and nanosensors, and chemiluminescence. |
Introduction
Quantitation of analytes of interest from real samples is important in many fields such as medical, environmental, material, pharmaceutical and food sciences.1,2 To develop highly sensitive and selective sensing systems, probes having high affinity toward analytes of interest and large signal changes induced by the analytes are required. Although a number of organic and biochemical probes have been applied successfully to quantitation of various analytes, they are sometimes limited by complicated preparation/fabrication processes of the probes, sophisticated and costly detection systems and/or the requirement of highly costly reagents. Therefore, simple, cost-effective, sensitive and selective probes for the quantitation of chemical and/or biological analytes from complicated samples are in demand.Nanomaterials have special physicochemical properties that offer a suitable platform for the design of sensitive and selective sensing systems for various analytes through integrating nanotechnology and biotechnology.3,4 Gold nanoparticles (Au NPs) have been extensively studied in many fields like analytical chemistry because of their unique optical, electrical and catalytic properties, which can be readily controlled by varying their size, shape, composition and surrounding environment.5 The simple preparation, excellent biocompatibility, wide surface conjugation chemistry, as well as the attractive plasmonic coupling of Au NPs make them excellent candidates for the development of probes for numerous analytes with desired properties.6–8 The high surface-to-volume ratio of Au NPs allows their multifunctionalization via assembling multiple organic or biological ligands onto their surfaces, which strengthens their interactions with target analytes and enhances their capability for real-time multiplex analysis of multiple targets.9,10 Various detection modes such as absorption, fluorescence, and surface enhanced Raman scattering (SERS) are commonly used in developing Au NP based sensing systems.11–13 These systems have shown that Au NPs are ideal for developing simple, sensitive, selective and cost-effective sensing systems for the quantitation of various target analytes in environmental and biological samples.12
Although many excellent review articles on Au NP based sensing systems have been published in the past decade,7–15 a tutorial review will facilitate junior researchers (potential users) to understand the fundamental principles and to select a proper system for the analysis of samples of interest. In this tutorial review, the syntheses and properties of Au NPs are discussed briefly. Optical sensing systems using Au NPs, including absorption, fluorescence and SERS, are provided, emphasizing their sensing strategies/mechanisms. Recent advances in Au NP based optical sensing systems for various analytes, including ions, small molecules and biopolymers, are provided to show their practicality in the analysis of biological and environmental samples. Owing to a space limit in this review article, only a few examples are selected to highlight the potential use of Au NPs in sensing and cell imaging. Current challenges and future outlooks of Au NP based sensors for real-time and high-throughput analysis of complex biological and environmental samples are discussed based on our experience in developing Au NP based optical systems.
Brief overview of Au NPs
Syntheses of Au NPs
Numerous physical and chemical methods for the synthesis of Au NPs have been reported, and much effort has been dedicated to the synthesis of Au NPs with controllable size, stability and functionality during the last two decades.5,16 Nevertheless, the wet chemical approach has overwhelmingly attracted growing attention because of their advantages over physical methods with respect to scalability, reproducibility, and versatility.17 The synthesis of Au NPs with different sizes and functionality is well established through wet-chemistry in both aqueous and organic solutions.Localized surface plasmon resonance of Au NPs
One of the most interesting optical properties of Au NPs is their localized surface plasmon resonance (LSPR), which results from the resonant oscillation of conduction electrons at the Au NP surface with the incident electromagnetic field.28 The LSPR contains absorption and scattering, and thus extinction instead of absorbance is used in most studies. The electromagnetic field at the Au NP surface allows enhanced optical properties, leading to an extremely high extinction coefficient of the LSPR band that is generally several orders of magnitude larger than that of traditional organic dyes.29 The LSPR resonance of Au NPs is located in the visible range, leading to their bright colors. The high extinction coefficient and the bright color lead to development of versatile Au NP based optical sensing systems.13The LSPR of Au NPs is largely affected by the particle size, interparticle distance and local environment.14 For instance, the maximum LSPR absorption wavelength is related to the diameter of Au NPs, mainly because increased Au NP size causes the decrease of discrete energy gaps.30 In addition, the number of conduction electrons increases with increasing particle size, leading to higher extinction coefficient and stronger scattering intensity of larger Au NPs. The interparticle distance dependence of LSPR is through the interparticle plasmon coupling effect. Therefore, aggregation of Au NPs results in dramatic red-shifts in their absorption spectra and thus changes in the solution color from red to blue. Mie et al. elucidated the origin of LSPR by solving Maxwell's electromagnetic equation based on a spherical particle model in a magnetic field.31 According to the equation, the refractive index can also affect the electromagnetic field appearing on the Au NP surface, resulting in changes in the LSRP properties of Au NPs. Thus, LSPR properties of Au NPs depend on the solution composition and ionic strength, as well as surface ligands. Consequently, Au NP based sensing systems are commonly developed through analyte induced changes in their particle size, interparticle distance and/or local environment.
Colorimetric assays
The aggregation of Au NPs results in color changes of the colloid solution from red to blue, while redispersion of the aggregated Au NPs leads to a color recovery from blue to red. The color change during Au NP aggregation or redispersion provides a general platform for absorption-based colorimetric detection of analytes that can induce the Au NP aggregation or redispersion directly or indirectly. Having an extremely high extinction coefficient and a strong interparticle distance-dependent LSPR absorption property, Au NPs are ideal for colorimetric assays. Moreover, the readout of Au NP based assays can be recorded with a UV-vis spectrophotometer or even observed by the naked eye. Accordingly, a lot of Au NP based colorimetric chemo/biosensors for the detection of inorganic ions, small molecules, oligonucleotides and proteins have been successfully fabricated based on three categories: aggregation induced colorimetric detection, disassembly mediated colorimetric sensing and colorimetric assays on single Au NPs.7,12,14Aggregation induced colorimetric detection
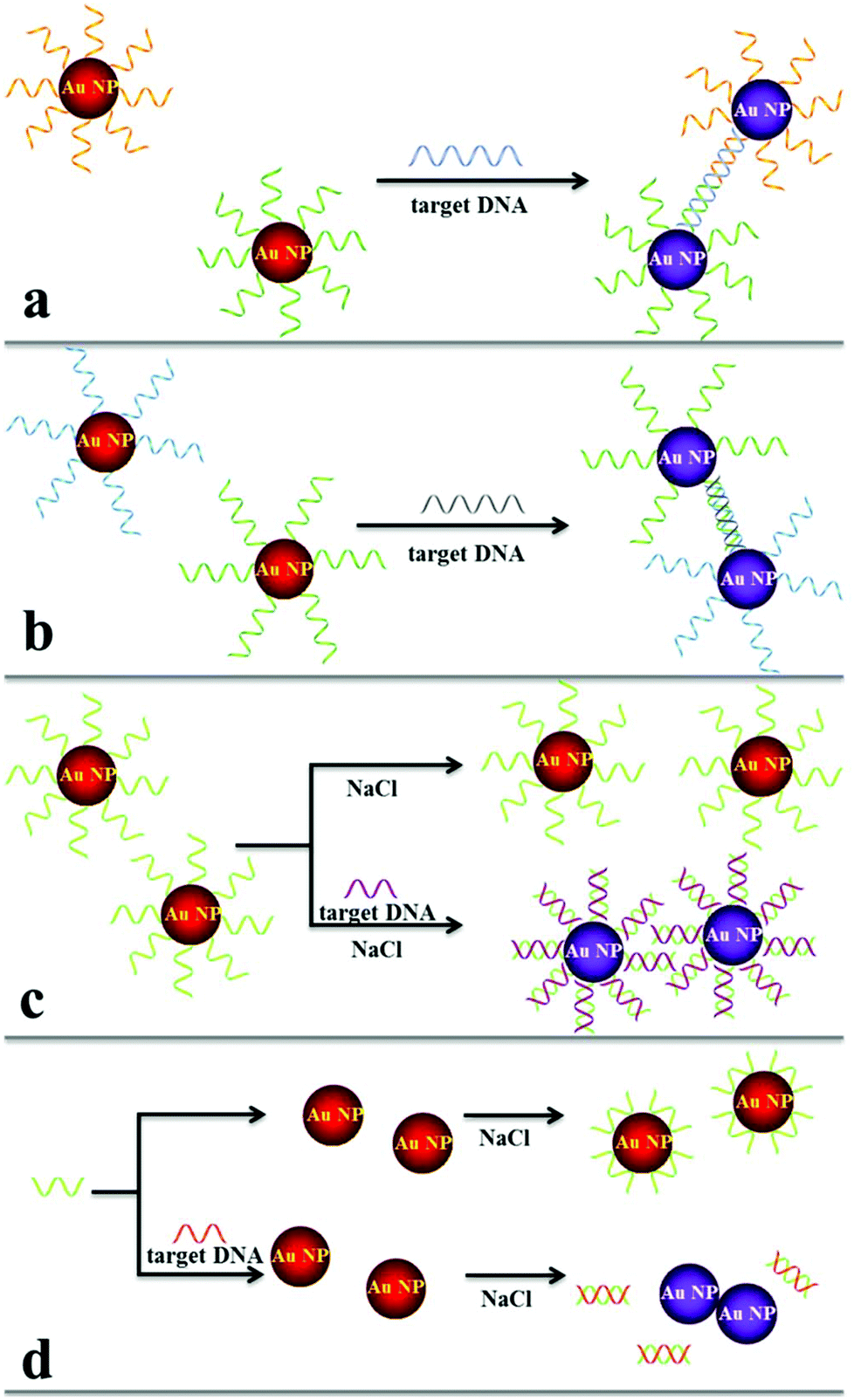 | ||
| Fig. 1 Schematics of colorimetric DNA hybridization detection systems. Duplex (a) and triplex (b) DNA crosslinking and non-crosslinking of (c) thiolated DNA–Au NPs and (d) non-thiolated DNA and Au NP. | ||
 | ||
| Fig. 2 Schematic illustration of attaching negatively charged thiolated DNA to negatively charged Au NPs using the low pH assisted method. Reprinted with permission from ref. 48. Copyright 2012, American Chemical Society. | ||
Having high specificity of base-pair hybridization and high extinction coefficient of Au NPs, DNA–Au NP based sensing systems provide selective DNA detection with LODs at the nanomolar and even subnanomolar levels. However, these assays are not sensitive enough for the analysis of disease related genes with concentrations usually ranging from attomolar to picomolar. To further improve the detection sensitivity, polymerase chain reaction (PCR) amplification is generally applied to amplify the concentration of the target gene.4 Typically, the target DNA serves as primers/templates for DNA replication in the presence of DNA polymerase. The resulting DNA number increases in an exponential manner with the amplification cycle, allowing highly sensitive DNA detection in femtomolar and even attomolar levels. In combination with PCR and Au NPs, many sensing systems have been developed for the quantitation of disease related genes, i.e., anthrax.54 A colorimetric assay in conjunction with a resonance light scattering technique provides a LOD of 10 pM for DNA, showing a potential for the analysis of single nucleotide polymorphisms in breast cancer.55
Another way to improve DNA detection sensitivity is through the control of the size of Au NP aggregates. Huge aggregates lead to precipitation of Au NPs and a decrease of absorbance, diminishing the sensitivity. To overcome this problem, Chen et al. designed a Janus Au NP based colorimetric system with high sensitivity and a wide dynamic range.56 The Janus Au NP possesses an asymmetrical surface that allows the formation of dimers rather than large aggregates and a reduction of the interparticle distance, providing picomolar sensitivity.
Proteases are special proteins that can selectively catalyze the hydrolysis or polymerization of their substrates, including peptides, proteins and DNA. Therefore, substrate stabilized Au NPs can act as reporters for the analysis of proteases such as phosphatase, thrombin and trypsin.73–75 Proteases catalyze the hydrolysis or polymerization of surface substrates and diminish the stability of Au NPs, thereby inducing aggregation of Au NPs and changes in the color of the solution from red to blue. For instance, Chang et al. disclosed a novel and label-free colorimetric assay for selective and sensitive detection of thrombin using fibrinogen modified Au NPs.72 Fibrinogen conjugates and stabilizes Au NPs through electrostatic and hydrophobic interactions, but adding thrombin in the presence of excess fibrinogen induced the formation of insoluble fibrillar fibrin–Au NP aggregates through the polymerization of the unconjugated and conjugated fibrinogen. The high catalytic performance allowed highly sensitive thrombin detection with a LOD of 0.04 pM. They also applied this system to plasmin detection based on plasmin mediated cleavage of fibrin structures, with a LOD of 0.4 nM in serum. The idea has been applied to the detection of other proteins, including adenosine deaminase, methyltransferase and acetyltransferase.76–78
Some of hydrolysis products can cause aggregation of citrate stabilized Au NPs directly, which can be transformed into a color change of Au NPs solution. In the light of this principle, acetylcholinesterase was easily detected using Au NPs.79 Acetylcholinesterase catalyzes acetylthiocholine to produce thiocholine, which binds to the citrate Au NP surface via Au–S interactions and induces Au NP aggregation through electrostatic attraction. Similarly, S-adenosylhomocysteine hydrolase was detected using fluorosurfactant-capped Au NPs and S-adenosylhomocysteine, with a LOD of 6 nM.80 Hydrolysis of S-adenosylhomocysteine in the presence of S-adenosylhomocysteine hydrolase yields homocysteine and subsequently causes the aggregation of the functional Au NPs.
To improve the sensitivity for the detection of proteins (enzymes), exonuclease-assisted cascaded recycling amplification has been applied for developing Au NP based sensing systems.81,82 Without the addition of exonuclease, two ssDNA probes hybridize and form dsDNA, resulting in the aggregation of Au NPs. Adding exonuclease digests the formed dsDNA and releases the individual Au NPs into solution. As a consequence, the solution color turns from blue to red. TATA binding protein can form a complex with dsDNA and inhibit its digestion. This colorimetric assay provides a LOD of 10 nM for TATA binding protein.81 With the assistance of magnetic nanoparticles, highly sensitive (LOD, 5.6 pM) thrombin detection was achieved by applying a similar strategy.82
Chemical reaction mediated Au NP assembly has been applied in sensing of small molecules, with advantages of sensitivity, selectivity and rapidity.98,99 Thiolated active ester and phenylboronic acid functionalized Au NPs were used for colorimetric detection of dopamine through analyte induced aggregation of Au NPs.100 More recently, thiolated azido derivatives and active ester cofunctionalized Au NPs (AE-Au NPs) were prepared for colorimetric detection of hydrogen sulfide (H2S), with a LOD of 0.2 μM.101 The azide groups on the surface of each AE-Au NP were reduced to form primary amines by H2S. The as-formed amines then interacted with the active esters through a crosslinking reaction, inducing the aggregation of Au NPs (Fig. 3). The AE-Au NP based nanosensors possess high selectivity towards H2S over other anions and thiols due to the specific azide–H2S chemistry.
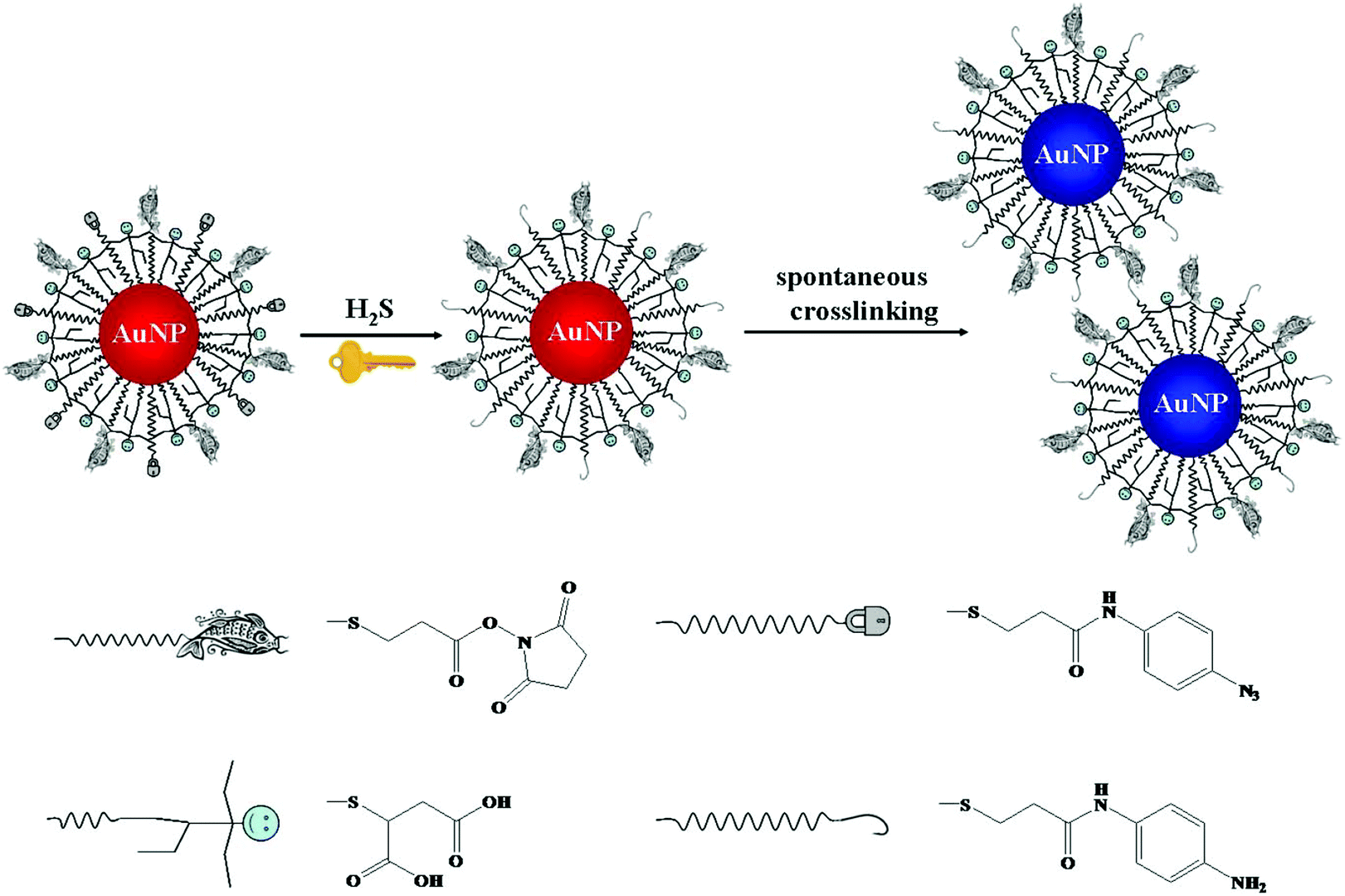 | ||
| Fig. 3 Schematic representation of the sensing principle of AE-Au NP probes for the detection of H2S. Reprinted with permission from ref. 101. Copyright 2015, American Chemical Society. | ||
Heavy metal ions such as mercury (Hg2+), lead (Pb2+) and copper (Cu2+) can cause damage to organisms and even nervous systems, and thus sensitive and selective approaches for their detection are of great importance. A large number of Au NP-based colorimetric systems have been developed for sensing of heavy metal ions based on coordination chemistry, aurophilic interactions and specific chemical reactions.6,102 For example, thiolated ligand or DNA modified Au NPs have been utilized for the colorimetric Hg2+, Pb2+ and Cu2+ detection through ligand–metal coordination.6 In particular, colorimetric detection of Hg2+ using DNA functionalized Au NPs based on thymine–Hg2+–thymine coordination chemistry has been widely studied.103 Without the addition of Hg2+, thymine rich ssDNA adsorbs onto the surfaces of Au NPs and stabilizes them in saline solution through electrostatic repulsions. However, introduction of Hg2+ induces the conformation change of thymine rich ssDNA and reduces the surface negative charged density, resulting in aggregation of the functional Au NPs. Combining this system with a paper device, an on-field colorimetric Hg2+ sensing platform was developed, with a LOD of 50 nM, which is ∼5 times lower than that obtained by using a conventional DNA–Au NP strategy.104 Ligands containing amino or carboxy groups have been used to functionalize Au NPs, which have been shown to be sensitive and selective for sensing of Hg2+via ligand–Hg2+ coordination.102,105,106 Similar sensing mechanisms have been applied to the detection of Pb2+ and Cu2+ using ligands such as glutathione and cysteamine that recognize the analytes to functionalize Au NPs, with LODs of 0.1 and 0.4 μM, respectively.107,108 Alternatively, by taking advantage of the high affinity of Hg2+ and Pb2+ toward Au NPs through the aurophilic interactions because of their similar electronic structures, a sensitive assay for the detection of the two analytes has been reported.109 Once the analytes adsorb onto the surface of each Au NP, surface ligands are released to the bulk solution and induce aggregation of the Au NPs. Colorimetric visualization of Cu2+ has been demonstrated by taking its catalytic activity (Cu+-catalyzed click chemistry) on the reaction of thiolated azide and alkyne functionalized Au NPs, with a LOD of 20 μM.110 The functional Au NPs form aggregates after the reaction, mainly due to Cu+-catalyzed cyclization of azide and alkyne. Similarly, azide and alkyne conjugated DNA modified Au NPs have also been applied to detect Cu2+ through Cu+-induced crosslinking between adjacent DNA, with a LOD of 20 μM.111 Although using DNA instead of a thiolate ligand to functionalize Au NPs is time-consuming, the resulting functional Au NPs provide high stability and reaction rate.
Disassembly mediated colorimetric sensing
Aggregation of Au NPs induces colloidal solution color changes from red to blue, while disassembly of aggregates leads to the reverse color variation. When compared to aggregation approaches, disassembly strategies show higher sensitivity due to the lower possibility of false positive signals. Several methods have been established for the colorimetric analysis of target analytes based on disassembly strategies.112,113 A colorimetric Pb2+ sensing system was proposed based on DNAzyme-directed assembly of Au NPs, with a LOD of 100 nM.114 The sensing system consists of DNA functionalized Au NPs, substrate ssDNA and DNAzyme. The substrate ssDNA contains complementary sequences in its head and tail ends to hybridize with the DNA on the surface of Au NPs, while its middle sequences hybridize DNAzyme. Mixing these three components leads to aggregation of Au NPs, leading to a blue solution color. The DNAzyme specifically interacts with Pb2+ to form a certain structure that catalyzes hydrolytic cleavage of substrate ssDNA, leading to disassembly of the Au NP aggregates and thus a solution color change to red after heating. This Pb2+ mediated detachment process can be accelerated (within 10 min) by adjusting the Au NPs size and distance between Au NPs.115 The cleavage speed is greatly accelerated by replacing the small Au NPs (13 nm) with larger ones (42 nm) and extending the distance between Au NPs, mainly because of reduced steric hindrance and aggregate size. Similarly, the rate of disassembly is further accelerated by using a short length of DNA to hybridize with the cleaved substrate ssDNA fragments, which reduces the amount of free ssDNA fragments and promotes the cleavage of the substrate.116,117An approach based on enzymatic disassembly of DNA–Au NP aggregates was developed for the colorimetric detection of the restriction endonuclease EcoRI.118 DNA–Au NP aggregates were formed through the classical sandwich DNA structure that contains a recognition site for EcoRI. EcoRI induced cleavage of the rigid dsDNA, leading to disassembly of the Au NP aggregates into monodisperse Au NP and thus a corresponding shift in the absorption maximum from 534 to 524 nm. In addition to the determination of the activity of enzymes, a similar approach has been applied to quantitation of DNA, with a LOD of 200 nM.119 To avoid the use of large size Au NP aggregates, dimers of aptamer functionalized Au NPs were prepared and used for sensing of microcystin-LR, a toxic peptide produced by cyanobacteria, with a LOD of 0.05 nM.120 Upon the specific interaction of the aptamer with microcystin-LR, the dimers of aptamer functional Au NPs dissociate to form monomers. Having a smaller size of the dimer compared to conventional Au NP aggregates, this assay is complete within ∼5 min, which is about 12 times faster than conventional approaches.
Analyte induced disassembly of aggregates of Au NPs functionalized with small ligands or polymers has also been reported.121,122 A colorimetric system using piperazine–bisdithiocarbamate and citrate–Au NPs was developed for sensing of glutathione.121 Bridge-linking coordination of piperazine–bisdithiocarbamate on the Au NP surface leads to aggregation of citrate–Au NPs. Through strong Au–S interactions between glutathione and Au NPs, surface replacement of the piperazinebisdithiocarbamate occurs, leading to disassembly of Au NP aggregates. Assembly of Au NPs through metal ion–ligand coordination can be disassembled after removal of the metal ions.122,123 Aggregates of thiolated naphthalimide functionalized Au NPs induced by Hg2+ were used for screening of biothiols including glutathione, cysteine and homocysteine.122 Addition of biothiols removes the Hg2+ and liberates individual Au NPs. The LODs of this approach for glutathione, cysteine and homocysteine are 17, 9 and 18 nM, respectively. To detect pyrophosphate anions, aggregates of thiolated dipycolylamine functionalized Au NPs induced by Mn2+ were used.123 The assay provides different sensitivities for the three metal ions tested: Mn2+ > Ni2+ > Cu2+, with a decreasing order that is in good agreement with their decreasing interaction strength with pyrophosphate anions.
Colorimetric assays on single Au NPs
Sensing systems based on the monitoring of the shift in the absorption wavelength have been applied for various analytes using functional Au NPs.124,125 Monitoring of the formation of dimers of DNA functional Au NPs by dark field microscopy provided a LOD of 0.02 pM for target DNA, which is ∼5 orders of magnitude lower than that obtained by conventional approaches.126 Formation of the Au NP dimer results in interparticle coupling, leading a red-shift of scattering spectra and a color change that was observed using dark field microscopy (Fig. 4). By recording the number ratio of the dimer of Au NPs to the monodisperse Au NPs, the assay is sensitive for the detection of target DNA, with a LOD of 100 fM.127 Quantitation of Cu2+ and monitoring of a click reaction were achieved by taking advantage of Cu+ catalyzing the formation of dimers of thiolated azide and alkyne functionalized Au NPs.128 The simple assay provides a LOD of 0.1 nM for Cu2+, based on the changes in the peak of scattering spectra of Au NPs. When using a laser instead of a white light source in a dark field microscopic system, a ∼10 fold increase in the sensitivity for Hg2+ detection was achieved.128 | ||
| Fig. 4 DNA sandwich assay on single Au NPs. Reprinted with permission from ref. 126. Copyright 2010, American Chemical Society. | ||
The scattering color and absorption wavelength of single Au NPs can be altered by changing its morphology and/or size. For example, the catalytic deposition of Cu2+ on single Au NPs by the reduced nicotinamide adenine dinucleotide cofactor results in an increase of scattering maxima and a color change in dark field microscopy.129 Such significant changes are mainly due to the change of the structure from sphere to core–shell after copper deposition onto the Au NPs. The assay holds potential for the study of reduced nicotinamide adenine dinucleotide dependent intracellular metabolic enzymatic pathways. To detect DNA, a single Au NP colorimetric system was developed based on the catalytic activity for the conversion of glucose and oxygen to form gluconic acid and hydrogen peroxide in the presence of glucose oxidase.130 The as-formed hydrogen peroxide then catalyzes the decomposition of Au3+ on the surface of single Au NPs, leading to a gradual size enlargement of Au NPs and thus the scattering color change can be monitored with a dark field microscopic system. The catalytic activity of Au NPs is significantly inhibited by ssDNA that possesses strong binding affinity toward the Au NP surface. Adding complementary target DNA induces the formation of dsDNA, leading to the release of surface ssDNA and thus an increase in the catalytic activity of Au NPs. Similarly, single Au NP colorimetric systems were developed for the detection of dopamine based on dopamine mediated surface Au3+ decomposition, with a LOD of 0.25 pM,131 and for the detection of Pb2+ based on Pb2+ accelerated Au NPs leaching in the presence of thiosulfate and 2-mercaptoethanol, with a LOD of 0.2 pM.132
Binding of small molecules to single Au NPs also causes the variation of the refractive index and changes in the scattering color of single Au NPs. By monitoring the changes in scattering light intensity of Au NP solutions, avidin was detected using biotin functionalized Au NPs, with a LOD of 1 nM.133 The sensitivity can be further improved by using Au@Ag core–shell NPs as probes, mainly because of its greater extinction coefficient when compared to that of Au NPs. In vivo and in vitro H2S detection was demonstrated using Au@Ag core–shell NPs, with LODs of 0.01 and 50 nM, respectively.134,135 Through the interactions of H2S with the surface Ag on the Au@Ag NPs, a Ag2S shell was formed on each core of Au NP, resulting in a red-shift of scattering maxima and color changes in dark field microscopy. Alternatively, a technique based on imaging observation of color changes and counting of individual Au NPs through SPR microscopy was applied for sensitive detection of melamine and ATP, with LODs of 1 and 5 nM, respectively.136
Fluorescence detection
Au NP based fluorescence sensing systems have become more popular in analytical chemistry, mainly because of their sensitivity.9,137,138 When compared to Au NP based colorimetric approaches, interference from the color sample matrix is minimized in the fluorescence system. The fluorescence approach also holds greater potential for cell imaging. Three kinds of Au NP based fluorescence sensing systems are discussed in this section: surface plasmon enhanced energy transfer, metal enhanced fluorescence and fluorescent Au NPs.Surface plasmon enhanced energy transfer
Fluorescence resonance energy transfer (FRET) is an electrodynamic phenomenon that occurs between a donor fluorophore in the excited state and an acceptor fluorophore in the ground state. The donor molecules typically emit at shorter wavelengths that overlap with the absorption spectrum of the acceptor. The efficiency of FRET depends on several parameters, such as the spectral overlap of the emission spectrum of the donor with the absorption spectrum of the acceptor, the quantum yield of the donor, the relative orientation of the donor and acceptor transition dipoles, and the distance between the donor and acceptor molecules. Au NPs possess high extinction cross-section and surface electron density, which allows efficient energy transfer of fluorophores to their surface through dipole–surface interactions when they become close.139 This so-called surface plasmon enhanced energy transfer (SPEET) has a higher efficiency than that of FRET, mainly because the isotropically distributed conducting electrons on the Au NP surface can interact strongly with the oscillating dipole of fluorophores.140 Because dipole–surface rather than dipole–dipole interactions occur in SPEET, spectral overlap between the emission spectrum of the donor and the absorption spectrum of Au NPs is not necessary.Over the past few years, Au NP based SPEET systems have been extensively used for in vitro and in vivo detection of analytes of interest.141,142 Compared with fluorescence turn-off approaches, target induced turn-on strategies have attracted growing interest because of their high sensitivity and low background. To achieve such a target induced fluorescence turn-on process, fluorophores are firstly adsorbed or immobilized on the Au NP surface to have maximum fluorescence quenching. The fluorescence recovers after the target interacts with the Au NPs/surface fluorophore as a result of increased distance between the fluorophore and the Au NP surface.143,144 Higher sensitivity is usually achieved if the fluorophore molecules are released from the surface to the bulk solution. Rhodamine B absorbed Au NPs were used for the detection of Hg2+.145 Because Hg2+ has a strong affinity toward Au NPs, it interacts strongly with Au NP through d10–d10 interactions, resulting in the release of rhodamine B from the surface to bulk solution and thus fluorescence restoration. This system allows selective Hg2+ detection over 50 fold that of other metal ions (e.g., Pb2+, Cd2+ and Co2+), with a LOD of 10 nM. Similar approaches have been applied to sensing of various analytes such as thiolates, iodide anions, glyphosate and melamine.146–149
A protein sensing system using six cationic Au NPs and an anionic poly(p-phenylene ethynylene) fluorescent polymer was designed for the detection of seven proteins.150 The electrostatic attraction between cationic Au NPs and the anionic polymer causes the fluorescence quenching of the fluorescent polymer. Proteins disrupt the Au NP–polymer complex via competitive binding and thus modulate the fluorescence quenching efficiency (Fig. 5). The fluorescence response pattern of individual proteins was characterized using linear discriminant analysis, providing a fingerprint for differentiation of seven proteins. Using the fluorophore conjugated DNA functionalized Au NP based sensing system, discrimination of eleven proteins was achieved.151 DNA can be detected using Au NPs functionalized with fluorophore derivatized DNA having a hairpin structure.152,153 In the hairpin structure on the Au NP surface, the fluorophore is close to the Au NP, leading to efficient fluorescence quenching. Once the constrained conformation changes in the presence of target DNA, the fluorophore is separated from Au NPs and the fluorescence is restored. A four-color nanobeacon sensing system using Au NPs conjugated with four different molecular beacons was developed for multiplexed detection and imaging of intracellular mRNAs (Fig. 6).154 The four molecular beacons were derivatized with four different fluorophores. Hybridization between target mRNA and molecular beacons opens the hairpin structure and restores the fluorescence of the fluorophore. Different mRNAs generate unique fluorescence spectra (colors), which facilitate the discrimination of mRNA. This nanobeacon shows rapid response, high specificity, nuclease stability and good biocompatibility. With the assistance of a duplex-specific nuclease enzyme, quantification of as little as 0.2 fmol of miRNA-203 was achieved.155 Direct quantification of cancer-related miRNA-203 and miRNA-21 in samples of extracted total RNA from cell cultures could be realized.
 | ||
| Fig. 5 Schematic depiction of the “chemical-nose” protein sensor array based on Au NP/fluorescent polymer conjugates. (a) Competitive binding between protein and Au NP–polymer induced fluorescence recovery. (b) The generation of fingerprint fluorescence response patterns with the combination of an array. (c) Canonical score plot for the first two factors of simplified response patterns of the Au NP–polymer array against proteins. Reprinted with permission from ref. 150. Copyright 2007, Nature Publishing Group. | ||
 | ||
| Fig. 6 Schematic diagram of multiple intracellular mRNA detection using four DNA–dye conjugate functionalized Au NP probes. Reprinted with permission from ref. 154. Copyright 2013, American Chemical Society. | ||
The fluorescence of fluorophores on the surface of Au NPs can be recovered by etching the Au core. Fluorescent polymer functionalized Au NP probes have been applied to sensing of cyanide, mainly because the cyanide anion is able to etch gold atoms to form Au(CN)2− complexes.156 Similarly, graphene oxide and amino pyrene-grafted Au NPs were used for the detection of Pb2+.157 In the absence of Pb2+, the fluorescence of amino pyrene-grafted Au NPs is quenched by graphene oxide. Through the oxidation of Au NPs accelerated by Pb2+ in the presence of both thiosulfate and 2-mercaptoethanol, amino pyrene was released into aqueous solution and fluorescence was recovered.
Metal enhanced fluorescence
Au NPs can be used to enhance the fluorescence intensity of fluorophores that do not directly contact with Au NPs through the so-called metal enhanced fluorescence (MEF) effect.158 SPR enhanced excitation and emission induced plasmon are responsible for the MEF effect.159 The former causes an increase of the absorption cross-section of the fluorophore near the metal surface and subsequently an increase of fluorescence intensity. The latter results in an increase of induced plasmon scattering around the metal surface by partial energy from fluorophores and thus enhances the fluorescence intensity. In MEF, the proximity of the metal surface is necessary to ensure fluorescence enhancement. The optimal distance should be larger than ∼11 nm to prevent the SPEET effect.160,161Fig. 7 shows the difference between SPEET and MEF. Since MEF is also based on the interaction between molecular dipole orientation and electric field around the metal surface, excitation wavelength and Au NP size show a magnificent influence on MEF efficiency.162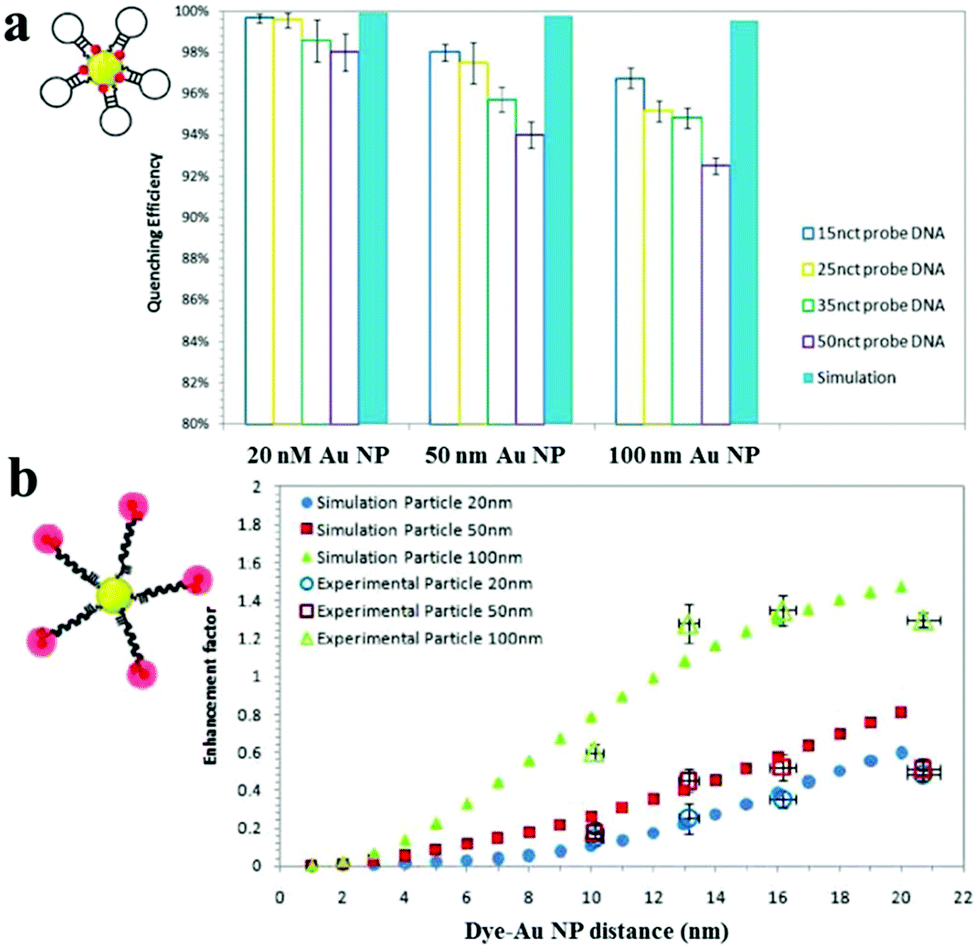 | ||
| Fig. 7 Fluorescence quenching and enhancement by Au NP size and dye–Au NP distance. (a) Quenching efficiency as a function of Au NP size and DNA length. (b) Enhancement factor as a function of particle size and dye–Au NP distance. Reprinted with permission from ref. 161. Copyright 2011, American Chemical Society. | ||
Using 100 nm of Au NPs as the substrate, MEF has been applied to the detection of DNA, with a LOD of 0.1 nM.161 Upon DNA hybridization, the formation of duplex DNA makes fluorophores stay away from the Au NP surface, resulting in an increase of fluorescence. Several MEF approaches have been developed for enhanced sensitivity in the detection of various analytes such as levofloxacin (LOD, 0.2 nM), Hg2+ (LOD, 4.5 nM) and β-galactosidase (LOD, 100 nM).163–165 Although MEF allows for improved sensitivity, the enhancement is usually limited to 10 fold.
Fluorescent Au NPs
Some Au NPs with sizes smaller than 3 nm emit fluorescence, which are called gold nanoclusters (Au NCs) or gold nanodots (Au NDs).26,166 They bridge the gap between individual atoms and large nanocrystals and display molecule-like properties. Because the size of fluorescent Au NPs is comparable to the Fermi wavelength of electrons, they show discrete electronic energy levels and unique physical and/or chemical properties different from large Au NPs. When compared with cadmium chalcogenide quantum dots and transition-metal-ion-doped quantum dots, fluorescent Au NPs show greater biocompatibility, but lower quantum yields.It is believed that the fluorescence of fluorescent Au NPs originates from the correlation between metal centered electron transition and ligand–metal/metal–metal charge transfer.167,168 Thus, the fluorescence of fluorescent Au NPs is primarily related to their size, structure, charge and surface ligand. Fig. 8 displays various colors of fluorescent Au NPs prepared using thiolates, polymers and biomolecules as ligands/scaffolds when excited with UV light.169,170 Some review articles on fluorescent Au NPs have been published in the last three years,21–23 thus only a brief discussion of their analytical applications is provided in this article. Most of the sensing methods using fluorescent Au NPs are based on a fluorescence turn-off strategy. Through the strong 5d10−5d10 interaction between Hg2+ and Au+, a sensitive and selective turn-off sensing system has been developed for the detection of Hg2+, with LOD at the nM level.171 Fluorescence quenching through the surface ligand–analyte interaction has also been utilized for the detection of Pb2+, Ag+, H2S or trinitrotoluene.172–175 Bovine serum albumin (BSA) stabilized fluorescent Au NPs have been applied for the detection of thiolates and acetylcholinesterase through a surface ligand exchange strategy.21,23 Both thiolate and protein stabilized fluorescent Au NPs have been used for the detection of hydrogen peroxide, with LODs of 27 and 30 nM, respectively.176,177 The sensing mechanism is mainly based on the oxidation of Au atoms in the fluorescent Au NPs, leading to fluorescence quenching. Fluorescent Au NPs containing cerium (Ce) were used for intracellular pH monitoring based on a ratiometric approach.170 The emission of the fluorescent Au NPs at 650 nm is insensitive to pH, while that for the BSA–Ce complex at 410 nm decreases with decreasing pH value. The fluorescent Au NPs containing Ce were further applied to sensing of CN−, with a LOD of 50 nM.178
 | ||
| Fig. 8 Photographs of fluorescent Au NP solution under UV light illumination. (a) Dual-thiolate ligand cofunctionalized fluorescent Au NPs, from 1 to 8: GSH/MUA, TSH/MUA, MH/MUA, MPA/MUA, MSA/MUA, MEN/MUA, MBA/MUA, and NSH/MUA, respectively (MUA: 11-mercaptoundecanoic acid, GSH: glutathione, TSH: 1-(10-mercaptodecyl)-5-methylpyrimidine-2,4-dione), MH: 6-mercapto-1-hexanol, MPA: 3-mercaptopropionic acid, MSA: mercaptosuccinic acid, MEN: (2-mercaptoethyl)amine, MBA: 4-mercaptobenzoic acid, and NSH: (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide). Reprinted with permission from ref. 169. Copyright 2015, American Chemical Society. (b) BSA protected fluorescent Au NPs containing Ce with different Ce/Au ratios, from I to VIII: 1000/0, 975/0.25, 950/0.5, 900/1.0, 800/2.0, 700/3.0, 500/5.0 and 0/10.0 μM/mM, respectively. Reprinted from ref. 170 with permission from the Royal Society of Chemistry. | ||
Fluorescent Au NPs have been used to detect biopolymers such as proteins, DNAs, cells and bacteria.23,179,180 BSA templated fluorescent Au NPs have also been used to determine the activity/concentration of enzymes, such as trypsin, phosphatase and proteinase K, with LODs of 86, 210 and 5 pM, respectively.181–183 The sensing mechanism is based on BSA degradation catalyzed by the enzymes, leading to fluorescence quenching. We note that the BSA template shell is very important to minimize fluorescence quenching by quenchers such as oxygen. Through different interactions of proteins with ligands on the surface of fluorescent Au NPs, fluorescent Au NPs conjugated with ligands such as proteins and thiolates have been used for the detection of proteins.169,184,185 For example, dual-ligand cofunctionalized fluorescent Au NPs were used for the differentiation of eight proteins.169 Using dual-ligand cofunctionalized fluorescent Au NP sensor arrays, eight proteins could be discriminated by analyzing the fluorescence response patterns via linear discriminant analysis. The protein induced fluorescence response of the fluorescent Au NPs is likely through the electrostatic as well as hydrophobic interactions between the surface thiolate ligands and protein amino acid residues.
SERS based sensing
Raman scattering is inelastic scattering of photons from a quantized vibrational signature. Both Raman shift and scattering intensity are sensitive to vibrational modes and provide feasibility for fingerprint sensing of target molecules. Unfortunately, direct use of Raman scattering for sensitive analyte detection is seriously restricted by the inherently weak Raman scattering intensity as a result of the small Raman scattering cross-section, which is 10–15 orders of magnitude smaller than that of fluorescence. The Raman scattering intensity can be enhanced dramatically (up to 14 orders of magnitude theoretically) by using plasmonic metal (e.g., silver and gold) nanoparticles or films.186,187 Such a physical phenomenon is called surface enhanced Raman scattering (SERS). Although the actual enhancement mechanism has not been completely understood, electromagnetic enhancement from LSPR/SPR and chemical enhancement through charge-transfer are generally accepted to be the two main contributions to SERS. The enhancement factor of SERS is related to the size, shape and surface properties of metal nanoparticles and also the interaction between the Raman reporter and metal nanoparticles.Using Au NPs as substrates, sensitive detection of inorganic/organic ions, small molecules and biopolymers has been achieved by SERS.188–193 For example, SERS was applied to the detection of CN− by monitoring the SERS signal at 2154 cm−1 of Au(CN)2− formed on the surface of Au NPs through etching of Au atoms by CN−.194 This approach provided a LOD of 100 ppt for CN−. Using Raman dye conjugated DNA functionalized Au NPs, sensitive detection of Hg2+ was obtained, with a LOD of 0.1 nM.195 Thymine–thymine mismatch in DNA templates hinders the formation of duplex DNA, which separates the Raman dye and Au NPs. In the presence of Hg2+, thymine–Hg2+–thymine coordination causes the hybridization of two DNA templates and promotes the formation of heterojunctions that can act as an excellent SERS platform.
SERS approaches using Au NPs have been applied to the detection of DNA.196 Both DNA probes and Raman tags were functionalized onto the Au NP surface first, hybridization with target DNA results in the variation of Raman tags-Au NPs and thus change in the SERS intensity. To enhance the sensitivity of SERS for DNA, surface silver coating on Au NPs was prepared as a SERS promoter to generate strong SERS signals of Cy3 dye molecules.197 This approach provided a LOD of 20 fM for target DNA. In conjunction with a chip device, this approach provided sensitive spectroscopic DNA fingerprinting for discrimination of six dissimilar DNA targets labeled with six Raman dyes. SERS approaches using Au NPs have also been demonstrated for the detection of proteins.198,199 Pearl necklace Au nanomaterials (Au PNNs) showing high enhancement factor of Raman signals of rhodamine 6G up to 9 orders of magnitude has been reported.199 Aptamer and 4-mercaptobenzoic acid (Raman reporter) functionalized Au NPs and Au PNNs (substrate) were used for sensitive detection of PDGF that is a potential marker of cancer diagnosis. The functionalized Au NPs first bind to PDGF through a specific aptamer–PDGF interaction, leading to purification and concentration of PDGF from the sample matrix as shown in Fig. 9. Then, PDGF containing Au NPs interact with Au PNNs through electrostatic attraction, and subsequently form aggregates with Au PNNs, leading to an enhanced Raman scattering signal of 4-mercaptobenzoic acid. Having a LOD of 0.5 pM for PDGF, this approach allowed quantitation of PDGF in urine samples.
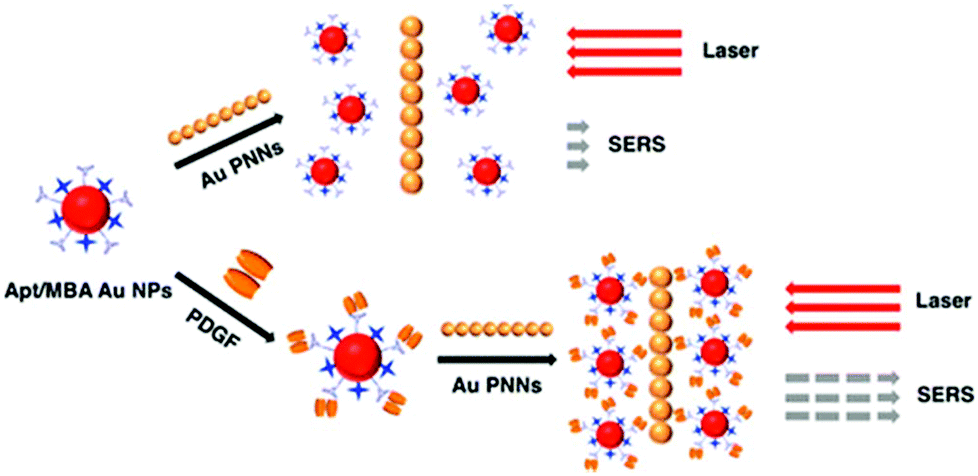 | ||
| Fig. 9 Schematic representation of the Au PNNs based SERS assay for the sensing of PDGF protein. Reprinted with permission from ref. 199. Copyright 2014, American Chemical Society. | ||
The distance (gap) between adjacent Au NPs plays an important role in determining the magnitude of the SERS signal; the SERS signal increases as the gap decreases.200,201 With ultrathin silica or alumina coating, shell-isolated Au NP probes provided enhanced SERS signals of pesticide residues.200 The ultrathin coating prevents the Au NPs from aggregation, separates them from direct contact with the probed material and allows the Au NPs to conform to different contours of substrates. A tip coated with a monolayer of shell-isolated Au NPs provided SERS signal of Si–H stretching vibration with 2–3 orders of magnitude higher than that of an Au tip. This dramatic enhancement is attributed to the association of single shell-isolated Au NPs; each of them acts as an individual Au tip. Alternatively, methoxy-mercaptopoly(ethylene glycol) was used to conjugate Au NPs to form dense monolayer films with 5 nm gaps, which provides a large SERS enhancement with an enhancement factor of ∼107.202 The gap between adjacent Au NPs can also be controlled by the modification of the surface of each Au NP with DNA. Compared to bare Au NPs, DNA–Au NPs as a substrate yield higher SERS signals for the tested analytes, including rhodamine 6G, trinitrotoluene and 4-aminothiophenol, leading to at least one order of magnitude lower LODs.203 When the gap between DNA–Au NP was shortened to 1–3 nm, SERS signals of 4-aminothiophenol with enhancement factors up to ∼109 were achieved.204 Such a high SERS active substrate provided sensitive 4-aminothiophenol detection with a LOD of 0.5 nM.
Because the enhancement factor of shell-isolated Au NPs is largely dependent on its core size and shell thickness,205 SERS signals of analytes can be further controlled by varying the sizes of the core and the shell of shell-isolated Au NPs. More importantly, the chemically inert shell avoids direct contact between the adsorbates and bare Au NPs, which allows observation of the true vibrational information of probed materials. Thus, the shell-isolated Au NP substrates provide more unfeigned Raman signal when compared to that of bare Au NP substrates. The shell-isolated Au NP substrates have been shown to be powerful for the analyses of cell walls and single molecules, food inspection, and monitoring of adsorption processes.206–208
Conclusion and outlooks
In this review, we have presented an overview of the progress of absorption, fluorescence and SERS-based optical sensing systems using Au NP probes. Most of these Au NP probes show enhanced sensitivity and selectivity toward chemical and biological agents, based on analyte induced changes in absorbance, fluorescence or Raman signals. These sensing systems are dependent on the analyte induced changes in the size or morphology of Au NPs or the distance between Au NPs. Most of the sensing systems provide LOD at the nM level for tested analytes. With signal amplification or special detection systems, some Au NP based optical systems allow detection of analytes down to pM and even fM levels.Although many Au NP based optical systems have been shown to be practical for the analysis of various samples, most reported systems can only be applied for the analysis of samples with low interference and ionic strength. Matrix and salt effects are still problematic for most developed sensing systems. False results occur commonly as a result of serious matrix interference. For the analysis of complicated samples, sample dilution and preconcentration of analytes are needed. They may however suffer from the change in the species of analytes, loss of the sample, contamination, and/or being time consuming. It is extremely difficult to achieve quantitation of trace amounts of analytes such as proteins in blood samples containing abundant proteins. For example, aptamer functionalized Au NPs cannot be applied to quantitation of PDGF in blood samples without sample pretreatment. Many signal amplification strategies applied in Au NP based sensing systems are time consuming or require the use of expensive enzymes. Special detection systems such as those used for monitoring single Au NPs require operators with a special skill.
Although single Au NP based approaches provide higher sensitivity than traditional Au NP based ones, sophisticated optical systems and Au NPs with a very narrow size distribution are needed. Thus, low-cost and automatic optical systems such as high-throughput microarrays and droplet-based microfluidics are in demand. Strategies such as using mild reducing agents with stronger capping strength than citrate for preparation of Au NPs with a narrow size distribution are worth trying. Cofunctionalization of Au NPs with ligands having different chemical/physical/recognition properties can be used to further improve the selectivity of the analyte, with minimum matrix interference. Functional magnetic nanoparticles have been shown to be effective for removal of the matrix and concentration of trace analytes before applying Au NPs for detection.209,210 However, the process is usually tedious and requires large sample volumes. It would be more effective if multifunctional Au NP composites can be prepared. For example, most of the matrix and salt are excluded by the shell and only analytes can interact with Au NPs inside the composites, leading to signal changes. It is also interesting to design Au NPs having enzyme-like properties and interesting optical properties as a new type of sensing system with enhanced selectivity and sensitivity.211–213 To speed up signal amplification, micro analysis systems in conjunction with Au NP based systems are a possible solution. Multichannel detection approaches (i.e., absorption/fluorescence, absorption/Raman or fluorescence/Raman) will be useful to further improve their reliability and practicability for the analysis of complicated biological and environmental samples.
Acknowledgements
This work was supported by the Ministry of Science and Technology of Taiwan under contracts NSC 104-2113-M-002-008-MY3 and 103-2923-M-002-002-MY3. This work was also supported by the National Basic Research Program of China (973 Program, 2014CB932103) and the National Natural Foundation of China (21375006 and 21575010), and the Free Exploration Project of Beijing University of Chemical Technology (ZY1625).Notes and references
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620 CrossRef CAS PubMed.
- H. Ma, J. Liu, M. M. Ali, M. A. I. Mahmood, L. Labanieh, M. Lu, S. M. Iqbal, Q. Zhang, W. Zhao and Y. Wan, Chem. Soc. Rev., 2015, 44, 1240 RSC.
- S. Song, Y. Qin, Y. He, Q. Huang, C. Fan and H.-Y. Chen, Chem. Soc. Rev., 2010, 39, 4234 RSC.
- Y. Song, W. Wei and X. Qu, Adv. Mater., 2011, 23, 4215 CrossRef CAS PubMed.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- Y.-W. Lin, C.-C. Huang and H.-T. Chang, Analyst, 2011, 136, 863 RSC.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849 RSC.
- Y. Sun and Y. Xia, Analyst, 2003, 128, 686 RSC.
- A. Heuer-Jungemann, P. K. Harimech, T. Brown and A. G. Kanaras, Nanoscale, 2013, 5, 9503 RSC.
- V. K. K. Upadhyayula, Anal. Chim. Acta, 2012, 715, 1 CrossRef CAS PubMed.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410 CrossRef CAS PubMed.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575 CrossRef CAS PubMed.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed.
- W. Zhao, M. A. Brook and Y. Li, ChemBioChem, 2008, 9, 2363 CrossRef CAS PubMed.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638 CrossRef CAS.
- T. Jain, Q. Tang, T. Bjørnholm and K. Nørgaard, Acc. Chem. Res., 2014, 47, 2 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143 RSC.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636 RSC.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216 CrossRef CAS PubMed.
- X. Su, H. Jiang and X. Wang, Anal. Chem., 2015, 87, 10230 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246 CrossRef CAS PubMed.
- C.-C. Huang, Z. Yang, K.-H. Lee and H.-T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824 CrossRef CAS PubMed.
- Y.-T. Tseng, Z. Yuan, Y.-Y. Yang, C.-C. Huang and H.-T. Chang, RSC Adv., 2014, 4, 33629–33635 RSC.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- H. Du, R.-C. A. Fuh, J. Li, L. A. Corkan and J. S. Lindsey, Photochem. Photobiol., 1998, 68, 141 CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215 CrossRef CAS PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377 CrossRef.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS PubMed.
- Z. Yuan, J. Cheng, X. Cheng, Y. He and E. S. Yeung, Analyst, 2012, 137, 2930 RSC.
- H. Yin, X. Huang, W. Ma, L. Xu, S. Zhu, H. Kuang and C. Xu, Biosens. Bioelectron., 2014, 52, 8 CrossRef CAS PubMed.
- E. Nourisaeid, A. Mousavi and A. Arpanaei, Physica E, 2016, 75, 188 CrossRef CAS.
- M. Mancuso, L. Jiang, E. Cesarman and D. Erickson, Nanoscale, 2013, 5, 1678 RSC.
- K. Kalidasan, J. L. Neo and M. Uttamchandani, Mol. BioSyst., 2013, 9, 618 RSC.
- O. Doluca, J. M. Withers and V. V. Filichev, Chem. Rev., 2013, 113, 3044 CrossRef CAS PubMed.
- C. Xiong, C. Wu, H. Zhang and L. Ling, Spectrochim. Acta, Part A, 2011, 79, 956 CrossRef CAS PubMed.
- Z. Chen, H. Zhang, X. Ma, Z. Lin, L. Zhang and G. Chen, Analyst, 2015, 140, 7742 RSC.
- A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Anal. Chem., 2007, 79, 6037 CrossRef CAS PubMed.
- G. Wang, Y. Akiyama, T. Takarada and M. Maeda, Chem. – Eur. J., 2016, 22, 258 CrossRef CAS PubMed.
- P. Valentini and P. P. Pompa, RSC Adv., 2013, 3, 19181 RSC.
- J. Liu, Phys. Chem. Chem. Phys., 2012, 14, 10485 RSC.
- K. M. Koo, A. A. I. Sina, L. G. Carrascosa, M. J. A. Shiddiky and M. Trau, Anal. Methods, 2015, 7, 7042 RSC.
- H. Li and L. J. Rothberg, J. Am. Chem. Soc., 2004, 126, 10958 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, Langmuir, 2012, 28, 3896 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266 CrossRef CAS PubMed.
- X. Zhang, T. Gouriye, K. Göeken, M. R. Servos, R. Gill and J. Liu, J. Phys. Chem. C, 2013, 117, 15677 CAS.
- P. Liu, X. Yang, S. Sun, Q. Wang, K. Wang, J. Huang, J. Liu and L. He, Anal. Chem., 2013, 85, 7689 CrossRef CAS PubMed.
- H. Deng, X. Zhang, A. Kumar, G. Zou, X. Zhang and X.-J. Liang, Chem. Commun., 2013, 49, 51 RSC.
- S. Abdul Rahman, R. Saadun, N. E. Azmi, N. Ariffin, J. Abdullah, N. A. Yusof, H. Sidek and R. Hajian, J. Nanomater., 2014, 2014, 5 Search PubMed.
- V. G. Joshi, K. Chindera, A. K. Singh, A. P. Sahoo, V. D. Dighe, D. Thakuria, A. K. Tiwari and S. Kumar, Anal. Chim. Acta, 2013, 795, 1 CrossRef CAS PubMed.
- H. Deng, Y. Xu, Y. Liu, Z. Che, H. Guo, S. Shan, Y. Sun, X. Liu, K. Huang, X. Ma, Y. Wu and X.-J. Liang, Anal. Chem., 2012, 84, 1253 CrossRef CAS PubMed.
- J. Gao, L. Ma, Z. Lei and Z. Wang, Analyst, 2016 10.1039/C5AN02510A.
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D.-H. Kim, J. Am. Chem. Soc., 2013, 135, 12338 CrossRef CAS PubMed.
- M. B. Pepys, G. M. Hirschfield, G. A. Tennent, J. Ruth Gallimore, M. C. Kahan, V. Bellotti, P. N. Hawkins, R. M. Myers, M. D. Smith, A. Polara, A. J. A. Cobb, S. V. Ley, J. Andrew Aquilina, C. V. Robinson, I. Sharif, G. A. Gray, C. A. Sabin, M. C. Jenvey, S. E. Kolstoe, D. Thompson and S. P. Wood, Nature, 2006, 440, 1217 CrossRef CAS PubMed.
- G. He, W. Luo, P. Li, C. Remmers, W. J. Netzer, J. Hendrick, K. Bettayeb, M. Flajolet, F. Gorelick, L. P. Wennogle and P. Greengard, Nature, 2010, 467, 95 CrossRef CAS PubMed.
- S. Sun, H. Shen, C. Liu and Z. Li, Analyst, 2015, 140, 5685 RSC.
- Y. T. Ho, B. Poinard, E. L. L. Yeo and J. C. Y. Kah, Analyst, 2015, 140, 1026 RSC.
- Y. Zhu, G. Wang, L. Sha, Y. Qiu, H. Jiang and X. Zhang, Analyst, 2015, 140, 1260 RSC.
- C. L. Schofield, B. Mukhopadhyay, S. M. Hardy, M. B. McDonnell, R. A. Field and D. A. Russell, Analyst, 2008, 133, 626 RSC.
- L. Otten, S.-J. Richards, E. Fullam, G. S. Besra and M. I. Gibson, J. Mater. Chem. B, 2013, 1, 2665 RSC.
- K. Aslan, C. C. Luhrs and V. H. Pérez-Luna, J. Phys. Chem. B, 2004, 108, 15631 CrossRef CAS.
- P. Chen, M. T. Chung, W. McHugh, R. Nidetz, Y. Li, J. Fu, T. T. Cornell, T. P. Shanley and K. Kurabayashi, ACS Nano, 2015, 9, 4173 CrossRef CAS PubMed.
- M. Bhagawati, C. You and J. Piehler, Anal. Chem., 2013, 85, 9564 CrossRef CAS PubMed.
- W.-J. Kim, H. Y. Cho, B. K. Kim, C. Huh, K. H. Chung, C.-G. Ahn, Y. J. Kim and A. Kim, Sens. Actuators, B, 2015, 221, 537 CrossRef CAS.
- X. Huang, Z. Xu, Y. Mao, Y. Ji, H. Xu, Y. Xiong and Y. Li, Biosens. Bioelectron., 2015, 66, 184 CrossRef CAS PubMed.
- Q. He, Z. Zhu, L. Jin, L. Peng, W. Guo and S. Hu, J. Anal. At. Spectrom., 2014, 29, 1477 RSC.
- C.-C. Huang, Y.-F. Huang, Z. Cao, W. Tan and H.-T. Chang, Anal. Chem., 2005, 77, 5735 CrossRef CAS PubMed.
- T.-E. Lin, W.-H. Chen, Y.-C. Shiang, C.-C. Huang and H.-T. Chang, Biosens. Bioelectron., 2011, 29, 204 CrossRef CAS PubMed.
- C.-K. Chen, C.-C. Huang and H.-T. Chang, Biosens. Bioelectron., 2010, 25, 1922 CrossRef CAS PubMed.
- R. Selegard, K. Enander and D. Aili, Nanoscale, 2014, 6, 14204 RSC.
- D. Huang, C. Niu, Z. Li, M. Ruan, X. Wang and G. Zeng, Analyst, 2012, 137, 5607 RSC.
- W. Xue, G. Zhang and D. Zhang, Analyst, 2011, 136, 3136 RSC.
- L. Zhang, J. Zhao, J. Jiang and R. Yu, Chem. Commun., 2012, 48, 10996 RSC.
- F. Cheng, Y. He, X.-J. Xing, D.-D. Tan, Y. Lin, D.-W. Pang and H.-W. Tang, Analyst, 2015, 140, 1572 RSC.
- E. Hutter and D. Maysinger, Trends Pharmacol. Sci., 2013, 34, 497 CrossRef CAS PubMed.
- D. Liu, Z. Wang, A. Jin, X. Huang, X. Sun, F. Wang, Q. Yan, S. Ge, N. Xia, G. Niu, G. Liu, A. R. Hight Walker and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 14065 CrossRef CAS PubMed.
- J.-H. Lin, C.-W. Chang, Z.-H. Wu and W.-L. Tseng, Anal. Chem., 2010, 82, 8775 CrossRef CAS PubMed.
- L.-J. Ou, P.-Y. Jin, X. Chu, J.-H. Jiang and R.-Q. Yu, Anal. Chem., 2010, 82, 6015 CrossRef CAS PubMed.
- S. Bi, X. Jia, J. Ye and Y. Dong, Biosens. Bioelectron., 2015, 71, 427 CrossRef CAS PubMed.
- J. Zhang, X. Xu and X. Yang, Analyst, 2012, 137, 1556 RSC.
- Z. Chen, Z. Wang, J. Chen, S. Wang and X. Huang, Analyst, 2012, 137, 3132 RSC.
- J. Zhang, X. Wang and X. Yang, Analyst, 2012, 137, 2806 RSC.
- Y. Jiang, H. Zhao, N. Zhu, Y. Lin, P. Yu and L. Mao, Angew. Chem., Int. Ed., 2008, 47, 8601 CrossRef CAS PubMed.
- X. Liang, H. Wei, Z. Cui, J. Deng, Z. Zhang, X. You and X.-E. Zhang, Analyst, 2011, 136, 179 RSC.
- F. Wang, X. Liu, C.-H. Lu and I. Willner, ACS Nano, 2013, 7, 7278 CrossRef CAS PubMed.
- Y.-Y. Yu, L. Zhang, X.-Y. Sun, C.-L. Li, Y. Qiu, H.-P. Sun, D.-Q. Tang, Y.-W. Liu and X.-X. Yin, Chem. Commun., 2015, 51, 8880 RSC.
- J. Peng, Y. Wang, L. Liu, H. Kuang, A. Li and C. Xu, RSC Adv., 2016, 6, 7798 RSC.
- S. Cheng, B. Zheng, M. Wang, M. H.-W. Lam and X. Ge, Talanta, 2013, 115, 506 CrossRef CAS PubMed.
- F. Li, J. Zhang, X. Cao, L. Wang, D. Li, S. Song, B. Ye and C. Fan, Analyst, 2009, 134, 1355 RSC.
- S.-J. Chen, Y.-F. Huang, C.-C. Huang, K.-H. Lee, Z.-H. Lin and H.-T. Chang, Biosens. Bioelectron., 2008, 23, 1749 CrossRef CAS PubMed.
- J. Hu, P. Ni, H. Dai, Y. Sun, Y. Wang, S. Jiang and Z. Li, Analyst, 2015, 140, 3581 RSC.
- J. Liu, W. Bai, S. Niu, C. Zhu, S. Yang and A. Chen, Sci. Rep., 2014, 4, 7571 CrossRef CAS PubMed.
- J. Xu, Y. Li, J. Bie, W. Jiang, J. Guo, Y. Luo, F. Shen and C. Sun, Microchim. Acta, 2015, 182, 2131 CrossRef CAS.
- M. Hoang, P.-J. J. Huang and J. Liu, ACS Sens., 2016 DOI:10.1021/acssensors.5b00147.
- D. Kato and M. Oishi, ACS Nano, 2014, 8, 9988 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, J. Lv and G. Li, Nano Res., 2015, 8, 920 CrossRef CAS.
- B. Kong, A. Zhu, Y. Luo, Y. Tian, Y. Yu and G. Shi, Angew. Chem., Int. Ed., 2011, 50, 1837 CrossRef CAS PubMed.
- Z. Yuan, F. Lu, M. Peng, C.-W. Wang, Y.-T. Tseng, Y. Du, N. Cai, C.-W. Lien, H.-T. Chang, Y. He and E. S. Yeung, Anal. Chem., 2015, 87, 7267 CrossRef CAS PubMed.
- W. Chansuvarn, T. Tuntulani and A. Imyim, Trends Anal. Chem., 2015, 65, 83 CrossRef CAS.
- J.-S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 4093 CrossRef CAS PubMed.
- G.-H. Chen, W.-Y. Chen, Y.-C. Yen, C.-W. Wang, H.-T. Chang and C.-F. Chen, Anal. Chem., 2014, 86, 6843 CrossRef CAS PubMed.
- G. Sener, L. Uzun and A. Denizli, Anal. Chem., 2014, 86, 514 CrossRef CAS PubMed.
- Y. Ma, L. Jiang, Y. Mei, R. Song, D. Tian and H. Huang, Analyst, 2013, 138, 5338 RSC.
- F. Chai, C. Wang, T. Wang, L. Li and Z. Su, ACS Appl. Mater. Interfaces, 2010, 2, 1466 CAS.
- L. Li, Z. Yuan, X. Peng, L. Li, J. He and Y. Zhang, J. Chin. Chem. Soc., 2014, 61, 1371 CrossRef CAS.
- Y.-R. Kim, R. K. Mahajan, J. S. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 292 CAS.
- Y. Zhou, S. Wang, K. Zhang and X. Jiang, Angew. Chem., Int. Ed., 2008, 47, 7454 CrossRef CAS PubMed.
- X. Xu, W. L. Daniel, W. Wei and C. A. Mirkin, Small, 2010, 6, 623 CrossRef CAS PubMed.
- X. Han, Y. Liu and Y. Yin, Nano Lett., 2014, 14, 2466 CrossRef CAS PubMed.
- M. Zhang, G. Qing, C. Xiong, R. Cui, D.-W. Pang and T. Sun, Adv. Mater., 2013, 25, 749 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2004, 126, 12298 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2005, 127, 12677 CrossRef CAS PubMed.
- J. H. Lee, Z. Wang, J. Liu and Y. Lu, J. Am. Chem. Soc., 2008, 130, 14217 CrossRef CAS PubMed.
- A. G. Kanaras, Z. Wang, M. Brust, R. Cosstick and A. D. Bates, Small, 2007, 3, 590 CrossRef CAS PubMed.
- I. A. Trantakis, S. Bolisetty, R. Mezzenga and S. J. Sturla, Langmuir, 2013, 29, 10824 CrossRef CAS PubMed.
- F. Wang, S. Liu, M. Lin, X. Chen, S. Lin, X. Du, H. Li, H. Ye, B. Qiu, Z. Lin, L. Guo and G. Chen, Biosens. Bioelectron., 2015, 68, 475 CrossRef CAS PubMed.
- Y. Li, P. Wu, H. Xu, H. Zhang and X. Zhong, Analyst, 2011, 136, 196 RSC.
- H. Xu, Y. Wang, X. Huang, Y. Li, H. Zhang and X. Zhong, Analyst, 2012, 137, 924 RSC.
- D. X. Li, J. F. Zhang, Y. H. Jang, Y. J. Jang, D. H. Kim and J. S. Kim, Small, 2012, 8, 1442 CrossRef CAS PubMed.
- L. Xiao and E. S. Yeung, Annu. Rev. Anal. Chem., 2014, 7, 89 CrossRef CAS PubMed.
- Y. Peng, B. Xiong, L. Peng, H. Li, Y. He and E. S. Yeung, Anal. Chem., 2015, 87, 200 CrossRef CAS PubMed.
- L. Xiao, L. Wei, Y. He and E. S. Yeung, Anal. Chem., 2010, 82, 6308 CrossRef CAS PubMed.
- T. Bu, T. Zako, M. Fujita and M. Maeda, Chem. Commun., 2013, 49, 7531 RSC.
- X. Cheng, D. Dai, Z. Yuan, L. Peng, Y. He and E. S. Yeung, Anal. Chem., 2014, 86, 7584 CrossRef CAS PubMed.
- L. Zhang, Y. Li, D.-W. Li, C. Jing, X. Chen, M. Lv, Q. Huang, Y.-T. Long and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 6789 CrossRef CAS PubMed.
- X. Zheng, Q. Liu, C. Jing, Y. Li, D. Li, W. Luo, Y. Wen, Y. He, Q. Huang, Y.-T. Long and C. Fan, Angew. Chem., Int. Ed., 2011, 50, 11994 CrossRef CAS PubMed.
- W. W. Qin, S. P. Wang, J. Li, T. H. Peng, Y. Xu, K. Wang, J. Y. Shi, C. H. Fan and D. Li, Nanoscale, 2015, 7, 15070 RSC.
- D. Dai, D. Xu, X. Cheng and Y. He, Anal. Methods, 2014, 6, 4507 RSC.
- Y. Liu and C. Z. Huang, Chem. Commun., 2013, 49, 8262 RSC.
- B. Xiong, R. Zhou, J. Hao, Y. Jia, Y. He and E. S. Yeung, Nat. Commun., 2013, 4, 1708 CrossRef PubMed.
- J. Hao, B. Xiong, X. Cheng, Y. He and E. S. Yeung, Anal. Chem., 2014, 86, 4663 CrossRef CAS PubMed.
- L. Yuan, X. Wang, Y. Fang, C. Liu, D. Jiang, X. Wo, W. Wang and H.-Y. Chen, Anal. Chem., 2016 DOI:10.1021/acs.analchem.5b04244.
- D. Liu, Z. Wang and X. Jiang, Nanoscale, 2011, 3, 1421 RSC.
- D. Ghosh and N. Chattopadhyay, J. Lumin., 2015, 160, 223 CrossRef CAS.
- P. C. Ray, Z. Fan, R. A. Crouch, S. S. Sinha and A. Pramanik, Chem. Soc. Rev., 2014, 43, 6370 RSC.
- C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115 CrossRef CAS PubMed.
- D. Huang, C. Niu, X. Wang, X. Lv and G. Zeng, Anal. Chem., 2013, 85, 1164 CrossRef CAS PubMed.
- J.-M. Liu, J.-T. Chen and X.-P. Yan, Anal. Chem., 2013, 85, 3238 CrossRef CAS PubMed.
- L. Li, J. Feng, Y. Fan and B. Tang, Anal. Chem., 2015, 87, 4829 CrossRef CAS PubMed.
- Z. Wu, G.-Q. Liu, X.-L. Yang and J.-H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829 CrossRef CAS PubMed.
- C.-C. Huang and H.-T. Chang, Anal. Chem., 2006, 78, 8332 CrossRef CAS PubMed.
- J. Xu, H. Yu, Y. Hu, M. Chen and S. Shao, Biosens. Bioelectron., 2016, 75, 1 CrossRef CAS PubMed.
- W. Li, Y. Dong, X. Wang, H. Li and D. Xu, Biosens. Bioelectron., 2015, 66, 43 CrossRef CAS PubMed.
- J. Guo, Y. Zhang, Y. Luo, F. Shen and C. Sun, Talanta, 2014, 125, 385 CrossRef CAS PubMed.
- X. Cao, F. Shen, M. Zhang, J. Guo, Y. Luo, X. Li, H. Liu, C. Sun and J. Liu, Food Control, 2013, 34, 221 CrossRef CAS.
- C. C. You, O. R. Miranda, B. Gider, P. S. Ghosh, I. B. Kim, B. Erdogan, S. A. Krovi, U. H. Bunz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 31 CrossRef PubMed.
- W. Sun, Y. Lu, J. Mao, N. Chang, J. Yang and Y. Liu, Anal. Chem., 2015, 87, 3354 CrossRef CAS PubMed.
- J. Xue, L. Shan, H. Chen, Y. Li, H. Zhu, D. Deng, Z. Qian, S. Achilefu and Y. Gu, Biosens. Bioelectron., 2013, 41, 71 CrossRef CAS PubMed.
- A. Giannetti, S. Tombelli and F. Baldini, Anal. Bioanal. Chem., 2013, 405, 6181 CrossRef CAS PubMed.
- W. Pan, T. Zhang, H. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581 CrossRef CAS PubMed.
- F. Degliangeli, P. Kshirsagar, V. Brunetti, P. P. Pompa and R. Fiammengo, J. Am. Chem. Soc., 2014, 136, 2264 CrossRef CAS PubMed.
- L. Shang, L. Zhang and S. Dong, Analyst, 2009, 134, 107 RSC.
- X. Shi, W. Gu, W. Peng, B. Li, N. Chen, K. Zhao and Y. Xian, ACS Appl. Mater. Interfaces, 2014, 6, 2568 CAS.
- J. R. Lakowicz, B. Shen, Z. Gryczynski, S. D'Auria and I. Gryczynski, Biochem. Biophys. Res. Commun., 2001, 286, 875 CrossRef CAS PubMed.
- J. Lakowicz, Plasmonics, 2006, 1, 5 CrossRef CAS PubMed.
- J. Zhang and J. R. Lakowicz, Opt. Express, 2007, 15, 2598 CrossRef CAS PubMed.
- Y. Cheng, T. Stakenborg, P. Van Dorpe, L. Lagae, M. Wang, H. Chen and G. Borghs, Anal. Chem., 2011, 83, 1307 CrossRef CAS PubMed.
- M. Li, Q. Wang, X. Shi, L. A. Hornak and N. Wu, Anal. Chem., 2011, 83, 7061 CrossRef CAS PubMed.
- S. H. Lee, S. M. Wabaidur, Z. A. Alothman and S. M. Alam, Luminescence, 2011, 26, 768 CrossRef CAS PubMed.
- Z. Cheng, G. Li and M. Liu, Sens. Actuators, B, 2015, 212, 495 CrossRef CAS.
- Z. Zeng, S. Mizukami, K. Fujita and K. Kikuchi, Chem. Sci., 2015, 6, 4934 RSC.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409 CrossRef CAS PubMed.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073 RSC.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- Z. Yuan, Y. Du, Y.-T. Tseng, M. Peng, N. Cai, Y. He, H.-T. Chang and E. S. Yeung, Anal. Chem., 2015, 87, 4253 CrossRef CAS PubMed.
- Y.-N. Chen, P.-C. Chen, C.-W. Wang, Y.-S. Lin, C.-M. Ou, L.-C. Ho and H.-T. Chang, Chem. Commun., 2014, 50, 8571 RSC.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961 RSC.
- Z. Yuan, M. Peng, Y. He and E. S. Yeung, Chem. Commun., 2011, 47, 11981 RSC.
- P.-C. Chen, T.-Y. Yeh, C.-M. Ou, C.-C. Shih and H.-T. Chang, Nanoscale, 2013, 5, 4691 RSC.
- A. Senthamizhan, A. Celebioglu and T. Uyar, Chem. Commun., 2015, 51, 5590 RSC.
- Z. Yuan, M. Peng, L. Shi, Y. Du, N. Cai, Y. He, H.-T. Chang and E. S. Yeung, Nanoscale, 2013, 5, 4683 RSC.
- H.-H. Deng, G.-W. Wu, D. He, H.-P. Peng, A.-L. Liu, X.-H. Xia and W. Chen, Analyst, 2015, 140, 7650 RSC.
- F. Wen, Y. Dong, L. Feng, S. Wang, S. Zhang and X. Zhang, Anal. Chem., 2011, 83, 1193 CrossRef CAS PubMed.
- C.-W. Wang, Y.-N. Chen, B.-Y. Wu, C.-K. Lee, Y.-C. Chen, Y.-H. Huang and H.-T. Chang, Anal. Bioanal. Chem., 2016, 408, 287 CrossRef CAS PubMed.
- L.-Y. Chen, C.-C. Huang, W.-Y. Chen, H.-J. Lin and H.-T. Chang, Biosens. Bioelectron., 2013, 43, 38 CrossRef CAS PubMed.
- Y.-T. Tseng, H.-T. Chang, C.-T. Chen, C.-H. Chen and C.-C. Huang, Biosens. Bioelectron., 2011, 27, 95 CrossRef CAS PubMed.
- L. Hu, S. Han, S. Parveen, Y. Yuan, L. Zhang and G. Xu, Biosens. Bioelectron., 2012, 32, 297 CrossRef CAS PubMed.
- W.-Y. Chen, L.-Y. Chen, C.-M. Ou, C.-C. Huang, S.-C. Wei and H.-T. Chang, Anal. Chem., 2013, 85, 8834 CrossRef CAS PubMed.
- Q. Wen, Y. Gu, L.-J. Tang, R.-Q. Yu and J.-H. Jiang, Anal. Chem., 2013, 85, 11681 CrossRef CAS PubMed.
- H. Kong, Y. Lu, H. Wang, F. Wen, S. Zhang and X. Zhang, Anal. Chem., 2012, 84, 4258 CrossRef CAS PubMed.
- S. Xu, X. Lu, C. Yao, F. Huang, H. Jiang, W. Hua, N. Na, H. Liu and J. Ouyang, Anal. Chem., 2014, 86, 11634 CrossRef CAS PubMed.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912 RSC.
- W. E. Smith, Chem. Soc. Rev., 2008, 37, 955 RSC.
- T. Senapati, D. Senapati, A. K. Singh, Z. Fan, R. Kanchanapally and P. C. Ray, Chem. Commun., 2011, 47, 10326 RSC.
- H. Wei, K. Rodriguez, S. Renneckar, W. Leng and P. J. Vikesland, Analyst, 2015, 140, 5640 RSC.
- M. Liu, Z. Wang, S. Zong, R. Zhang, D. Zhu, S. Xu, C. Wang and Y. Cui, Anal. Bioanal. Chem., 2013, 405, 6131 CrossRef CAS PubMed.
- V. V. Thacker, L. O. Herrmann, D. O. Sigle, T. Zhang, T. Liedl, J. J. Baumberg and U. F. Keyser, Nat. Commun., 2014, 5, 3448 Search PubMed.
- L. Perez-Mayen, J. Oliva, A. Torres-Castro and E. De la Rosa, Nanoscale, 2015, 7, 10249 RSC.
- A. M. Paul, Z. Fan, S. S. Sinha, Y. Shi, L. Le, F. Bai and P. C. Ray, J. Phys. Chem. C, 2015, 119, 23669 CAS.
- D. Senapati, S. S. R. Dasary, A. K. Singh, T. Senapati, H. Yu and P. C. Ray, Chem. – Eur. J., 2011, 17, 8445 CrossRef CAS PubMed.
- X. Ding, L. Kong, J. Wang, F. Fang, D. Li and J. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 7072 CAS.
- L. Sun, C. Yu and J. Irudayaraj, Anal. Chem., 2007, 79, 3981 CrossRef CAS PubMed.
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536 CrossRef CAS PubMed.
- Y. Li, X. Qi, C. Lei, Q. Yue and S. Zhang, Chem. Commun., 2014, 50, 9907 RSC.
- C.-W. Wang and H.-T. Chang, Anal. Chem., 2014, 86, 7606 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, Y. WuDe, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- W. Cai, X. Tang, B. Sun and L. Yang, Nanoscale, 2014, 6, 7954 RSC.
- X. Zhou, F. Zhou, H. Liu, L. Yang and J. Liu, Analyst, 2013, 138, 5832 RSC.
- L. Zhang, H. Ma and L. Yang, RSC Adv., 2014, 4, 45207 RSC.
- H. Ma, D. Lin, H. Liu, L. Yang, L. Zhang and J. Liu, Mater. Chem. Phys., 2013, 138, 573 CrossRef CAS.
- J. F. Li, X. D. Tian, S. B. Li, J. R. Anema, Z. L. Yang, Y. Ding, Y. F. Wu, Y. M. Zeng, Q. Z. Chen, B. Ren, Z. L. Wang and Z. Q. Tian, Nat. Protoc., 2013, 8, 52 CrossRef CAS PubMed.
- J.-F. Li, S.-Y. Ding, Z.-L. Yang, M.-L. Bai, J. R. Anema, X. Wang, A. Wang, D.-Y. Wu, B. Ren, S.-M. Hou, T. Wandlowski and Z.-Q. Tian, J. Am. Chem. Soc., 2011, 133, 15922 CrossRef CAS PubMed.
- Z. Liu, S.-Y. Ding, Z.-B. Chen, X. Wang, J.-H. Tian, J. R. Anema, X.-S. Zhou, D.-Y. Wu, B.-W. Mao, X. Xu, B. Ren and Z.-Q. Tian, Nat. Commun., 2011, 2, 305 CrossRef PubMed.
- J.-F. Li, J. R. Anema, T. Wandlowski and Z.-Q. Tian, Chem. Soc. Rev., 2015, 44, 8399 RSC.
- J. Yang, Y. Zhao, J. Zhang and C. Zheng, Environ. Sci. Technol., 2014, 48, 14837 CrossRef CAS PubMed.
- J. Wu, N. Wang, Y. Zhao and L. Jiang, Nanoscale, 2015, 7, 2625 RSC.
- C.-W. Lien, Y.-C. Chen, H.-T. Chang and C.-C. Huang, Nanoscale, 2013, 5, 8227 RSC.
- C.-W. Lien, Y.-T. Tseng, C.-C. Huang and H.-T. Chang, Anal. Chem., 2014, 86, 2065 CrossRef CAS PubMed.
- Y.-S. Wu, F.-F. Huang and Y.-W. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1503 CAS.
| This journal is © The Royal Society of Chemistry 2016 |