
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of the class II lanthipeptide protease LicP for sequence-specific, traceless peptide bond cleavage†
Weixin
Tang‡
a,
Shi-Hui
Dong‡
b,
Lindsay M.
Repka
a,
Chang
He
a,
Satish K.
Nair
*bc and
Wilfred A.
van der Donk
 *ab
*ab
aDepartment of Chemistry and Howard Hughes Medical Institute, University of Illinois at Urbana-Champaign, 600 S. Mathews Ave., Urbana, IL 61801, USA. E-mail: vddonk@illinois.edu; Fax: +1 217 244 8533; Tel: +1 217 244 5360
bDepartment of Biochemistry, University of Illinois at Urbana-Champaign, 600 S. Mathews Ave., Urbana, IL 61801, USA
cCenter for Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, 600 S. Mathews Ave., Urbana, IL 61801, USA. E-mail: s-nair@life.illinois.edu; Fax: +1 217 244 5858; Tel: +1 217 333 0641
First published on 2nd September 2015
Abstract
The final step of lanthipeptide biosynthesis involves the removal of leader peptides by dedicated proteases. In vitro characterization of LicP, a class II LanP protease involved in the biosynthesis of the lantibiotic lichenicidin, revealed a self-cleavage step that removes 100 amino acids from the N-terminus. The 2.35 Å resolution crystal structure provides insights into the active site geometry and substrate specificity, and unveiled an unusual calcium-independent maturation mechanism of a subtilisin family member. LicP processes LicA2 peptides with or without post-translational modifications, but dehydrated and cyclized LicA2 is favored. Investigation of its substrate specificity demonstrated that LicP can serve as an efficient sequence-specific traceless protease and may have great utility in basic research and biotechnology. Encouraged by these findings for LicP, we identified 13 other class II LanPs, ten of which were previously unknown, and suggest that these proteins may serve as a pool of proteases with diverse recognition sequences for general traceless tag removal applications, expanding the current toolbox of proteases.
Introduction
Proteases are ubiquitous in most organisms.1 They have been the focus of intense research not only because of their pivotal physiological functions but also because of their potential applications. The use of proteases has led to advances in analytical chemistry, proteomics, medicine, and the food, detergent and leather industries.2–4 Although much effort has been spent on engineering of proteases with desired sequence specificity using both rational design and high throughput screening,2,5,6 few such efforts have reached the stage of commercial applications.3 Major challenges are the sacrifice of efficiency and stability when engineering new substrate specificity, or the loss of sequence specificity when focusing on improving protein robustness.7–9 As a result, nature is still the major source of proteases with novel recognition sequences. The biosynthetic machinery responsible for the production of ribosomally synthesized and post-translationally modified peptides (RiPPs)10 is a promising area for discovering new sequence-specific proteases, as dedicated proteolytic enzymes are employed to remove leader peptides with highly diverse amino acids occupying the P and P′ positions. Lanthipeptides are a large family of RiPPs, with a subfamily that exhibits antimicrobial activities termed lantibiotics. The biosynthesis of lanthipeptides initiates from a linear precursor peptide with an N-terminal leader peptide that is important for the recognition by the corresponding post-translational modification machinery.10,11 The C-terminus of the precursor peptide, termed the core peptide, is where the post-translational modifications take place.10 Typically, Ser and Thr residues are dehydrated to generate dehydroalanine (Dha) and dehydrobutyrine (Dhb), respectively. Subsequently, a cyclase catalyzes the attack of thiols of cysteine residues on the Dha and Dhb residues to form the characteristic thioether cross-links called lanthionine (Lan) and methyllanthionine (MeLan) (Fig. 1A and B).12,13 With all post-translational modifications installed, the leader peptide is removed by proteases to yield the mature lanthipeptide (Fig. 1A).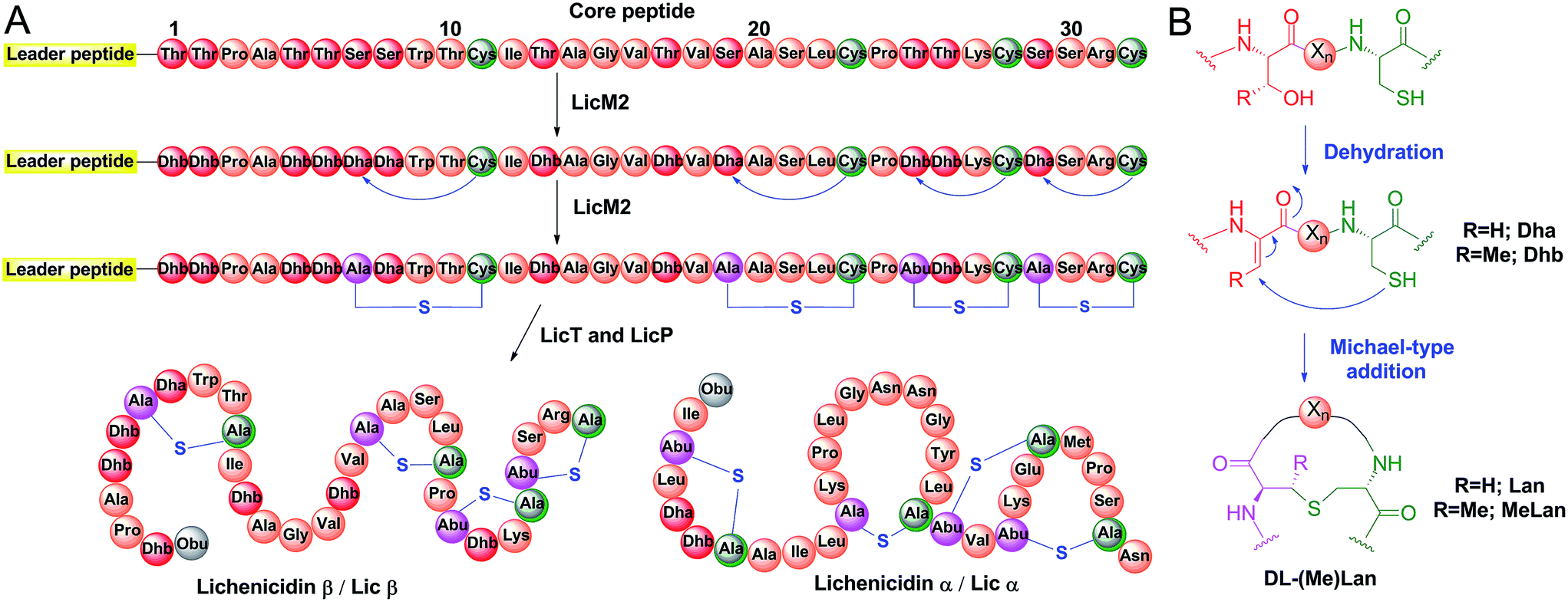 | ||
| Fig. 1 Biosynthesis of class II lanthipeptides. (A) Biosynthetic pathway of class II lanthipeptides with lichenicidin β (Licβ) shown as an example. The precursor peptide LicA2 is dehydrated and cyclized by the class II lanthipeptide synthetase LicM2. Then, the leader peptide is removed in a two-step process by LicT and LicP (see Fig. S1†). Also shown is the structure of the α component that together with Licβ makes up the two-component lantibiotic lichenicidin. Obu, 2-oxobutyryl group resulting from hydrolysis of an N-terminal Dhb; Abu, α-aminobutyric acid. (B) Dehydration and cyclization reactions that take place during the lanthipeptide post-translational modification process. For the sequence of the leader peptide, see Fig. S1.† | ||
Lanthipeptides are currently classified according to the synthetases that install the thioether cross-linked amino acids (Fig. S1 in the ESI†).12 Compared to the well-characterized lanthionine synthetases, the proteases responsible for leader peptide removal are much less studied.14–22 Two types of proteases have been reported for the maturation of class I and II lanthipeptides (Fig. S1†) – the subtilisin-like serine protease LanP employed by both classes and the papain-like cysteine protease domain of the LanT transporter protein involved exclusively in the biosynthesis of class II lanthipeptides.14–20 The cysteine protease domain is located at the N-terminus of LanT proteins and typically cleaves its substrate after a double Gly-type motif (GG/GA/GS).17,18 In contrast to LanT protease domains, the sequence specificity of subtilisin-like LanP proteins remains mostly elusive. Thus far, two class I LanP proteases (NisP and ElxP) have been heterologously expressed and reconstituted in vitro with their native substrates,23–25 and the substrate specificity has been characterized in vitro for ElxP. The substrate specificity of class II LanPs, which fall into a different phylogenetic clade,23 has not been explored.
The biosynthesis of only a few class II lanthipeptides involves LanP proteases.12 These enzymes remove a short N-terminal oligopeptide after a LanT protein detaches the majority of the leader peptide at a double Gly-type cleavage site. For example, LicP is an extracellularly located serine protease expressed by some strains of Bacillus licheniformis and is required for the production of the two-component lantibiotic lichenicidin (Fig. 1A).20 After installation of the thioether rings in the precursor peptides LicA1 and LicA2, LicT removes the leader peptide of modified LicA1 to generate Licα. LicT also cleaves off the majority of the leader peptide from modified LicA2 to generate NDVNPE-Licβ (hereafter LicA2′) (Fig. S1†).20 The maturation of Licβ requires one more cleavage step outside the cell, where LicP trims off the six remaining amino acids at the N-terminus of LicA2′ (Fig. S1†).
In this study, we describe the in vitro characterization of LicP, demonstrate that the protein self-cleaves, and show that it recognizes a specific cleavage sequence but otherwise is very tolerant of the peptide sequence. In addition, prompted by the lack of structural information for class II lanthipeptide leader proteases, we solved a 2.35 Å resolution structure of the enzyme, revealing an unanticipated intramolecular activation strategy that stabilizes the mature form of the enzyme in a calcium-independent manner. LicP has potential utility for sequence-specific proteolysis because it does not leave a scar.3,26 Encouraged by these observations, we identified ten new class II LanP proteins with diverse recognition sequences, which may expand the current protease toolbox for specific removal of expression tags in protein chemistry or leader peptides in RiPP biosynthesis.
Results
Expression of LicP reveals a self-cleavage maturation process
The licP gene was amplified from genomic DNA of B. licheniformis ATCC 14580 (ref. 27) and cloned into an expression vector. A hexa-histidine tag was installed at its N-terminus, and the first 24 amino acids of LicP, which correspond to a secretion signal peptide, were omitted. Upon expression in Escherichia coli BL21 (DE3) and purification using immobilized metal affinity chromatography, two bands were observed by gel electrophoresis (Fig. 2A). Analysis by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) demonstrated masses of 9923 Da and 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 431 Da (Fig. 2B). These molecular weights are in agreement with two fragments of LicP, an N-terminal portion His6-LicP-25-100 with a calculated mass of 9924 Da and a C-terminal portion LicP-101-433 with a calculated mass of 37449 Da, suggesting that a cleavage event occurred during the expression of His6-LicP-25-433 (hereafter referred to as wild type LicP). Expression of LicP-101-433 on its own resulted in protein that was only observed in inclusion bodies.
431 Da (Fig. 2B). These molecular weights are in agreement with two fragments of LicP, an N-terminal portion His6-LicP-25-100 with a calculated mass of 9924 Da and a C-terminal portion LicP-101-433 with a calculated mass of 37449 Da, suggesting that a cleavage event occurred during the expression of His6-LicP-25-433 (hereafter referred to as wild type LicP). Expression of LicP-101-433 on its own resulted in protein that was only observed in inclusion bodies.
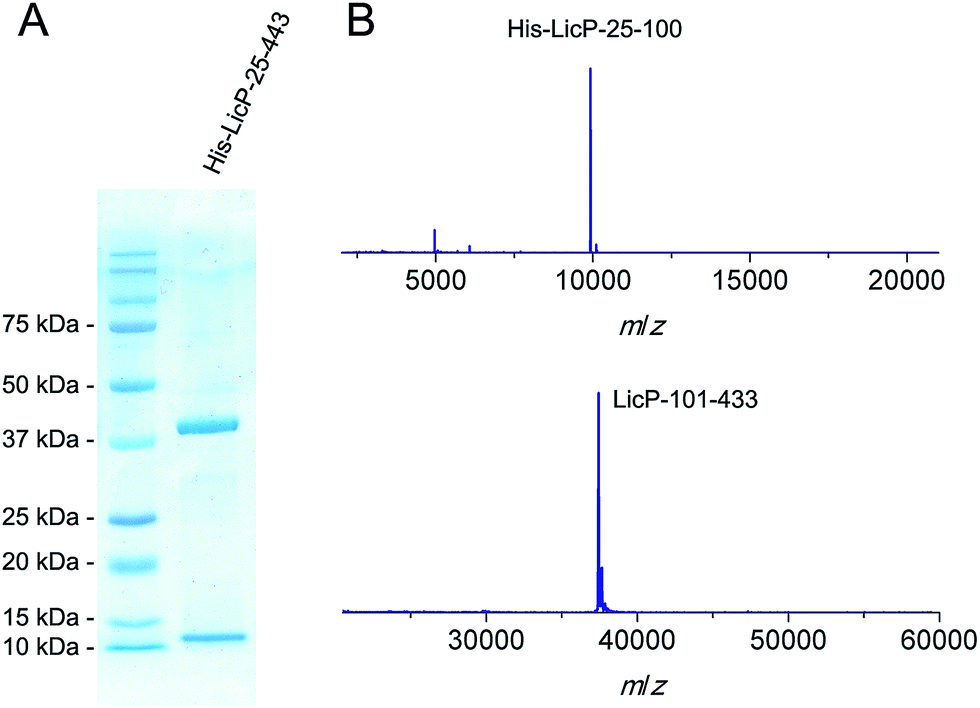 | ||
| Fig. 2 SDS-PAGE (A) and MALDI-TOF MS (B) analysis of His6-LicP-25-433 expressed in E. coli. His6-LicP-25-100, calculated M: 9924, average mass (i.e. not monoisotopic); observed M + H+: 9923, average mass. LicP-101-433, calculated M: 37431, average mass; observed M + H+: 37449, average mass. | ||
Such proteolytic processing has been reported for several extracellular class I LanP proteases and was suggested to be autocatalytic14,15 like other subtilisins.28 To test whether this mechanism applied to the class II enzyme LicP, we substituted the predicted catalytic Ser376 with Ala. Unfortunately, His6-LicP-25-433-S376A was expressed almost exclusively in the insoluble fraction. We were able to obtain a very small amount of soluble His6-LicP-25-433-S376A, demonstrating that indeed the proteolytic cleavage after Glu100 was abolished (Fig. S2†). The observation that an inactivated LicP was expressed as the full length protein indicates that the cleavage event is catalyzed by LicP rather than E. coli proteases. Our findings mirror those of a very recent report on a protease that has sequence homology with the lanthipeptide protease EpiP. This homolog employs an autocatalytic mechanism of cleavage between Lys98 and Thr99.29 Using His6-LicP-25-433 and its S376A mutant, we showed that the proteolysis can take place intermolecularly, albeit slowly (Fig. S3†). To obtain a form of LicP with the pro-sequence still covalently attached, we substituted Glu100 at the native cleavage site with Ala. Surprisingly, His6-LicP-25-433-E100A was again expressed and purified as two fragments (Fig. S4†). MS analysis showed that the resulting fragments corresponded to a shifted cleavage site from residue Ala100 to Glu102 (Fig. S4†). Further mutation of Glu102 to Ala abolished the production of soluble protein.
In vitro characterization of LicP
We next tested LicP activity against the substrate peptide. It has been suggested that LicP trims off the 6-residue oligopeptide NDVNPE from LicA2′ to generate mature Licβ.20 In this work, dehydrated and cyclized LicA2 was obtained by co-expressing LicA2 with its cognate lanthionine synthetase LicM2 in E. coli (Fig. 1A and S5†).30 Instead of using the membrane-bound protein LicT to produce LicA2′, we employed the commercial protease AspN to generate DVNPE-Licβ. Upon incubation with wild type LicP, the 5-residue oligopeptide was successfully removed (Fig. 3A), confirming LicP's anticipated proteolytic activity.20 When the full length modified LicA2 was incubated with LicP, the peptide was also consumed, resulting in two fragments corresponding to the leader peptide and Licβ (Fig. S6†). This observation suggests that LicP does not require prior proteolysis by LicT to produce Licβ. We next incubated linear LicA2 with LicP to test whether unmodified LicA2 was also a substrate. Cleavage of the unmodified LicA2 peptide was indeed observed (Fig. S6†), indicating that post-translational modifications are not required for substrate recognition by LicP.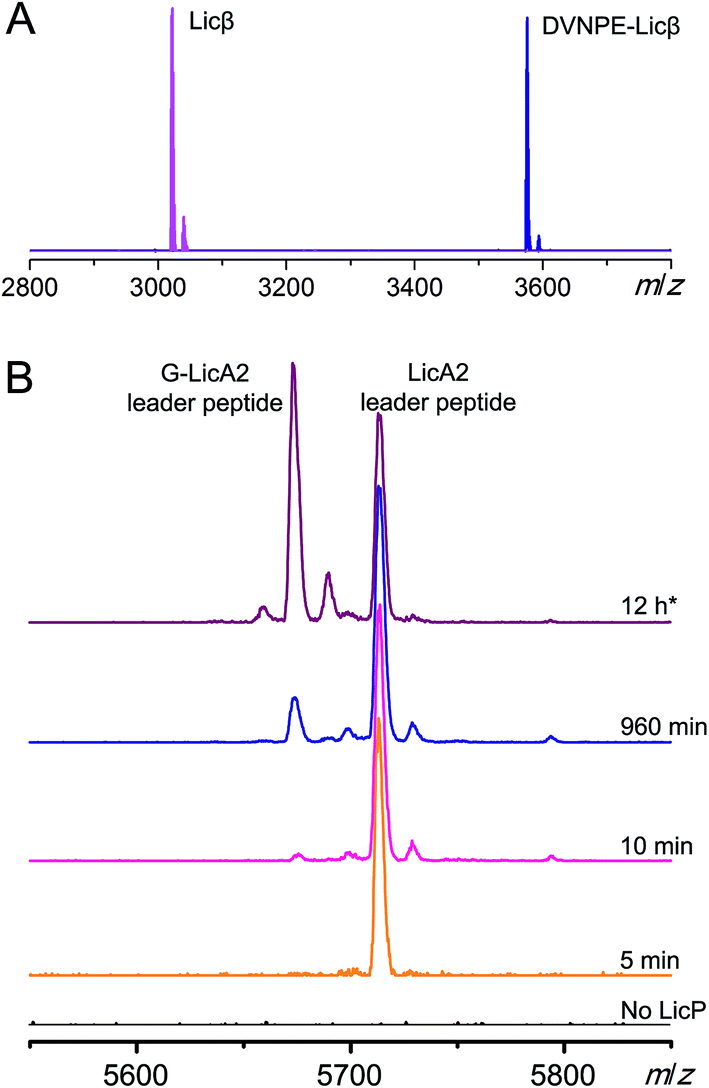 | ||
| Fig. 3 In vitro activity of LicP. (A) MALDI-TOF mass spectra of DVNPE-Licβ peptide with (magenta) or without (blue) incubation with LicP. (For DVNPE-Licβ, calculated M: 3572.6, monoisotopic; observed M + H+: 3573.6, monoisotopic. For Licβ, calculated M: 3019.4, monoisotopic; observed M + H+: 3020.5, monoisotopic.) (B) Time-dependent MALDI-TOF MS analysis of modified LicA2 and linear G-LicA2 treated with LicP, monitoring the production of the leader peptides. For all traces, both peptides were each supplied with a final concentration of 17 μM. For the purple trace, 2.1 μM of His6-LicP-25-433 was added and the reaction was incubated at room temperature for 12 h (asterisk); for the other traces, 21 nM His6-LicP-25-433 was employed. | ||
We further investigated whether the enzyme displays a preference for modified or linear LicA2. Liquid chromatography-based kinetic analysis of the concentration dependence of the cleavage reactions was hampered by the poor solubility of the LicA2 and Licβ peptides. Instead, we monitored the proteolysis of linear LicA2 and dehydrated and cyclized LicA2 at one concentration by HPLC. The time dependence of the disappearance of the substrates and appearance of the leader peptide clearly shows that modified LicA2 is a much better substrate than linear LicA2 (Fig. S7†). Modified LicA2 (100 μM) was consumed by 0.4 μM LicP in 5 min, corresponding to a turnover rate of at least 50 min−1, whereas it took much longer for consumption of 100 μM linear LicA2 (Fig. S7†). We also employed a competitive MALDI-TOF MS assay at lower substrate concentration, in which LicP was supplied to an equimolar mixture of modified and linear LicA2 and the production of leader peptides was monitored over time. In order to differentiate the otherwise identical leader peptides after proteolysis, we introduced a Pro to Gly mutation between the hexa-histidine tag and the precursor peptide in linear LicA2 (G-LicA2). The leader peptides obtained by complete proteolysis of equimolar amounts of modified and linear LicA2 exhibited comparable signal intensities when monitored by MALDI-TOF MS, confirming that the Pro to Gly mutation does not alter the ionization efficiency significantly (Fig. 3B). LicP was incubated with an 800-fold excess of modified and linear LicA2 (i.e. enzyme:combined substrates = 1:1600), and MALDI-TOF MS analysis illustrated complete consumption of modified LicA2 peptide within 10 min, corresponding to a rate of at least 80 min−1, whereas the cleavage of linear LicA2 only started after the modified LicA2 had been consumed and required more enzyme to be completed (Fig. 3B and S7†). Collectively, our observations indicate that although both are substrates for the enzyme, LicP prefers modified LicA2.
LicP can serve as a sequence-specific traceless protease
The observation that LicP removes the oligopeptide NDVNPE and the entire leader peptide from modified or linear LicA2 suggests that it specifically recognizes the NDVNPE sequence but is rather tolerant of other regions of the peptides. We decided to test this hypothesis in the lanthipeptide family, as site-specific removal of leader peptides is critical for producing lanthipeptides in vitro and this step is often challenging as only a limited choice of proteases is available.31–33 ProcA1.7 and NisA, the precursor peptides of the lanthipeptides prochlorosin 1.7 and nisin, were mutated to substitute the last six residues of their leader peptides with the NDVNPE sequence (Fig. S8†). NDVNPE-ProcA1.7 and NDVNPE-NisA were incubated with LicP, resulting in successful removal of their leader peptides (Fig. S8†). Encouraged by these observations, we further explored LicP's potential of removing an expression tag and compared its activity with that of the widely used sequence-specific Tobacco Etch Virus (TEV) protease. The methyltransferase BamL was expressed with a maltose-binding protein (MBP) fused at the N-terminus34 and the protease recognition sequences were installed between MBP and BamL. LicP was capable of removing the MBP-tag in front of the BamL protein with similar efficiency as TEV protease (Fig. S9†), confirming that the substrate scope of LicP is not limited to peptides.The experiments with linear and modified LicA2 as well as NDVNPE-ProcA1.7, NDVNPE-NisA and MBP-BamL demonstrated that LicP tolerates Dhb, Thr, Ser and Ile in the P1′ position. To further evaluate its tolerance, we altered the P1′ position in NDVNPE-NisA from Ile to eight other amino acids (Gly, Cys, Thr, Leu, Phe, Trp, Glu and Lys; Fig. S10†). All these mutants were accepted by LicP as in all cases removal of the NisA leader peptide was observed when substrate peptides were supplied in 30 to 300 fold excess over the enzyme (Fig. S11†). The highest activity was observed when the P1′ position was occupied by Thr or Cys. The latter is noteworthy because traceless removal of tags in front of Cys is highly valuable for cysteine-based native chemical ligation-type chemistry.35,36 The removal of the NisA leader peptide in front of a Gly or Ile residue was slightly less efficient, but complete consumption of precursor peptides was still observed. NDVNPE-NisA analogs with Trp, Leu, and Lys at the P1′ position were also accepted by LicP, although some substrate still remained after 30 hours of incubation. Peptides with Glu or Phe in the P1′ position turned out to be poor substrates. Collectively, these results show that LicP serves as a sequence-specific protease for non-native substrates and that its activity is highly portable with respect to the P1′ position (Table 1).
| Substrate sequences | P and P′ positions | [LicP] | [Substrate] | Incubation time at RT | Complete reaction |
|---|---|---|---|---|---|
| a Performed at 4 °C. RT: room temperature. | |||||
| LicP-95-105 | NTAVNE|TESVI | — | — | — | Y |
| LicP-E100A-97-107 | AVNATE|SVISG | — | — | — | Y |
| Modified LicA2 | NDVNPE|DhbDhbPADhb | 0.4 μM | 100 μM | 5 min | Y |
| Linear LicA2 | NDVNPE|TTPAT | 0.4 μM | 100 μM | 2 h | Y |
| NDVNPE-ProcA1.7 | NDVNPE|TIGGT | 0.2 μM | 180 μM | 4 h | Y |
| NDVNPE-NisA | NDVNPE|ITSIS | 2 μM | 65 μM | 30 h | Y |
| MBP-BamL | NDVNPE|SGSEN | 0.5 μM | 50 μM | 12a h | Y |
| NDVNPE-NisA-I1T | NDVNPE|TTSIS | 0.2 μM | 65 μM | 20 h | Y |
| NDVNPE-NisA-I1C | NDVNPE|CTSIS | 0.2 μM | 65 μM | 20 h | Y |
| NDVNPE-NisA-I1G | NDVNPE|GTSIS | 2 μM | 65 μM | 30 h | Y |
| NDVNPE-NisA-I1W | NDVNPE|WTSIS | 2 μM | 65 μM | 30 h | N |
| NDVNPE-NisA-I1L | NDVNPE|LTSIS | 2 μM | 65 μM | 30 h | N |
| NDVNPE-NisA-I1K | NDVNPE|KTSIS | 2 μM | 65 μM | 30 h | N |
| NDVNPE-NisA-I1F | NDVNPE|FTSIS | 2 μM | 65 μM | 30 h | N |
| NDVNPE-NisA-I1E | NDVNPE|ETSIS | 2 μM | 65 μM | 30 h | N |
X-ray structure of LicP and substrate selectivity
The 2.35 Å resolution structure of LicP reveals a peptidase_S8 family serine protease fold common among subtilisin and related enzymes (Fig. 4A).37 Although the polypeptide was cleaved, the structure reveals a non-covalent complex of the resultant two fragments, consisting of the N-terminal prodomain and the C-terminal catalytic protease domain. Continuous electron density was observed for residues Glu30 through Glu90 (prodomain) and residues Val104 through Met440 (catalytic domain). The peptide sequence that includes Glu100, where cleavage occurs, was not observed. Interestingly, the N-terminus of the catalytic domain (Val104) is positioned more than 50 Å away from the prodomain (Fig. S12†), and movement of a large loop encompassing residues Val104 through Leu117 facilitates this displacement. Within the protease domain, the putative catalytic triad, consisting of Asp151/His186/Ser376, is situated with distances and orientations that are similar to those found in the triad of subtilisin BPN′.38 Based on the structural homology with subtilisin BPN′, the backbone amide of Ser376 and the amide side chain of Asn275 comprise the oxyanion hole in LicP.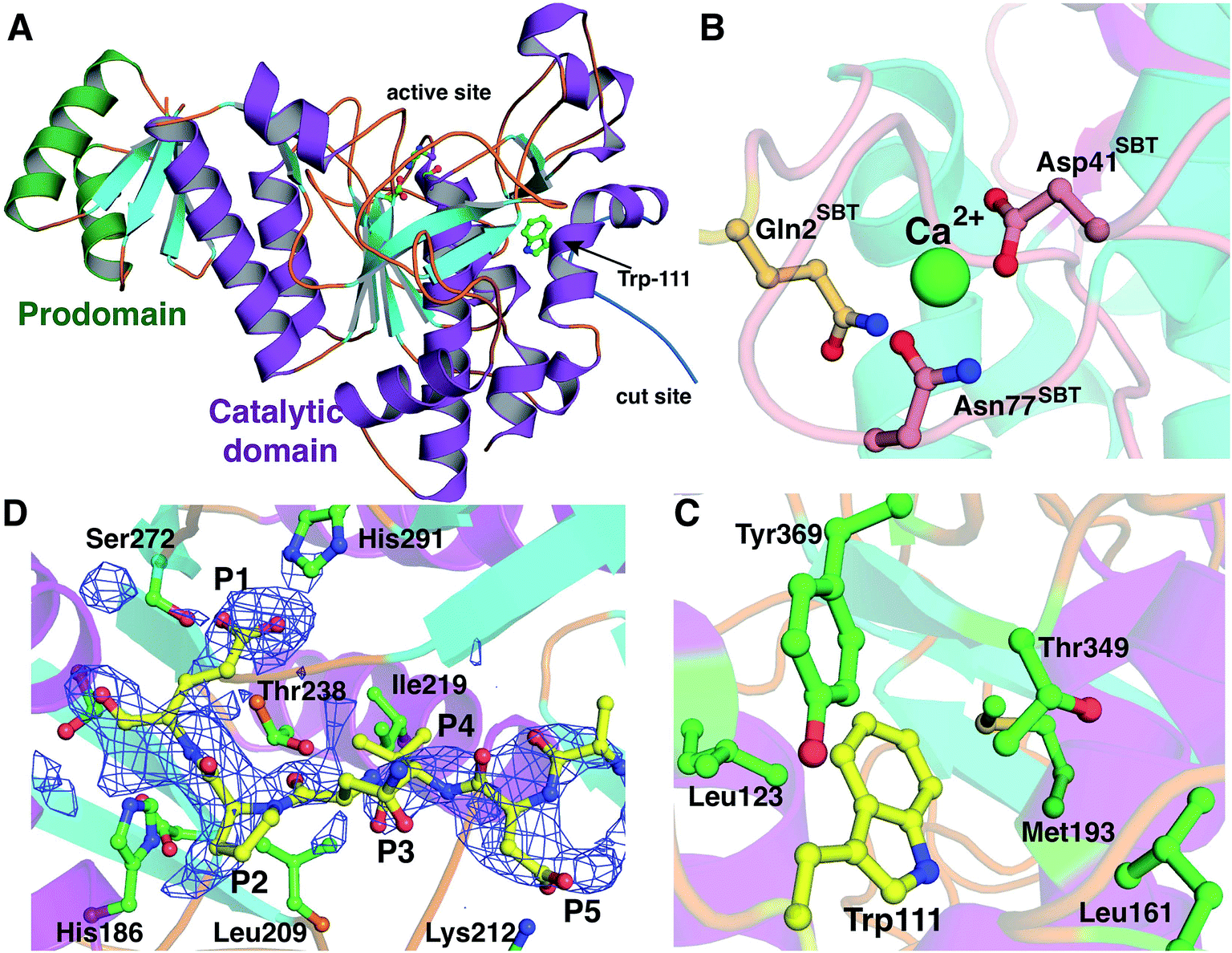 | ||
| Fig. 4 Crystal structure of LicP. (A) Cartoon representation of the overall structure of LicP with the prodomain colored in green and the catalytic domain colored in purple and light blue (PDB ID: 4ZOQ). See also Fig. S12.† (B and C) Comparison of the structures of LicP (C) and subtilisin BPN′ (B) in the vicinity of the calcium-binding region, which in LicP is replaced by a hydrophobic pocket into which Trp111 from the linker region binds. In panel B, the residues numbering refers to the subtilisin (SBT) catalytic domain, whereas in panel C and elsewhere LicP numbering is based on the full length protein. (D) Omit electron density map superimposed on a model of LicP bound to a peptide substrate. The density corresponds to residual features of the linker into which the substrate has been modeled. | ||
The prodomain folds into a single compact domain consisting of four antiparallel β-strands interconnected with two helices. The length of the helices is larger than those found in subtilisin BPN′, and consequently, the LicP prodomain has only limited structural similarity to the prodomain of subtilisin BPN′ (Z-score = 5.7; RMSD of 2.4 Å over 57 aligned Cα atoms). The LicP prodomain has an overall fold that is more reminiscent of the PII proteins that facilitate signal transduction in response to build-up of primary metabolites (PDB code = 3DFE; Z-score = 8.8; RMSD = 1.6 Å over 60 aligned Cα atoms).39 In the subtilisin BPN′ structure, two long α-helices from the catalytic domain were proposed to stabilize the prodomain during the folding process.40 A similar interaction between the prodomain and the catalytic domain is preserved in LicP (Fig. 4A).
A second central feature in subtilisin maturation is that once the prodomain has been cleaved, the N-terminus of the catalytic domain is displaced to a high-affinity calcium-binding loop, where it provides additional residues for coordinating the metal (Fig. 4B).41 In the LicP crystal structure, this calcium-binding site is not conserved, and the long loop that comprises this site in subtilisin BPN′ is shortened. Additionally, the helix that precedes this loop is lengthened in LicP, and these rearrangements result in the formation of a hydrophobic pocket. In the mature form of LicP observed in the crystal structure, the excised N-terminus of the catalytic domain (Val104 through Leu117) is situated in the same place as the calcium-binding site, and Trp111 from this linker inserts into this hydrophobic pocket (Fig. 4C). Several hydrophobic residues, including Leu123, Phe125, Leu161, Met193, Thr349, and Tyr369, define the contours of this pocket (Fig. 4C), and insertion of Trp111 into this cavity creates favorable van der Waals interactions that may be a necessary step in LicP maturation. Perhaps the observed difficulty of obtaining soluble mutants of LicP that prevent proteolytic processing can be attributed to the inability of Trp111 to access this hydrophobic pocket.
All eight crystallographically independent copies of LicP in the asymmetry unit contain spurious but persistent electron density in the vicinity of the active site (Fig. 4D), which may be due to partial occupancy of the cleaved linker within the active site. Because of the paucity of well-defined features, no attempts were made to model residues from the cleaved linker region into this density. Nonetheless, we utilized the contours of this density to derive an informed model of how LicP may interact with a NDVNPE peptide substrate (Fig. 4D). According to this model, the specificity for the P1 Glu of the substrate peptide is dictated by interactions with the side chains of Ser272 and His291 of LicP, as well as weak interactions with the hydroxyl groups of Thr238 and Tyr241. Likewise, a pocket that consists of Leu209, Ile219, and Thr238 appears to dictate the P4 specificity for a small hydrophobic residue such as Val. Lastly, the Asp at the P5 position of the substrate is positioned to make ionic interactions with Lys212 of LicP. The paucity of additional interactions with the enzyme is consistent with the tolerance of LicP for the linker that connects the prodomain and the catalytic domain (sequence NTAVNE) and its substrate peptide (NDVNPE).
The substrate specificity of LicP was further evaluated by an SDS-PAGE-based assay monitoring the time-dependent cleavage of mutants of linear LicA2. The assay was first validated by analyzing samples by both HPLC and SDS-PAGE (Fig. S7†), which showed that the gel-based assay provides a good read-out of relative activity. The presence of a Glu at the P1 position was critical for LicP activity as LicA2-E–1A was not a substrate under the assay conditions and even substitution with structurally related amino acids Gln and Asp was not tolerated (Fig. S13 and S14†). The importance of the P5 position was tested by mutating Asp to Lys, Ala or Val. We found that all three P5 mutants were processed much more slowly, suggesting that the P5 position of the substrate is also important for LicP's activity (Fig. S14–S16†). The P4 position of LicA2 was substituted with three hydrophobic amino acids of varying size, Ala, Leu and Phe, and with a polar amino acid, Asn. LicA2-V–4A, LicA2-V–4L and LicA2-V–4N were still accepted by LicP with slightly reduced cleavage efficiency (Fig. S16 and S17†). However, LicA2-V–4F was not processed (Fig. S15†), indicating that only relatively small amino acids are tolerated at the P4 position of the substrate. The P2 and P3 positions (Pro and Asn, respectively) are not critical for LicP recognition as alanine substitution at both sites did not alter the processing efficiency of LicP significantly (Fig. S18†). The observations that P1 plays a critical role and that D–5V and V–4N mutants are still processed by LicP, albeit at a slower rate, are consistent with the shift in proteolysis site for LicP(-E100A) resulting in cleavage after AVNATE (Table 1). Collectively, our biochemical data are in good agreement with the model based on the LicP crystal structure with respect to the substrate specificity.
Class II LanP proteins: a pool of sequence-specific proteases
LanP genes are not often found in class II lanthipeptide gene clusters. Only four class II LanPs have been reported to date – LicP, CylA, CerP and CrnP, which have been suggested to remove six-residue sequences at the N-terminus of Licβ,20 cytolysin,19 cerecidins42 and carnolysin,43 respectively. Among them, the proteolytic activity, and hence the identity of the cleavage sites, has been confirmed for CylA,19 CerP42 and LicP (this work). To identify additional class II LanP proteins and potentially identify additional recognition sequences that might be useful, we performed a search of the UniProtKB database with the LicP protease domain (LicP-101-433) as a query and of the non-redundant protein sequence database with LicA2 as a query using the default BLAST parameters for proteins provided by the National Center for Biotechnology Information (NCBI) website. The first 250 hits were subjected to further analysis and several were correlated to class II lanthipeptide biosynthesis by the observation of nearby genes encoding LanM proteins. Ten additional representative class II lanthipeptide gene clusters with LanP genes are shown in Fig. 5 (see also Fig. S19 and Table S4†), all of which contain multiple genes for LanA substrates. These LanP proteins share a minimal sequence identity of 30% with the LicP protease domain with E values lower than e−26. The putative cleavage sites for these LanP enzymes are proposed to locate immediately C-terminal to the double Gly-type motif used by LanT enzymes and upstream of the Thr/Ser/Cys rich core peptides (Fig. S19†). The predicted cleavage sites were confirmed for two representative examples (Bacillus licheniformis 9945A and Bacillus cereus VD156) by incubating the purified LanP and an associated LanA substrate (Fig. S20†). Interestingly, all putative LanP recognition sequences consist of six residues with the exception of the A1–A3 peptides from Bacillus cereus FRI-35, which contain eight residues with two additional amino acids at the N-terminus. An Asp at the P5 position and a Val at the P4 position are conserved among most clusters with the precursor peptides from Bacillus cereus VD156 and VD045 being exceptions. Pro and Ala are frequently found at the P2 position, whereas the P1 position is almost exclusively occupied by polar/charged residues, such as Asp, Glu, His or Arg, but the putative P1 position of the A1 peptide from Bacillus thuringiensis DB27 is occupied by Ala. | ||
| Fig. 5 Representative class II lanthipeptide biosynthetic gene clusters containing LanP genes. Genes with unknown functions are indicated with X. For substrate peptides and their proposed recognition sequences, please see Fig. S19.† Clusters are annotated using the standard lanthipeptide biosynthesis nomenclature: LanM proteins catalyze the dehydration and cyclization reactions, LanA peptides are the lanthipeptide precursors, LanT proteins are transporters with an N-terminal Cys protease domain, LanEFGHI proteins are immunity conferring proteins, LanJ are dehydrogenases, and LanHR are regulatory proteins. The cytolysin cluster is annotated differently: the substrates are CylLL and CylLS, CylB is a transporter with a protease domain, and CylA is the class II LanP. | ||
Discussion
We report here the heterologous expression and in vitro characterization of a class II LanP protease, a protein class that has been underexplored. LicP was obtained from E. coli as two fragments that bound tightly to each other in a 1:1 ratio. A self-catalyzed cleavage process has been reported for two class I LanPs, NisP and EpiP, which lack the first 195 and 99 amino acids in their mature forms, respectively,14,15 and was recently confirmed to be intramolecular for a homolog of EpiP.29 Here we show that for LicP, such cleavage can proceed intermolecularly, but because hours of incubation are required for full cleavage it is likely that the proteolysis also occurs intramolecularly under physiological conditions. Interestingly, the E100A mutant was still expressed as two fragments with a shift in the cleavage site by two residues to Glu102. This observation strongly suggests that a Glu at the P1 position is essential for LicP activity. The cleavage at Glu102 was surprising because residues 97–102 in LicP-E100A do not appear to be homologous to residues 95–100 in LicP, but the observed alternative cleavage is consistent with our demonstration that individual mutations of the P2–P5 positions do not abolish processing. In addition, the linker sequence for autocatalytic processing is presumably positioned sufficiently close to the active site to maintain a high effective concentration such that cleavage at alternative Glu sites is still relatively efficient.
Comparing the two class I LanP proteases for which a structure has been reported (NisP and an EpiP homolog)24,29 with the class II LicP protein in the current study shows that there are similarities and differences. Both the EpiP homolog and LicP purified with the prodomain non-covalently bound to the catalytic domain. Furthermore, neither the prodomain nor the catalytic domain of the EpiP homolog could be obtained individually,29 which we also observed for LicP, suggesting that prodomain binding and/or the folding process induced by prodomain cleavage is critical for obtaining a stable protein. Conversely, NisP was obtained as an active protein in the absence of the prodomain, which is also not present in its reported structure.24 Although mechanistic details for protease maturation have not been described for either NisP or the EpiP homolog, structural considerations provide a plausible framework. The hydrophobic cavity stabilized by Trp111 in the LicP structure is enclosed by a long loop in the catalytic domains of NisP and the EpiP homolog, which is absent in LicP. This loop superimposes upon the calcium-binding site of subtilisin BPN′, which is missing in both the NisP and EpiP homolog structures. Notably, amino-terminal residues from the N-terminus of the catalytic domain that is generated following proteolysis buttress this loop, suggesting that stabilization of the loop may be involved in class I LanP protease maturation.
In addition to its physiological role of trimming a 6-residue oligopeptide from LicA2′, LicP removed the entire leader peptides from modified and linear LicA2. An estimated substrate consumption rate of 50–80 min−1 was observed for LicA2 with the post-translational modifications installed, indicating LicP is a highly efficient protease. NisP, the class I LanP responsible for the maturation of the lantibiotic nisin, in vivo accepts modified NisA as its substrate but not the linear version.44,45 Our observations suggest that LicP also favors modified substrate in vitro. Such substrate selectivity by LanP proteins is beneficial in the lanthipeptide producing organism as it prevents undesired consumption of unmodified precursor peptides.
Although LicP favors modified LicA2 over linear LicA2, which indicates that post-translational modifications in the core peptide contribute to LicP's substrate recognition in addition to the NDVNPE sequence, our observations with substrate analogs demonstrate its application as a sequence-specific protease for traceless removal of leader peptides and an MBP expression tag. The substrate specificity of LicP was identified using both structural information and biochemical characterizations. The P5, P4 and P1 residues of LicA2 were found to be important for LicP recognition. These three sites were also suggested as the origin of specificity for a class I LanP, ElxP, as determined by kinetic analysis based on LC quantification.23 The similarity in the important positions may suggest a general substrate recognition mechanism by the entire subtilisin-like LanP family.
The thermostability of subtilisin BPN′ and related proteases is enhanced significantly in the presence of calcium ions, which are necessary for maturation, and subsequent stabilization of a large loop in the catalytic domain.46 However, the calcium dependence constitutes a drawback for industrial utility, e.g. in detergents containing metal chelators. Therefore, much effort has been spent on engineering thermostable mutants of subtilisin that function in a calcium-independent manner.47,48 The structural and biochemical analysis of LicP reveals an elegant solution to this limitation, as maturation and subsequent stabilization of the enzyme is facilitated not by metal ions, but rather by the insertion of Trp111, liberated by cleavage of the linker between the prodomain and the catalytic domain, into a hydrophobic pocket located in the same vicinity as the calcium-binding site in subtilisin BPN′. The recent structure of the class I lanthipeptide protease NisP also demonstrated loss of a calcium binding site, although unlike the structure of LicP, the prodomain was not present in the NisP structure and the alternative mechanism of stabilization in the absence of a calcium binding site was not discussed.24 Based on structural comparisons with LicP, we speculate that stabilization of the region in the catalytic domain corresponding to the calcium-binding site of subtilisin BPN′ by the new N-terminus after proteolytic processing may prove to be a general mechanism for LanP maturation.
Over the past several decades, the toolbox of useful proteases has been significantly enlarged. Several proteases with strict recognition sequences have been commercialized for biochemical or industrial applications, including Factor Xa, enterokinase, and TEV protease. Factor Xa and enterokinase exhibit P1 preferences similar to trypsin and cleave after an Arg or Lys. TEV protease recognizes a larger motif and exhibits better reliability in terms of specificity, but TEV protease requires either a Gly or Ser at the P1′ position for efficient cleavage. LicP is complementary in that it specifically cleaves after a Glu in the NDVNPE sequence, and is quite tolerant of various residues in the P1′ position. LicP accepted a range of residues at the P1′ site (Table 1) such as glycine, small polar residues (Ser, Thr, Cys) and large aliphatic residues (Ile). LicP also processed peptides with Leu, aromatic (Phe, Trp) and charged (Lys, Glu) residues at the P1′ position albeit with reduced efficiency. Also noteworthy is the ability to cleave adjacent to post-translationally modified residues such as Dhb. In previous work, commercial proteases have shown considerable reluctance to cleave next to such a post-translationally modified amino acid.31–33,49 Additional favorable properties include the enzyme's stability demonstrated by the persistent activity of LicP after 12 weeks at 4 °C, and no obvious decrease in activity after multiple rounds of freeze–thaw procedures.
This study also identified ten new class II lanthipeptide gene clusters containing lanP genes, suggesting they are more widely distributed than previously expected.12 Although the putative recognition sequences of these newly identified LanPs show a certain level of homology, they also exhibit considerable diversity. Similar to other proteases, most of these LanPs are predicted to cleave after charged residues such as arginine, glutamate or aspartate, but a few appear to cleave after unusual P1 residues such as histidine or alanine that are rarely the site of cleavage for other proteases (Fig. S19†). We confirmed the predicted sites for two examples that have a His and an Arg in the P1 position (Fig. S20†). Hence, this naturally occurring protease family may serve as a basis to construct a general protease pool for traceless tag removal purposes.5,50,51
Methods
For details regarding molecular biology procedures, protein expression and purifications, and enzyme assays, see the ESI.† The LicP structure has been deposited in the Protein Data Bank under ID: 4ZOQ.Acknowledgements
This work was supported by National Institutes of Health Grants GM58822 (to W. A. v. d. D.), GM079038 (to S. K. N.) and F32 GM108275 (to L. M. R). We thank Dr Xiao Yang (UIUC) for constructing the expression plasmids for the enzyme and substrate from B. cereus VD156. A Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer used for this work was purchased in part with a grant from the National Institutes of Health (S10 RR027109). The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.Notes and references
- M. B. Rao, A. M. Tanksale, M. S. Ghatge and V. V. Deshpande, Microbiol. Mol. Biol. Rev., 1998, 62, 597–635 CAS.
- M. Pogson, G. Georgiou and B. L. Iverson, Curr. Opin. Biotechnol., 2009, 20, 390–397 CrossRef CAS PubMed.
- Q. Li, L. Yi, P. Marek and B. L. Iverson, FEBS Lett., 2013, 587, 1155–1163 CrossRef CAS PubMed.
- C. S. Craik, M. J. Page and E. L. Madison, Biochem. J., 2011, 435, 1–16 CrossRef CAS PubMed.
- L. Yi, M. C. Gebhard, Q. Li, J. M. Taft, G. Georgiou and B. L. Iverson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7229–7234 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618–5622 CrossRef CAS.
- D. T. Berg, B. Gerlitz, J. Shang, T. Smith, P. Santa, M. A. Richardson, K. D. Kurz, B. W. Grinnell, K. Mace and B. E. Jones, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4423–4428 CrossRef CAS PubMed.
- L. Hedstrom, J. J. Perona and W. J. Rutter, Biochemistry, 1994, 33, 8757–8763 CrossRef CAS PubMed.
- D. N. Rubingh, Curr. Opin. Biotechnol., 1997, 8, 417–422 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- X. Yang and W. A. van der Donk, Chem.–Eur. J., 2013, 19, 7662–7677 CrossRef CAS PubMed.
- P. J. Knerr and W. A. van der Donk, Annu. Rev. Biochem., 2012, 81, 479–505 CrossRef CAS PubMed.
- G. Bierbaum and H. G. Sahl, Curr. Pharm. Biotechnol., 2009, 10, 2–18 CAS.
- J. R. van der Meer, J. Polman, M. M. Beerthuyzen, R. J. Siezen, O. P. Kuipers and W. M. de Vos, J. Bacteriol., 1993, 175, 2578–2588 CrossRef CAS PubMed.
- S. Geissler, F. Götz and T. Kupke, J. Bacteriol., 1996, 178, 284–288 CrossRef CAS PubMed.
- J. E. Velásquez, X. Zhang and W. A. van der Donk, Chem. Biol., 2011, 18, 857–867 CrossRef PubMed.
- M. Nishie, K. Shioya, J. Nagao, H. Jikuya and K. Sonomoto, J. Biosci. Bioeng., 2009, 108, 460–464 CrossRef CAS PubMed.
- L. A. Furgerson Ihnken, C. Chatterjee and W. A. van der Donk, Biochemistry, 2008, 47, 7352–7363 CrossRef CAS PubMed.
- M. C. Booth, C. P. Bogie, H. G. Sahl, R. J. Siezen, K. L. Hatter and M. S. Gilmore, Mol. Microbiol., 1996, 21, 1175–1184 CrossRef CAS PubMed.
- T. Caetano, J. M. Krawczyk, E. Mosker, R. D. Süssmuth and S. Mendo, Chem. Biol., 2011, 18, 90–100 CrossRef CAS PubMed.
- G. H. Völler, B. Krawczyk, P. Ensle and R. D. Süssmuth, J. Am. Chem. Soc., 2013, 135, 7426–7429 CrossRef PubMed.
- Y. Ike and D. B. Clewell, J. Bacteriol., 1992, 174, 8172–8177 CrossRef CAS PubMed.
- M. A. Ortega, J. E. Velásquez, N. Garg, Q. Zhang, R. E. Joyce, S. K. Nair and W. A. van der Donk, ACS Chem. Biol., 2014, 9, 1718–1725 CrossRef CAS PubMed.
- Y. Xu, X. Li, R. Li, S. Li, H. Ni, H. Wang, H. Xu, W. Zhou, P. E. Saris, W. Yang, M. Qiao and Z. Rao, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1499–1505 CrossRef CAS PubMed.
- A. Abts, M. Montalban-Lopez, O. P. Kuipers, S. H. Smits and L. Schmitt, Biochemistry, 2013, 52, 5387–5395 CrossRef CAS PubMed.
- C. L. Young, Z. T. Britton and A. S. Robinson, Biotechnol. J., 2012, 7, 620–634 CrossRef CAS PubMed.
- M. Begley, P. D. Cotter, C. Hill and R. P. Ross, Appl. Environ. Microbiol., 2009, 75, 5451–5460 CrossRef CAS PubMed.
- S. D. Power, R. M. Adams and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3096–3100 CrossRef CAS.
- M. L. Kuhn, P. Prachi, G. Minasov, L. Shuvalova, J. Ruan, I. Dubrovska, J. Winsor, M. Giraldi, M. Biagini, S. Liberatori, S. Savino, F. Bagnoli, W. F. Anderson and G. Grandi, FASEB J., 2014, 28, 1780–1793 CrossRef CAS PubMed.
- Q. Zhang, M. Ortega, Y. Shi, H. Wang, J. O. Melby, W. Tang, D. A. Mitchell and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12031–12036 CrossRef CAS PubMed.
- N. A. Bindman and W. A. van der Donk, J. Am. Chem. Soc., 2013, 135, 10362–10371 CrossRef CAS PubMed.
- W. Tang and W. A. van der Donk, Biochemistry, 2012, 51, 4271–4279 CrossRef CAS PubMed.
- A. Plat, L. D. Kluskens, A. Kuipers, R. Rink and G. N. Moll, Appl. Environ. Microbiol., 2011, 77, 604–611 CrossRef CAS PubMed.
- J. Lee, Y. Hao, P. M. Blair, J. O. Melby, V. Agarwal, B. J. Burkhart, S. K. Nair and D. A. Mitchell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12954–12959 CrossRef CAS PubMed.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- J. Kraut, Annu. Rev. Biochem., 1977, 46, 331–358 CrossRef CAS PubMed.
- R. Bott, M. Ultsch, A. Kossiakoff, T. Graycar, B. Katz and S. Power, J. Biol. Chem., 1988, 263, 7895–7906 CAS.
- A. J. Ninfa and P. Jiang, Curr. Opin. Microbiol., 2005, 8, 168–173 CrossRef CAS PubMed.
- T. Gallagher, G. Gilliland, L. Wang and P. Bryan, Structure, 1995, 3, 907–914 CrossRef CAS PubMed.
- P. Bryan, P. Alexander, S. Strausberg, F. Schwarz, W. Lan, G. Gilliland and D. T. Gallagher, Biochemistry, 1992, 31, 4937–4945 CrossRef CAS PubMed.
- J. Wang, L. Zhang, K. Teng, S. Sun, Z. Sun and J. Zhong, Appl. Environ. Microbiol., 2014, 80, 2633–2643 CrossRef PubMed.
- C. T. Lohans, J. L. Li and J. C. Vederas, J. Am. Chem. Soc., 2014, 136, 13150–13153 CrossRef CAS PubMed.
- A. Kuipers, E. de Boef, R. Rink, S. Fekken, L. D. Kluskens, A. J. Driessen, K. Leenhouts, O. P. Kuipers and G. N. Moll, J. Biol. Chem., 2004, 279, 22176–22182 CrossRef CAS PubMed.
- O. Koponen, M. Tolonen, M. Qiao, G. Wahlstrom, J. Helin and P. E. J. Saris, Microbiology, 2002, 148, 3561–3568 CrossRef CAS PubMed.
- C. A. Smith, H. S. Toogood, H. M. Baker, R. M. Daniel and E. N. Baker, J. Mol. Biol., 1999, 294, 1027–1040 CrossRef CAS PubMed.
- S. L. Strausberg, P. A. Alexander, D. T. Gallagher, G. L. Gilliland, B. L. Barnett and P. N. Bryan, J. Biotechnol., 1995, 13, 669–673 CAS.
- S. L. Strausberg, B. Ruan, K. E. Fisher, P. A. Alexander and P. N. Bryan, Biochemistry, 2005, 44, 3272–3279 CrossRef CAS PubMed.
- N. Garg, W. Tang, Y. Goto and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5241–5246 CrossRef CAS PubMed.
- R. J. Siezen, W. M. de Vos, J. A. Leunissen and B. W. Dijkstra, Protein Eng., 1991, 4, 719–737 CrossRef CAS PubMed.
- J. J. Perona and C. S. Craik, Protein Sci., 1995, 4, 337–360 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular biology procedures, protein expression and purifications, procedures for enzyme assays, supplementary figures and tables. See DOI: 10.1039/c5sc02329g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |