
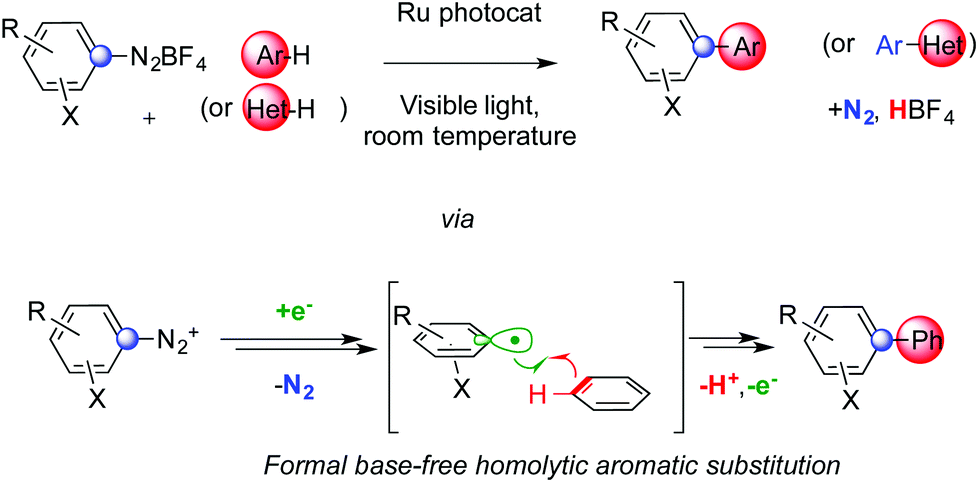
Formal base-free homolytic aromatic substitutions via photoredox catalysis†
Filipe
Gomes‡
a,
Vanessa
Narbonne‡
a,
Florent
Blanchard
a,
Giovanni
Maestri
*ab and
Max
Malacria
*ac
aInstitut de Chimie des Substances Naturelles, ICSN-CNRS UPR 2301, Gif/Yvette Cedex 91198, France. E-mail: max.malacria@cnrs.fr
bDipartimento di Chimica, Università di Parma, Parma 43124, Italy. E-mail: giovanni.maestri@unipr.it
cInstitut Parisien de Chimie Moléculaire, UPMC Univ-Paris 06 – Sorbonne Université, UMR 8232, Paris Cedex 75005, France
First published on 26th March 2015
Abstract
We developed a simple and convenient method to assemble biaryls exploiting a photoredox catalyst and visible light. Diazonium salts generate aryl radicals that could then add on unactivated (hetero)arenes and the sequence eventually delivers products via formal homolytic aromatic substitutions. The direct C–H arylation of these usually unreactive substrates is achieved at room temperature using low catalyst loadings and shows broad functional group tolerance.
Introduction
The development of simple synthetic routes that can form biaryls from simple building blocks represents a long-standing goal for organic chemists because of the broad domain of application of functional molecules that present this structural motif. Together with catalytic methods that can activate C–H bonds,1 in particular those of simple, unactivated arenes,2 homolytic aromatic substitutions (HAS) are suitable free radical processes to achieve this synthetic goal. Their major pitfall is the often-required need of stoichiometric, hazardous, tin derivatives as radical mediators.3 This issue can be addressed using stoichiometric amounts of strong bases such as alkoxides to assist chain propagation.4A complementary strategy uses photoredox catalysts that harvest visible light5 to mildly activate onium species generating aryl radicals.6 Diazonium and iodonium derivatives can indeed regioselectively arylate various heterocycles at the 2-position via photoredox catalysis.7
Merging these two approaches, Li used a strongly reducing Ir photocatalyst to activate aryl iodides at room temperature and trigger base-assisted HAS on arenes.8
We report a HAS cascade that does not require the presence of any base (Scheme 1). This minimizes consumption of reagents and production of waste, therefore increasing the practical viability of the method. It allows the C–H arylation of simple arenes and heteroarenes at room temperature with remarkable functional group tolerance. Aryl radicals are readily generated from diazonium salts using a suitable Ru photoredox catalyst,6 as originally introduced in an intramolecular fashion by Deronzier.6a The desired products form together with a nitrogen molecule and an equivalent of non-toxic HBF4. The presence of protic acidic species does not inhibit the reaction and is tolerated by the photocatalyst. Under these conditions, sufficiently basic species, even in traces, could have an impact on the outcome of these experiments. The presence of water molecules and/or suitable anions could indeed vary the time required to achieve full conversion of the substrate and the selectivity towards the desired product or the reduced starting material.
 | ||
| Scheme 1 Base-free HAS to synthesise biaryls. | ||
Results and discussion
We discovered this process while attempting to develop a synthesis of biaryls merging photoredox and gold catalysis. Reasoning on the feasibility of Ru/Pd dual catalytic cascades6b and on the possibility to access unusual spin states via addition of aryl radicals on suitable transition metal complexes,6c we wondered whether it would have been possible to develop an alternative access to biaryls via Ru/Au joint catalysis (see ESI† for details on our initial idea).9In an initial experiment (Table 1, entry 1), 4-nitrophenyldiazonium tetrafluoroborate reacted with toluene (40 equiv., 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with acetonitrile) at room temperature for 3 hours in the presence of 0.5 mol% of Ru(bpy)3Cl2 as a photocatalyst, 10 mol% of JP Au(MeCN)SbF6 (JP = JohnPhos, (2-biphenyl)di-tert-butylphosphine) and 2 equiv. of K2CO3 as a base under irradiation of a blue LED. To our delight, 81% of the desired biaryl product 1a was recovered as a mixture of its three regioisomers. Toluene was arylated in a 4:1.3:1 ratio at its ortho-, meta- and para-positions. Direct reduction of the diazonium salt to nitrobenzene was the main side product (17%).
:1 mixture with acetonitrile) at room temperature for 3 hours in the presence of 0.5 mol% of Ru(bpy)3Cl2 as a photocatalyst, 10 mol% of JP Au(MeCN)SbF6 (JP = JohnPhos, (2-biphenyl)di-tert-butylphosphine) and 2 equiv. of K2CO3 as a base under irradiation of a blue LED. To our delight, 81% of the desired biaryl product 1a was recovered as a mixture of its three regioisomers. Toluene was arylated in a 4:1.3:1 ratio at its ortho-, meta- and para-positions. Direct reduction of the diazonium salt to nitrobenzene was the main side product (17%).
|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Additive (mol%) | Yield (%) |
| a Reaction conditions: ArN2BF4 0.45 mmol, 0.225 M, isolated yields. b Without irradiation for 24 h. c For 72 h without a photocatalyst. d With 0.9 mmol of K2CO3, 2 equiv., as a base. e JP = JonhPhos, (2-biphenyl)di-tert-butylphosphine. | |||
| 1d | Ru(bpy)3Cl2, 0.5 | JPe Au(MeCN)SbF6, 10 | 81 |
| 2d | Ru(bpy)3Cl2, 0.5 | PPh3AuCl, 10 | 63 |
| 3 | Ru(bpy)3Cl2, 0.5 | PPh3AuCl, 10 | 62 |
| 4 | Ru(bpy)3Cl2, 0.5 | — | 66 |
| 5 | Ru(bpy)3(PF6)2, 0.5 | — | 71 |
| 6 | Ru(bpy)3(PF6)2, 0.5 | JPe Au(MeCN)SbF6, 10 | 70 |
| 7 | Ru(bpy)3(PF6)2, 0.5 | PPh3AuCl, 10 | 68 |
| 8 | Ru(bpy)3(SbF6)2, 0.5 | — | 79 |
| 9b | Ru(bpy)3(SbF6)2, 2.5 | — | Traces |
| 10c | — | — | 10 |
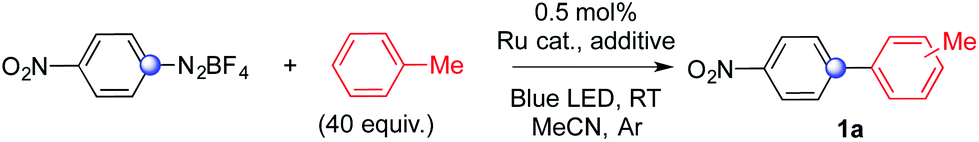
However, successive experiments showed us that the desired products were formed through a mechanism different from our originally envisioned one. In other words, neither gold nor the inorganic base was necessary to deliver biaryls in high yields.
The yield of 1a was lower when the gold complex was replaced with PPh3AuCl (63%, entry 2). We soon found that a comparable selectivity towards 1a was observed on repeating the reaction without K2CO3 (62%, entry 3). In both cases we observed complete conversion of the diazonium salt and nitrobenzene as the main byproduct. The limited difference in yields with and without K2CO3 let us question our initial idea that the sequence could involve a base-assisted C–H activation by gold.9 We then realized the importance to immediately check whether a gold salt had or not an impact on the outcome of the reaction. Biaryl 1a formed in 66% yield in the presence of 0.5 mol% of Ru(bpy)3Cl2 without any other additive (entry 4).
This result suggested that 1a could in principle form under these conditions via a HAS-like mechanism (vide infra) and not through our originally supposed pathway.
We were however puzzled by the difference observed on comparing the yield of 1a with and without a gold complex featuring a SbF6− counteranion (81% for entry 1 and 66% for entry 4). We then wondered whether counteranions of metal salts might play a role, especially in the absence of stoichiometric bases, and affect therefore the outcome of the sequence. The reaction with a commercially available Ru(bpy)3(PF6)2 catalyst provided 71% of 1a (entry 5). For sake of comparison, similar results were achieved with the addition of either 10 mol% of JP Au(MeCN)SbF6 (70%, entry 6) or 10 mol% of the PPh3AuCl complex (68%, entry 7). We then synthesized Ru(bpy)3(SbF6)2 seeking to take advantage of the solubility properties of a non-coordinating SbF6− anion. The switch proved important to increase the yield of 1a by reducing the formation of nitrobenzene as a byproduct. We were indeed delighted that a selectivity comparable with our best result could be achieved performing the arylation of toluene with 0.5 mol% Ru(bpy)3(SbF6)2 as a photocatalyst (entry 8, 79%).
This result suggested that the high yield observed in our preliminary experiment using both Au and Ru salts (entry 1, 81%) might originate from anion metathesis between gold and ruthenium complexes.
Performing the reaction in the dark delivered only traces of 1a, thus portraying the role of light irradiation (entry 9). The latter alone is similarly unable to allow significant conversion of the diazonium salt, 1a being retrieved in 10% yield without Ru (entry 10). This back-ground reactivity is a relatively common feature in photoredox reactions.6b,7a Formation of products is inhibited by TEMPO. The radical scavenger traps the initially generated aryl radical (see ESI†). Lower yields were observed performing our experiments in air (see Table S1 in ESI†). This effect is usually not observed in conceptually analogous photoredox reactions, which do not involve intermediates that could easily react with dioxygen.5–8 Reactions became faster using wet solvents. Despite our best efforts, reproducibility issues were invariably observed. This problem was addressed using dry solvents and adding a precise amount of water to the reaction mixture. Full conversion of the diazonium salt was observed in one hour upon addition of 200 μL of water, but the yield of 1a dropped to 63% because of more pronounced direct reduction of the onium reagent. We then decided to continue the study using anhydrous solvents and degassed mixtures.
It is worth noting that the regioselectivity of the process did not change significantly among all these reactions. The ratios of o/m/p arylation of toluene ranged between 3.6:1.3:1 and 4:1.4:1. These observations are consistent with a negligible impact of a catalytic amount of gold and/or a stoichiometric inorganic base on the sequence. Furthermore, the efficient formation of 1a suggested that the reaction with a photoredox catalyst did not require the presence of a stoichiometric base and confirmed the stability of Ru(bpy)32+ cations in acidic media.6b Slight differences in yields were thus most likely due to the effects displayed by anions present in solution.10 It is worth noting that their role is expectedly marginal in most photoredox cascades5–8 and HAS reactions3,4,8 as these processes usually occur in basic and neutral media.
We then examined the scope of this mild arylation by screening various unactivated arenes (Table 2). On selected examples we performed reactions with different photocatalysts, varying their counterions, and adding a hexafluoroantimonate gold salt as an additive to test whether they might play a role using different substrates.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar–H | Time (h) | Major isomer | Yield (%) | Regio selectivityb |
| a Reaction conditions: as in Table 1, entry 8. Reaction time determined by analyzing samples via1H NMR. Isolated yields based on the average of at least two runs. b Determined by NMR, spectra in the ESI. c With 0.5 mol% of Ru(bpy)3Cl2 hexahydrate as a photocatalyst. d With 10 mol% of JP Au(MeCN)SbF6. | |||||
| 1 |
|
23 |
|
80 | — |
| 2c | 8 | 79 | — | ||
| 3 |

|
7 |
|
69 | — |
| 4c | 3 | 61 | — | ||
| 5 |
|
4 |
|
84 | 3.3:1.2:1 (o![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :m:p) :m:p) |
| 6c | 4 | 83 | 4:1.3:1 |
||
| 7 |
|
24 |
|
91 | — |
| 8c | 13 | 73 | — | ||
| 9c,d | 4 | 86 | — | ||
| 10 |
|
48 |

|
32 | 4.5:1.6:1 |
| 11 |
|
96 |
|
37 | — |
| 12 |
|
24 |
|
86 | 5.8:1 (α:β) |
| 13 |
|
48 |
|
35 | — |
| 14 |
|
24 |
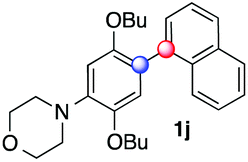
|
50 | 4.2:1 (α:β) |
| 15 |
|
36 |
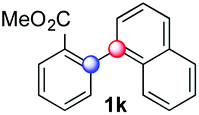
|
54 | 4.1:1 (α:β) |
| 16 |
|
12 |
|
71 | 1.8:1.3:1 (o:m:p) |
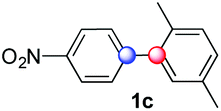
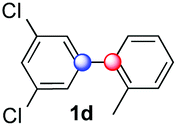
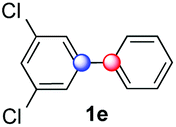
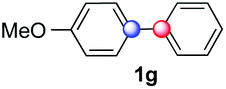
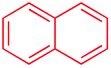
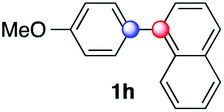
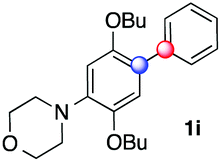
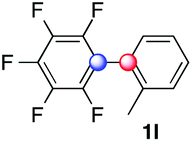
Beside toluene, nitro phenyldiazonium smoothly reacts with benzene and p-xylene to provide biaryls 1b and 1c (61–80% yields, entries 1–4). While yields remain comparable, reactions with Ru(bpy)3Cl2 hexahydrate as a catalyst were faster (entries 2 and 4). Chlorine substituents are well tolerated by the cascade, delivering the corresponding products 1d and 1e in excellent yields (73–91%, entries 5–9). The most selective reaction with benzene was the slowest one (entry 7, 91%, 24 hours). The use of Ru(bpy)3Cl2 hexahydrate as a catalyst allowed full conversion of the diazonium reagent in 13 hours and the yield of 1e dropped to 73% (entry 8). The addition of 10 mol% of a gold salt along with the non-coordinating SbF6− counterion provided full conversion of the substrate in 4 hours and delivered 86% of 1e (entry 9). Electron rich diazonium salts were less efficient and required longer reaction times (entries 10 and 11, 32–37%). Employing naphthalene as an acceptor the coupling occurred smoothly and delivered 1h in 86% yield with good regioselectivity (entry 12, 5.8:1 ratio). Diazonium salts bearing ortho substituents could be more prone to undergo direct reduction rather than the desired product, usually retrieved in moderate to good yields (see also Table S5 in ESI†). Among selected examples, an onium reagent with a bulky n-butoxy chain can be readily coupled delivering arylated products 1i and 1j in 35% and 50% respectively, entries 13 and 14. The sequence tolerated esters as witnessed by formation of 1k in 54% yield (entry 15). A perfluorodiazonium could be employed, which delivered the corresponding biaryl 1l in 71% yield, although with poor regiocontrol (1.8:1.3:1 mixture of isomers, entry 16).
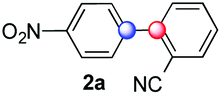
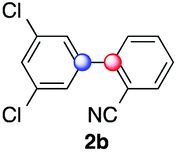
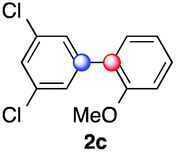
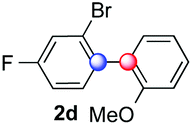
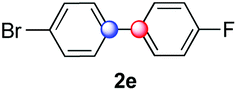
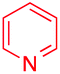
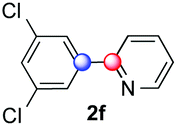
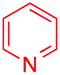
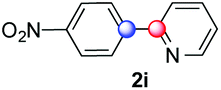
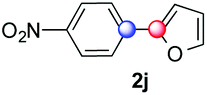
We next examined the scope of the reaction using various functionalized arenes and heterocycles as radical acceptors (Table 3). Benzonitrile can be arylated preferentially at the 2-position by an electron poor diazonium salt (2a, 67%, entry 1). Remarkably, yields could range from poor to excellent with subtle changes in the catalytic system (37%–87%, entries 2–4). The use of Ru(bpy)3(SbF6)2 as a catalyst provided the highest yield of 2b (87%, entry 2) and 24 hours were required to achieve full conversion of the diazonium reagent. Full conversion of the latter was attained in 7 hours with Ru(bpy)3Cl2 hexahydrate, but a significantly lower yield was observed in this case (37%, entry 3). The addition of 10 mol% of JP Au(MeCN)SbF6 provided full conversion in 7 hours and 82% of 2b (entry 4). Anisole too is an efficient coupling partner towards various polyhalogenated diazonium salts, delivering 2c and 2d in 92% and 72% yield respectively in one hour (entries 5–7). The method is suitable towards functionalization of fluorinated arenes, biaryl 2e being isolated in 71% yield (entry 8). We then tested pyridine as a reagent. The reaction with a chlorinated diazonium salt delivered biaryl 2f in 55% yield with good regioselectivity (7:3 ratio of isomers, entry 9). A lower selectivity was observed using Ru(bpy)3Cl2 hexahydrate (39% yield, entry 10). Surprisingly, the favored isomer of the product became the meta-arylated pyridine when the reaction was carried out with 2 equivalents of potassium carbonate to neutralize in situ generated HBF4 (73%, 1:8 ratio of isomers, entry 11). The yield was limited using a diazonium salt with a para-bromo substituent (2g, 21% and 18% yield respectively, entries 12 and 13). Selective ortho-arylation prevails with a donating methoxy group too, product 2h being retrieved in 63% yield (7:3 mixtures of isomers, entry 14). A 15.6:1 ratio was observed in the presence of a base (72%, entry 15). Complete regioselectivity towards ortho-arylation was observed with a nitro-substituted diazonium salt (entry 16, 41% yield). The preference for β functionalisation is usually not observed in homolytic aromatic substitutions (vide infra).7,11 Taken together these results suggest that the behavior of pyridine and a protonated pyridine could differ in these sequences. We could not manage yet to fully control and predict the reactivity of nitrogen heterocycles. Switching to electron rich heterocycles, our method selectively arylated furan at the 2-position, albeit in moderate yields (47% and 34% for entries 17 and 18 respectively).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar–H | Time (h) | Major isomer | Yield (%) | Regio selectivityb |
| a Reaction conditions: as in Table 1, entry, 8. Reaction time determined by analyzing samples via1H NMR. Isolated yields based on the average of at least two runs. b Determined by NMR, spectra in the ESI. c With 0.5 mol% of Ru(bpy)3Cl2 hexahydrate as a photocatalyst. d With 10 mol% of JP Au(MeCN)SbF6. e 72% yield on a 2.25 mmol scale (20 h). f With 2 equiv. of K2CO3 to neutralize in situ generated HBF4. | |||||
| 1 |
|
8 |
|
67 | 3.5:1:1.4 (o:m:p) |
| 2 |
|
24 |
|
87 | 5.5:1:2.3 |
| 3c | 7 | 37 | 5.8:1:2.1 |
||
| 4c,d | 7 | 82 | 5.7:1:2.2 |
||
| 5 |
|
1 |
|
91 | 4.1:1:1.2 |
| 6c | 1 | 92 | 4.6:1:1.2 |
||
| 7e |
|
1 |
|
72 | 3.3:1:1.5 |
| 8 |
|
12 |
|
71 | 1:3.2:6 |
| 9 |
|
1 |
|
55 | 7:3 (α:β) |
| 10c | 1 | 39 | α | ||
| 11f | 1 | 73 | 1:8 |
||
| 12 |
|
1 |
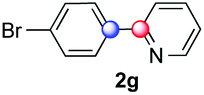
|
21 | 2:1 |
| 13c | 1 | 18 | α | ||
| 14 |
|
5 |
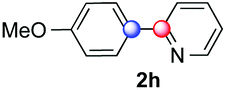
|
63 | 7:3 |
| 15f | 5 | 72 | 15.6:1 |
||
| 16 |
|
2 |
|
41 | — |
| 17 |
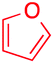
|
1 |
|
47 | — |
| 18 |
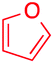
|
1 |
|
34 | — |
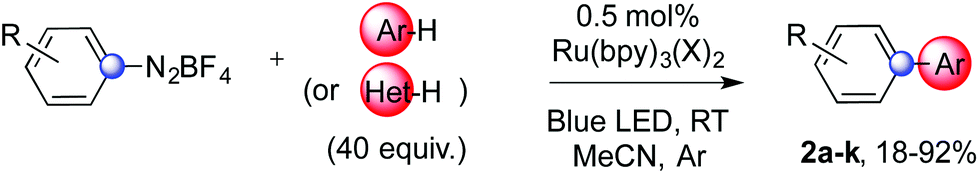
In all cases halogenated reagents successfully react leaving their C–X bonds untouched and suitable for further functionalisation.
A possible rationale for the mechanism of this cascade is presented in Scheme 2.5,6 Visible light allows the Ru(II) complex Ired to convert to its excited form I*. The latter can deliver an electron to diazonium salt 3. This SET provides the Ru(III) complex Iox together with radical 4 that can then liberate aryl radical 5 and N2. The former could then be added to an arene yielding cyclohexadienyl radical 6. At this stage, two competitive manifolds could be imagined. On the one hand, Iox can conclude the photocatalytic cycle by oxidizing radical 6, which provides biaryl 1 upon deprotonation. However, analogous mechanisms reported in the literature suggest that a radical chain reaction might compete (highlighted in pink in Scheme 2).7a In this scenario, its propagation step would feature the electron transfer between 6 and another molecule of 3, generating Wheland-type cation 7 together with 4 that could then add on to another arene molecule. Rearomatization of the former would eventually provide product 1. Whether a radical chain mechanism operates in these sequences, its termination step is likely ensured by an oxidizing Ru species rather than via free radical recombination. We did not observe indeed any traces of homocoupling products. It is worth noting that, even if the concentration in solution of both 6 and I* is most likely low, aryl radicals are weaker reducing agents than dRu(bpy)33+ and thus might not be sufficient to support chain propagation.12 This hypothesis is confirmed by calculating the ΔG of these two competitive redox reactions. Oxidation of radical 6 by I* is accompanied by a large negative ΔG (−26.4 kcal mol−1). The SET between 6 and cation 3 to generate a diazoyl radical has instead a positive ΔG of +0.6 kcal mol−1. The most energetically favored pathway appears and therefore the oxidation of radical 6 by ruthenium.
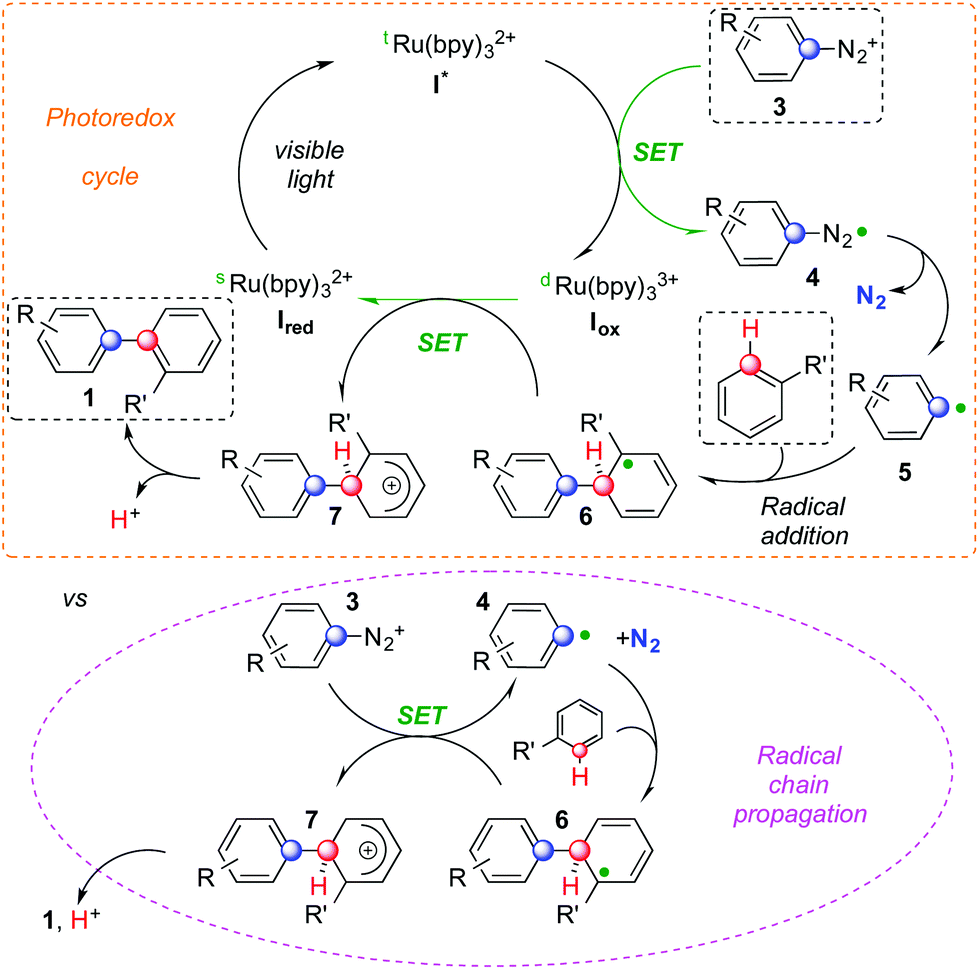 | ||
| Scheme 2 Plausible reaction mechanisms. | ||
Diazonium salts bearing electron withdrawing groups led to higher yields than their peers with electron donating substituents. This can correlate with the relatively higher reduction potential of the former compared to the latter. Furthermore, upon loss of N2, the resulting aryl radical is more electrophilic and could thus better add on electron rich arenes.3 Among these reagents, substituted ones such as toluene and naphthalene react faster and provide better yields than benzene. Besides heterocycles,7,9 the control of regiochemistry is limited and the distribution of isomers is not statistical. When multiple regioisomers could form, the favored one usually features substituent ortho to the aryl–aryl bond. Remarkably, electronic effects do not affect this preference for ortho-arylation.1 A comparable regioselectivity is indeed achieved replacing toluene or anisole with benzonitrile. This is consistent with the relative extra stabilization of the ortho-substituted cyclohexadienyl radical 6 compared to its isomers. The regioselectivity of the process should be indeed determined by the relative stability of different isomers of cyclohexadienyl radical 6. All these features are hallmarks of homolytic aromatic substitutions3,4 and our base-free photoredox cascade might be considered an example of HAS that mildly generates aryl radicals triggering the C–H arylation of simple arenes.
These reactions are often performed with stoichiometric bases but could remain very efficient processes in neutral and acidic media. In this case, it has been possible to observe differences by subtle tuning conditions. Remarkably, traces of different counterions and/or water molecules could change the time necessary to achieve full conversion of the onium reagent and the yield of the biaryl product (compare entries 7–9, Table 2 and 2–4, Table 3). Reactions with nitrogen heterocycles showed significant changes in regioselectivity depending on the acidity of the media. In contrast, no significant effect on regioselectivity is observed in reactions involving unactivated arenes. This could result from the formal deprotonation of Wheland-type cation 7 that delivers a molecule of product 1. This step is generally assisted by a sufficiently basic species. In acidic media, chloride anions and molecules of water or acetonitrile, which we used as solvents, could become the most basic species and might thus have an impact on time necessary to consume starting material and on the selectivity towards either the desired biaryls or (undesired) reduced onium reagents.
Conclusions
We developed a catalytic method for the synthesis of biaryls exploiting a suitable Ru photocatalyst and visible light. The study of these formal C–H arylations revealed several analogies with homolytic aromatic substitutions, a prototypical free-radical process. Simple arenes could be readily activated under very mild conditions and with broad functional group tolerance.Acknowledgements
We thank CNRS, UPMC and MIUR for funding.References
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; G. P. Chiusoli, M. Catellani, M. Costa, E. Motti, N. Della Ca’ and G. Maestri, Coord. Chem. Rev., 2010, 254, 456–469 CrossRef PubMed.
- Selected examples: (a) A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–157132 CrossRef CAS PubMed; (b) R. J. Phipps and M. J. Gaunt, Science, 2011, 323, 1593–1597 CrossRef PubMed. For reviews: (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- S. E. Vaillard and A. Studer, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. A. Studer and C. Chatgilialoglu, Wiley, Chichester, 2012, vol. 2, p. 1059 Search PubMed.
- For a recent tin-free and base-promoted HAS method see: (a) A. Dewanji, S. Murarka, D. P. Curran and A. Studer, Org. Lett., 2013, 15, 6102–6105 CrossRef CAS PubMed. For a meaningful discussion on factors governing HAS and base-promoted HAS reactions see ref. 3 and (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5021 CrossRef CAS PubMed.
- (a) J. J. Douglas, J. D. Nguyen, K. P. Cole and C. R. J. Stephenson, Aldrichimica Acta, 2014, 47, 15–24 CAS; (b) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (e) M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463–4466 CrossRef CAS PubMed.
- (a) H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517–5520 CrossRef CAS; (b) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed; (c) G. Maestri, M. Malacria and E. Derat, Chem. Commun., 2013, 49, 10424–10427 RSC; (d) A. Baralle, L. Fensterbank, J.-P. Goddard and C. Ollivier, Chem. – Eur. J., 2013, 19, 10809–10813 CrossRef CAS PubMed; (e) S. Donck, A. Baroudi, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2013, 355, 1477–1482 CrossRef CAS PubMed.
- (a) D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed; (b) D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J., 2014, 20, 2960–2965 CrossRef CAS PubMed; (c) Y.-X. Liu, D. Xue, J.-D. Wang, C.-J. Zhao, Q.-Z. Zou, C. Wang and J. Xiao, Synlett, 2013, 507–513 Search PubMed.
- Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664–2667 CrossRef CAS PubMed.
- For recent examples that inspired our initial strategy: (a) G. C. Lloyd-Jones and L. T. Ball, Science, 2014, 345, 381–382 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254–264 CrossRef CAS PubMed; (c) M. Font, F. Acuna-Pares, T. Parella, J. Serra, J. M. Luis, J. Lloret-Fillol, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 4373 CAS; (d) D.-H. Zhang, L.-Z. Dai and M. Shi, Eur. J. Org. Chem., 2010, 5454–5459 CrossRef CAS PubMed. For joint Au/Ru catalysis see: (e) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed; (f) X. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed.
- As correctly suggested by a reviewer, we could not rule out that these differences in yields could be the result of standard operator errors. To minimize this risk, experiments presented herein were always independently carried out by both FG and VN and provided comparable and reproducible results.
- (a) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed; (b) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed. For mechanistic studies see: (c) A. Gansauer, M. Seddiqzai, T. Dahmen, R. Sure and S. Grimme, Beilstein J. Org. Chem., 2013, 9, 1620–1629 CrossRef PubMed.
- For instance, the reduction potential of 4-nitrophenyldiazonium tetrafluoroborate is +0.2 V vs. SCE while that of the oxidizing photocatalyst is +1.29 V; see ref. 6 and P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthesis of photocatalysts, characterization of products, copies of NMR spectra. CCDC 1015490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00031a |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2015 |