
An ortho-directed C–H borylation/Suzuki coupling sequence in the formation of biphenylbenzylic amines†
Lillian V. A.
Hale
,
David G.
Emmerson
,
Emma F.
Ling
,
Andrew J.
Roering
,
Marissa A.
Ringgold
and
Timothy B.
Clark
*
Department of Chemistry and Biochemistry, University of San Diego, 5998 Alcala Park, San Diego, California 92110, USA. E-mail: clarkt@sandiego.edu
First published on 17th February 2015
Abstract
A C–H borylation/Suzuki–Miyaura coupling sequence is reported using benzylic amines to direct the C–H borylation to the ortho C–H bond. Both aryl bromides and iodides were coupled to the resulting arylboronate esters. A series of biphenylbenzylic amines are reported, requiring a single purification in the two-step reaction sequence.
Metal-catalysed C–H borylation reactions have received significant attention over the past 15 years,1–3 advancing early stoichiometric reactions of metal boryl complexes with a C–H bond of the solvent4–6 to catalytic methods that require 1–2 equivalents of the substrate. Early stoichiometric and catalytic reactions provided sterically-controlled selectivity, resulting in functionalization of primary over secondary C–H bonds for alkanes and selectivity for the meta or para position over the ortho position with arenes.2,7,8 Since 2008, several approaches to substrate-directed C–H borylation have resulted in a number of methods that provide ortho-selective C–H borylation of arenes.9,10 The extent of functional groups that can now be used to direct arene C–H borylation includes silanes,9,11 halides,12 esters,12–15 amides,12,14 carbamates,16,17 ethers,12,14 amines,18–20 imines,21–23 and phosphines.24 The ability to utilize nitrogen directing groups in C–H borylation is of particular interest to the synthetic community due to the prevalence of nitrogen in biologically relevant targets.
The synthetic utility of metal-catalysed C–H borylation reactions lies in the versatility of the C–B bond, which is readily converted into C–O, C–N, and C–C bonds.25–31 Many examples of C–H borylation reactions in target-directed syntheses have now appeared in the literature,3 demonstrating the power of converting an otherwise unreactive C–H bond into a variety of C–Y bonds. Although transformations involving C–B bonds are generally robust and functional group tolerant, the presence of an adjacent directing group can cause significant complications in the formation of new C–Y bonds. These limitations are particularly apparent with Lewis base directing groups that can significantly alter the reactivity of a metal-catalysed transformation utilizing a C–B bond. With this limitation in mind, the Suzuki–Miyaura coupling of arylboronates27,32–34 derived from the iridium-catalysed C–H borylation of benzylic amines is reported herein. This transformation utilizes our recently reported amine-directed C–H borylation of benzylic amines using readily available hemi-labile diamine ligands,19,35 ultimately providing biphenylbenzylic amines upon subjecting the resulting arylboronate esters to Suzuki–Miyaura coupling conditions.36–39 Biphenylbenzylic amines are prevalent in a number of biologically active compounds, including anacetrapib (cholesterol ester transfer protein inhibitor in phase III clinical trials)40 and allocolchicine41 (binds to tubulin and disrupts tubulin polymerization) (Fig. 1).
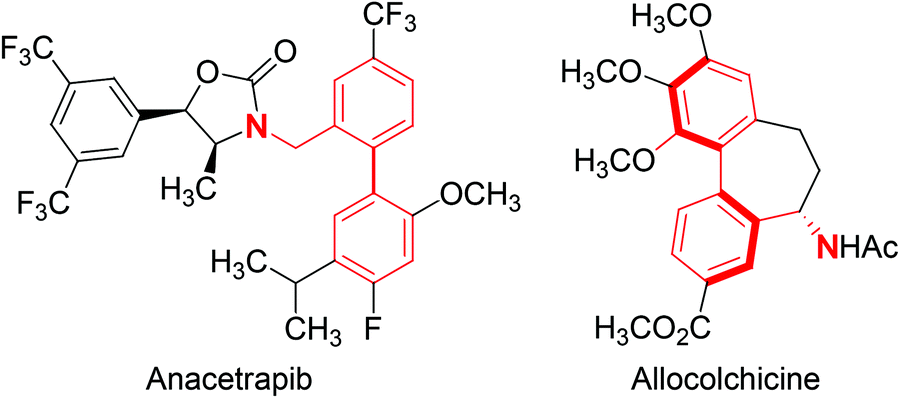 | ||
| Fig. 1 Structures of anacetrapib and allocolchicine with the biphenylbenzylic amine highlighted in red. | ||
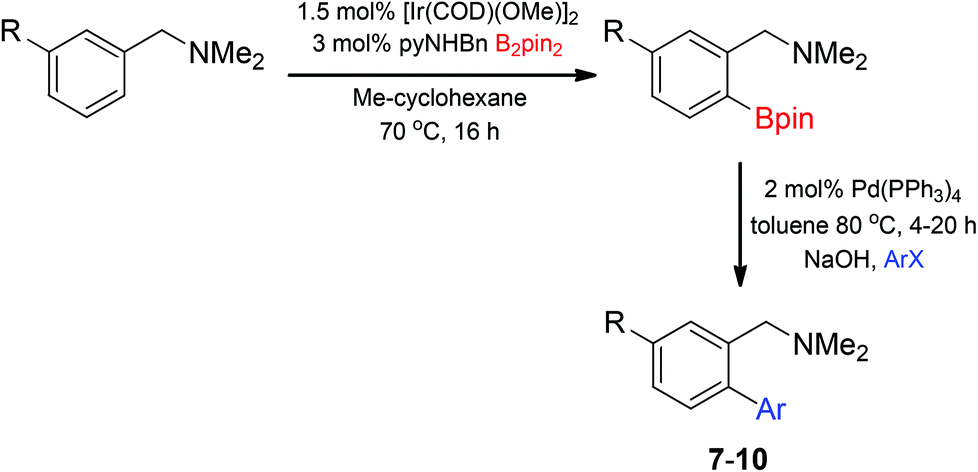
Access to the desired biphenylbenzylic amines was initiated by combining the amine-directed C–H borylation to Suzuki–Miyaura reaction conditions that are known for related boronic acids with an ortho-situated benzylic amine.42 2-Fluoro-N,N-dimethylbenzylamine was used initially since only one ortho C–H bond is available for functionalization. C–H borylation of 2-fluoro-N,N-dimethylbenzylamine was accomplished with 1.5 mol% [Ir(μ-OMe)(COD)]2, 3 mol% picolylamine (picNH2) and 0.83 equivalent of B2pin2 (a small excess of benzylic amine improves selectivity for the mono-ortho borylation product).19 Addition of iodobenzene to the unpurified C–H borylation product in the presence of a catalytic amount of Pd(PPh3)4 and aqueous sodium hydroxide provided the desired biphenylbenzylic amine 1 in 68% yield over the two-step reaction sequence (Scheme 1, Table 1).43 Encouraged by this result, additional benzylic amines were subjected to these reaction conditions and coupled to aryl halides. Additional 2-substituted benzylic amines were effective in the reaction sequence, providing 2 and 3 in good yield. Aryl bromides were shown to react under identical conditions to provide 2 and 4. Borylation and coupling at the sterically encumbered C–H bond of the mesitylene-derived benzylic amine provided 5 in good yield. A substituent in the benzylic position was also tolerated under the reaction conditions, providing 6 in high yield.
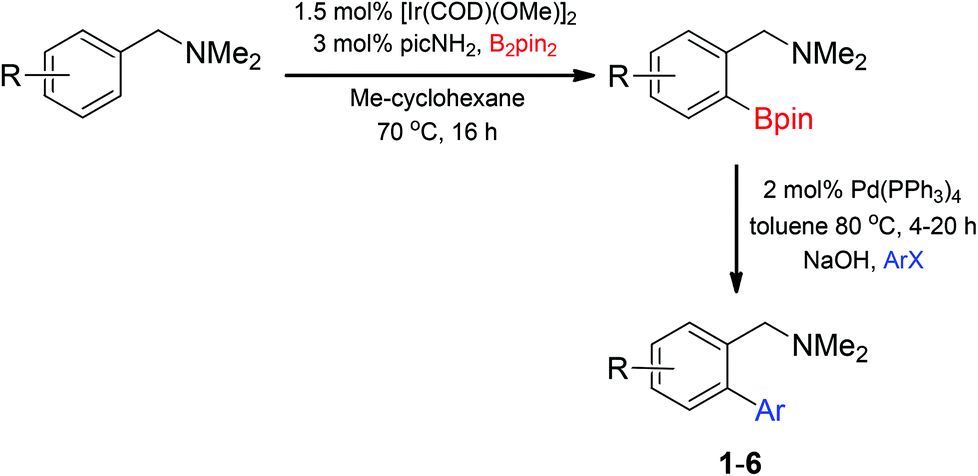 | ||
| Scheme 1 C–H borylation/Suzuki–Miyaura coupling sequence. | ||
| R | ArX | Product | Yield |
|---|---|---|---|
| a 0.83 equivalent of B2pin2 to benzylamine in C–H borylation; 1.1 equivalents of ArX to arene used in Suzuki coupling. Yield based on stoichiometry of the benzylamine substrate used in C–H borylation. | |||
| 2-F | C6H5I |
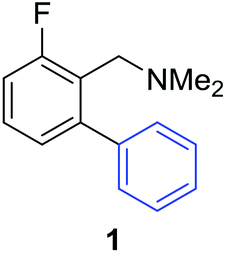
|
68% |
| 2-F | 4-MeC6H4Br |
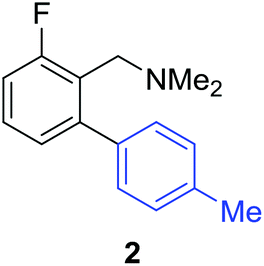
|
52% |
| 2-Me | 4-CF3C6H4I |
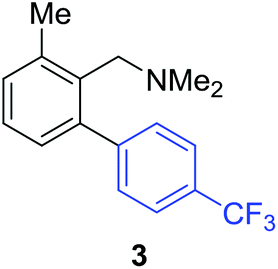
|
59% |
| 4-Me | 4-MeOC6H4Br |
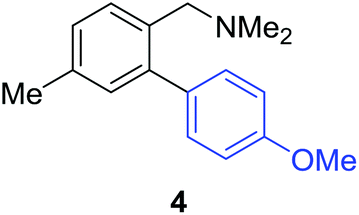
|
55% |
| 1′-Me | 3-CF3C6H4I |
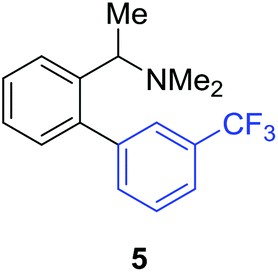
|
57% |
| 3,5-Me2 | C6H5I |
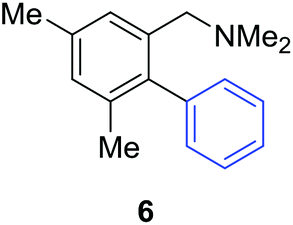
|
79% |
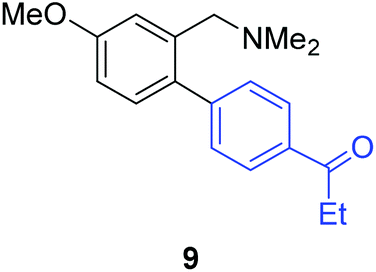
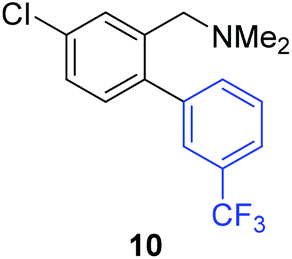
Unsymmetrical 3-substituted benzylic amines were investigated next to access highly-substituted biphenylbenzylic amines (Scheme 2). These benzylic amines are challenging substrates in the C–H borylation reaction due to the possibility of 3 different ortho borylation products (borylation at the 6-position-major, borylation at the 2-position, and bis-ortho borylation). We recently reported a study on the role of the ligand in C–H borylation and found that 2-(N-benzylamino)pyridine (pyNHBn) provides improved yields and selectivity over picolylamine for these substrates.35 The C–H borylation/Suzuki coupling sequence was examined using pyNHBn in place of picolylamine, providing high yields of the desired biphenylbenzylic amines (Scheme 2, Table 2). Electron-withdrawing aryl bromides provided significantly higher yield (7) than the corresponding electron-rich aryl bromide (8). The 3-methoxy-substituted benzylic amine was shown to undergo both transformations, providing 9 in high yield. Notably, the presence of the 3-chloro substituent was tolerated during both metal-catalysed reactions in spite of the propensity of C–Cl bonds to undergo oxidative addition, providing 10 in 62% yield. The presence of this reactive C–Cl bond in 10 provides an additional synthetic handle for further transformations in target-directed synthesis.
 | ||
| Scheme 2 C–H borylation/Suzuki coupling with 3-substituted benzylic amines. | ||
| R | ArX | Product | Yield |
|---|---|---|---|
| a 0.83 equivalent of B2pin2 to benzylamine in C–H borylation; 1.1 equivalents of ArX to arene used in Suzuki coupling. Yield based on stoichiometry of the benzylamine substrate used in C–H borylation. | |||
| 3-Me | 3-MeCOC6H4Br |

|
84% |
| 3-Me | 4-MeOC6H4Br |

|
40% |
| 3-OMe | 4-EtCOC6H4Br |
|
79% |
| 3-Cl | 3-CF3C6H4I |
|
62% |
Conclusions
Efficient synthesis of biphenylbenzylic amines, a common core structure in biologically active compounds, has been reported. The iridium-catalysed C–H borylation provides reactive arylboronate esters that were subjected to Suzuki–Miyaura coupling conditions without purification of the arylboronate ester intermediates. A wide range of functional groups are tolerated under the reaction conditions, providing highly-functionalized biphenylbenzylic amines.Experimental section
General methods
All procedures involving air- or moisture-sensitive reagents were performed in oven-dried glassware under purified nitrogen, either in an inert atmosphere glovebox or by standard Schlenk techniques. All solvents were dried and degassed by standard procedures unless employed for reaction work-up or purification. TLC analysis was performed on 60 Å silica layer fluorescence UV plates. Flash column chromatography was carried out on hand-packed columns of 70–230 mesh silica gel, 40–63 microns. NMR spectra were collected on a Unity Inova spectrometer at 500 MHz for 1H NMR, 125 MHz for 13C NMR or on a Mercury spectrometer at 100 MHz for 13C NMR. 1H NMR spectra are referenced to CDCl3 at 7.26 ppm, or to an internal tetramethylsilane (TMS) standard at 0.00 ppm. The 1H NMR spectral data are reported as follows: chemical shift ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, hex = hextet, sep = septet, oct = octet, m = multiplet), coupling constants (Hz), and integration. 13C NMR spectra are referenced to CDCl3 at 77.0 ppm. Attenuated total reflection IR (ATR-IR) spectra were obtained with a Jasco FT-IR-480 Plus and absorptions were reported in cm−1.Chloroform-d, bis(pinacolato)diboron, [Ir(μ-OMe)(COD)]2, 2-(aminomethyl)pyridine, 2-(N-benzylamino)pyridine, Pd(PPh3)4, and all required benzylbromides and aryl halides were purchased and used without purification. All benzylamines were known and synthesized by the same procedure.19
General procedure for the C–H borylation35/Suzuki–Miyaura coupling of benzylic amines42
To a 50 mL PTFE-valved reaction tube, equipped with a stir bar and charged with [Ir(μ-OMe)(COD)]2 (0.020 g, 0.030 mmol), was added 2-(aminomethyl)pyridine (0.0062 mL, 0.060 mmol) or 2-(N-benzylamino)pyridine (0.011 g, 0.060 mmol), B2pin2 (0.423 g, 1.67 mmol), benzylamine (2.00 mmol), and 4.0 mL of methylcyclohexane. After 16 h at 70 °C, the volatiles were removed in vacuo.To a round bottom flask charged with Pd(PPh3)4 (0.046 g, 0.040 mmol) was added aryl halide (2.20 mmol) and toluene (10 mL). After 20 minutes at 22 °C the crude ortho-borylated N,N-dimethylbenzylamine (2.00 mmol) and aqueous sodium hydroxide (5.0 M, 2.00 mL, 10 mmol) were added and the round bottom flask was fitted with a reflux condenser and flushed with N2 gas. After 4–20 h at 100 °C, the reaction mixture was diluted with diethyl ether (50 mL), filtered through a pad of Celite with additional diethyl ether (200 mL) and concentrated in vacuo. Purification by silica gel flash column chromatography provided the biphenylbenzylic amine.
Acknowledgements
Financial support from the National Science Foundation (CHE-1151092), the University of San Diego, Research Corporation for Science Advancement, and the National Science Foundation for NMR (0417731) facilities at USD. Dr John Greaves (University of California, Irvine) for mass spectrometry.Notes and references
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed.
- K. M. Waltz, X. He, C. Muhoro and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11357–11358 CrossRef CAS.
- C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1999, 121, 7696–7697 CrossRef CAS.
- S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS.
- C. S. Wei, C. A. Jiménez-Hoyos, M. F. Videa, J. F. Hartwig and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 3078–3091 CrossRef CAS PubMed.
- H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505–3515 RSC.
- T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed.
- A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068–4069 CrossRef CAS PubMed.
- S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058–5059 CrossRef CAS PubMed.
- T. Ishiyama, H. Isou, T. Kikuchi and N. Miyaura, Chem. Commun., 2010, 46, 159–161 RSC.
- B. Ghaffari, S. M. Preshlock, D. L. Plattner, R. J. Staples, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr. and M. R. Smith III, J. Am. Chem. Soc., 2014, 136, 14345–14348 CrossRef CAS PubMed.
- S. Kawamorita, H. Ohmiya and M. Sawamura, J. Org. Chem., 2010, 75, 3855–3858 CrossRef CAS PubMed.
- P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka Jr. and M. R. Smith III, J. Am. Chem. Soc., 2012, 134, 11350–11353 CrossRef CAS PubMed.
- K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978–3981 CrossRef CAS PubMed.
- S. Kawamorita, T. Miyazaki, H. Ohmiya, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2011, 133, 19310–19313 CrossRef CAS PubMed.
- A. J. Roering, L. V. A. Hale, P. A. Squier, M. A. Ringgold, E. R. Wiederspan and T. B. Clark, Org. Lett., 2012, 14, 3558–3561 CrossRef CAS PubMed.
- S. Konishi, S. Kawamorita, T. Iwai, P. G. Steel, T. B. Marder and M. Sawamura, Chem. – Asian J., 2014, 9, 434–438 CrossRef CAS.
- A. Ros, B. Estepa, R. López-Rodríguez, E. Álvarez, R. Fernández and J. M. Lassaletta, Angew. Chem., Int. Ed., 2011, 50, 11724–11728 CrossRef CAS PubMed.
- A. Ros, R. López-Rodríguez, B. Estepa, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 4573–4576 CrossRef CAS PubMed.
- I. Sasaki, T. Amou, H. Ito and T. Ishiyama, Org. Biomol. Chem., 2014, 12, 2041–2044 CAS.
- K. M. Crawford, T. R. Ramseyer, C. J. A. Daley and T. B. Clark, Angew. Chem., Int. Ed., 2014, 53, 7589–7593 CrossRef CAS PubMed.
- H. C. Brown, G. W. Kramer, A. B. Levy and M. M. Midland, Organic Syntheses via Boranes, Wiley-Interscience, New York, 1975 Search PubMed.
- H. C. Brown and M. Zaidlewicz, Organic Syntheses via Boranes-Recent Developments, Aldrich Chemical Company, Milwaukee, 2001 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS.
- Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, vol. 1, 2011 Search PubMed.
- Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, vol. 2, 2011 Search PubMed.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- L. V. A. Hale, K. A. McGarry, M. A. Ringgold and T. B. Clark, Organometallics, 2015, 34, 51–55 CrossRef CAS.
- G. A. Chotana, V. A. Kallepalli, R. E. Maleczka Jr. and M. R. Smith III, Tetrahedron, 2008, 64, 6103–6114 CrossRef CAS PubMed.
- D. W. Robbins and J. F. Hartwig, Org. Lett., 2012, 14, 4266–4269 CrossRef CAS PubMed.
- I. Ahmed and D. A. Clark, Org. Lett., 2014, 16, 4332–4335 CrossRef CAS PubMed.
- P. Harrisson, J. Morris, P. G. Steel and T. B. Marder, Synlett, 2009, 147–150 CAS.
- A. M. J. Gotto, U. Kher, M. S. Chatterjee, Y. Liu, X. S. Li, S. Vaidya, C. P. Cannon, E. A. Brinton, J. E. Moon, S. Shah, H. M. Dansky, Y. Mitchel and P. Barter, J. Cardiovasc. Pharmacol. Ther., 2014, 19, 543–549 CrossRef CAS PubMed.
- B. Pérez-Ramírez, M. J. Gorbunoff and S. N. Timasheff, Biochemistry, 1998, 37, 1646–1661 CrossRef PubMed.
- P.-L. Boudreault, S. Cardinal and N. Voyer, Synlett, 2010, 2449–2452 CAS.
- Attempts to conduct the Suzuki–Miyaura coupling reaction without switching solvents resulted in decreased reaction yields. For examples of a C–H borylation/Suzuki–Miyaura coupling sequence without changing the solvent, see ref. 38 and 39.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and 1H and 13C NMR spectra for compounds 1–10. See DOI: 10.1039/c4qo00348a |
| This journal is © the Partner Organisations 2015 |