
Remarkable electron-withdrawing effect of the Ph2P(O)-ethynyl group: Ph2P(O)-ethynyl-substituted aryl halides and copper acetylides for tailor-made Sonogashira couplings†‡
Lifen
Peng
a,
Feng
Xu
a,
Kenta
Shinohara
a,
Takanori
Nishida
a,
Kan
Wakamatsu
b,
Akihiro
Orita
*a and
Junzo
Otera
a
aDepartment of Applied Chemistry, Okayama University of Science, 1-1 Ridai-cho, Kita-ku, Okayama 700-0005, Japan. E-mail: orita@dac.ous.ac.jp; Fax: +81 86 256 4292
bDepartment of Chemistry, Okayama University of Science, 1-1 Ridai-cho, Kita-ku, Okayama 700-0005, Japan
First published on 29th January 2015
Abstract
The electronic effect of the Ph2P(O)-ethynyl group in Sonogashira couplings was investigated. Ph2P(O)-ethynylphenyl iodide showed higher reactivity than diiodobenzene because of the electron-withdrawing effect of the Ph2P(O) group. Copper acetylide, which was prepared in situ from Ph2P(O)-ethyne, exhibited poor nucleophilicity and reacted with aryl iodides in Sonogashira couplings, but did not react with aryl bromides. This high selectivity enabled practical syntheses of a cyclic phenyleneethynylene and unsymmetrically substituted 9,10-bis(ethynyl)anthracenes.
Phenyleneethynylenes can be used as organic materials for electroluminescence (EL),1 for organic transistors2 and for dye-sensitized solar cells.3 For tuning the physical properties of phenyleneethynylenes that have rigid structures and highly expanded π-systems, it is necessary to change the length of their π-conjugated systems. To this end, Sonogashira coupling is one of the powerful technologies used to synthesize arylalkynes using the corresponding aryl halide and terminal alkyne as starting compounds.4 Although the trimethylsilyl (TMS) group is a routinely-used protecting group for terminal ethynes, we developed the phosphoryl group (Ph2P(O)) as a new protecting group.5 The high polarity of the phosphoryl group enabled easy separation of the desired products from the less polar hydrocarbon byproducts.5c More recently we succeeded in a one-pot synthesis of arylethynes by invoking dephosphorylation followed by transition metal-catalyzed coupling reactions such as the Sonogashira and Migita–Kosugi–Stille couplings.5a,b Although the usability of the polar phosphoryl protecting group was exhibited, the electronic effect of the phosphorylethynyl group and the resulting reactivity and selectivity in the coupling reactions have not been explored.
We are intrigued by these issues and investigated the reactivities of Ph2P(O)-substituted alkynes and aryl halides in Sonogashira coupling. We report herein that the strong electron-withdrawing effect of the Ph2P(O) group enables tuning of the reactivities of Ph2P(O)-substituted substrates and reagents.
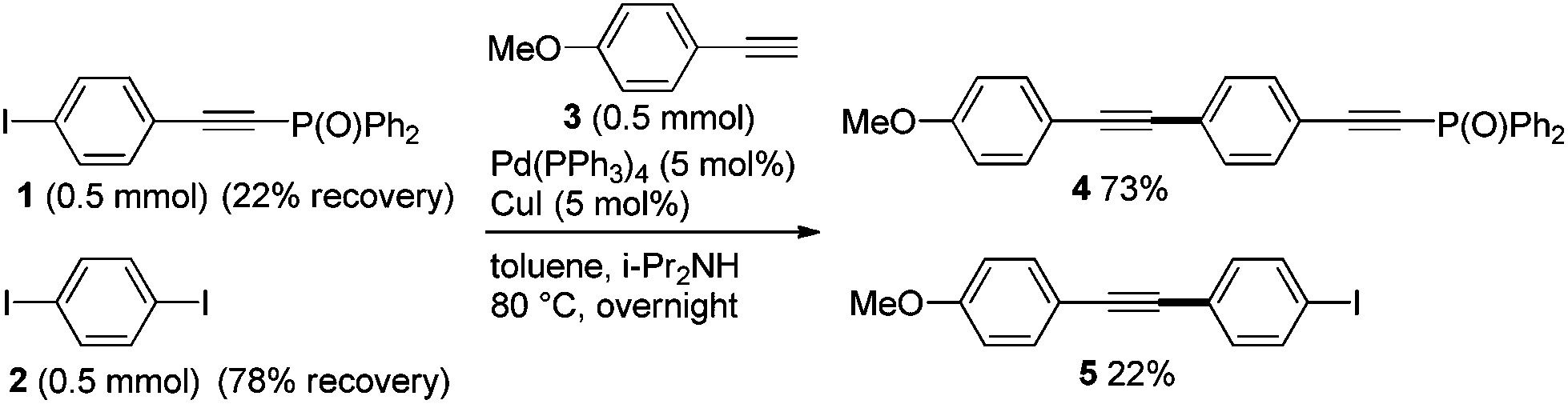
Firstly, we carried out the competitive Sonogashira coupling of phenyl iodides 1vs. 2 with terminal ethyne 3 (Scheme 1). When a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and 2 was heated with 3 at 80 °C in the presence of Pd and Cu catalysts, the desired couplings occurred to give 4 (73% yield) and 5 (22% yield). The ratio of 4 to 5 (77:23) reflects the comparable reactivity of 1 and 2 in the coupling reaction, and this result indicates that the phosphorylethynyl-substituted iodide 1 reacts more rapidly with 3.
:1 mixture of 1 and 2 was heated with 3 at 80 °C in the presence of Pd and Cu catalysts, the desired couplings occurred to give 4 (73% yield) and 5 (22% yield). The ratio of 4 to 5 (77:23) reflects the comparable reactivity of 1 and 2 in the coupling reaction, and this result indicates that the phosphorylethynyl-substituted iodide 1 reacts more rapidly with 3.
 | ||
| Scheme 1 Competitive Sonogashira coupling of 1vs. 2 with 3. | ||
Another competitive reaction of bromides 6vs. 7 provided a similar result, and the corresponding coupling products 4 and 8 were obtained in 76% and 10% yields, respectively (Scheme 2). The high reactivities of phosphorylethynyl-substituted halides 1 and 6 observed in these competitive reactions could be explained in terms of an acceleration of the couplings by the electron-withdrawing effect of the phosphorylethynyl group.6
 | ||
| Scheme 2 Competitive Sonogashira coupling of 6vs. 7 with 3. | ||
In order to gain further insights into the enhanced reactivities of Ph2P(O)-substituted ethynylphenyl halides 1 and 6, DFT calculations were performed on 1, 2, 6 and 7 at the B3LYP/6-31G(d) level (I: LANL2DZ). In Fig. 1 the LUMO energy levels of 1 and 2 are shown. The LUMO of 1 is located at a potential level lower than that of 2 by 0.38 eV, and this result supports that 1 could undergo oxidative addition of the nucleophilic Pd(0) species more rapidly than 2.7 DFT calculation demonstrated that the LUMO of 1 is expanded over the iodobenzene moiety as well as the Ph2P(O)-ethynyl groups, and thus the electron-withdrawing effect of the phosphoryl group serves effectively in leading to the lower potential energy of the LUMO in 1 (Fig. 1 inset).8
 | ||
| Fig. 1 LUMO energy diagrams of 1 and 2, and a diagram for the LUMO of 1 (inset). | ||
In the competitive coupling of para- and meta-bromophosphorylethynylbenzenes 6vs. 9 with 3, the para-adduct 4 was furnished in 56% yield as the major product while 10 was obtained in 20% yield (Scheme 3). In agreement with this, DFT calculations on 6 and 9 presented that the electron-withdrawing effect of the phosphorylethynyl group served more efficiently in 6 than in 9 to provide the lower LUMO energy level of 6 (Fig. 2).9
 | ||
| Scheme 3 Competitive Sonogashira coupling of 6vs. 9 with 3. | ||
 | ||
| Fig. 2 LUMO energy diagrams of 6 and 9. | ||
Secondly, the unique selectivity of iodide vs. bromide in Sonogashira coupling was investigated. When a mixture of phenyl iodide 11 and bromide 12 was subjected to Sonogashira coupling with 13, the iodide 11 selectively underwent the desired coupling (14: 70%, 15: not detected), and 12 was recovered quantitatively (Scheme 4).
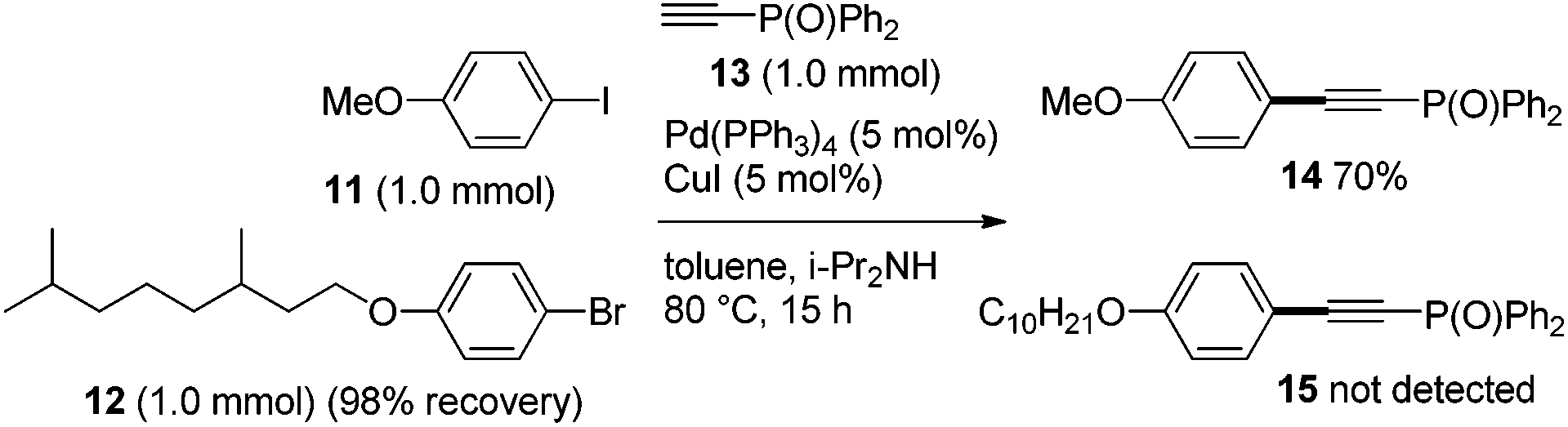 | ||
| Scheme 4 Competitive Sonogashira coupling of 11vs. 12 with 13. | ||
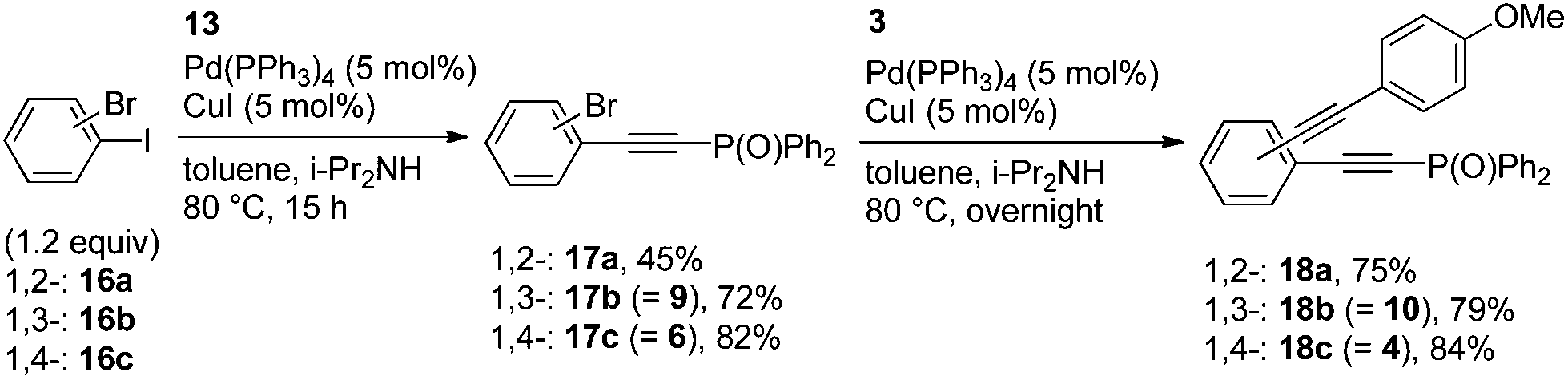
In the intramolecular Br vs. I-competitive couplings of 16a–c, only the phenyl iodide moieties reacted with 13 to afford the corresponding phosphorylethynyl-substituted bromobenzenes 17a–c in moderate to good yields (Scheme 5). The bromides 17a–c thus obtained reacted with 3 in the consecutive coupling to provide 18a–c, respectively.
 | ||
| Scheme 5 Preferential Sonogashira coupling of the iodide moiety in 16 with 13. | ||
When two equivalents of 13 were used in the Sonogashira coupling of 16c, 17c was obtained in 96% yield while bis(Ph2P(O)-ethynyl) adduct 19 was formed in only 2% yield (Scheme 6). These results demonstrate that phenyl iodide undergoes Sonogashira coupling with phosphorylethyne 13, but that the bromide does not.
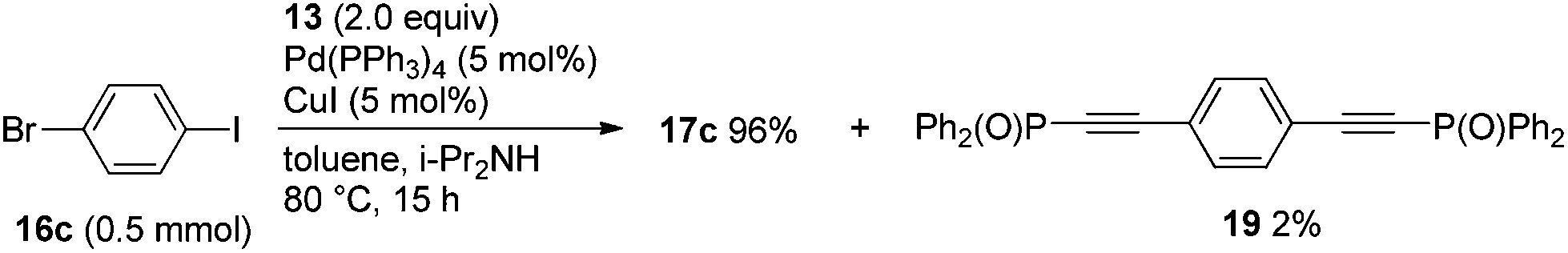 | ||
| Scheme 6 Sonogashira coupling of 16c with an excess of 13. | ||
When phosphorylethyne 13 and 4-phosphorylethynylphenylethyne 20 were subjected to a competitive coupling with iodide 11, the diyne 20 reacted more rapidly, and 4 and 14 were provided in 69% and 18% yields, respectively (Scheme 7).
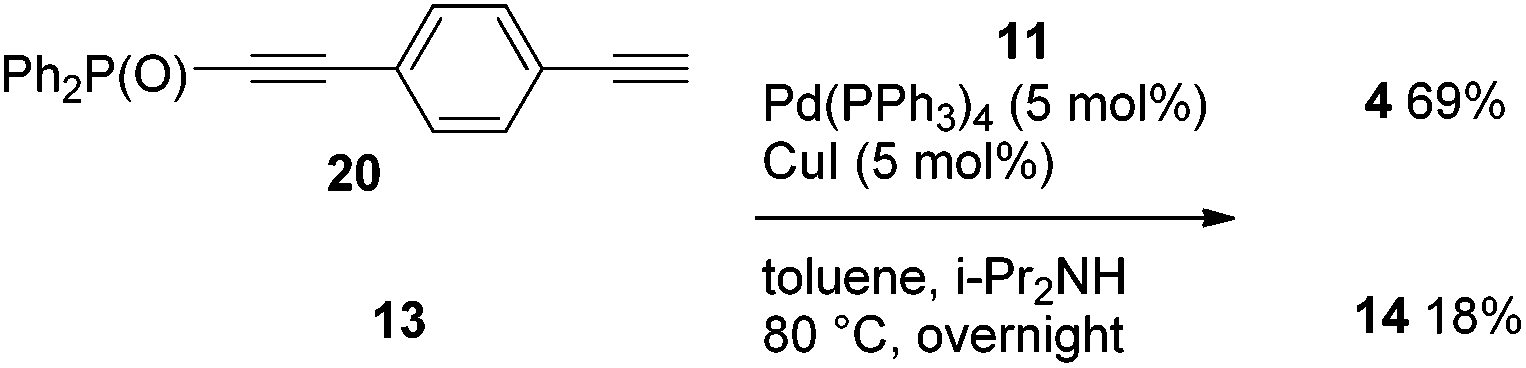 | ||
| Scheme 7 Competitive Sonogashira coupling of 20vs. 13 with 11. | ||
In order to rationalize the low reactivity of 13 in Sonogashira coupling, the HOMO energy levels of the copper acetylides (3-Cu, 20-Cu and 13-Cu), which might be produced in situ in the coupling, were calculated at the B3LYP/6-31G(d) (Cu: LANL2DZ) level (Fig. 3). The low HOMO energy level of 13-Cu indicated its low nucleophilicity which enabled transmetallation only with the reactive iodopalladium species which was produced in situ from the oxidative addition of Pd(0) to the aryl iodide.
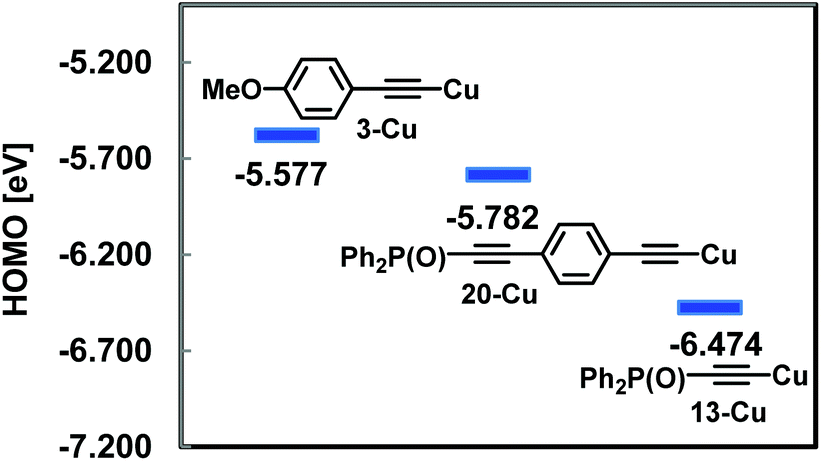 | ||
| Fig. 3 HOMO energy diagrams of 3-Cu, 20-Cu and 13-Cu. | ||
Although the high energy level of copper acetylide promoted Sonogashira coupling as shown in Fig. 3, a similar electronic effect was observed in a click reaction. For instance, 21 reacted rapidly with benzyl azide 22 to give triazole 23 (79%, 2 h). In contrast to this, 13 produced 24 sluggishly (73% (6 h), 42% (2 h)) (Scheme 8). The difference in reaction rates of these click reactions was also rationalized in terms of the HOMO energy levels of the corresponding Cu acetylides which could be obtained from 21 and 13, respectively; HOMO energy levels: −5.909 eV for 21-Cu; −6.474 eV for 13-Cu.10
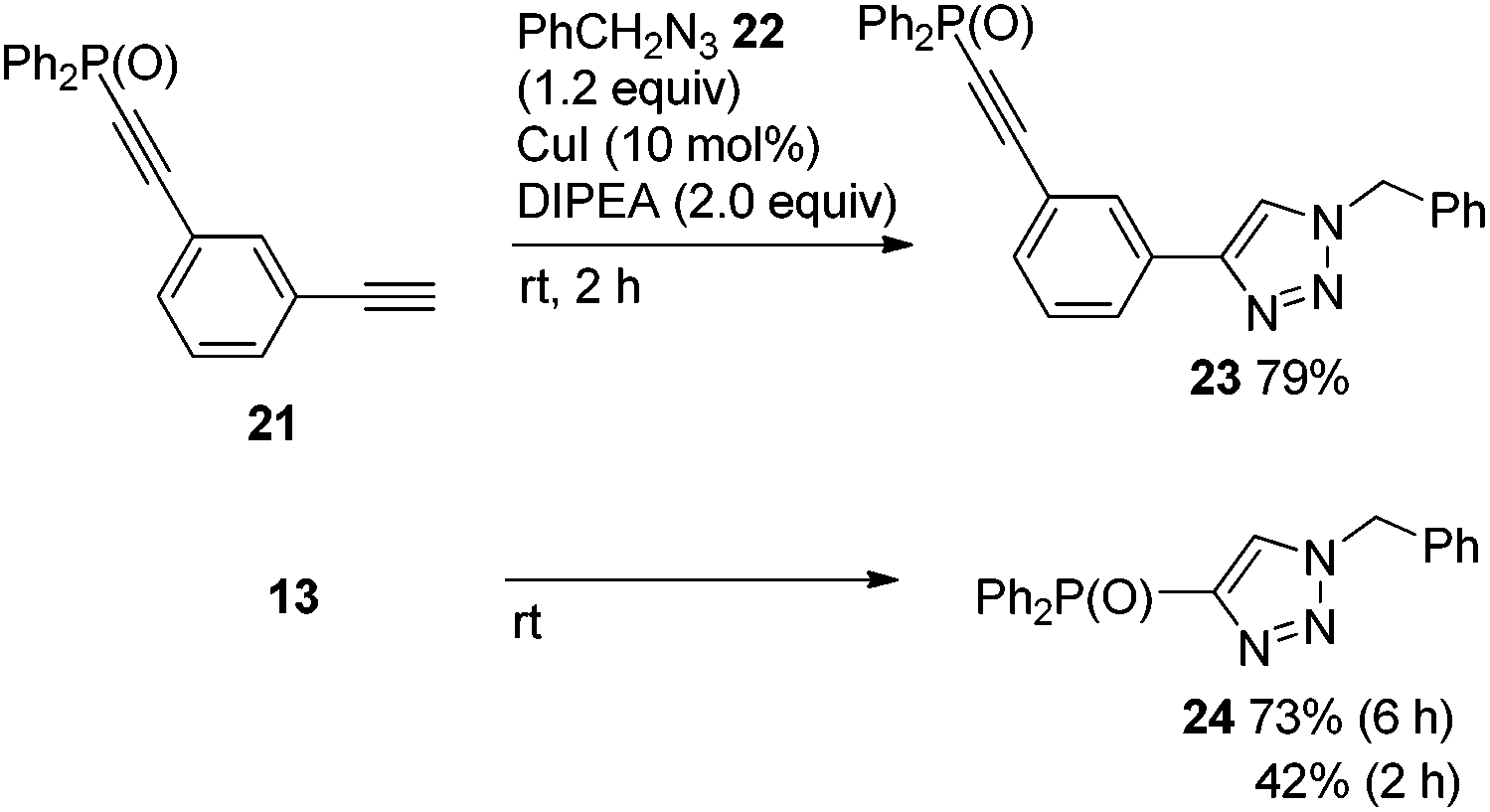 | ||
| Scheme 8 Competitive click reaction of 21vs. 13 with 22. | ||
Finally, we performed syntheses of cyclic phenyleneethynylene 25 and unsymmetrically substituted 9,10-bis(ethynyl)anthracenes 26 and 27 by making use of the high iodo-preference of phosphorylethyne 13 in Sonogashira coupling (Schemes 9 and 10). In the synthesis of 25, a key starting compound 17a, phosphorylethynyl bromide, was obtained easily from the Sonogashira coupling of 16a with 13 as shown in Scheme 5. The bromide 17a was transformed into 28 by a one-pot dephosphorylation/Sonogashira coupling with 29 in 62% yield. Subjection of 28 to the same one-pot deprotection–coupling provided 30. When 30 was treated with t-BuOK and then Pd/Cu catalysts, the desired intramolecular cyclization took place to afford 25 in 68% yield.
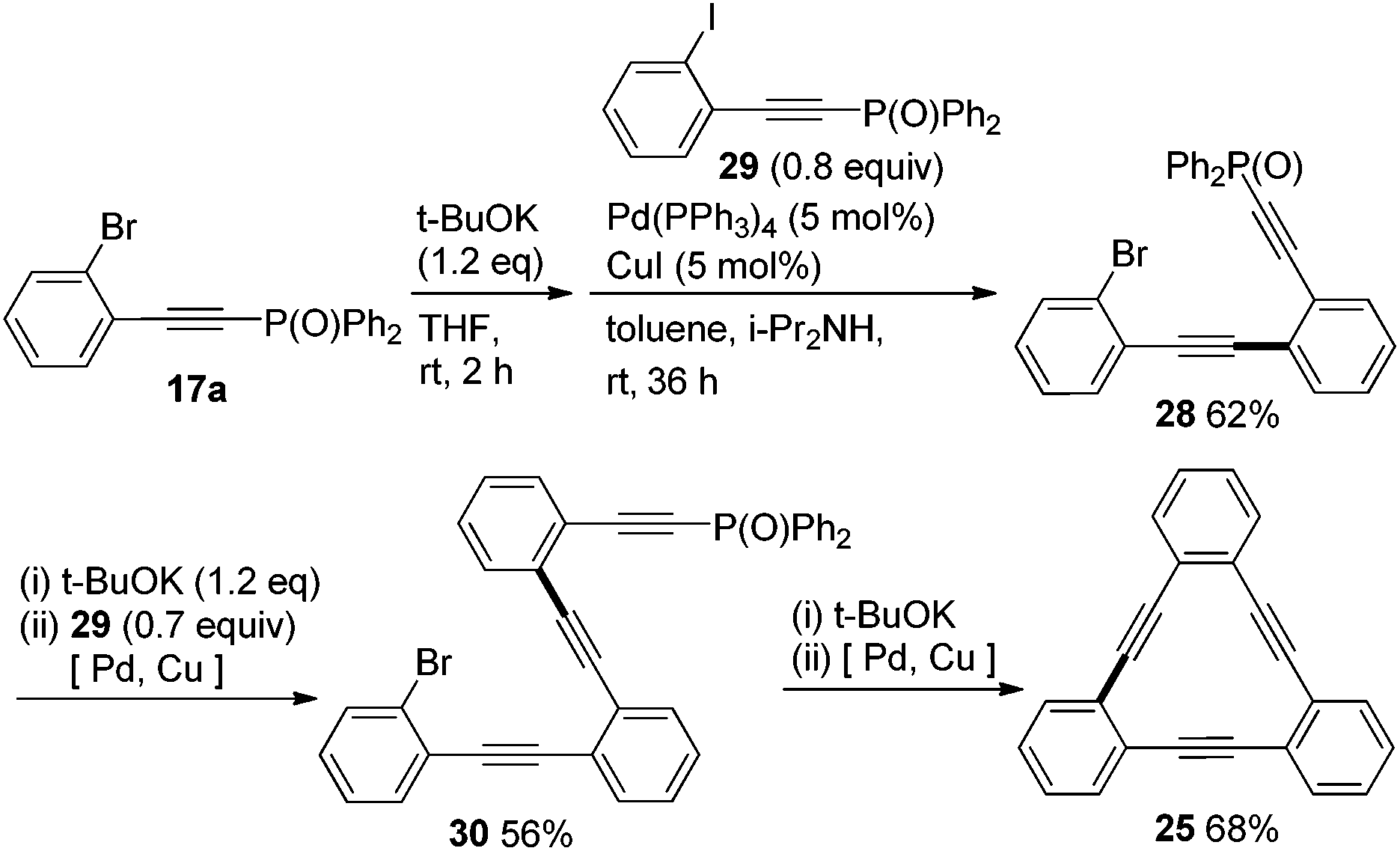 | ||
| Scheme 9 Synthesis of cyclic phenyleneethynylene 25. | ||
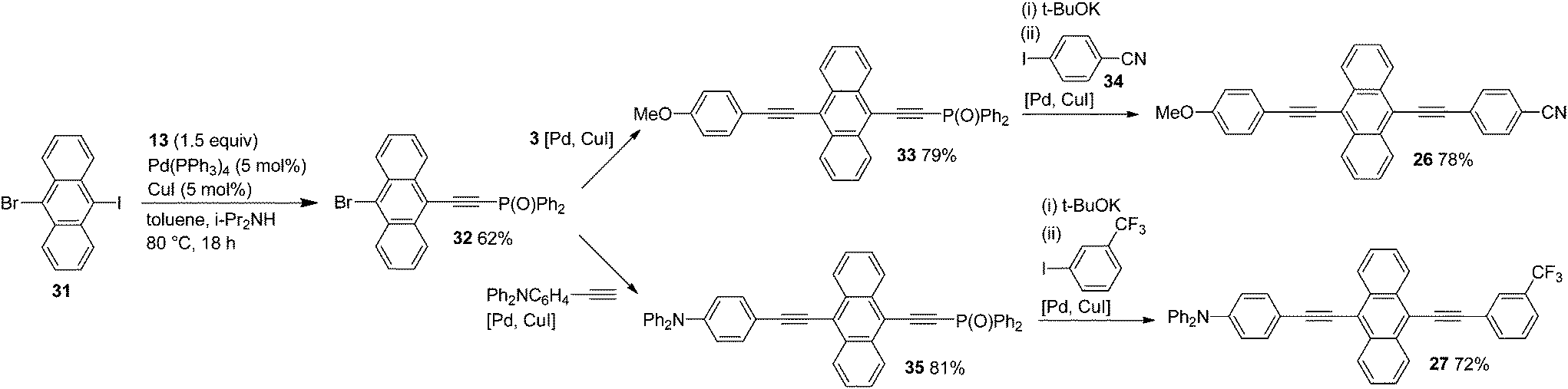 | ||
| Scheme 10 Synthesis of unsymmetrically substituted anthracenes. | ||
Sonogashira coupling of 31 with phosphorylethyne 13 gave 32 exclusively in 62% yield, and 32 was transformed into 33 in 79% yield by coupling with 4-methoxyphenylethyne (3) (Scheme 10). In these couplings, the high polarity of the Ph2P(O) group enabled easy isolation of the desired coupling products.5
Successive subjection of 33 to t-BuOK-promoted dephosphorylation and coupling with 34 provided 26 in 78% yield. Similarly, Ph2P(O)-ethynylanthranyl bromide 32 was converted to 27 through the phosphorylethynylanthracene intermediate 35.
Conclusions
Phosphorylethynyl-substituted aryl iodides and bromides revealed high reactivity in Sonogashira coupling, and the high reactivity was explained by the electron-withdrawing nature of the phosphorylethynyl group leading to the lower LUMO potential energy of the corresponding aryl halides. When phosphorylethyne was used in Sonogashira coupling with aryl halides, a unique iodide preference was observed, and only aryl iodides reacted with phosphorylethyne, with aryl bromides remaining intact even though an excess of phosphorylethyne was used. This iodide preference was explained in terms of the sluggish transmetallation of the copper acetylides, which were prepared in situ from the phosphorylethynes during the Sonogashira coupling, because of their low HOMO energy levels. By invoking this iodo-preference of phosphorylethyne, syntheses of cyclic phenyleneethynylene and unsymmetrically substituted 9,10-bis(ethynyl)anthracenes were achieved. Further application of the electron-withdrawing effect of the phosphorylethynyl group is under investigation.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Organic Synthesis based on Reaction Integration. Development of New Methods and Creation of New Substances” (no. 2105), matching fund subsidy for private universities from the Ministry of Education, Culture, Sports, Science and Technology, Japan, the Japan Society for the Promotion of Science (JSPS) through its “Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program)” and Okayama Prefecture Industrial Promotion Foundation.Notes and references
- (a) T.-L. Wu, H.-H. Chou, P.-Y. Huang, C.-H. Cheng and R.-S. Liu, J. Org. Chem., 2014, 79, 267 CrossRef CAS PubMed; (b) M. Porz, F. Paulus, S. Höfle, T. Lutz, U. Lemmer, A. Colsmann and U. H. F. Bunz, Macromol. Rapid Commun., 2013, 34, 1611 CrossRef CAS PubMed; (c) K. R. J. Thomas, N. Kapoor, M. N. K. P. Bolisetty, J.-H. Jou, Y.-L. Chen and Y.-C. Jou, J. Org. Chem., 2012, 77, 3921 CrossRef CAS PubMed; (d) Z. Zhao, S. Ye, Y. Guo, Z. Chang, L. Lin, T. Jiang, J. W. Y. Lam, P. Lu, H. Qiu, Y. Liu and B. Z. Tang, Org. Electron., 2011, 12, 2236 CrossRef CAS PubMed; (e) H. Aihara, T. Tanaka, M. Satou and T. Yamakawa, Trans. Mater. Res. Soc. Jpn., 2010, 35, 675 CrossRef CAS.
- (a) J. Kim, A.-R. Han, J. Hong, G. Kim, J. Lee, T. J. Shin, J. H. Oh and C. Yang, Chem. Mater., 2014, 26, 4933 CrossRef CAS; (b) H.-J. Yun, H. H. Choi, S.-K. Kwon, Y.-H. Kim and K. Cho, Chem. Mater., 2014, 26, 3928 CrossRef CAS; (c) P. Deria, C. D. Von Bargen, J.-H. Olivier, A. S. Kumbhar, J. G. Saven and M. J. Therien, J. Am. Chem. Soc., 2013, 135, 16220 CrossRef CAS PubMed; (d) T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898 CrossRef CAS PubMed; (e) Y. Li, Y. Wu, P. Liu, Z. Prostran, S. Gardner and B. S. Ong, Chem. Mater., 2007, 19, 418 CrossRef CAS.
- (a) C.-L. Mai, T. Moehl, Y. Kim, F.-Y. Ho, P. Comte, P.-C. Su, C.-W. Hsu, F. Giordano, A. Yella, S. M. Zakeeruddin, C.-Y. Yeh and M. Grätzel, RSC Adv., 2014, 4, 35251 RSC; (b) Y.-Z. Lin, C.-W. Yeh, P.-T. Chou, M. Watanabe, Y.-H. Chang, Y. J. Chang and T. J. Chow, Dyes Pigm., 2014, 109, 81 CrossRef CAS PubMed; (c) T. Sakurada, Y. Arai and H. Segawa, RSC Adv., 2014, 4, 13201 RSC; (d) C. Teng, X. Yang, C. Yang, H. Tian, S. Li, X. Wang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 11305 CrossRef CAS; (e) C. Teng, X. Yang, C. Yang, S. Li, M. Cheng, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 9101 CrossRef CAS.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS; (b) Y. Tohda, K. Sonogashira and N. Hagihara, Synthesis, 1977, 777 CrossRef CAS; (c) S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS. For a recent reviews: (d) R.-J. Song and J.-H. Li, Copper-Catalyzed Alkynylation, Alkenylation, and Allylation Reactions of Aryl Derivatives, In Copper-MediatedCross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley, Hoboken, NJ, 2013 Search PubMed; (e) M. O. Sydnes, Curr. Org. Chem., 2014, 18, 312 CrossRef CAS; (f) R. Chinchilla and C. Nájera, Sonogashira Reactions Using Nanocatalysts, In Nanocatalysis: Synthesis and Applications, ed. V. Polshettiwar and T. Asefa, Wiley, Hoboken, NJ, 2013 Search PubMed; (g) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- (a) L. Peng, F. Xu, K. Shinohara, A. Orita and J. Otera, Chem. Lett., 2014, 43, 1610 CrossRef CAS; (b) L. Peng, F. Xu, Y. Suzuma, A. Orita and J. Otera, J. Org. Chem., 2013, 78, 12802 CrossRef CAS PubMed; (c) X. Yang, D. Matsuo, Y. Suzuma, J.-K. Fang, F. Xu, A. Orita, J. Otera, S. Kajiyama, N. Koumura and K. Hara, Synlett, 2011, 2402 Search PubMed. A direct Sonogashira-type coupling using TMS-substituted ethynes has been reported: (d) K. Ikegashira, Y. Nishihara, K. Hirabayashi, A. Mori and T. Hiyama, Chem. Commun., 1997, 1039 RSC; (e) Y. Nishihara, K. Ikegashira, A. Mori and T. Hiyama, Chem. Lett., 1997, 1233 CrossRef CAS; (f) Y. Nishihara, K. Ikegashira, K. Hirabayashi, J. Ando, A. Mori and T. Hiyama, J. Org. Chem., 2000, 65, 1780 CrossRef CAS PubMed.
- It was reported that an electron-withdrawing group on aryl halides accelerated the oxidative addition of a transition-metal catalyst to aryl halides to promote the coupling reaction. See also Principles and Applications of Organotransition Metal Chemistry, ed. J. P. Collman, L. S. Hegedus, J. R. Norton amd R. G. Finke, University Science Books, Mill Valley, California, 1987, ch. 3. Search PubMed.
- In sharp contrast to 1, 1-iodo-4-(trimethylsilylethynyl)benzene exhibited a higher LUMO potential level (−1.208 eV) in comparison with 2.
- The DFT calculations indicated similarly that the LUMO energy level of 6 was lower than that of 7: −1.626 eV for 6; −1.482 eV for 7.
- The LUMO energy level calculated for bromobenzene is −0.343 eV, and this value is much higher than those of 6 and 9.
- Although the mechanism of the click reaction is ambiguous, the strong electron-withdrawing effect of the Ph2P(O) group in 13 might lead to the sluggish formation of a triazole ring through addition of the less nucleophilic acetylenic carbon to the azide terminal nitrogen. (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (b) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed; (c) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and the characterization data of new compounds. See DOI: 10.1039/c4qo00325j |
| This journal is © the Partner Organisations 2015 |