
Co-catalysis between DABCO and a Brønsted acid in the catalytic [4 + 2] annulation of isatin with but-3-yn-2-one and mechanistic investigations†
Qiang
Wang
a,
Qin
Xu
*a and
Min
Shi
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Mei Long Road, Shanghai 200237, China. E-mail: qinxu@ecust.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Lu, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn; Fax: +86-21-64166128
First published on 6th January 2015
Abstract
Catalytic amounts of the base catalyst DABCO in cooperation with a proton source (732 cation exchange resin) afford the [4 + 2] cycloadducts of isatins with but-3-yn-2-one in moderate to good yields. Moreover, related plausible mechanisms have been proposed in detail based on control and deuterium labeling experiments.
Structures that contain spirooxindole exist in many natural and unnatural compounds that exhibit diverse biological activities.1 Although a number of useful methods have been developed to access these interesting motifs over the past years,2,3 efficient synthetic strategies towards spirooxindole motifs are still in high demand at the present stage. Tandem reactions serve as a powerful tool for the rapid and efficient assembly of complex structures from simple starting materials with minimized production of waste. Organocatalytic tandem processes are even more appealing because of their operational simplicity and environmental friendliness.4,5 Recently, we have reported that the nitrogen-containing Brønsted base 1,4-diazabicyclo[2,2,2]octane (DABCO, 100 mol%, 1.0 eq.) mediates [4 + 2] annulations of isatins 1 with activated ketones such as but-3-yn-2-one 2a, which proceed smoothly to give the corresponding spiro[indoline-3,2′-pyran]-2,4′(3′H)-diones or 2,3-dihydropyran-4-ones in good to excellent yields under mild conditions (Scheme 1).6 In this paper, we wish to report the catalytic version of this transformation with DABCO (20 mol%, 0.2 eq.) in the presence of the 732 cation exchange resin, and mechanistic investigations based on our control and deuterium labeling experiments (Scheme 1).
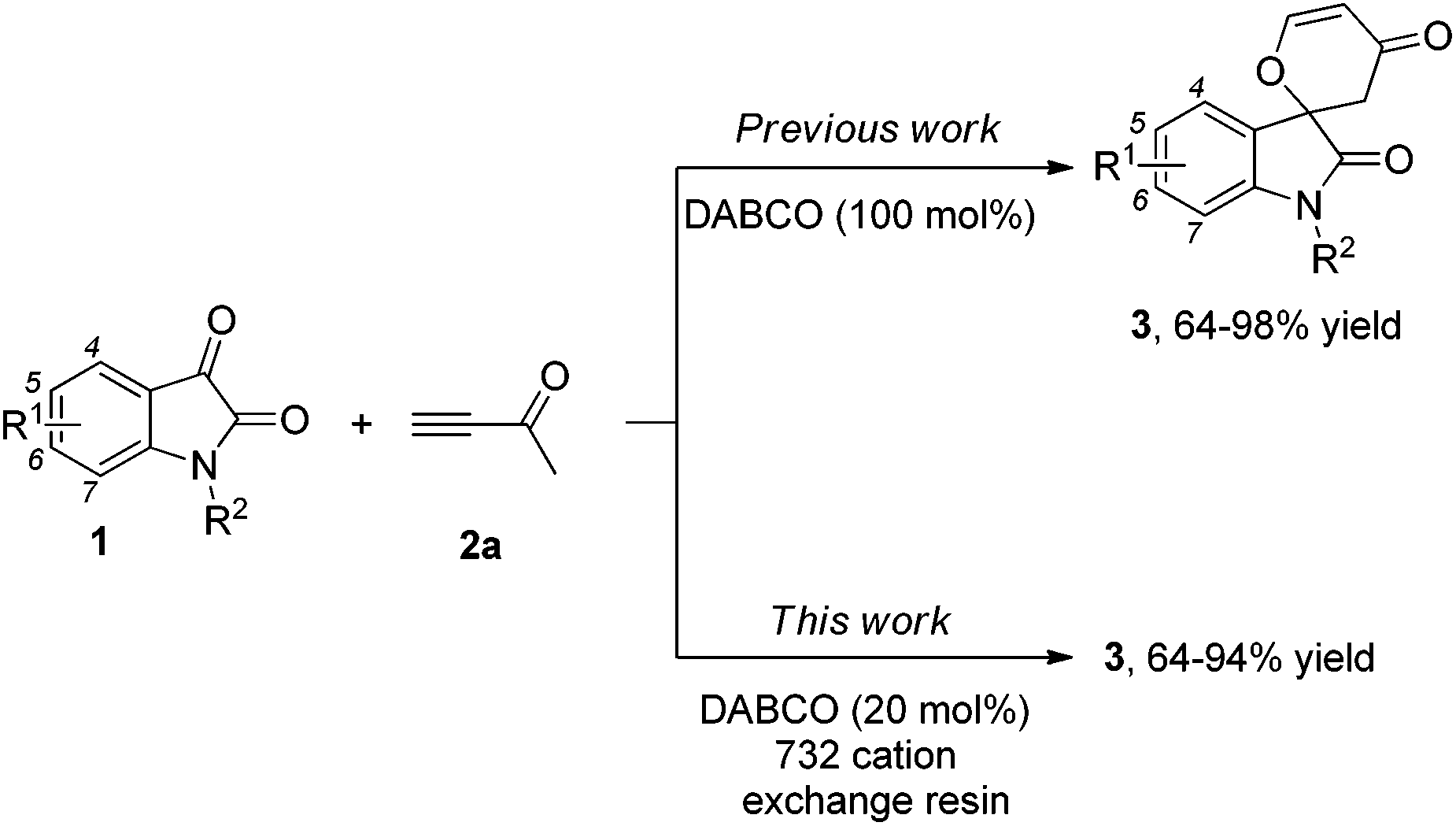 | ||
| Scheme 1 DABCO-mediated [4 + 2] annulations of isatins with but-3-yn-2-one. | ||
According to our previous findings,6 in the presence of catalytic amounts of DABCO (20 mol%), the desired product 3a was formed in only 17% yield along with an etherified product 5a (see Table 2 and Fig. 1) in traces and a self-condensation product 7a7 (see Scheme 3) of but-3-yn-2-one 2a in 19% yield. Inspired by these findings, we envisaged that if we could inhibit the self-condensation of 2a using some additives such as a proton source, the catalytic reaction could perform more efficiently. Moreover, the mechanistic aspect of this reaction might also be clarified along with developing the catalytic variant.
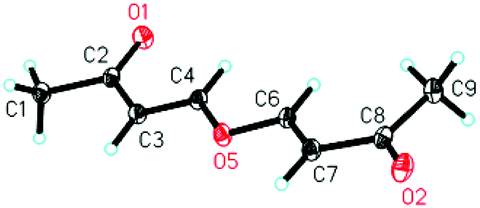 | ||
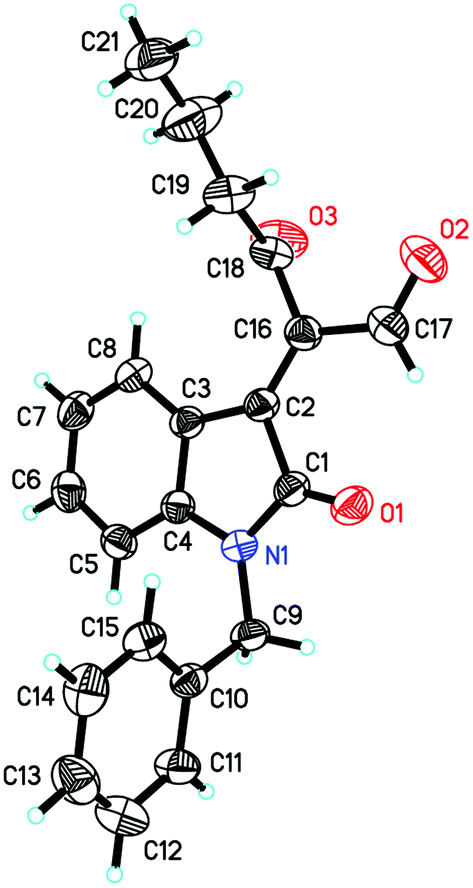
| Fig. 1 The ORTEP drawing of 5a. | ||
Results and discussion
We initially utilized isatin 1a (0.2 mmol, 1.0 equiv.) and but-3-yn-2-one 2a (0.3 mmol, 1.5 equiv.) as the substrates in the presence of catalytic amounts of DABCO and the 732 cation exchange resin in THF at room temperature to examine the reaction outcomes. The results are shown in Table 1. To our delight, we found that 3a was obtained in 78% yield in the presence of 732 cation exchange resin (60 mg) using DABCO (20 mol%, 0.2 equiv.) as the catalyst (Table 1, entry 1). 732 cation exchange resins can provide H+ acting as a proton source similar to Brønsted acids. Increasing the employed amount of the 732 cation exchange resin to 100 mg afforded 3a in 85% yield (Table 1, entry 2). However, further increasing the employed amount of the 732 cation exchange resin to 120 mg or decreasing the employed amount of DABCO to 0.1 equiv. did not improve the yield of product 3a (Table 1, entries 3 and 4).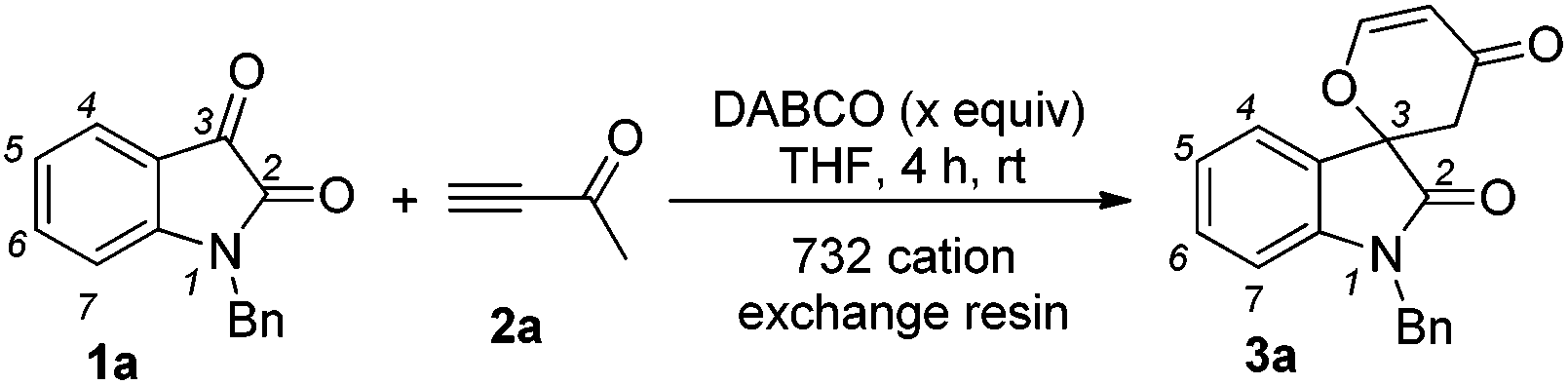
On the other hand, to further improve the reaction outcome, we also screened other proton sources (1.0 equiv.) such as PTSA, benzoic acid, phenol, benzyl alcohol, and acetic acid for this reaction and the results are shown in Table 2. However, the desired product 3a was obtained in lower yields in all cases along with some other oxa-Michael addition products 4 (anions of proton sources added to but-3-yn-2-one 2a) and the etherified product 5a (self-condensation of but-3-yn-2-one with an O-atom from H2O 2a) in large amounts. The structure of 5a has also been confirmed using X-ray diffraction. Its ORTEP drawing is shown in Fig. 1 and the CIF data are summarized in the ESI.†
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R3OHb | Yieldc (%) | Yieldc (%) | Yieldc (%) | Recovered starting materialc (%) |
| 3a | 4 | 5a | |||
a The reactions were carried out on a 0.2 mmol scale in solvent (2.0 mL).
b 1.0 eq. of the additive was used. The ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a was 1:1.5.
c Isolated yields. ND = not detected. :2a was 1:1.5.
c Isolated yields. ND = not detected.
|
|||||
| 1 | TsOH | ND | — | — | — |
| 2 | PhCO2H | Trace | 4b, 55 | 13 | 29 |
| 3 | PhOH | 15 | 4c, 47 | 8.5 | 52 |
| 4 | PhCH2OH | 19 | 4d, 52 | Trace | 57 |
| 5 | CH3CO2H | Trace | 4e, trace | Trace | 55 |
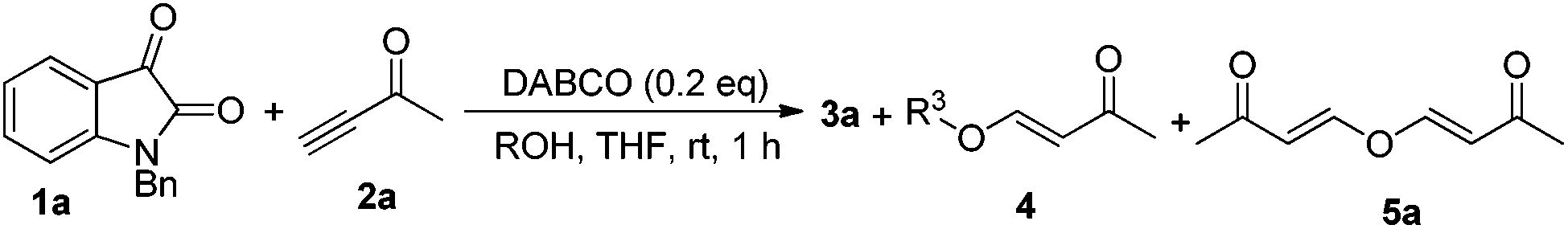
Having established the optimal reaction conditions for the formation of 3a, we next surveyed the substrate scope of the reaction by varying the structure of isatin 1 in the presence of DABCO (20 mol%) and the 732 cation exchange resin (100 mg). As shown in Table 3, regardless of whether electron-donating or electron-withdrawing groups were introduced at the 4-, 5-, 6- or 7-positions of isatin 1, these reactions proceeded smoothly, giving the corresponding products 3b–3n in 64–78% yield (Table 3, entries 1–15), suggesting that the electronic properties of the substituent on the benzene ring does not have a significant impact on the reaction outcome. The N-protecting groups such as methyl, allyl or Ph were equally well tolerated in the reaction, furnishing the desired cycloadducts 3o–3q in moderate to good yields ranging from 71–94% under the standard conditions (Table 3, entries 14–16). It should be noted that the use of 4-phenylbut-3-yn-2-one in this reaction only afforded the corresponding aldol reaction product without formation of the desired annulation product.8
|
|
||||
|---|---|---|---|---|
| Entrya | No. | R1 | R2 | Yieldb (%) |
| a Compound 1 (0.2 mmol) and the catalyst were dissolved in the solvent (2 mL) and then compound 2a (0.3 mmol) and the 732 cation exchange resin (100 mg) were added to the reaction solution. The resulting reaction mixture was stirred for 4.0 h. b Isolated yields. | ||||
| 1 | 1b | 4-Cl | Bn | 3b, 78 |
| 2 | 1c | 4-Br | Bn | 3c, 68 |
| 3 | 1d | 5-F | Bn | 3d, 78 |
| 4 | 1e | 5-CH3 | Bn | 3e, 75 |
| 5 | 1f | 5-Cl | Bn | 3f, 68 |
| 6 | 1g | 5-Br | Bn | 3g, 64 |
| 7 | 1h | 5,7-CH3 | Bn | 3h, 68 |
| 8 | 1i | 6-Cl | Bn | 3i, 66 |
| 9 | 1j | 6-Br | Bn | 3j, 65 |
| 10 | 1k | 6-CH3 | Bn | 3k,69 |
| 11 | 1l | 7-Cl | Bn | 3l, 66 |
| 12 | 1m | 7-Br | Bn | 3m, 65 |
| 13 | 1n | 7-F | Bn | 3n, 65 |
| 14 | 1o | H | CH3 | 3o, 87 |
| 15 | 1p | H | Allyl | 3p, 94 |
| 16 | 1q | H | Ph | 3q, 71 |

While examining the effect of the proton source, another interesting finding was that we identified the formation of (E)-2-(1-benzyl-2-oxoindolin-3-ylidene)-3-oxohexanal 6a in 23% yield in the presence of acetic acid (1.0 eq.) at room temperature when hex-1-yn-3-one 2b instead of but-3-yn-2-one 2a was used as a Michael acceptor (Scheme 2), and the corresponding [4 + 2] annulation product was not observed. Its structure has been unambiguously determined using X-ray analysis of its single crystals. The ORTEP drawing of 6a is shown in Fig. 2 and the CIF data are presented in the ESI.† A similar product was identified in traces in the [4 + 2] annulation of isatin 1a with but-3-yn-2-one 2a, on the basis of the 1H NMR spectroscopic data, under the same conditions along with trace amounts of the other byproducts as shown in entry 5 of Table 2.
 | ||
| Fig. 2 The ORTEP drawing of 6a. | ||
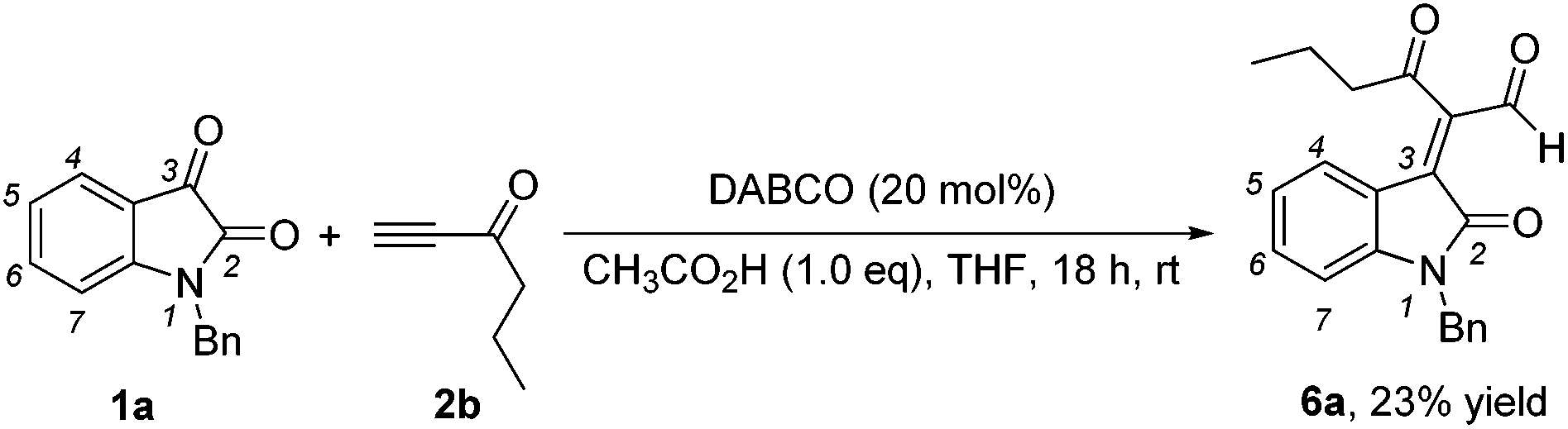 | ||
| Scheme 2 The reaction of isatin with hex-1-yn-3-one 2b in the presence of acetic acid. | ||
In our previous work, we proposed that the formation of 3 might undergo a stepwise aldol/intramolecular oxa-Michael addition pathway.8 In order to gain more mechanistic insights, we performed several isotopic labeling experiments by adding D2O (1.0 equiv.) to the reaction systems. In the first experiment, we conducted the self-condensation of but-3-yn-2-one 2a catalyzed by DABCO (0.1 equiv.) in the presence of D2O (1.0 equiv.), affording the deuterium containing product 7a in 29% yield as indicated by the 1H NMR spectroscopic analysis (eqn (1), Scheme 3). The self-condensation of oct-1-yn-3-one 2c was conducted under similar conditions, affording the deuterium containing product 8a in 65% yield, as shown in eqn (2) of Scheme 3 (also see ESI†). This result clearly clarified that there is no proton exchange at the α-position of the carbonyl group as well as the terminal olefin in product 7a. Next, we carried out the [4 + 2] annulation of isatin 1a with but-3-yn-2-one 2a by adding D2O (5.0 equiv.) under the standard conditions, giving the corresponding deuterium containing product [D]-3a in 78% yield (eqn (3), Scheme 3). Moreover, we also performed the reaction of 4-phenylbut-3-yn-2-one 2d in the presence of DABCO (1.0 equiv.) and D2O (2.0 equiv.) in THF at room temperature. It was found that the corresponding deuterium containing product [D]-2d was recovered in 81% yield along with 26% D content at the α-position of the carbonyl group (eqn (4), Scheme 3).
 | ||
| Scheme 3 Deuterium labeling experiments. | ||
According to our previous work,8 we also synthesized the possible deuterated terminal alkyne intermediate 10 in 98% yield along with 66% D content via desilylation with KHF2 in MeOD after silica gel column chromatography, and it should be noted that the hydroxyl group has been completely protonated (Scheme 4, also see ESI†). Under the standard conditions, 10 could be transformed into [D]-3a in 75% yield in the presence of DABCO (20 mol%) along with the acetylenic deuterium shift to the α-position of the carbonyl group (Scheme 4).
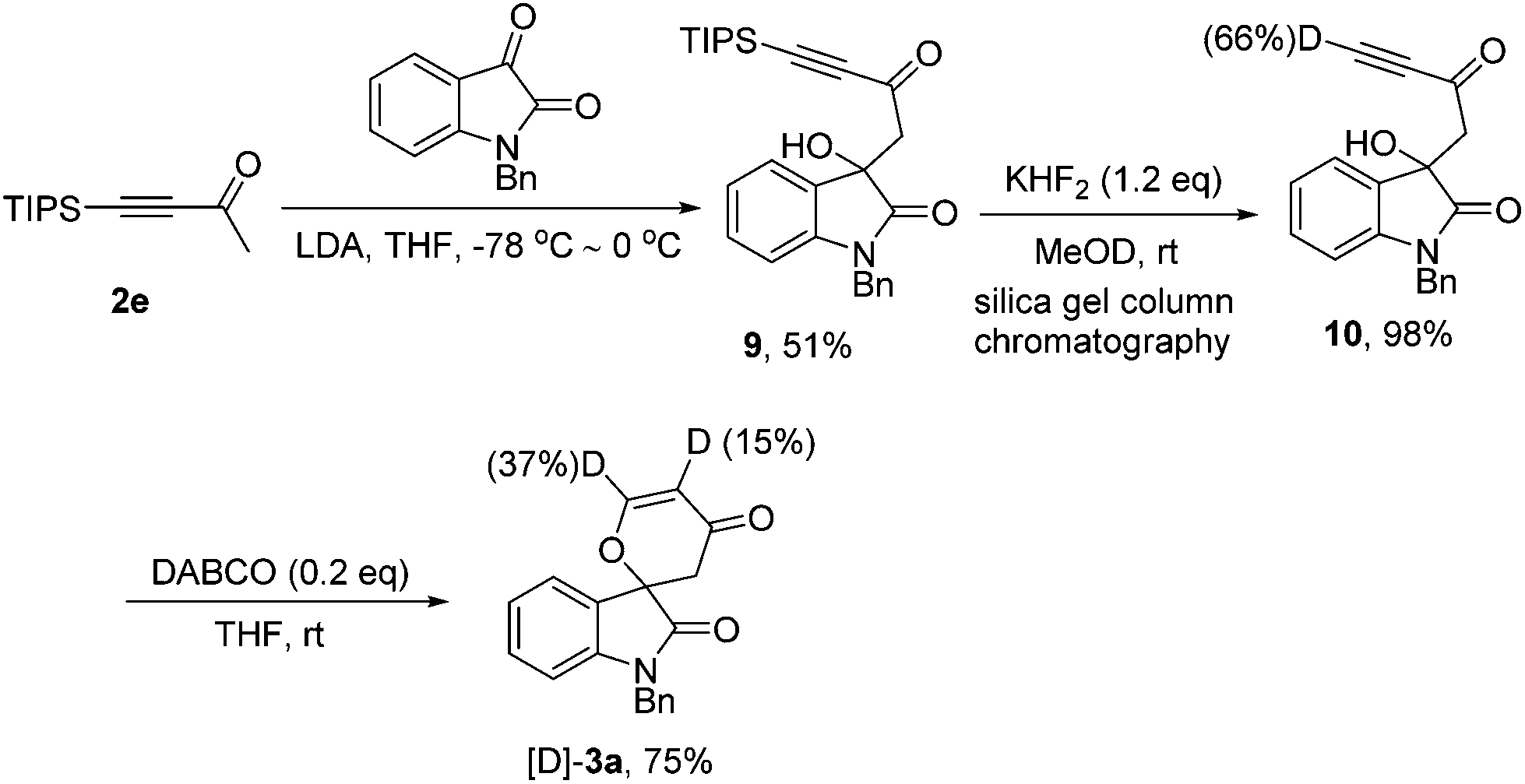 | ||
| Scheme 4 Synthesis of the possible intermediate 10 and isotopic labeling experiment. | ||
Based on the deuterium labeling experiments described above, we proposed a more specific mechanism for the formation of the self-condensation product 7a, which is slightly different from the previous mechanism proposed by Ramachandran (Scheme 5).7 The base catalyst deprotonates but-3-yn-2-one 2a to generate an enolate intermediate 2a-1 or 2a-2, which undertakes an oxonucleophilic attack of another molecule of 2a to produce the corresponding allenic intermediate 2a-3. Protonation of 2a-3 by the acetylenic proton of 2a gives self-condensation product 7a and the corresponding acetylenic anion 2a-4, which abstracts a proton from the water or the protonated base catalyst (R3N+H) to regenerate the base catalyst as shown in path b. In addition, allenic intermediate 2a-3 might abstract a proton from a third alkynone molecule, regenerating enolate intermediate 2a-1 to continue the catalytic cycle as shown in path a.
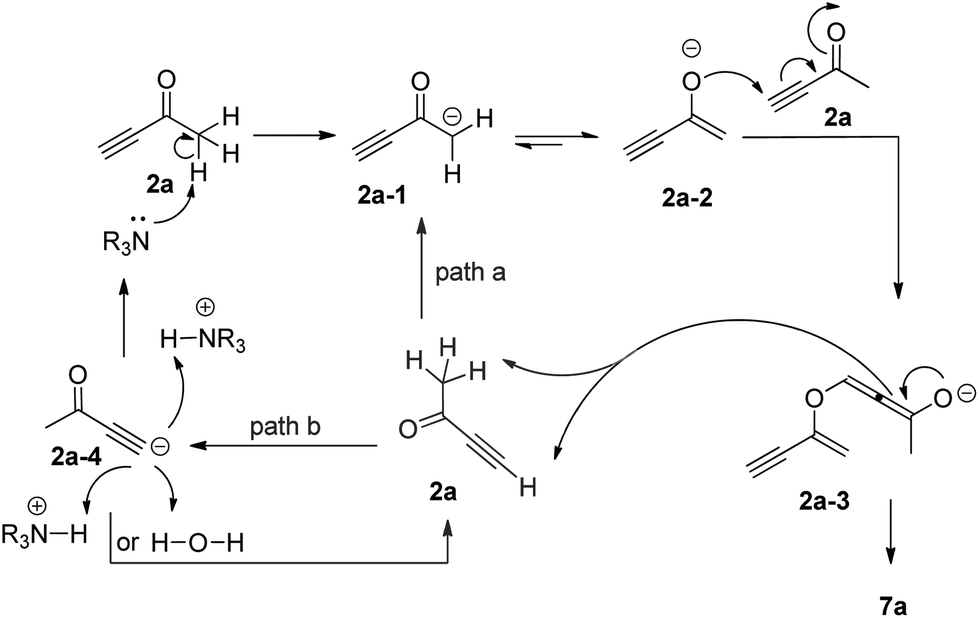 | ||
| Scheme 5 Proposed mechanism for DABCO-catalyzed self-condensation of but-3-yn-2-one. | ||
Similar to the proposed mechanism for the formation of self-condensation product 7a, a plausible stepwise mechanism for the catalytic [4 + 2] annulation of isatin with but-3-yn-2-one 2a in the presence of DABCO is shown in Scheme 6. Initially, DABCO deprotonates but-3-yn-2-one 2a to generate the enolate intermediate 2a-2,9 followed by a nucleophilic addition at the 3-carbonyl group of isatin 1a to give intermediate 3a-1, which abstracts a proton from the protonated DABCO to afford the corresponding key intermediate 3a-2 as mentioned above. Subsequently, a Michael addition of DABCO to the alkynyl group of 3a-2 gives the intermediate 3a-3, which obtains a proton from the acetylenic proton in 3a-2, 2a, or 3a-4 or from the protonated base (R3N+H) to give intermediate 3a-5. This vinylogous oxonucleophile wins over the ambient carbon nucleophile. The oxoanion of intermediate 3a-5, generated from the deprotonation of 3a-4, undergoes a Michael addition to form the six-membered cyclic intermediate 3a-6. The release of the base catalyst produces the corresponding spirooxindole 3a. According to our previous work, product 3a may also be deprotonated by the base catalyst to form the ionic pair 3a-7, which can be dissociated using the 732 cation exchange resin to generate product 3a and the base catalyst.
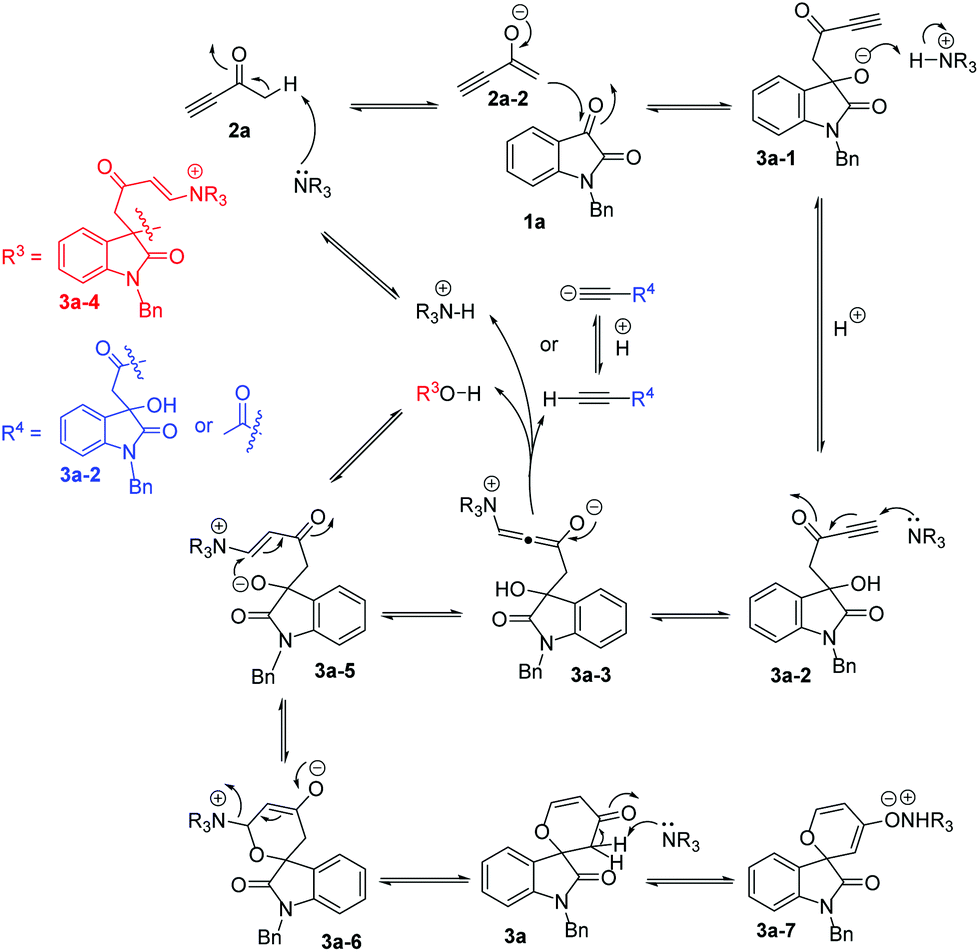 | ||
| Scheme 6 Proposed mechanism for the [4 + 2] annulation of isatin with but-3-yn-2-one. | ||
In order to clarify the formation of 6a, we also proposed a plausible mechanism in Scheme 7. Initially, DABCO attacks the alkynyl moiety of 2b to give a vinylogous intermediate, followed by a nucleophilic addition to the 3-carbonyl group of isatin 1a to give intermediate 6a-1. Subsequently, the ambient H2O attacks the alkenyl moiety in intermediate 6a-2 along with the elimination of H2O in the presence of acetic acid to deliver intermediate 6a-3. The release of the base catalyst in 6a-3 produces the corresponding product 6a.
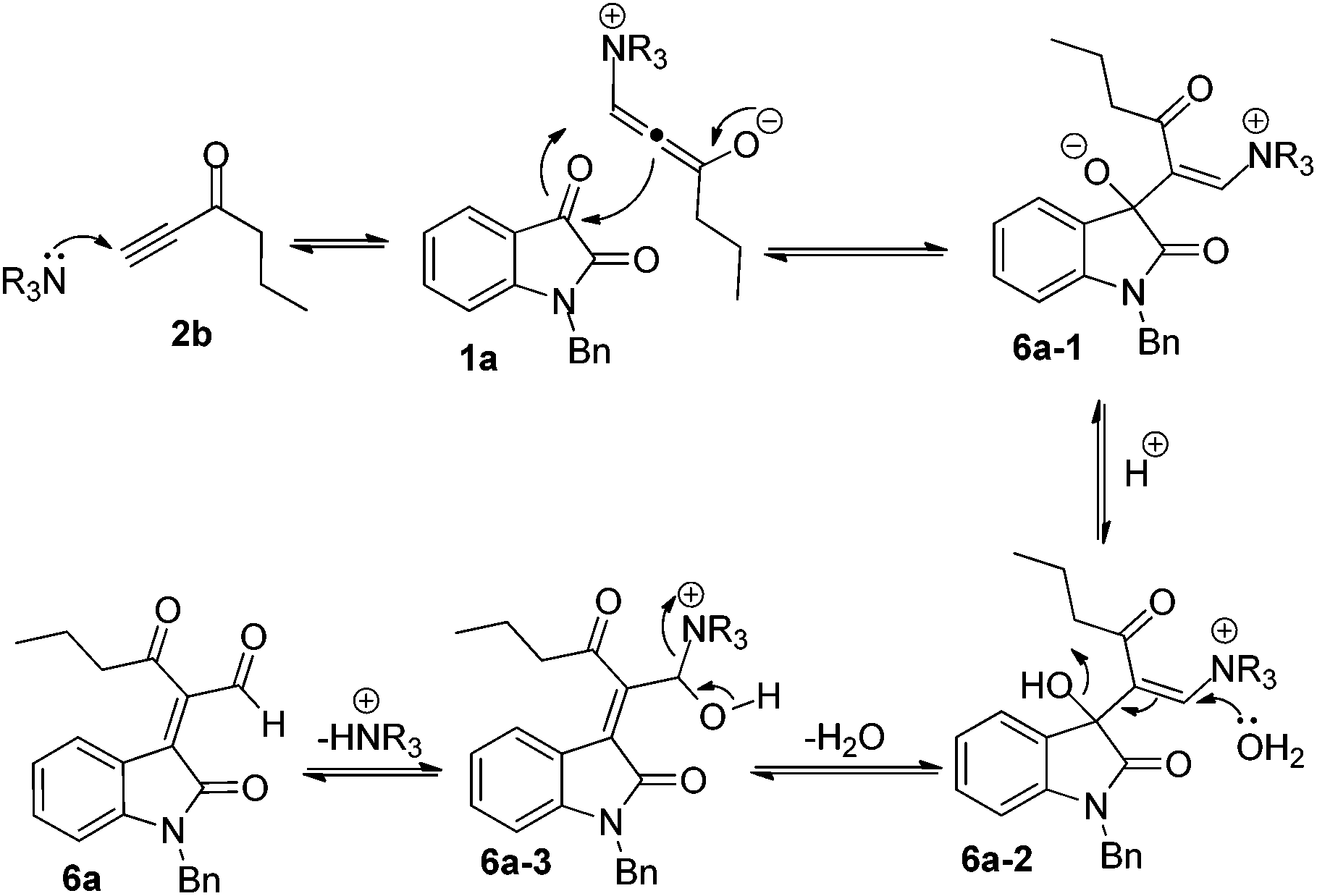 | ||
| Scheme 7 Proposed mechanism for the formation of 6a. | ||
In conclusion, we have developed a novel strategy in which a base catalyst in cooperation with a proton source can afford the [4 + 2] cycloadducts of the isatins 1 with but-3-yn-2-one10 in moderate to good yields in a catalytic manner. Moreover, three plausible mechanisms have been proposed in detail on the basis of previous literature and deuterium labeling experiments. The identification of new and more efficient catalytic systems to access spiro-compounds beyond oxindoles with high stereoselectivity and to apply those new methodologies to synthesize biologically active products is the focus of current efforts in our laboratories.
Acknowledgements
We are grateful for financial support from the National Basic Research Program of China (973)-2015CB856603, Shanghai Municipal Committee of Science and Technology (11JC1402600), and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21102166, 21121062, 21302203, 20732008, 21421091 and 21372250).Notes and references
- (a) For a review, see: C. V. Galliford and K. A. Scheidt, Angew. Chem., 2007, 119, 8902–8912 ( Angew. Chem., Int. Ed. , 2007 , 46 , 8748–8758 ) CrossRef; (b) For a textbook, see: Indole and biogenetically related alkaloids, ed. J. D. Phillipson and M. H. Zenk, Academic press inc., London, 1980 Search PubMed; (c) S. M. Hande and Y. Takemoto, Org. Lett., 2011, 13, 1828–1831 CrossRef CAS PubMed.
- (a) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003–3025 CrossRef CAS; (b) H. Lin and S. J. Danishefsky, Angew. Chem., 2003, 115, 38–53 ( Angew. Chem., Int. Ed. , 2003 , 42 , 36–51 ) CrossRef; (c) R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127–139 CrossRef CAS PubMed; (d) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS; (e) R. Rojas-Duran, G. González-Aspajo, C. Ruiz-Martel, G. Bourdy, V. H. Doroteo-Ortega, J. Alban-Castillo, G. Robert, P. Auberger and E. Deharo, J. Ethnopharmacol., 2012, 143, 801–804 CrossRef CAS PubMed; (f) V. Milata, Acta Chim. Slovaca, 2008, 1, 221–237 Search PubMed; (g) H. Venkatesan, M. C. Davis, Y. Altas, J. P. Snyder and D. C. Liotta, J. Org. Chem., 2001, 66, 3653–3661 CrossRef CAS PubMed.
- (a) X. H. Chen, Q. Wei, H. Xiao, S. W. Luo and L. Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819–13825 CrossRef CAS PubMed; (b) G. Bencivenni, L. Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M. P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., 2009, 121, 7336–7339 ( Angew. Chem., Int. Ed. , 2009 , 48 , 7200–7203 ) CrossRef; (c) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y. C. Chen, Chem. – Eur. J., 2010, 16, 2852–2856 CrossRef CAS PubMed; (d) Q. Wei and L. Z. Gong, Org. Lett., 2010, 12, 1008–1011 CrossRef CAS PubMed; (e) R. Shintani, S. Y. Hayashi, M. Murakami, M. Takeda and T. Hayashi, Org. Lett., 2009, 11, 3754–3756 CrossRef CAS PubMed; (f) M. P. Castaldi, D. M. Troast and J. A. Porco, Org. Lett., 2009, 11, 3362–3365 CrossRef CAS PubMed; (g) M. Gicquel, C. Gomez, P. Retailleau, A. Voituriez and A. Marinetti, Org. Lett., 2013, 15, 4002–4005 CrossRef CAS PubMed; (h) X. Y. Guan, Y. Wei and M. Shi, Chem. – Eur. J., 2011, 16, 13617–13621 CrossRef PubMed; (i) X.-C. Zhang, X.-H. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 1548–1550 RSC; (j) X. C. Zhang, X. Cao, Y. Wei and M. Shi, Org. Lett., 2011, 13, 1142–1145 CrossRef CAS PubMed; (k) Z. Lian, Y. Wei and M. Shi, Tetrahedron, 2012, 68, 2401–2408 CrossRef CAS PubMed; (l) H. B. Yang, X.-Y. Guan, Y. Wei and M. Shi, Eur. J. Org. Chem., 2012, 2792–2800 Search PubMed; (m) H. B. Yang, Y. Wei and M. Shi, Tetrahedron, 2013, 69, 4088–4097 CrossRef CAS PubMed; (n) X.-N. Zhang, G.-Q. Chen, X. Dong, Y. Wei and M. Shi, Adv. Synth. Catal., 2013, 355, 3351–3357 CrossRef CAS.
- For some reviews of tandem reactions, see: (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., 2006, 118, 7292–7344 ( Angew. Chem., Int. Ed. , 2006 , 45 , 7134–7186 ) CrossRef; (b) H. Guo and J. Ma, Angew. Chem., 2006, 118, 362–375 ( Angew. Chem., Int. Ed. , 2006 , 45 , 354–366 ) CrossRef; (c) H. Pellissier, Tetrahedron, 2006, 62, 2143–2173 CrossRef CAS PubMed; (d) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed; (e) J. Zhou, Chem. – Asian J., 2010, 5, 422–434 CrossRef CAS PubMed; (f) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Acc. Chem. Res., 2012, 45, 1278–1293 CrossRef CAS PubMed; (g) F.-L. Hu and M. Shi, Org. Chem. Front., 2014, 1, 587–598 RSC.
- For selected examples of organocatalytic tandem reactions, see: (a) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Angew. Chem., 2003, 115, 4365–4369 ( Angew. Chem., Int. Ed. , 2003 , 42 , 4233–4237 ) CrossRef; (b) M. Marigo, T. Schulte, J. Franzen and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 15710–15711 CrossRef CAS PubMed; (c) J. W. Yang, M. T. Hechavarria Fonseca and B. List, J. Am. Chem. Soc., 2005, 127, 15036–15037 CrossRef CAS PubMed; (d) Z. Mao, Y. Jia, Z. Xu and R. Wang, Adv. Synth. Catal., 2012, 354, 1401–1406 CrossRef CAS; (e) Z.-Q. He, Q. Zhou, L. Wu and Y.-C. Chen, Adv. Synth. Catal., 2010, 352, 1904–1908 CrossRef CAS; (f) Y.-L. Liu, X. Wang, Y.-L. Zhao, F. Zhu, X.-P. Zeng, L. Chen, C.-H. Wang, X.-L. Zhao and J. Zhou, Angew. Chem., 2013, 125, 13980–13984 ( Angew. Chem., Int. Ed. , 2013 , 52 , 13735–13739 ) CrossRef; (g) Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 5053–5061 CrossRef CAS PubMed; (h) Z.-X. Jia, Y.-C. Luo and P.-F. Xu, Org. Lett., 2011, 13, 832–835 CrossRef CAS PubMed; (i) K. Albertshofer, B. Tan and C. F. Barbas III, Org. Lett., 2012, 14, 1834–1837 CrossRef CAS PubMed.
- Z. Lian and M. Shi, Eur. J. Org. Chem., 2012, 581–586 CrossRef CAS.
- P. V. Ramachandran and M. T. Rudd, Tetrahedron Lett., 1999, 40, 3819–3822 CrossRef CAS.
- Q. Wang, Z. Lian, Q. Xu and M. Shi, Adv. Synth. Catal., 2013, 355, 3344–3350 CrossRef CAS.
- Z. Lian, Q. Zhao, Y. Wei and M. Shi, Eur. J. Org. Chem., 2012, 3338–3341 CrossRef CAS.
- For selected papers on ynone utilization in organocatalysis, see: (a) Z. Lian, Y. Wei and M. Shi, Tetrahedron, 2012, 68, 2401–2408 CrossRef CAS PubMed; (b) L. Yang, P. Xie, E. Li, X. Lin, Y. Huang and R. Chen, Org. Biomol. Chem., 2012, 10, 7628–7634 RSC; (c) Z. Lian and M. Shi, Org. Biomol. Chem., 2012, 10, 8048–8050 RSC; (d) J. E. Wilson, J. Sun and G. C. Fu, Angew. Chem., 2010, 122, 165–167 ( Angew. Chem., Int. Ed. , 2010 , 49 , 161–163 ) CrossRef; (e) L.-G. Meng, P. Cai, Q. Guo and S. J. Xue, J. Org. Chem., 2008, 73, 8491–8496 CrossRef CAS PubMed; (f) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (g) H. Kuroda, I. Tomita and T. Endo, Org. Lett., 2003, 5, 129–131 CrossRef CAS PubMed; (h) D. B. Ramachary, C. Venkaiah and R. Madhavachary, Org. Lett., 2013, 15, 3042–3045 CrossRef CAS PubMed; (i) D. B. Ramachary, C. Venkaiah and P. M. Krishna, Chem. Commun., 2012, 48, 2252–2254 RSC; (j) F. Silva, M. Sawicki and V. Gouverneur, Org. Lett., 2006, 8, 5417–5419 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 914026 and 936172. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c4qo00323c |
| This journal is © the Partner Organisations 2015 |