
Transition metal-catalyzed direct remote C–H functionalization of alkyl groups via C(sp3)–H bond activation
Guanyinsheng
Qiu
a and
Jie
Wu
*bc
aCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn; Fax: +86 21 6564 1740; Tel: +86 21 6510 2412
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, China
First published on 1st December 2014
Abstract
This review is focused on the recent advances in the transition metal-catalyzed direct remote C–H-functionalization of alkyl groups via C(sp3)–H bond activation. In general, carboxamide/ester-chelated β-functionalization reactions are summarized, which mainly include direct β-arylation, β-alkylation, β-alkynylation, etc. Additionally, some biologically interesting architectures are constructed by using these direct β-functionalization reactions.
 Guanyinsheng Qiu | Guanyinsheng Qiu was born in Jiangxi (China) in 1984. He received his BS (2008) and MS (2011) from the Department of Chemistry at Jiangxi Normal University. In the year 2011, he moved to Fudan University, pursuing his Ph.D. under the guidance of Professor Jie Wu. After obtaining his Ph.D. degree in 2014, he now works in Jiaxing University as a lecturer. His current research interest mainly focuses on methodological development for the synthesis of natural product-like compounds under palladium catalysis. |
 Jie Wu | Jie Wu received his B.Sc. in chemistry from Jiangxi Normal University in 1995 and he pursued his Ph.D. studies at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the supervision of Prof. Xue-Long Hou (1995–2000). After conducting research as a postdoctoral fellow at Harvard University (2000–2001, Prof. Zhen Yang), he worked as a visiting scientist at the Aaron Diamond AIDS Research Center (2001–2002) and as a staff scientist in VivoQuest, Inc. (2002–2004). In 2004, he moved to the Department of Chemistry at Fudan University and held the Professor rank two years later. He received the Thieme Chemistry Journals Award in 2010 and Dow Innovation Challenge Award in 2013. He also serves as a member of Editorial Advisory Board of ACS Combinatorial Science. |
1 Introduction
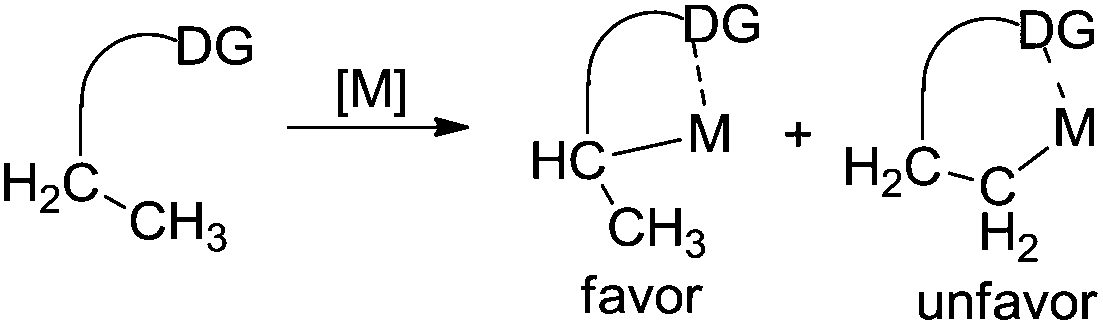
It is well known that the direct C–H functionalization has remarkable advantages in organic synthesis.1 Consequently, tremendous efforts have been made towards the direct C–H functionalization for the formation of C–X bonds (X = C, N, O, P, etc.). In general, exploration of C–H functionalization is mainly focused on addressing the following two issues: (i) reaction efficiency and (ii) site selectivity.With the development of organometallic chemistry (especially palladium chemistry), the reaction efficiency of C–H functionalization has been achieved in various transformations. Many reactions related to C–H functionalization could be successfully realized through the employment of appropriate catalytic systems such as transition metal catalysts, ligands, oxidants, and additives. As for selective functionalization of a single C–H bond within a complex substrate, ortho selectivity generally relies on the assistance of directing groups in substrates, where the σ-directing groups ensure the formation of conformationally rigid five-membered, six-membered, or seven-membered cyclic pre-transition states (such as cyclopalladium species) through the insertion of a transition metal (such as palladium) into the C–H bond at the ortho position.2 As we know, many heteroatom-containing groups (such as pyridine, amide, ester, etc.) have been demonstrated as efficient directing groups so far. The selectivities on the meta- and para-positions are often accomplished by controlling the electronic effect of substrates.3 Recently, by controlling the weak “end-on” interaction between a linear nitrile group and a metal centre, Yu and co-workers achieved a direct β-alkenylation of a remote meta-C–H bond in a tethered arene.3h–i
The above established achievements mainly focused on the direct functionalization of the C(sp2)–H bond. There is no doubt that transition metal-catalyzed C(sp2)–H functionalization has made unprecedented progress in the past decade, and many related summaries have been published.1,2 Compared with C(sp2)–H functionalization, method development with C(sp3)–H functionalization has made a breakthrough after cross-dehydrogenative-coupling (CDC) reactions were developed. Related reviews have been reported by Li and others.4 However, these accounts usually described the direct C(sp3)–H bonds activation adjacent to nitrogen/oxygen in heterocycles. Unlike CDC reactions and C–H functionalization occurring at the α position of directing groups, remote C–H functionalization of alkyl groups is still a challenge due to the inert nature of a remote Csp3–H bond and thus the cyclometallic intermediate is not readily formed (Fig. 1). To the best of our knowledge, reviews on site-selective functionalization of a remote C(sp3)–H bond remain rare.5 As a result, this review summarizes the β-functionalization of alkyl groups via a remote C(sp3)–H bond activation. Both synthetic applications and mechanistic aspects of these transformations are presented where appropriate. This review is organized as a sequence of the bond types being formed, and it is mainly focused on the direct β-functionalization of carboxamide/ester-chelated alkyl groups.
 | ||
| Fig. 1 Challenge on remote C–H functionalization. | ||
2 C–C bond formation via direct β-arylation
As we know, direct C–H activation reactions have recently emerged as powerful tools for the formation of C–C bonds. Early studies involving direct β-functionalization were initiated by the direct β-functionalization of 2-substituted pyridines. An elegant example was reported by Daugulis and co-workers, where the β-arylative reaction of 2-ethylpyridine 1 was investigated.6 As illustrated in Scheme 1, aryl iodide 2 was used as an efficient aryl source by employing a combination of palladium acetate (5 mol%) and a stoichiometric amount of silver acetate. The corresponding β-arylative 2-ethylpyridines 3 were provided in moderate yields, although 2-ethylpyridine 1 was the only substrate that underwent β-arylation of the unactivated C(sp3)–H bond. The result reported by Yu and co-workers disclosed the same phenomenon in the methylative reaction of 2-ethylpyridine 1 with methylboroxine 4.7 According to their findings, the pyridinyl group in the substrate would saturate the coordination site on the palladium species, thus suppressing β-hydride elimination from the palladium intermediate while improving β-functionalization. Based on these two achievements, it was thought that the generality of the reaction could be improved by introducing pyridinyl or other directing groups as removable auxiliaries.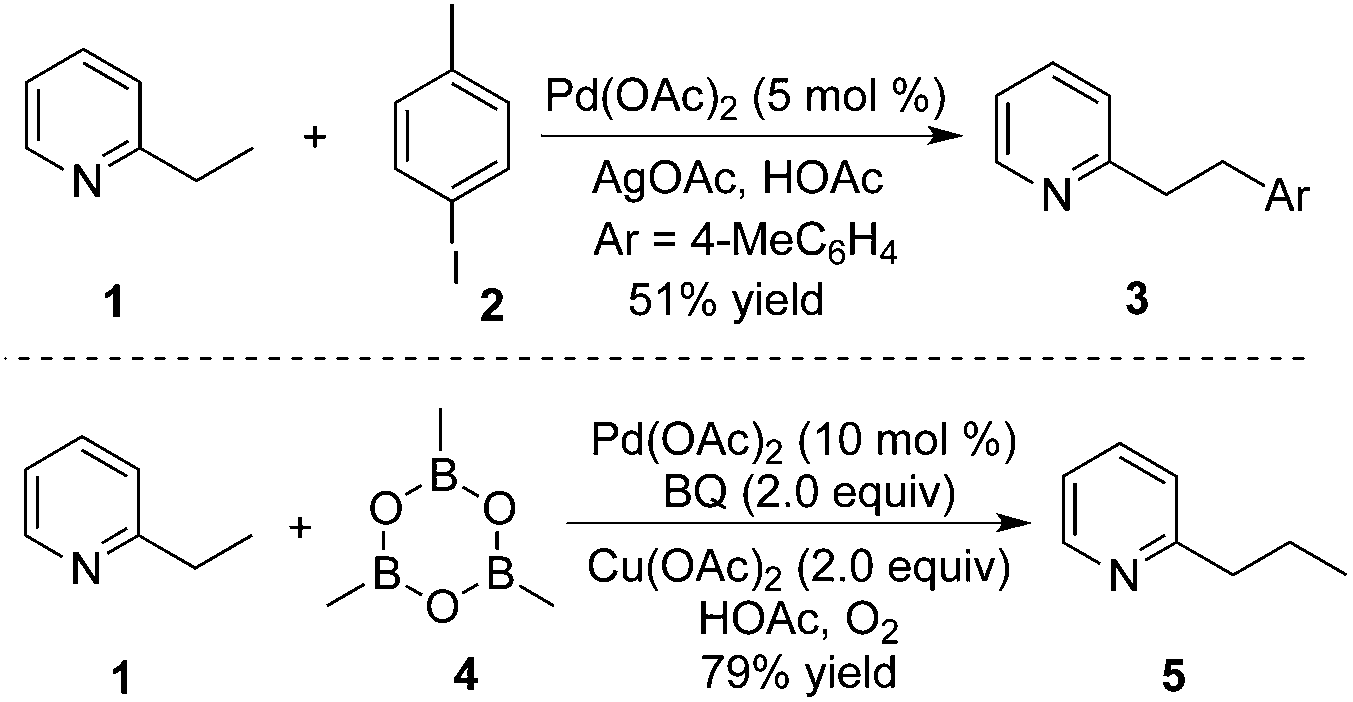 | ||
| Scheme 1 β-Arylation of 2-ethylpyridine. | ||
2.1 Direct β-arylation of alkyl groups
One of the best-recognized removable auxiliaries was 8-aminoquinoline. Various 8-aminoquinoline-based amides were employed to study the β-functionalization reactions, especially the β-arylation.
A notable example was developed by Daugulis and co-workers.8 As expected, the β-arylative reaction of N-(quinolin-8-yl)butyramide 6 with 4-methoxylphenyl iodide 7 proceeded smoothly in the presence of a palladium catalyst at 120 °C, resulting in β-arylative product 8 with excellent regioselectivity and high reaction efficiency (Scheme 2). However, under standard conditions, the reaction of cyclohexyl-substituted amide produced diarylative product 8a in 61% yield, and isobutyric amide 6b was tetra-arylated. These results indicated that the secondary C–H bond could be activated as well. Surprisingly, direct β-arylation reaction could not be observed by changing the auxiliary to 2-aminomethylpyridine, which was presumably due to the susceptibility of the cyclopalladium intermediate. From the mechanistic studies, it was believed that the regioselectivity resulted from the formation of the dicyclopalladium species intermediate 10.
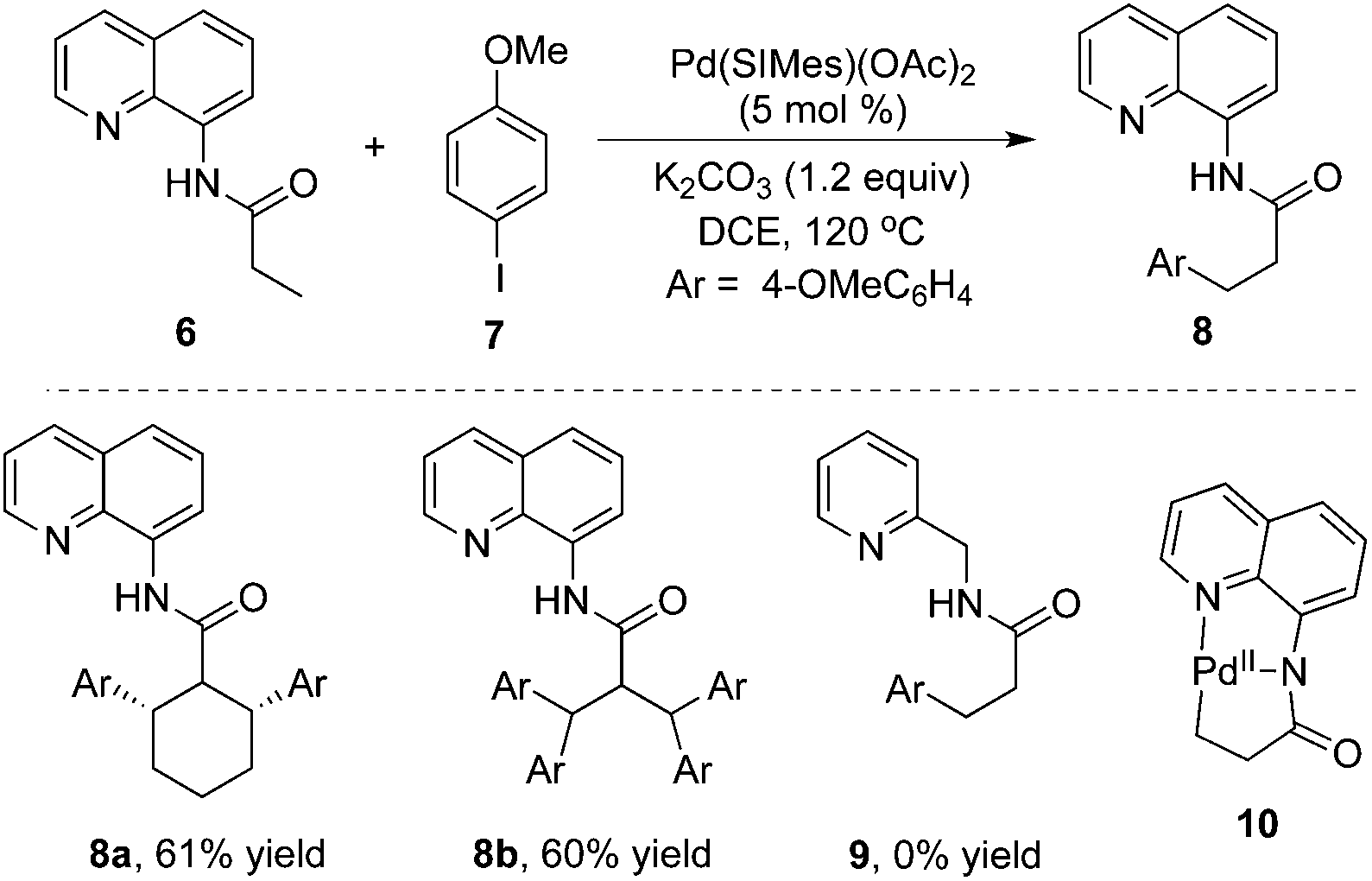 | ||
| Scheme 2 8-Aminoquinoline-assisted β-arylation. | ||
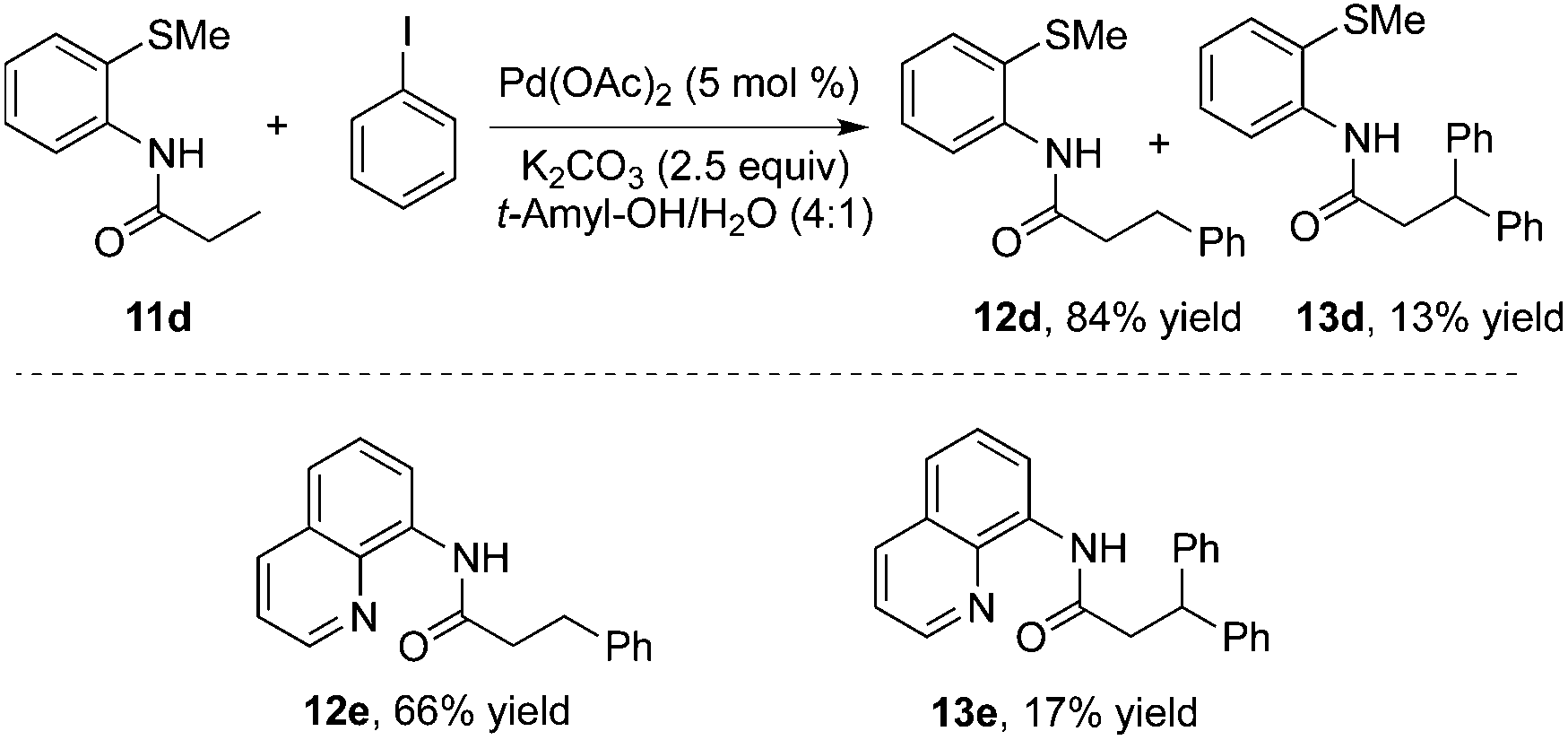
To realize selective monoarylation, the same group described 2-thiomethylaniline as a successful auxiliary to predominantly produce β-monoarylative propoinamide 12 (Scheme 3).9 The co-solvent combining tert-amyl alcohol and water was favourable for the β-monoarylation reactions. The size of the substituent R made a significant impact on both the β-arylative reaction efficiency and the ratio of monoarylative product 12 and diarylative product 13. Lower reaction yields and ratios of selectivity were observed with larger sizes of the substituent R. After the reaction, the auxiliary of 2-thiomethylaniline could be readily removed by NaOH–MeOH. These results indicated that 2-thiomethylaniline was a better auxiliary in the β-arylation process with regard to reaction efficiency and selectivity ratios between monoarylation and diarylation (Scheme 4). For example, the direct β-arylation of 8-aminoquinoline-assisted substrate 11e gave rise to β-monoarylative product 12e in 66% yield and β-diarylative product 13e in 17% yield, suggesting both poorer reactivities and chemoselectivity of β-monoarylation/diarylation than that of 2-thiomethylaniline.
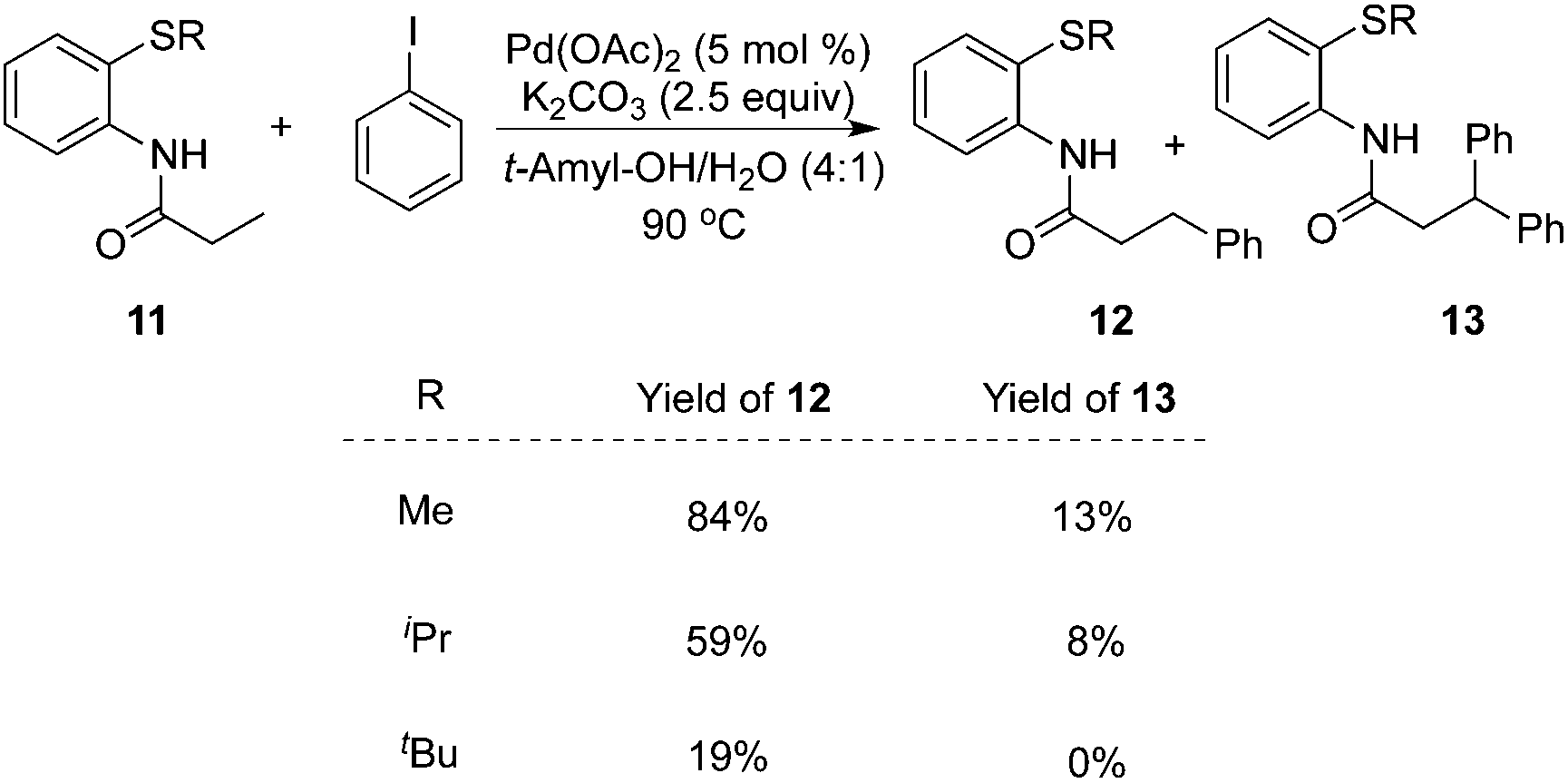 | ||
| Scheme 3 2-Thiomethylaniline-assisted β-arylation. | ||
 | ||
| Scheme 4 Compared results of two different auxiliaries. | ||
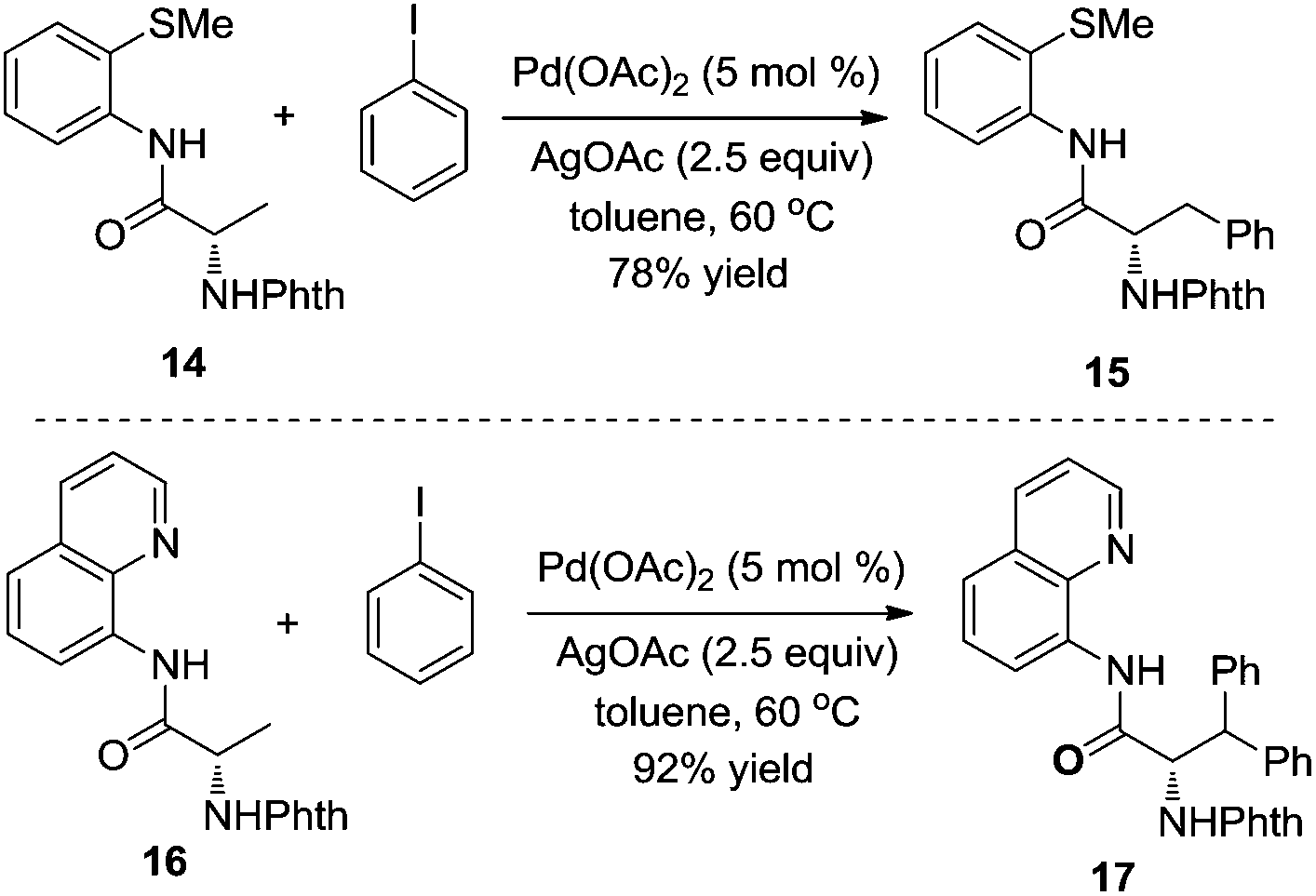
According to the recent report by Daugulis,10 the different chelating reactivities between 8-aminoquinoline and 2-thiomethylaniline allowed the selective monoarylation and diarylation of protected non-natural amino acids 14 and 16 under the same conditions. As shown in Scheme 5, under the same conditions, the substrate containing 2-thiomethylaniline 14 was prone to produce monoarylative product 15 in 78% yield, and the reaction of the substrate 16 was diarylated, forming the product 17 in 92% yield. Interestingly, the retention of enantioselectivity in the non-natural amino acid was observed.
 | ||
| Scheme 5 Selective monoarylation and diarylation. | ||
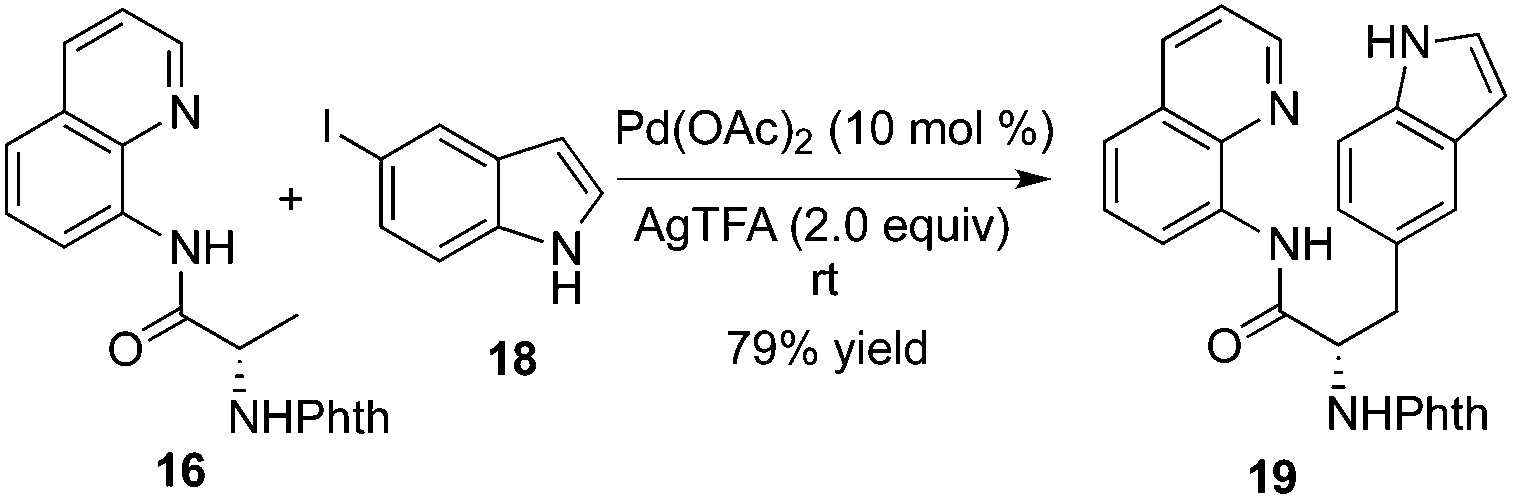
Chen and co-workers found that substrate 16 tended to be β-monoarylated at room temperature with the assistance of silver trifluoroacetate (Scheme 6).10b The mechanistic studies revealed that the trifluoroacetate ion served as an unusual ligand to stabilize the cyclopalladium intermediate or facilitated the oxidative addition of aryl halides. The unactivated 3-position of 8-aminoquinoline-modified proline derivatives could be arylated via β-functionalization of remote C–H bond activation, which provided an efficient and practical alternative to the small yet complex enantiopure cis-2,3-disubstituted pyrrolidines.10c The total synthesis of Celogentin C could be achieved through the above β-monoarylation.11
 | ||
| Scheme 6 β-Monoarylation with an assistance of silver trifluoroacetate. | ||
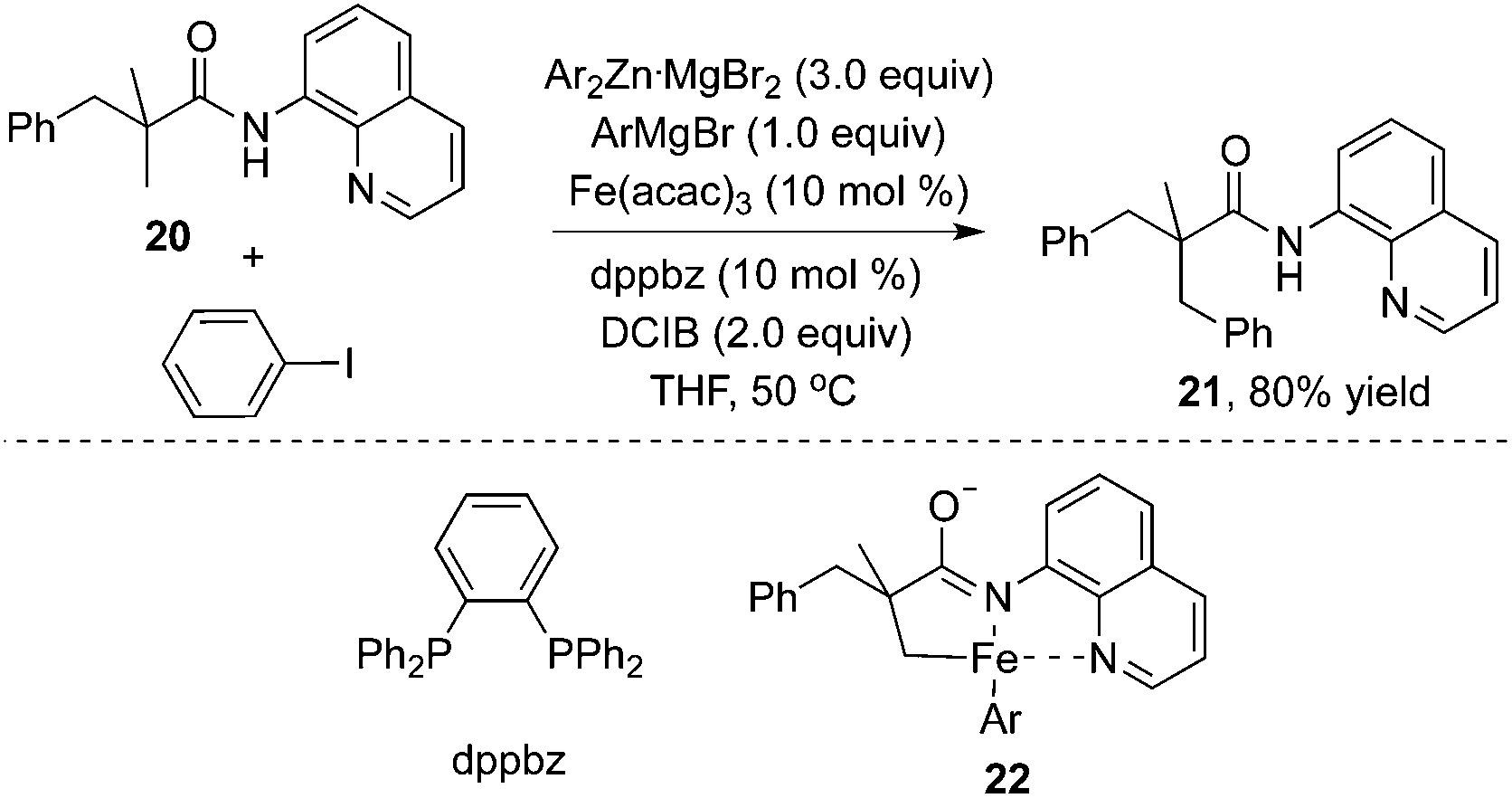
Additionally, an iron catalyst could be applied in the β-arylation of 8-aminoquinoline-modified 2,2-disubstituted propoinamide 20 (Scheme 7).12a In the transformation, an arylzinc reagent was used as the aryl source, and 1,2-dichloroisobutane (DCIB) acted as an organic oxidant. Mechanistic studies indicated that the reaction did not undergo a radical process, and the double cycloferrum species 22 was proposed as the key intermediate. The phosphine ligand (dppbz) was found to be effective for the reactions. The exploration of the substrate scope revealed that the substituent of propionamide 20 at the α position was critical for the β-arylation process. From Chatani's finding, nickel salt was also an efficient catalyst in β-arylation of 8-aminoquinoline-modified 2,2-disubstituted propoinamide 20, although β-diarylation was mainly observed during the process.12b
 | ||
| Scheme 7 Iron-catalyzed β-monoarylation. | ||
Shi and co-workers reported that diarylhyperiodium 24 was an efficient aryl source in the palladium-catalyzed β-arylation of 8-aminoquinoline-modified propoinamide 23 (Scheme 8).13a The reactions worked well with a broad substrate scope, and the substrates with α-H were compatible in the reactions. Compared with Daugulis's report, Shi's work employed K2CO3 as a base, devoid of the use of silver acetate. An N-heterocyclic carbene-coordinated palladium complex was used as the catalyst. Based on the mechanistic studies, it seemed that C–H activation was the rate-determining step in the process. The substrate scope suggested that the substituent R2 made a great impact on the chemoselectivity of the transformation. When R2 was replaced by a non-hydrogen group, the reaction was prone to afford the β-monoarylative product, while the reaction furnished a mixture of β-monoarylation and β-diarylation (25c, 84% total yield) when R2 was replaced by a hydrogen atom. Interestingly, the β-diarylation reaction of cyclic substrate 23b was observed, yielding product 25b in 52% yield in a cis fashion. A similar study was reported by Chatani and co-workers, where a nickel complex was used as the catalyst.13b
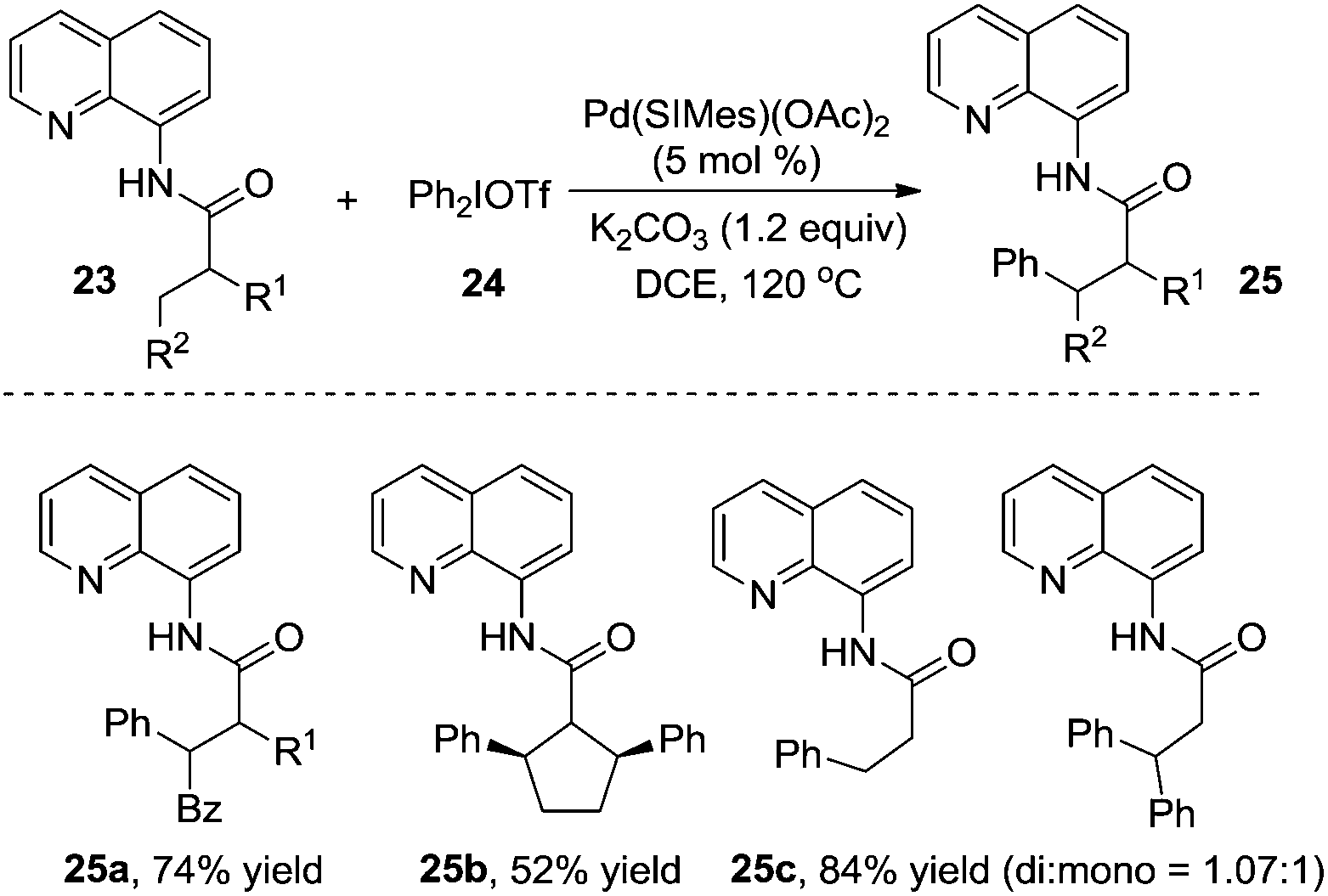 | ||
| Scheme 8 β-Arylation using diarylhyperiodium as the aryl source. | ||
According to Wu and Zeng's studies, the aryl source could be expanded to less reactive aryl bromides (Scheme 9).14 A mechanistic study of DFT calculations revealed that the oxidative addition of a substrate was the rate-determining step, and an energetically feasible C(sp3)–H bond activation-led pathway was proposed. Similar to previous studies, the substituent of R imposed a significant effect on the final outcome of reactions, especially on the selectivity between monoarylation and diarylation. As illustrated in Scheme 9, the β-monoarylation reactions were achieved when R was equal to non-hydrogen groups. Cyclic alkyl amide 26b and 8-aminoquinoline-assisted amide 26c were diarylated, leading to the β-diarylative products 28b and 28c in 59% and 91% yields, respectively. Aryl bromides including heterocyclic aryl bromides were suitable in the transformation.
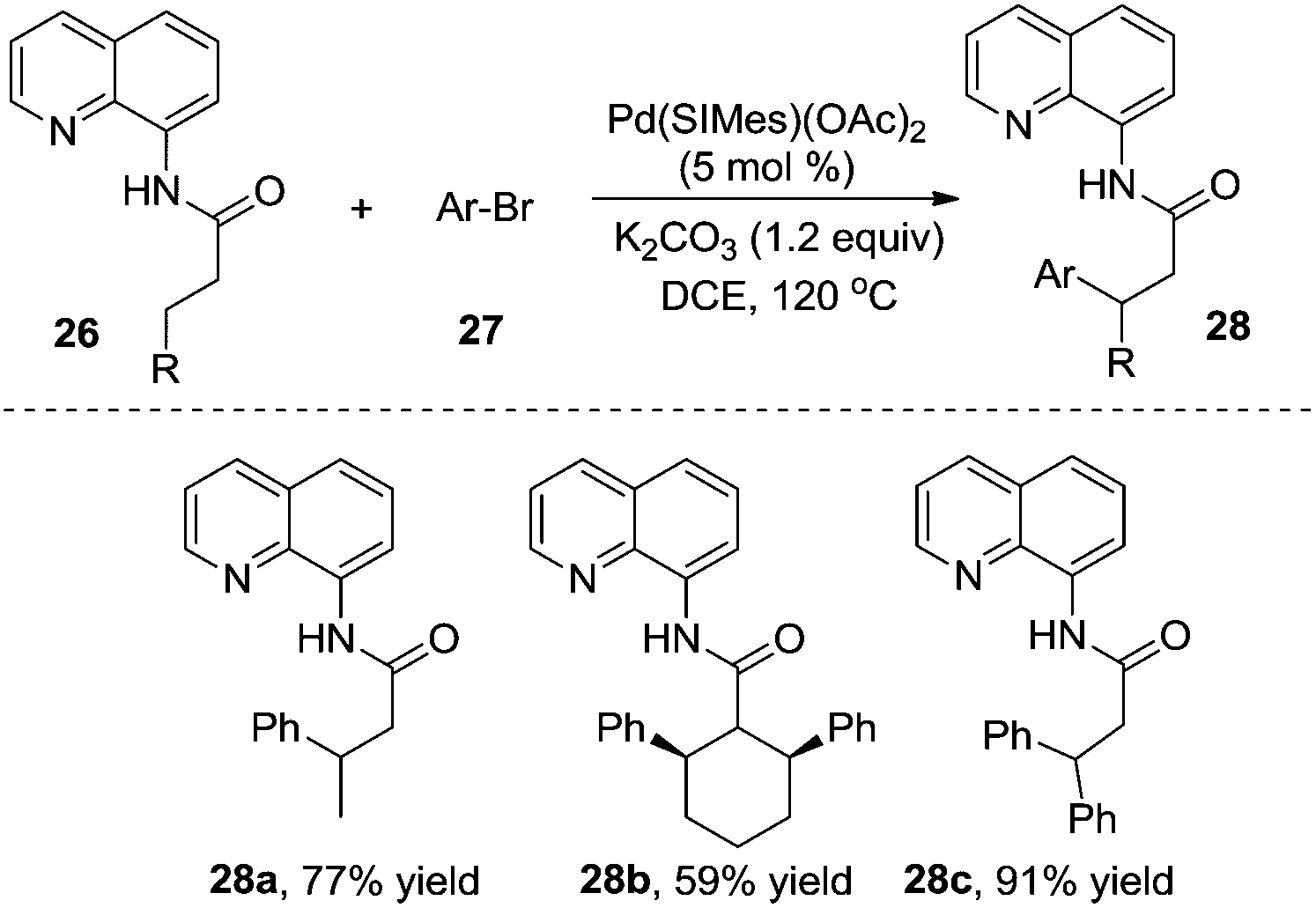 | ||
| Scheme 9 β-Arylation using aryl bromide as the aryl source. | ||
Intramolecular β-arylation of the C(sp3)–H bond in aliphatic amides was developed by Chen's group.15 8-Aminoquinoline was still selected as a removable auxiliary, and sterically hindered 2-phenylbenzoic acid was used as an additive (Scheme 10). During the reaction process, a PdII/PdIV catalytic cycle was proposed.
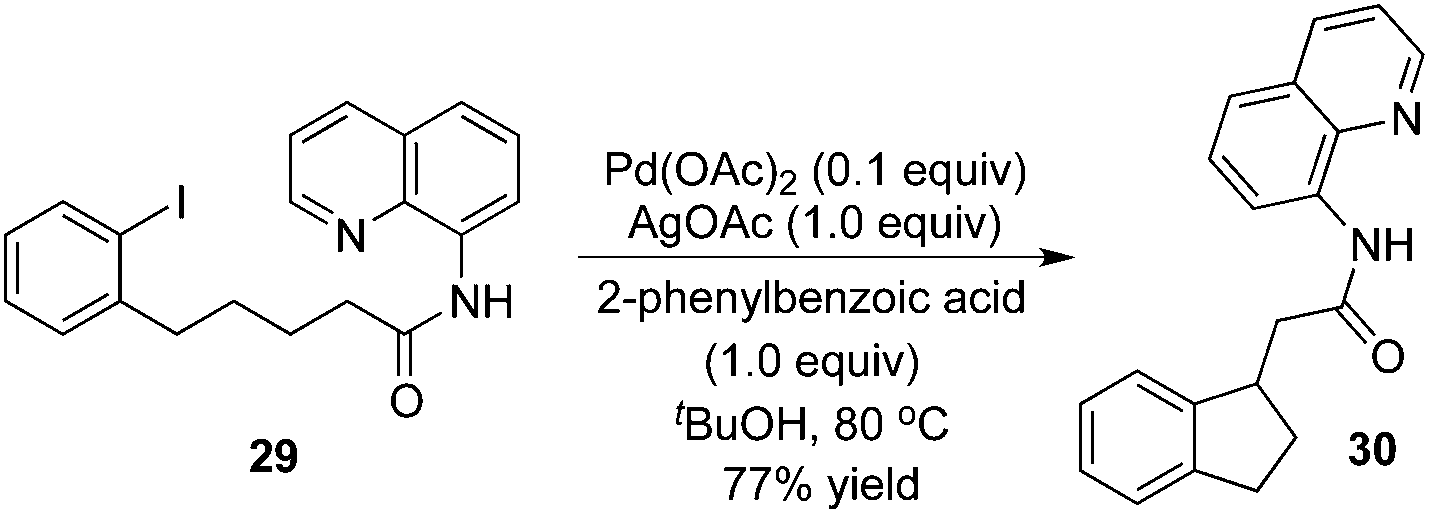 | ||
| Scheme 10 Intramolecular β-arylation. | ||
Based on Yu's results, the β-arylation of pivalic acid 31 with phenylboronic reagent 32 proceeded smoothly without the assistance of auxiliaries, producing the desired β-arylated carboxylic acid 33 in 38% yield (Scheme 11).16 In the process, the in situ formation of carboxylate directing groups was crucial for the β C–H bond activation and subsequent coupling with phenyl boronic reagent 32.
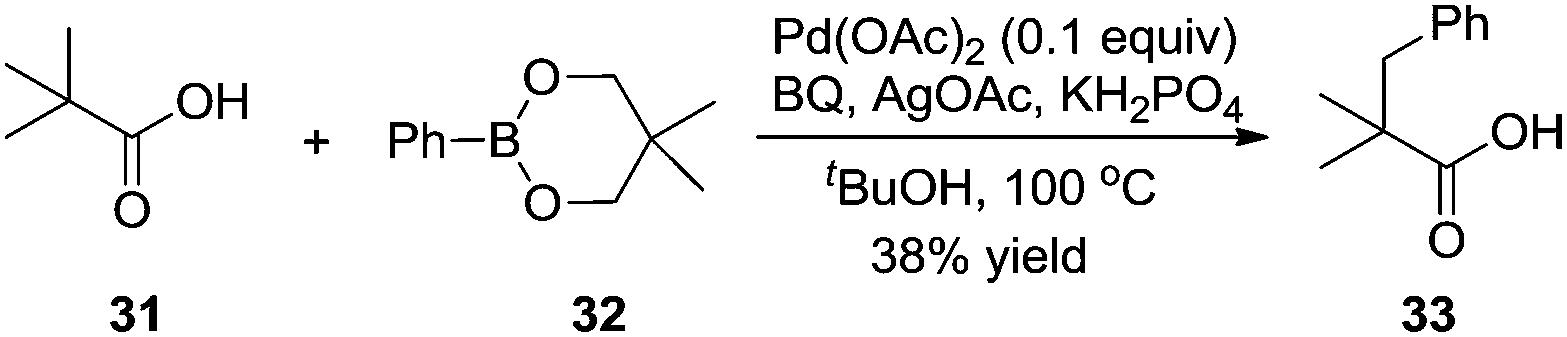 | ||
| Scheme 11 β-Arylation of carboxylic acid without the directing group. | ||
By introducing a stronger binding O-methyl hydroxamic acid, the yield of the transformation could be increased to 85% under milder reaction conditions.17 As shown in Scheme 12, the reaction proceeded by using silver oxide/BQ as the oxidant. Aryl and alkyl boronic acids were all compatible for the β-functionalization of the alkyl group. Since the –CONHOMe group could be readily converted into ester, amide, and hydrogen the synthetic utility of this reaction could be expected.
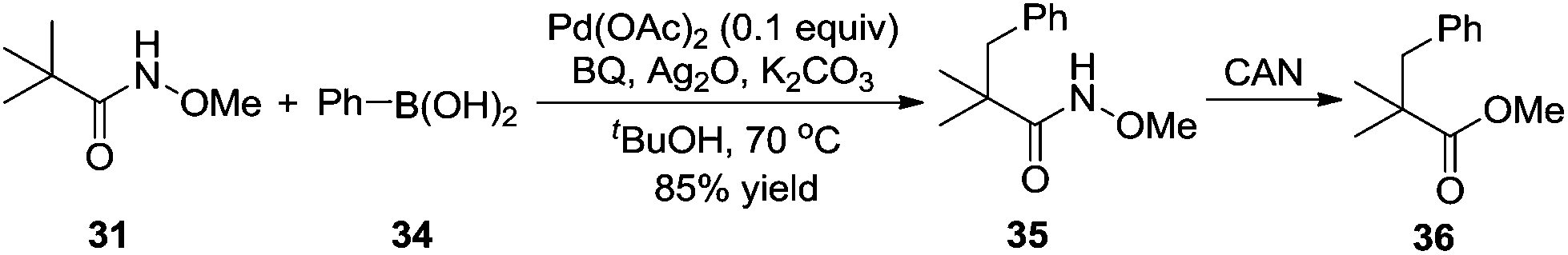 | ||
| Scheme 12 β-Arylation of carboxylic acid directed by O-methylhydroxamic acid. | ||
Surprisingly, the reaction of O-methyl hydroxamic acid 31 catalyzed by Pd(0)/PR3 did not release the desired β-arylated products by changing aryl boronic acid to aryl halide as the aryl source. The products from Buchwald–Hartwig amination were obtained instead. To minimize this competitive pathway, the pentafluorophenyl group was introduced into carboxamide 37. As expected, the reactions of carboxamide 37 with aryl iodides in the presence of Pd(OAc)2 and Cy-JohnPhos afforded β-arylated products in good yields. From the optimization conditions, CsF was the effective base in the β-arylation of carboxamides containing α-hydrogen, while the substrates without α-hydrogen worked well in the presence of Cs2CO3. The scope exploration indicated that the reactivity of the primary C–H bond was higher than that of the secondary one, thus the β-arylated product was not obtained from the methylene C(sp3)–H bond. An insight into the mechanism suggested that acidic N–H in the directing group was essential for the reactivity. The intermediate 39 was proposed as the key precursor for C–H bond activation (Scheme 13).18
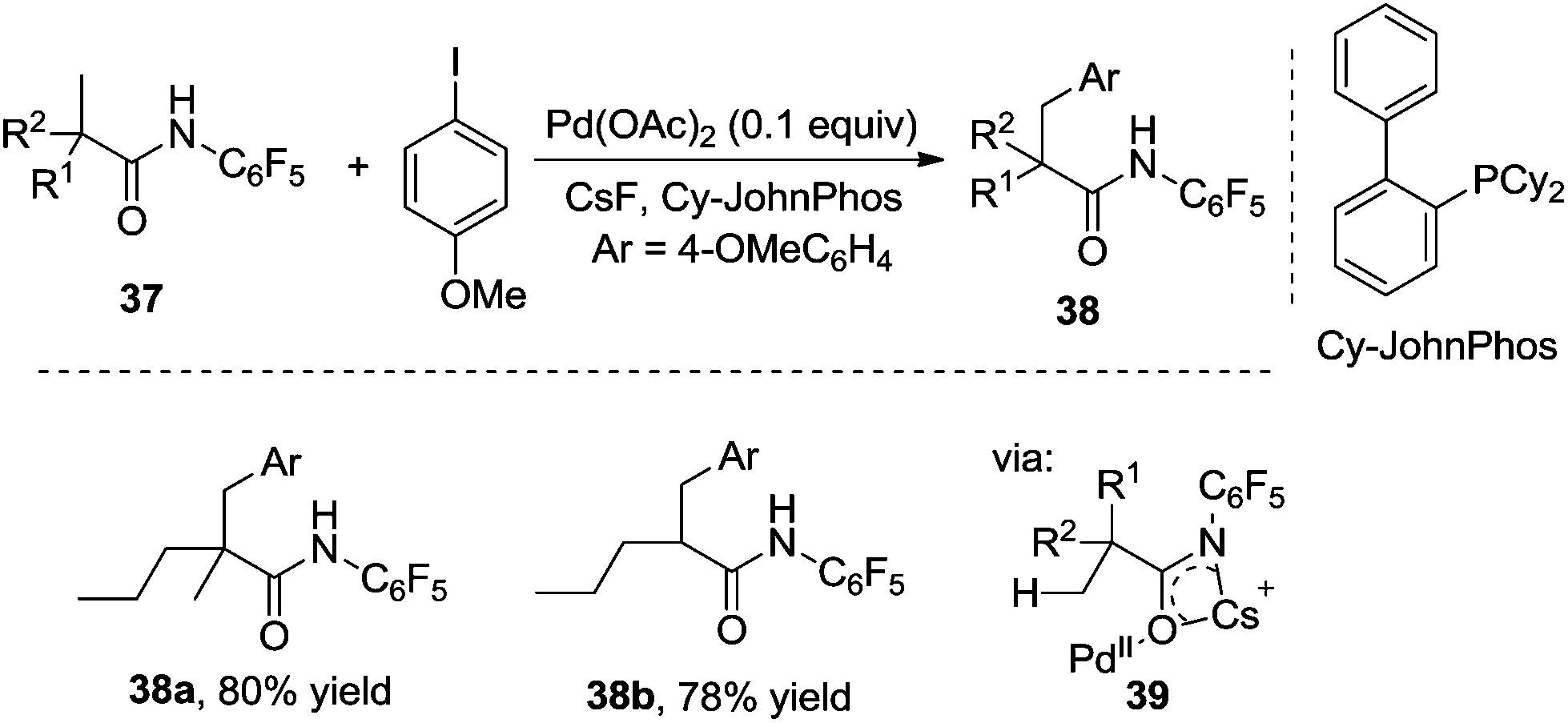 | ||
| Scheme 13 β-Arylation of carboxylic acid directed by the pentafluorophenyl group. | ||
By tuning the ligand and further increasing the acidity of the N–H bond in carboxamide, this reaction could be extended to β-arylation of the methylene C(sp3)–H bond in carboxamide 41. According to Yu's finding, 2-isobutoxylquinoline 42 was the optimized ligand in direct β-arylation with full conversion and a high diastereotopic ratio of monoarylation and diarylation. Particularly, a single diastereomer was obtained when carboxamide 41 took part in the β-arylation reactions (Scheme 14).19a The finding from Yu's group indicated that a weak ligand such as 42 was crucial for such β-functionalization of the unactivated Csp3–H bond. Encouraged by these findings, Yu and co-workers demonstrated that substituted pyridines were excellent ligands for β-arylation,19b,c olefination,19d and methylation (Fig. 2).19e Additionally, amino acid was also an excellent ligand in β-functionalization of the unactivated Csp3–H bond.19f,g For example, enantioselective β-arylation of cyclopropanecarboxylic acid derivatives was accomplished by employing chiral amino acid as a ligand.19f
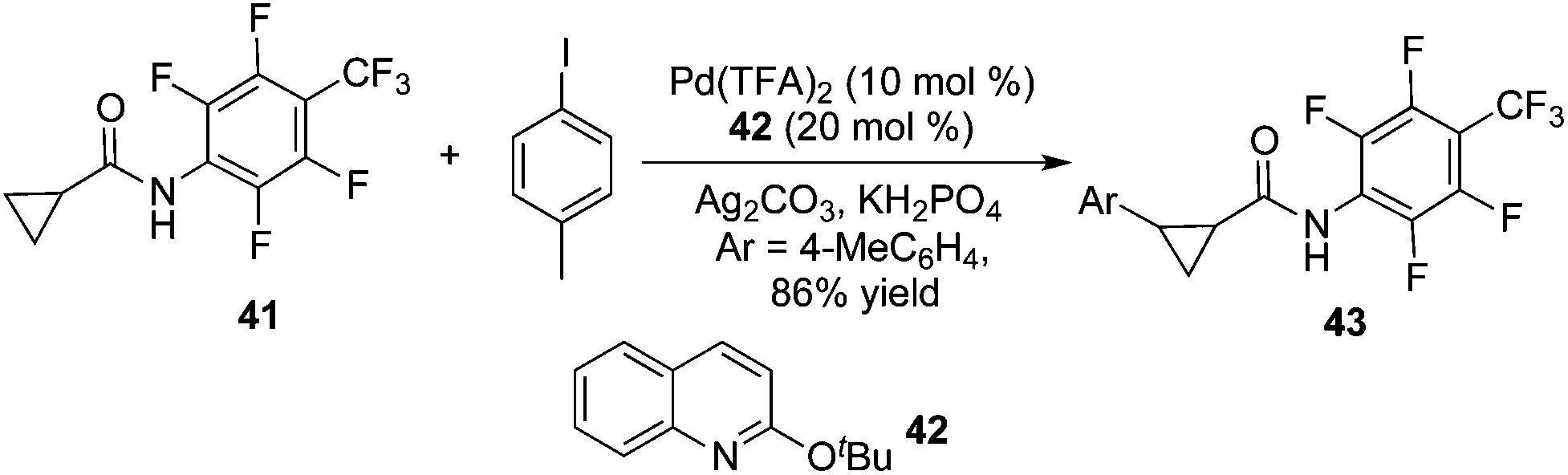 | ||
| Scheme 14 β-Arylation of the methylene C(sp3)–H bond directed by the pentafluorophenyl group. | ||
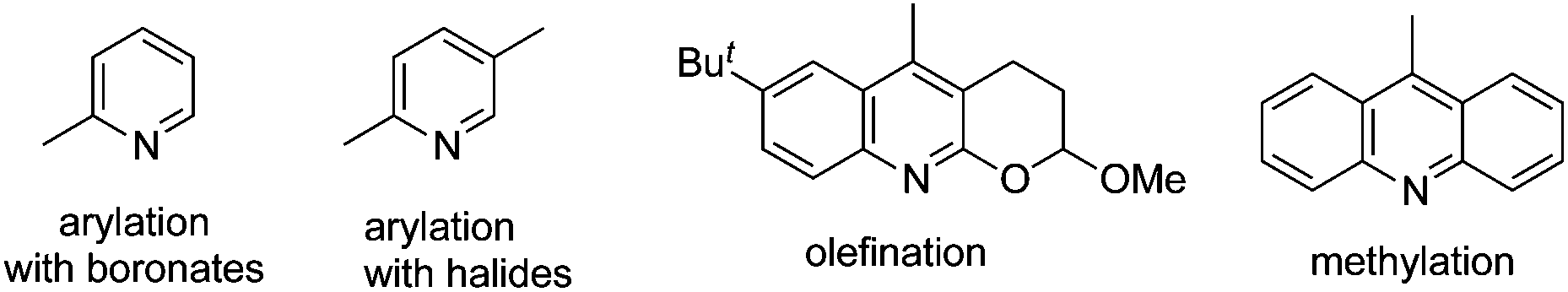 | ||
| Fig. 2 Pyridine-based ligands. | ||
Intramolecular C(sp3)–H bond activation of the alkyl group in carboxamide 44 was developed by Shi and co-workers.20 In the transformation, various 3,4-dihydroquinolinones were produced in moderate to good yields (Scheme 15). Mechanistic studies of the kinetic isotope effect revealed that C(sp3)–H bond activation was the rate-determining step, and a Pd(0)/Pd(II) process was proposed. The pallacycle 46 was demonstrated as the “real” catalyst.
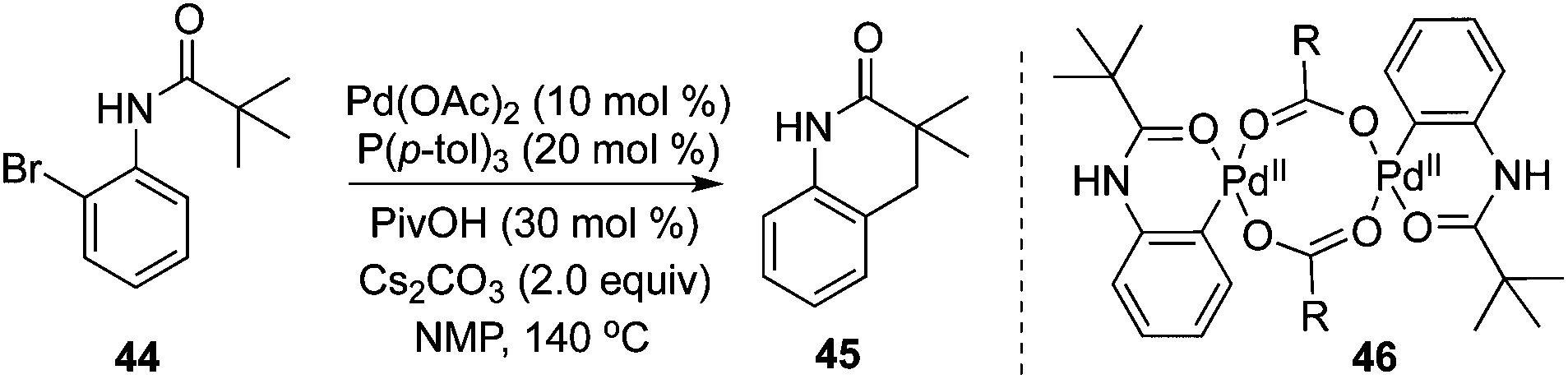 | ||
| Scheme 15 Intramolecular C(sp3)–H bond activation of the alkyl group in carboxamide. | ||
In 2010, Clot and Baudoin developed a mild and efficient intermolecular β-arylation of the unactivated C(sp3)–H bond in carboxylic ester 47, affording a series of synthetically useful functionalized ester enolates 48 (Scheme 16).21 Evaluation of the substrate scope revealed that the reaction was limited to aryl halides containing electronegative substituents at the ortho position. meta-Substituted substrates gave a mixture of α-arylation and β-arylation. Computational studies suggested that oxidative addition, α C–H bond insertion, β-hydride elimination, C–C double bond rotation, β-hydride insertion, and reductive elimination were involved in the process.
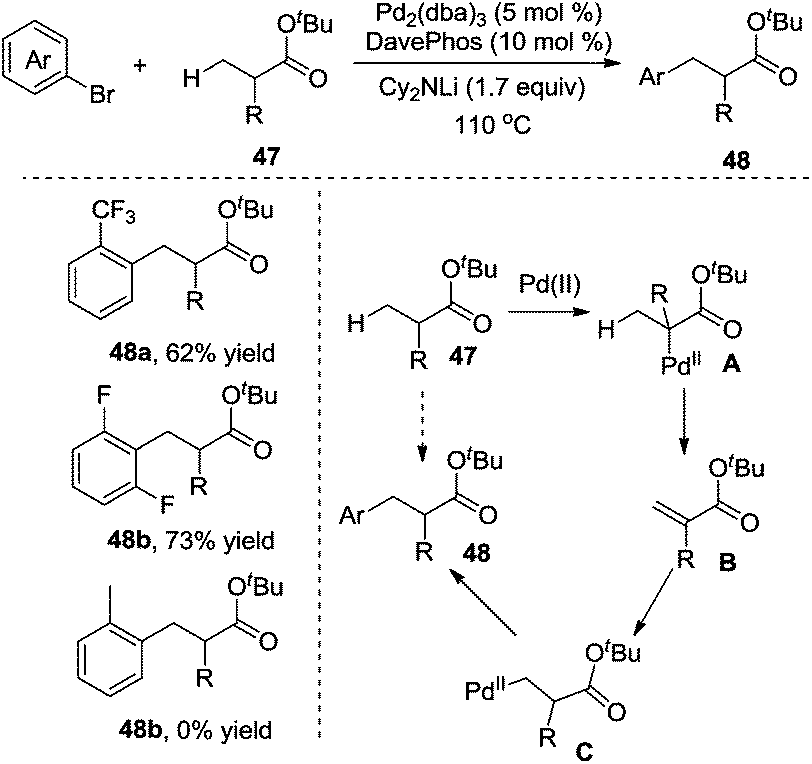 | ||
| Scheme 16 Intramolecular β-arylation of ester with aryl bromide. | ||
To address the generality of the transformation, the same group further explored the direct β-arylation of protected alanine esters 49 (Scheme 17).22N-Protecting groups were critical in the reactions. N-Benzyl-protected alanine ester gave the desired product in 78% yield, while no reaction occurred for N-methyl protected alanine esters. meta-Substituted alanine esters were suitable for the β-arylation.
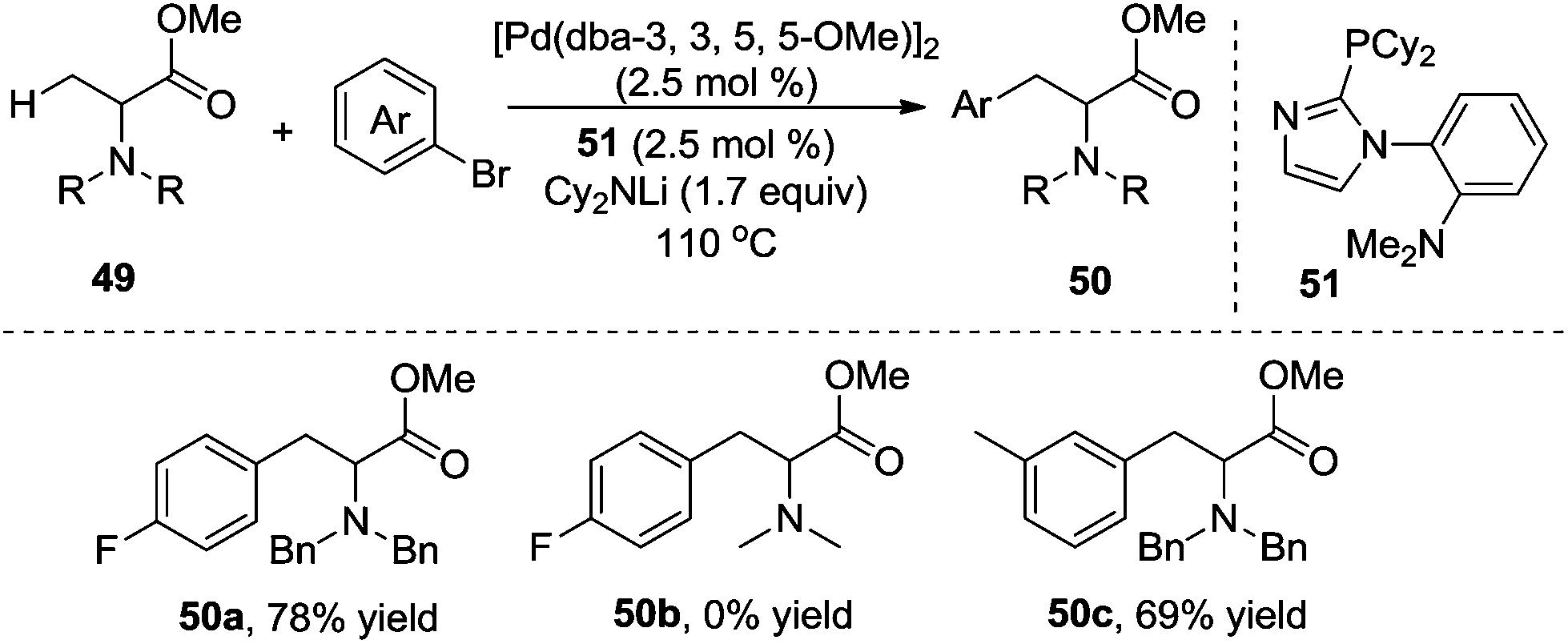 | ||
| Scheme 17 Intramolecular β-arylation of ester with aryl bromide. | ||
Additionally, β-indolylation of keto ester 52 was studied by Pihko and co-workers.23 As presented in Scheme 18, β-functionalization of keto ester 52 was achieved. Mechanistic studies indicated that an indole-assisted dehydrogenation was involved (Scheme 19).
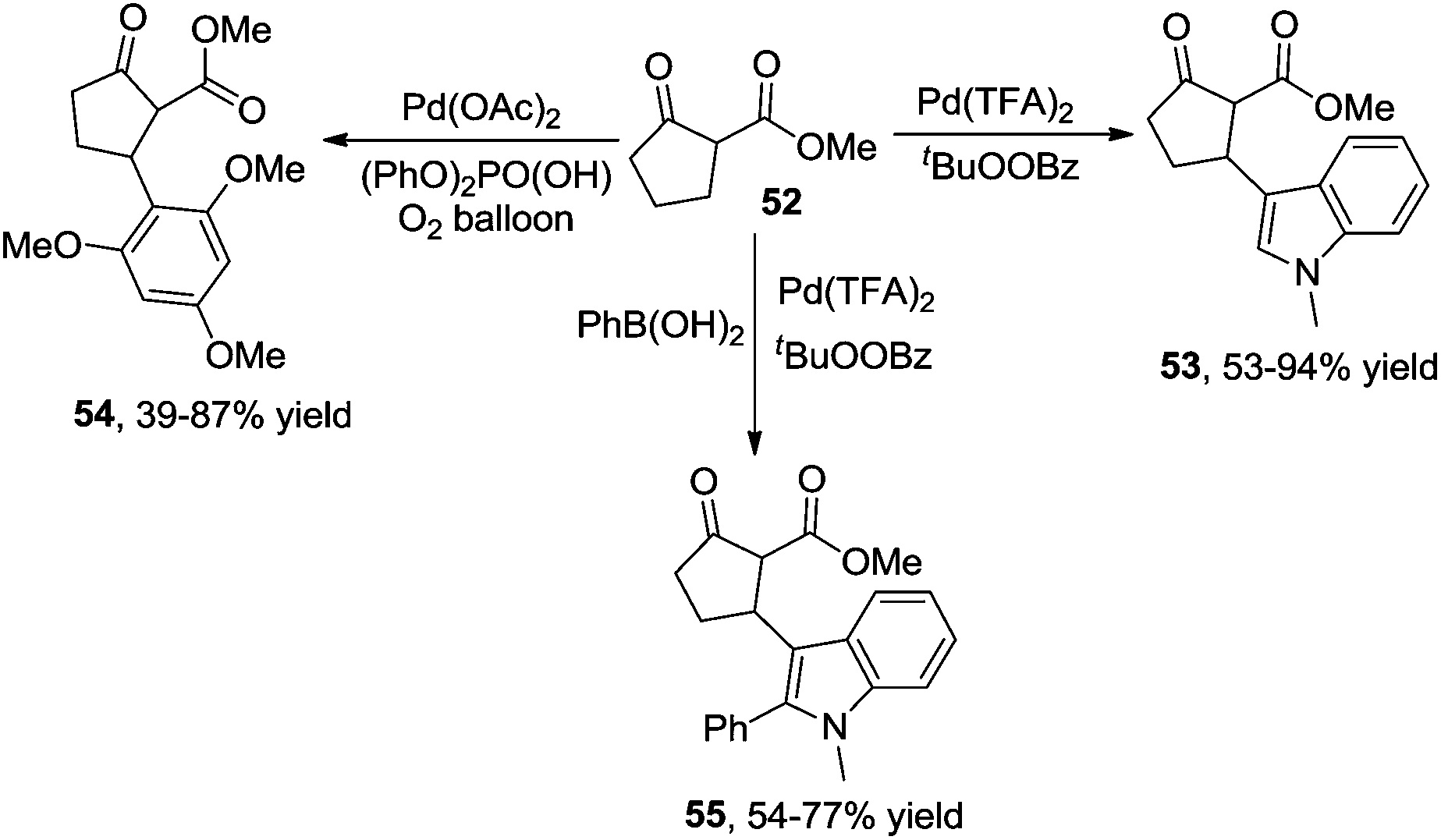 | ||
| Scheme 18 β-Functionalization of keto ester. | ||
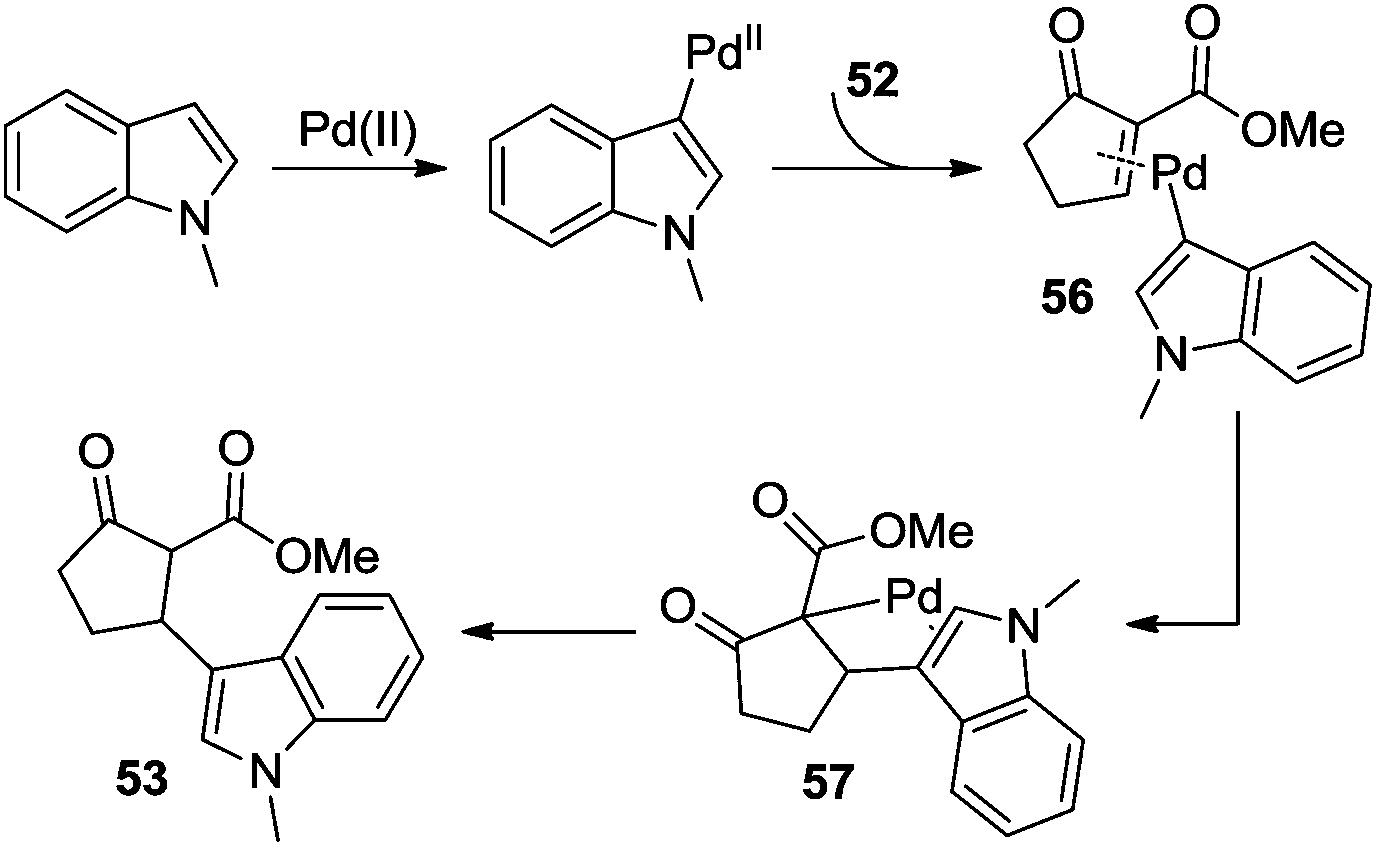 | ||
| Scheme 19 Mechanism for the β-indolylation of keto ester. | ||
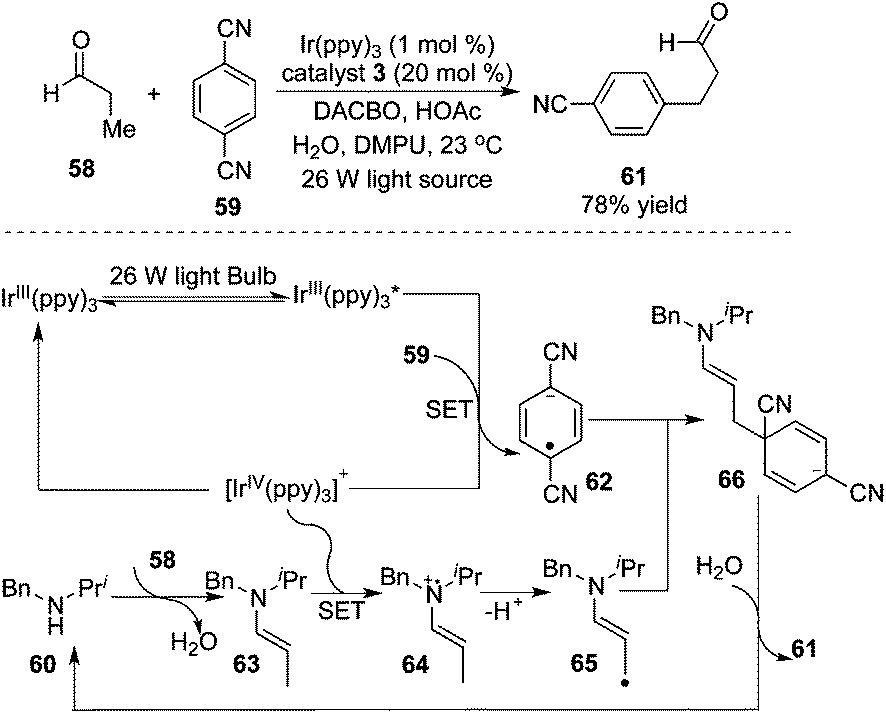
As illustrated in Scheme 20, MacMillan and co-workers devoted themselves to the understanding of “accelerated serendipity”.24 The photoredox strategy was initially employed to realize β-arylation of saturated aldehyde 58. The optimized procedure revealed that the transformation proceeded smoothly in the presence of a 26 W fluorescent light bulb by using 1,4-dicyanobenzene 59 as the aryl source and [Ir(ppy)3] as the photoredox catalyst. In this process, an unprecedented 5πe− activation mode that capitalized on a synergistic merger of photocatalysis and amine organocatalysis was proposed. The formation of enamines was critical for the generation of 5πe− intermediate 65, thus determining the practicality of the transformation. The optimal photocatalyst of [Ir(ppy)3] was shown to be indispensable. Interestingly, the generality and scope of this photo-catalyzed β-arylation reaction were electron effect-sensitive. Specifically, aldehydes with electron-donating groups at the β-position such as p-methoxylphenyl were favourable to β-arylation coupling, while the use of the substrates with an electron-deficient nature (such as 4-cyanophenyl) did not provide the desired products, probably due to the low nucleophilicity of the corresponding enaminyl radical. This observation could account for why mono-arylation of aldehydes and ketones took place exclusively. The cyclic ketones could be amenable to this β-arylation coupling also by switching the organocatalyst to azepane, and an excellent diastereoselectivity of the product was observed.
 | ||
| Scheme 20 β-Arylation of aldehyde via photocatalysis. | ||
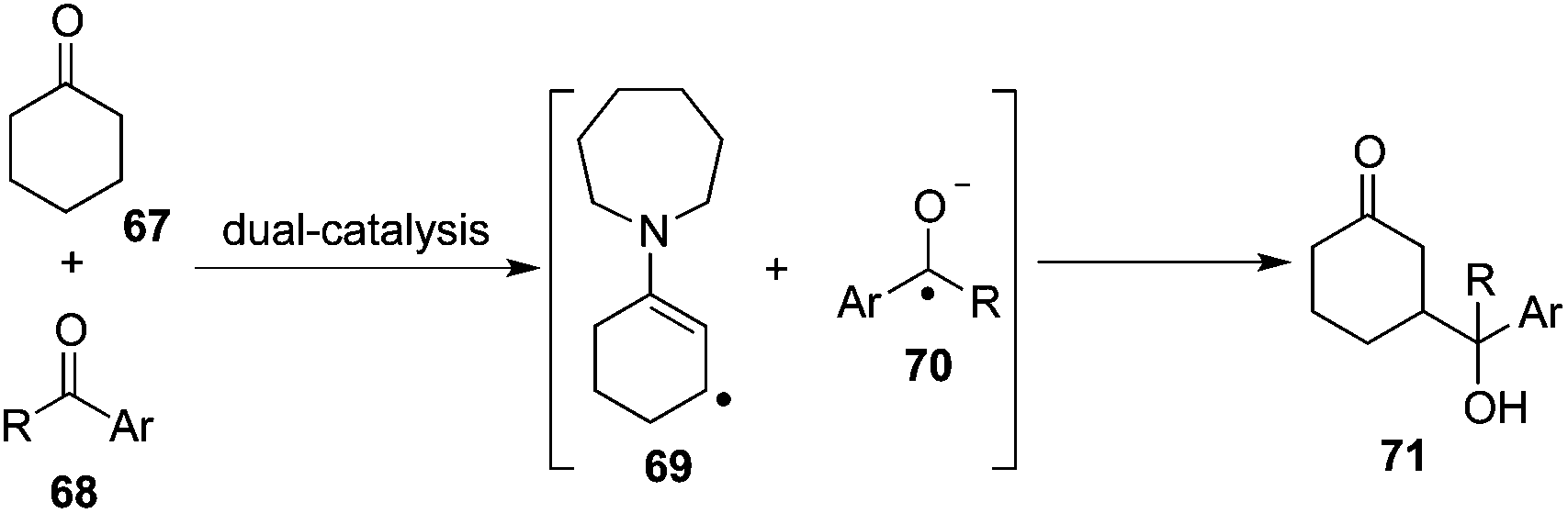
This dual-catalysis design mentioned above relies on the coupling of transiently formed β-enaminyl radical 65 with a radical anion generated from aryl nitriles 61. As expected, the reaction of cyclic ketone 67 with aryl ketone 68 involving the interception of nucleophilic β-enaminyl radical 69 with ketyl radical 70 was postulated to complement this β-functionalization reaction via two concurrently catalytic cycles of photocatalysis and organocatalysis, delivering γ-hydroxyketone architecture 71 (Scheme 21).25
 | ||
| Scheme 21 β-Functionalization under photocatalysis and organocatalysis. | ||
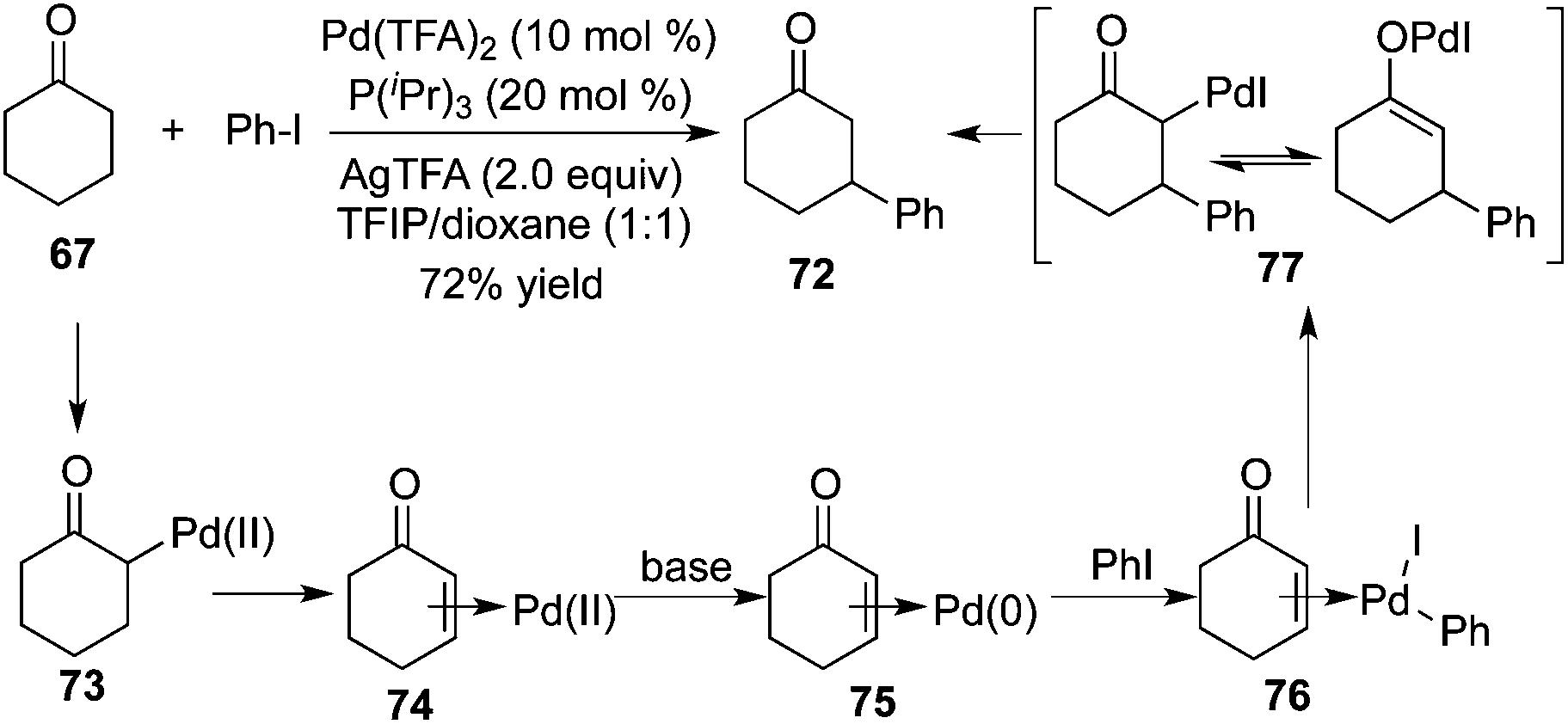
Prompted by the photoredox strategy, Dong and co-workers reported a procedure allowing the direct β-arylation of ketone 67 in the presence of palladium catalysis (Scheme 22).26 This method renders the combination of the palladium-catalyzed ketone oxidation, aryl halide activation, and conjugate addition reliable. The mechanistic insight revealed that each of the ligands, bases, and solvents played a positive role in this β-arylation of cyclic ketones as well as acyclic ketones. Moreover, an electron-rich mono-dentate ligand P(iPr)3 could inhibit the radical dimerization. The presence of a base (AgTFA) could enhance the halide–carboxylate exchange, and a mildly acidic solvent (TFIP) was crucial for the regeneration of the palladium catalyst by protonation of Pd-enolate 72.
 | ||
| Scheme 22 β-Arylation of ketones. | ||
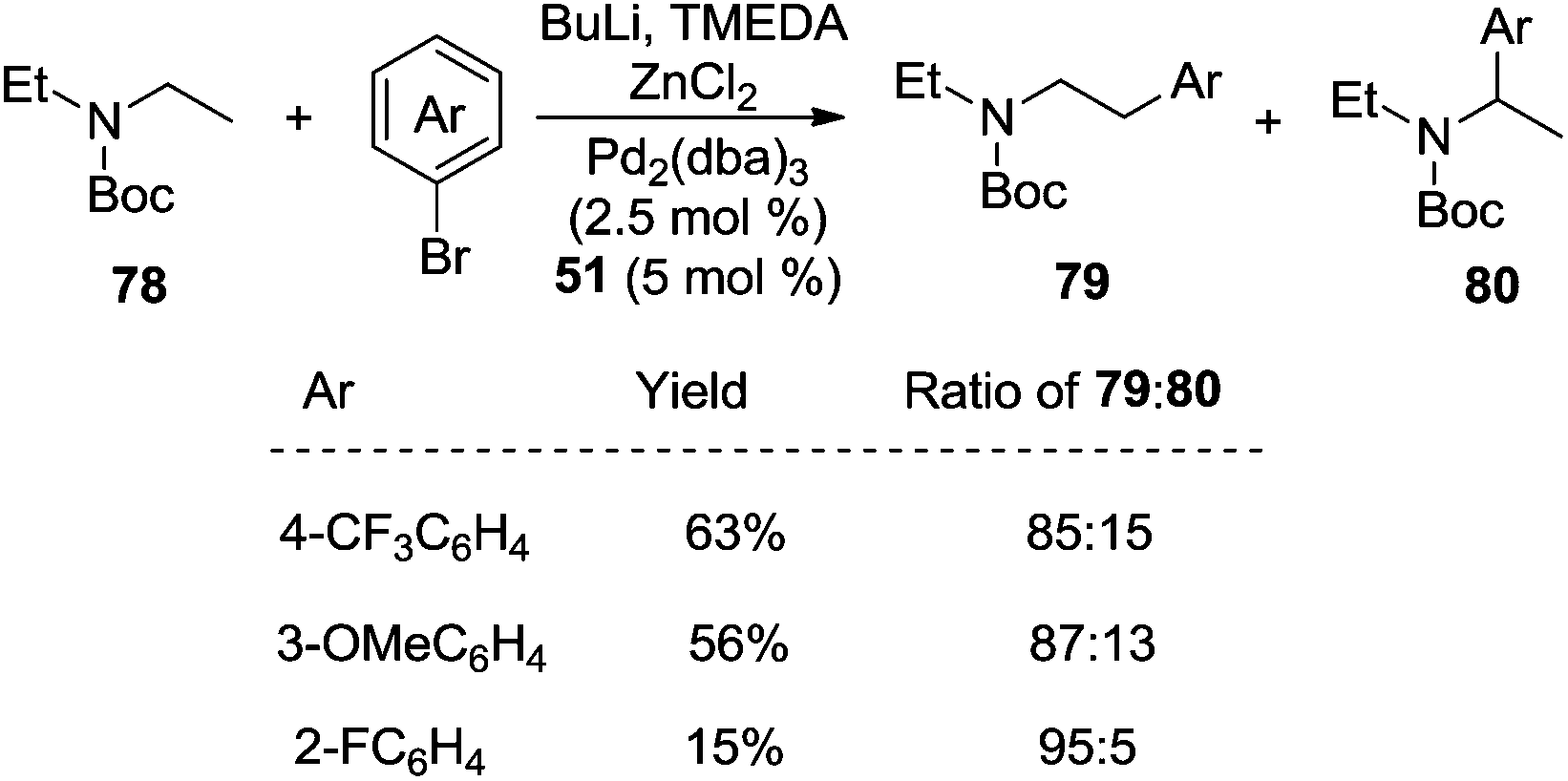 | ||
| Scheme 23 β-Arylation of N-protected amines. | ||
2.2 Direct alkynylation/alkylation at the β position of alkyl groups
In addition to the direct β-arylation of alkyl groups, direct β-alkylation and β-alkynylation were also developed. The breakthrough of aliphatic C–H alkynylation was accomplished by Chatani and co-workers in 2011.29 In the reactions, 8-aminoquinoline was selected as a removable auxiliary to conduct β-alkynylation. The reactions worked well for electrophilic alkynylation of the secondary C–H bond and tolerated a broad range of substituents including alkyl, aryl, methoxyl, and trifluoromethyl (Scheme 24). A Pd(II)/Pd(IV) catalytic cycle was believed to be involved in the process.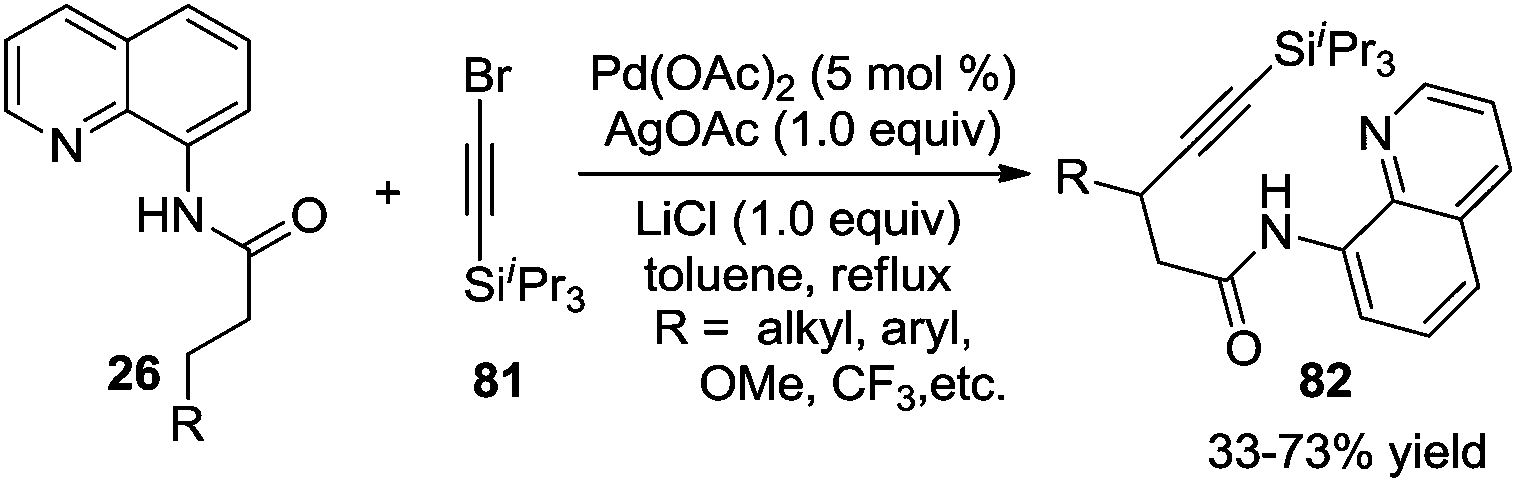 | ||
| Scheme 24 β-Alkynylation of carboxamide. | ||
Subsequently, Yu and co-workers reported a Pd(0)-catalyzed β-alkynylation of the C(sp3)–H bond, which was different from previous studies. They used CON(4-CF3C6F4) as a directing group. The direct β-alkynylation reaction occurred through primary C–H bond activation, and a distinctive pathway involving a Pd(0)/Pd(II) was proposed (Scheme 25).30
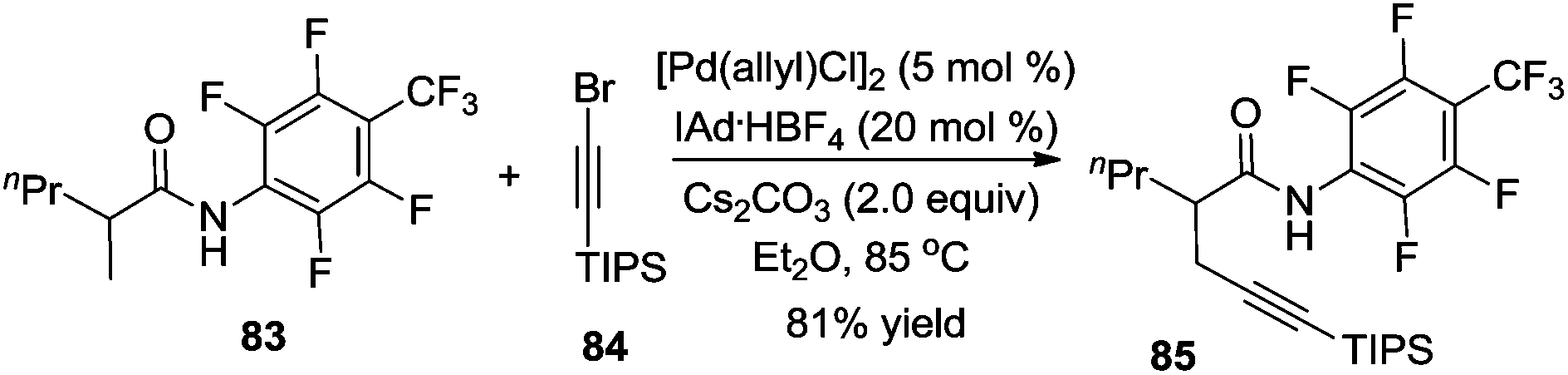 | ||
| Scheme 25 β-Alkynylation of carboxamide using CON(4-CF3C6F4) as a chelating group. | ||
8-Aminoquinoline-assisted direct β-alkylation of alkyl groups was highlighted by Daugulis and Ge, respectively (Scheme 26).31a,b In Daugulis's work, palladium-catalyzed direct β-benzylation could be suitable for the substrates with the α C–H bond, while the substrates without the α C–H bond were compatible in Ge's process. Recently, Shi and co-workers found that the substrates with α C–H bonds could be employed in the alkylation of unactivated methylene Csp3–H bonds when sodium cyanate and 4-chlorobenzenesulfonamide were used as the additives.31c
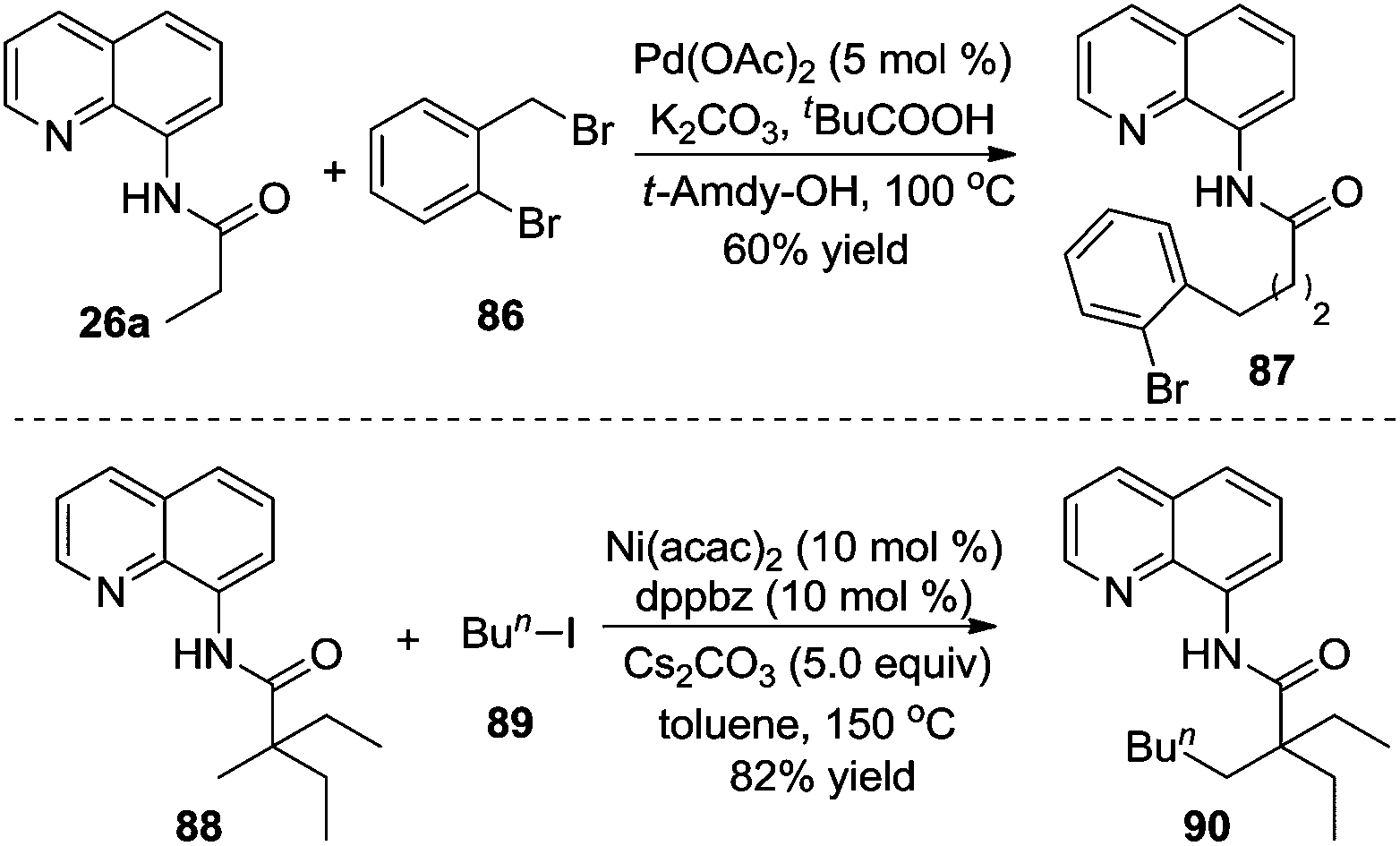 | ||
| Scheme 26 Palladium and nickel-catalyzed β-alkylation of carboxamide. | ||
3 Formation of the C–O bond via direct β-functionalization
In 2004, Sanford and co-workers used O-methyl oxime as a directing group to investigate the β-acetoxylation of pinocalone 91 (Scheme 27).32 The reaction proceeded efficiently in the presence of Pd(OAc)2 (5 mol%), PhI(OAc)2 (1.1 equiv.), and HOAc or Ac2O (50 mol%) at 100 °C. However, the scope exploration revealed that the direct β-acetoxylation only worked well for the substrates with the primary β C–H bond. According to the results from Sukbok and co-workers, direct β-amidation of the unactivated methyl C–H bonds in substrate 91 could be realized when an iridium complex was used as the catalyst and azides were employed as the amidyl source. The reactions provided a protocol for late-stage C–H functionalization of complex molecules in synthetic and medicinal chemistry.33 | ||
| Scheme 27 β-Acetoxylation of pinocalone. | ||
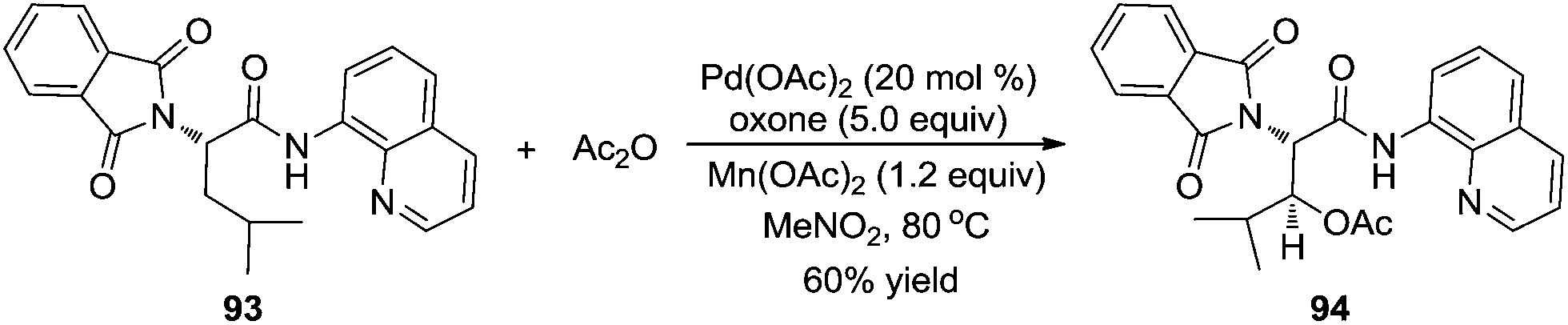
Subsequently, Corey and co-workers described a direct β-acetoxylation of N-phthaloyl-α-alanine derivative 93, where 8-aminoquinoline was employed as the directing group (Scheme 28).34 Compared with Sanford's work, this direct β-acetoxylation used Mn(OAc)2 as a promoter. In the presence of Pd(OAc)2, oxone, acetic anhydride, and Mn(OAc)2 in CH3NO2 at 80 °C, the acetoxylative N-phthaloyl-α-alanine derivative 94 was afforded efficiently.
 | ||
| Scheme 28 β-Acetoxylation of the N-phthaloyl-α-alanine derivative. | ||
By switching the oxidant to hypervalent iodides, sequential double C–H alkoxylation of unactivated methyl groups was developed by Rao and co-workers. Evaluation of reaction conditions indicated that the addition of silver carbonate was crucial for improving the reaction efficiency.35
4 Conclusions and outlook
This review is focused on the recent advances in transition metal-catalyzed direct remote C–H functionalization of alkyl groups via C(sp3)–H activation. In general, directing groups in the molecules play a crucial role in the reactions. From a mechanistic perspective, 8-aminoquinoline has been described as an efficient auxiliary in remote C–H bond activation, probably because of the readily formed dicyclometallic species. Amides with electron-drawing groups in combination with weak ligands such as substituted pyridines are also demonstrated as promising directing groups in the β-Csp3–H bond activation. To date, the efficiency and regioselectivity of remote C–H activation have been achieved, and some biologically interesting architectures are constructed by using these direct β-functionalization reactions. The high efficiency with good selectivity would make this type of transformation attractive in organic synthesis. It is believed that more general, efficient, and facile procedures for direct C–H functionalization at the β-position of alkyl groups will appear in the near future, which will be widely applied for the formation of carbon–carbon bonds and carbon–heteroatom bonds.Notes and references
- For selected reviews, see (a) W. Song, S. Kozhushkov and L. Achermann, Angew. Chem., Int. Ed., 2013, 52, 6576 CrossRef CAS PubMed; (b) Y. Deng, A. Persson and J.-E. Bäckvall, Chem. – Eur. J., 2012, 18, 11498 CrossRef CAS PubMed; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (d) J. Wencel-Dclord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (e) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- For selected reviews, see (a) M.-L. Louillat and F. W. Patureau, Chem. Rev., 2014, 43, 901 RSC; (b) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (c) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed.
- For selected reviews, see (a) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed; (b) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS PubMed; (c) D. W. Robbins and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 933 CrossRef CAS PubMed; (d) S. Zhang, L. Shi and Y. Ding, J. Am. Chem. Soc., 2011, 133, 20218 CrossRef CAS PubMed; (e) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed; (f) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS PubMed; (g) F. Juliá-Hernández, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458 CrossRef PubMed; (h) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed; (i) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed.
- For selected reviews, see (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- For selected reviews, see (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (b) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (c) G. Yan and A. J. Borah, Org. Chem. Front., 2014, 1, 838 RSC; (d) Z. Huang and G. Dong, Tetrahedron Lett., 2014, 55, 5869 CrossRef CAS PubMed; (e) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC.
- D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657 CrossRef CAS PubMed.
- X. Chen, C. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- (a) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (b) B. Wang, W. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3952 RSC; (c) D. Affron, O. Davis and J. Bull, Org. Lett., 2014, 16, 4956 CrossRef CAS PubMed.
- Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958 CrossRef CAS PubMed.
- (a) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030 CrossRef CAS PubMed; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898 CrossRef CAS PubMed.
- (a) F. Pan, P.-X. Shen, L.-S. Zhang, X. Wang and Z.-J. Shi, Org. Lett., 2013, 15, 4758 CrossRef CAS PubMed; (b) M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014 DOI:10.1021/JO501691f.
- Y. Wei, H. Tang, X. Cong, B. Rao, C. Wu and X. Zeng, Org. Lett., 2014, 16, 2248 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS PubMed.
- R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed.
- D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed.
- M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS PubMed.
- (a) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570 CrossRef CAS PubMed; (b) J. He, S. Li, Y. Deng, H. Fu, B. Laforteza, J. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (c) Y. Deng, W. Gong, J. He and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 6692 CrossRef CAS PubMed; (d) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS PubMed; (e) R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194 CrossRef CAS PubMed; (f) M. Wasa, K. Engle, D. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef CAS PubMed; (g) K.-J. Xiao, D. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed.
- J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 4945 CrossRef CAS PubMed.
- (a) A. Renaudat, L. Jean-Gerard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261 CrossRef CAS PubMed; (b) P. Larini, C. E. Kefalidis, R. Jazzar, A. Re audat, E. Clot and O. Baudoin, Chem. – Eur. J., 2012, 18, 1932 CrossRef CAS PubMed.
- S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808 CrossRef CAS PubMed.
- (a) M. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750 CrossRef CAS PubMed; (b) R. Nimje, M. Leskinen and P. M. Pihko, Angew. Chem., Int. Ed., 2013, 52, 4818 CrossRef CAS PubMed; (c) M. Leskinen, A. Madarasz, K.-T. Yip, A. Vuorinen, I. Papai, A. Neuvonen and P. M. Pihko, J. Am. Chem. Soc., 2014, 136, 6453 CrossRef CAS PubMed; (d) K.-T. Yip, R. Nimje, M. Leskinen and P. M. Pihko, Chem. – Eur. J., 2012, 18, 12590 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed.
- F. Petronijević, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323 CrossRef PubMed.
- Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747 CrossRef CAS PubMed.
- A. Millet, D. Dailler, P. Larini and O. Baudoin, Angew. Chem., Int. Ed., 2014, 53, 2678 CrossRef CAS PubMed.
- T. Watanbe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2008, 10, 1759 CrossRef PubMed.
- Y. Ano, M. Tohisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed.
- J. He, M. Wasa, K. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed.
- (a) E. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689 CrossRef CAS PubMed; (b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789 CrossRef CAS PubMed; (c) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed.
- L. Desai, K. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed.
- T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed.
- B. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed.
- Y. Zong and Y. Rao, Org. Lett., 2014, 16, 5278 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |