
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A γ-cyclodextrin duplex connected with two disulfide bonds: synthesis, structure and inclusion complexes†
Sergey
Volkov
a,
Lukáš
Kumprecht
a,
Miloš
Buděšínský
a,
Martin
Lepšík
a,
Michal
Dušek
b and
Tomáš
Kraus
*a
aInstitute of Organic Chemistry and Biochemistry AS CR, v.v.i., Flemingovo nám. 2, 166 10 Prague 6, Czech Republic. E-mail: kraus@uochb.cas.cz
bInstitute of Physics, AS CR, v.v.i., Na Slovance 2, 182 21 Praha 8, Czech Republic
First published on 12th January 2015
Abstract
Per(2,3,6-tri-O-benzyl)-γ-cyclodextrin was debenzylated by DIBAL-H to produce a mixture of C6I,C6IV and C6I,C6V isomeric diols, which were separated and isolated. The C2-symmetrical C6I,C6V diol was transformed into dithiol and dimerized to produce a γ-cyclodextrin duplex structure. A crystal structure revealed tubular cavity whose peripheries are slightly elliptically distorted. The solvent accessible volume of the cavity of the γ-CD duplex is about 740 Å3. Due to this large inner space the duplex forms very stable inclusion complexes with steroids; bile acids examined in this study show binding affinities to the γ-cyclodextrin duplex in the range of 5.3 × 107 M−1–1.9 × 108 M−1.
Introduction
Complexation of molecules and ions, both organic and inorganic, by receptors with high affinity and selectivity in an aqueous environment plays a key role in various biological processes. Chemists discovered a broad range of synthetic analogues that were designed to mimic these processes. Within macrocyclic receptors operating in aqueous media, cyclodextrins1 (CDs) play important roles due to their abilities to form host–guest complexes with a wide range of organic molecules. The binding of guest molecules inside CDs is assumed to be driven mainly by the hydrophobic effect (favorable solvation entropy due to the release of host-bound water molecules upon complexation) and van der Waals interactions between the guest and non-polar parts of the cavity.2 It was suggested that the free energy of binding in such host–guest systems is proportional to the surface area of the guest buried upon binding.3 Since cavities of CDs are relatively shallow (≈6–7 Å) they do not allow complete inclusion of molecules longer than one benzene ring preventing thus a strong binding unless additional attractive interactions, such as ionic4 or coordination5 bonding with suitably modified CD hosts, are involved. Consequently, binding affinities of native CDs mostly fall in the 102–103 M−1 interval3,6 limiting the scope of their applications in fields such as drug delivery systems or in supramolecular analytical chemistry,7 namely in fluorescent indicator displacement assays,8 operating at low concentrations in a complex biological environment.9,10In order to improve binding affinities of CDs, dimeric constructs were prepared with the enlarged inner cavity by means of a connection of two CD macrocycles together via single or multiple linkers. Singly-bridged dimers11 revealed binding affinities significantly higher as compared to native CDs in some cases, yet the cavities acted independently12,13 in other cases due to the large flexibility of the single connection, yielding 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with usual stabilities known for native CDs. To prevent flexibility, rotational freedom of the two CDs was restricted by the introduction of additional linking groups. These molecules, termed duplex CDs14 (or CD duplexes15), were composed of α- or β-cyclodextrin units connected with two bridges of variable length and with varying mutual orientations of the bridged macrocycles.14–25 In addition, α-cyclodextrin duplexes connected with three26 and six27 linkers have been reported. Nevertheless, only few examples of significantly increased binding capabilities are known,25,26,28 supposedly due to non-optimal spacing of the two macrocyclic cavities or low solubilities of the duplexes.
:1 complexes with usual stabilities known for native CDs. To prevent flexibility, rotational freedom of the two CDs was restricted by the introduction of additional linking groups. These molecules, termed duplex CDs14 (or CD duplexes15), were composed of α- or β-cyclodextrin units connected with two bridges of variable length and with varying mutual orientations of the bridged macrocycles.14–25 In addition, α-cyclodextrin duplexes connected with three26 and six27 linkers have been reported. Nevertheless, only few examples of significantly increased binding capabilities are known,25,26,28 supposedly due to non-optimal spacing of the two macrocyclic cavities or low solubilities of the duplexes.
We have recently described the syntheses and properties of new host systems composed of two α-CD or β-CD macrocycles linked with disulfide bonds.25,26,29–31 These tube-like molecules showed unusually high binding affinities (Ka up to 1010 M−1) to various organic compounds from hydrophobic α,ω-alkanediols25,26 to fairly hydrophilic medium-sized molecules such as imatinib.31 Now, we have turned our attention to a larger γ-CD homologue. In this work, we report the synthesis, crystal structure and binding properties of a novel tubular receptor consisting of two γ-CDs linked with two disulfide bonds on their primary rims in a head-to-head manner designed for the complexation of larger organic molecules, such as steroids, in aqueous media.
Results and discussion
Synthesis
In our previous studies25,31 on dimerization of α-CD or β-CD macrocycles, the C6I,C6IV-disulfanyl α-CD or β-CD precursors were prepared by a sequence of synthetic transformations starting from perbenzylated α-CD or β-CD that were selectively debenzylated at C6I and C6IV positions by a DIBAL-H promoted procedure.32,33 This method allowed cleavage of benzylic groups in positions C6I,C6IV of perbenzylated α-CD or β-CD macrocycles with high selectivities and yields. However, preparation of pure selectively bis(de-O-benzylated) γ-CD homologue(s) has not been reported yet. Thus, in order to prepare the required C2-symmetrical C6I,C6V-disulfanyl γ-CD we needed to develop a procedure for the preparation of C6I,C6V-debenzylated γ-CD.Starting perbenzyl-γ-CD 1 (Scheme 1) was prepared by exhaustive benzylation of anhydrous γ-CD with benzyl chloride in DMSO using sodium hydride as a base. Next, we investigated the course of the debenzylation reaction at various concentrations of DIBAL-H as we had observed earlier29,31 with smaller α-CD or β-CD homologues that more concentrated solutions allow smoother cleavage at or below room temperature. We found that 3 M solution of DIBAL-H in toluene allowed partial control over the extent of debenzylation allowing the isolation of a mixture of products containing perbenzyl-γ-CD diols de-O-benzylated at C6I,C6IV and C6I,C6V positions, respectively, which could be separated by combined column and HPLC chromatographies. Thus, the treatment of perbenzyl-γ-CD 1 with 3 M solution of DIBAL-H in toluene for 41 hours at 0 °C gave a mixture of two isomeric diols 2a and 2b, which could be resolved by TLC on silica in a dichloromethane–acetone mixture. On a preparative scale, the symmetrical isomer 2a was partly separated from the mixture by column chromatography on silica using a mixture of dichloromethane–acetone 98:2, which allowed isolation of 2a in 14% yield. The residual mixture was separated by preparative HPLC using the same solvent system allowing another crop of 2a (10% yield) and 2b (10% yield). In total, pure compounds 2a and 2b were isolated in 24% and 10% yields, respectively. The identification of the constitution of the isomers was then achieved by NMR methods; compound 2a possesses C2 symmetry axis, hence 1H NMR spectrum reveals four signals of H-1 protons (Fig. 1a), whilst the C1-symmetrical isomer 2b shows eight doublets, one for each H-1 proton (Fig. 1b). The C6I–C6IV substitution pattern in C1-symmetrical isomer 2b, which was proposed with help of “hex-5-enose method” in the first report32 on selective de-O-benzylation of CDs, is consistent with the full NMR assignment achieved in this work. For further transformations, only the isomer 2a was used. Conversion of the free hydroxyl groups of 2a to bromides was achieved by the action of triphenylphosphane and tetrabromomethane in DMF allowing the isolation of the corresponding dibromide 3 in 76% yield. Subsequently, benzyl groups were removed by hydrogenation using palladium on charcoal as a catalyst in a DMF–ethanol mixture25 and compound 4 was isolated in 71% yield. Reaction of dibromide 4 with potassium thioacetate in DMF at room temperature for 12 hours gave rise to the acetylated disulfanyl derivative 5 in 73% yield. Subsequent alkaline hydrolysis of 5 under an argon atmosphere allowed isolation of the disulfanyl derivative 6 in 66% yield.
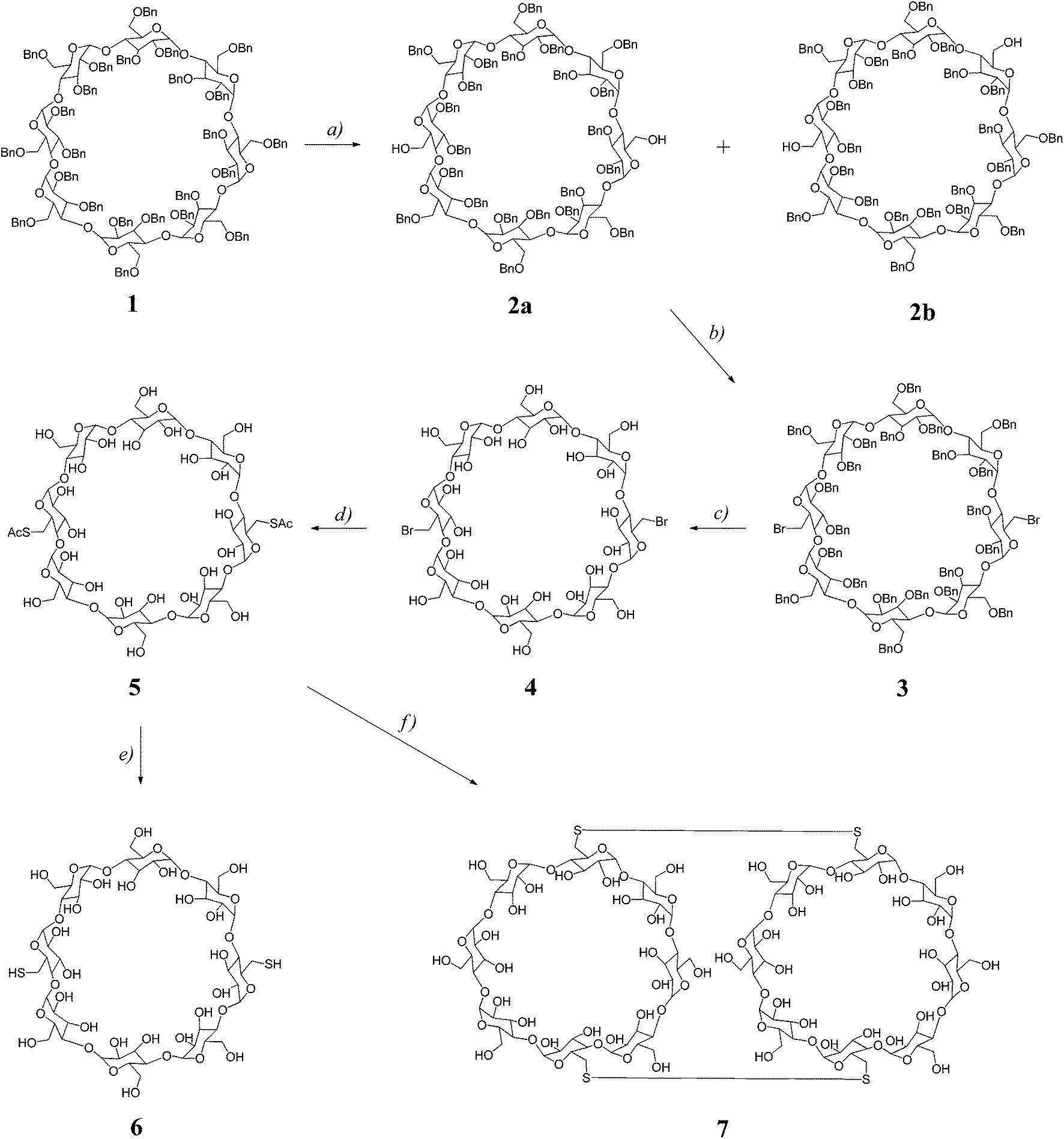 | ||
| Scheme 1 Reaction conditions: (a) DIBAL-H, toluene, 0 °C, 41 hours, yield – see text; (b) CBr4, Ph3P, DMF, 65 °C, 4 hours, 76%; (c) Pd/C, 40 bar, DMF–EtOH, 6 hours, r.t., 71%; (d) CH3COSK, DMF, 12 hours, r.t., 73%; (e) NaOH, H2O, 5 hours, r.t., 66%; (f) (1) NaOH, H2O, 5 hours, r.t., (2) NaHCO3–Na2CO3 aq. buffer, pH 9, air atmosphere, 72 hours, 82%. | ||
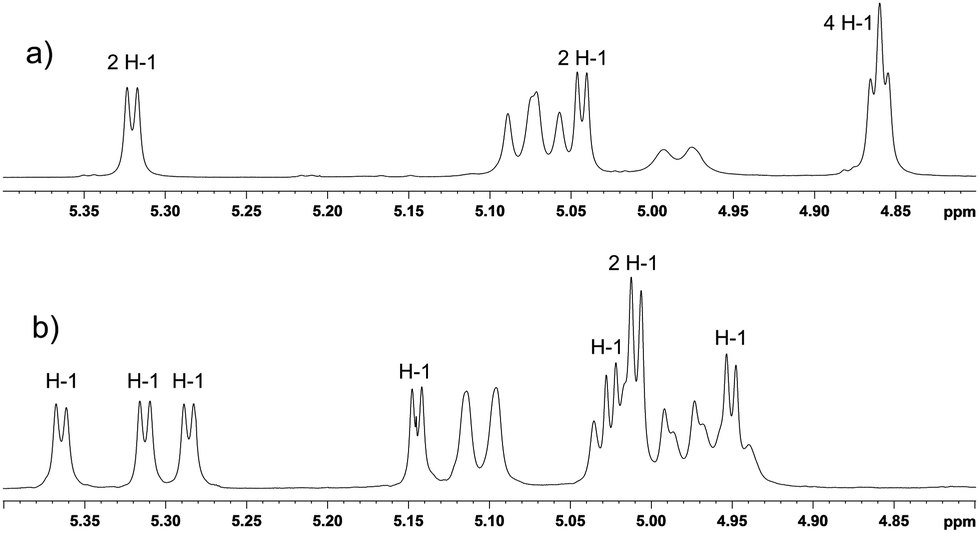 | ||
| Fig. 1 Part of 1H NMR (600 MHz) spectra showing H-1 protons of: (a) compound 2a, (b) compound 2b. Non-labeled signals belong to benzylic protons. | ||
The oxidative dimerization experiments were performed under the conditions described in our previous reports,25,31i.e., in aqueous solution at pH ∼ 9, in a concentration range 0.1–10 mM. For practical reasons the acetylated disulfanyl derivative 5 was used as the starting material for dimerization and it was deprotected in situ by treatment with aqueous solution of sodium hydroxide (0.17 M) under an inert atmosphere after which the solution was diluted to the required concentration. The pH of the solution was adjusted to ∼9 by the introduction of gaseous carbon dioxide and the mixture was exposed to air. Monitoring of the oxidation reaction by reversed phase TLC revealed the formation of one product accompanied – above approximately one millimolar concentration – with another material, presumably oligomeric/polymeric by-products, which showed no mobility on an RPTLC plate. The analysis of the major product isolated by reversed phase chromatography revealed its dimeric structure. The structure was confirmed by MALDI–HRMS, NMR and X-ray analysis of a single crystal. In contrast to our previous studies carried out with smaller α-CD or β-CD homologues the presence of a monomeric product (intramolecular disulfide) was not detected, or this by-product remained hidden with inseparable polymeric material. The preparative run was carried out at 0.8 mM concentration of the starting thioacetate 5. The product precipitated out of the solution upon neutralization and could be isolated by simple centrifugation in 82% yield. The isolation was possible due to the relatively low solubility of the product – approx. 0.1 mM in water or in a phosphate buffer at pH 7.
The above described procedure allowed the isolation of both C6I,C6V and C6I,C6IV debenzylated isomers 2a and 2b, the former being then converted to the required C2-symmetrical dithiol 6. In addition to this strategy for the synthesis of duplex 7, we have explored two alternative pathways avoiding somewhat laborious separation of the isomers 2a and 2b. The first approach relies on the use of the mixture of isomers 2a and 2b for the subsequent two synthetic steps and separation of isomeric 6I,6V- and 6I,6IV-dibromo-γ-CDs by means of reversed-phase chromatography. Alternatively, the whole synthesis was carried out with the unseparated mixture of 6I,6V and 6I,6IV derivatives which gave rise to a mixture of duplexes from which compound 7 was isolated by reversed phase chromatography. The separation of the mixture of the three other possible non-symmetrical duplexes was not successful. Both approaches allowed somewhat a more efficient separation of the isomeric derivatives at later stages of synthesis than that of diols 2a and 2b. On the other hand, the use of a mixture of isomers precludes an appropriate analysis and characterization of the intermediate compounds (especially by NMR methods) in the course of the synthesis. For this reason, these alternative approaches are not described in detail here, nevertheless the individual synthetic steps are analogous to those described for the transformation of a pure isomer 2a.
Crystal structure
A single crystal suitable for X-ray analysis was grown by the hanging drop method from aqueous dimethyl sulfoxide solutions. Its analyses revealed that the geometry of the parent γ-CD is preserved in the duplex, nevertheless the macrocycles, especially their wider rims, are somewhat elliptically distorted. The lengths of the longer and shorter axes of the pseudoellipse measured between the most proximal opposite C-2 atoms (“a” and “b” in Fig. 2) are 15.3 Å and 12.2 Å, respectively. The length of the duplex expressed as the average internuclear distance (“c” in Fig. 2) between the closest H-3 and H-3′ protons located on the opposite rims of the duplex is 12.1 Å, an analogous distance between O-3 to O-3′ is about 13.5 Å.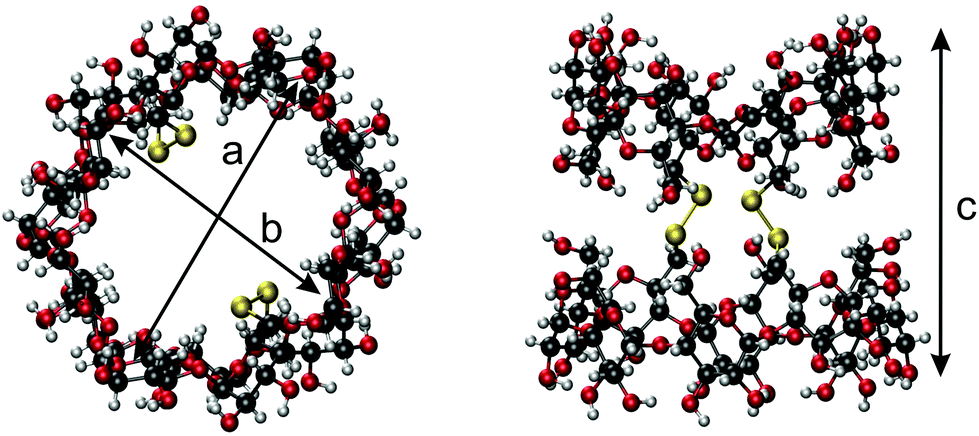 | ||
| Fig. 2 Dimensions of the duplex 7 derived from its crystal structure; the diameters of the elliptically distorted γ-CD macrocycle (a = 15.3 Å3, b = 12.2 Å3) and the length (c = 13.5 Å3) of the duplex structure; see crystal structure discussion for details. | ||
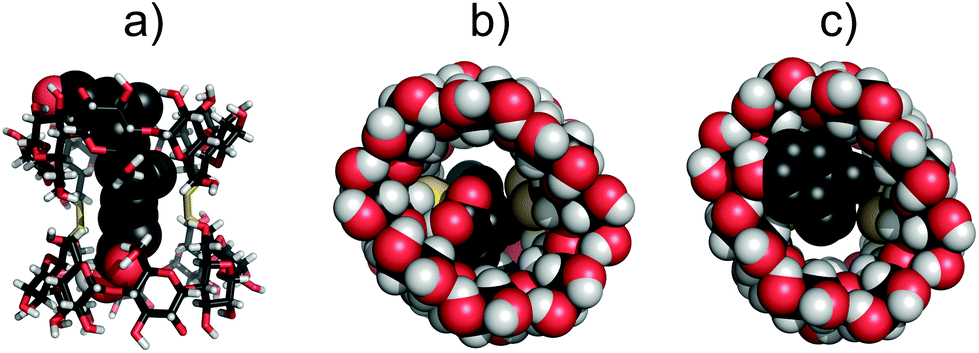
The solvent accessible volume of the inner cavity of duplex 7 in the crystal structure was computed using the Surfnet algorithm34 to be 740 Å3 (Fig. 3). Volumes of the smaller β- and α-CD homologues25,26,31 reported earlier by us were also calculated and compared with that of duplex 7. Two disulfide bond-connected β- and α-CD duplexes25,31 reveal cavities with volumes of 426 Å3 and 296 Å3, respectively. Interestingly, three disulfide bonds in triply-connected α-CD duplex26 close the cavity in the center of the duplex forming two smaller cavities with volumes of 114 Å3.
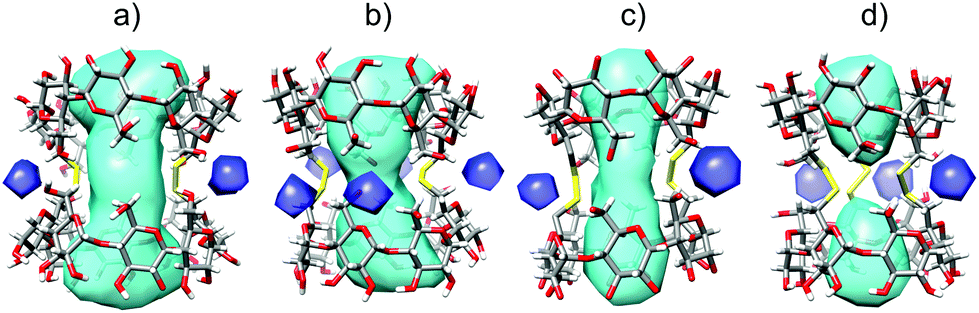 | ||
| Fig. 3 Solvent accessible cavities (cyan) of duplex 7 as calculated using Surfnet. Comparison with smaller homologues. (a) γ-CD duplex 7 (740 Å3); (b) β-CD duplex (426 Å3); (c) α-CD duplex (296 Å3); (d) α-CD duplex connected with three disulfide bonds (2 × 114 Å3). The dark blue-colored spots are small solvent-accessible voids outside the cavity. | ||
Inclusion complexes
The above discussed size parameters of the cavity of duplex 7 indicate that it should be able to efficiently bind larger compounds than its smaller α-CD or β-CD homologues. Thus, the ability to form inclusion complexes was investigated by means of isothermal titration calorimetry in aqueous phosphate buffer at pH 7 using a series of twelve guest compounds that had been used in our previous study31 on analogous β-CD duplexes. Out of that series of compounds, a satisfactory fit to 1:1 model could only be obtained for deoxycholic acid (9) and hexadecafluorodecane-1,10-dioic acid (12, Table 1 and Fig. 4). For other compounds, complexes with more complicated stoichiometry were apparent, for which we were unable to find models allowing a convincing fit. Therefore, we expanded the series of investigated structurally analogous salts35 of bile acids (Fig. 4) to see whether subtle structural changes translate into apparent binding affinities. These were found to be comparable across the series, being in the range of 5.3 × 107 M−1–1.9 × 108 M−1. Although the differences in binding affinities expressed as free energy changes are quite small (0.8 kcal mol−1 between the largest and smallest value), there is a clear distinction between lithocholic acid (8) and the remaining bile acids when comparing their thermodynamic parameters; the complex of 7 with lithocholic acid (8) is strongly enthalpy driven (−11 kcal mol−1) whilst complexes with the other bile acids, in particular with deoxycholic acid (9) and chenodeoxycholic acid (10), show significantly larger entropic components. Lithocholic acid (8) is known36 to form a strong complex with native β-CD (∼106 M−1) that exhibits significantly higher stability than that with 5α-cholanic acid (∼104 M−1), the difference being explained by cis fusion of A and B rings of the former which is apparently more favorable for inclusion into the β-CD macrocycle than trans AB fused 5α-cholanic acid. In our series all bile acids are cis AB fused and thus other structural features must affect the observed behavior. Interestingly, lithocholic acid (8) appears to be the most hydrophobic compound of this series having only one hydroxyl group, whilst deoxycholic (9) and chenodeoxycholic (10) acids have two hydroxyls, and dehydrocholic acid (11) possesses three keto groups on the steroid skeleton. These oxygen atoms could participate in hydrogen bonding with hydroxyl groups on the cyclodextrin skeleton and surrounding water molecules affecting the solvation free energy which would in turn change the thermodynamic signature of the binding process. The duplex 7 thus appears to have some ability to differentiate subtle structural changes in steroid structures, number of hydroxyl groups in particular. The molecular model of the inclusion complex of duplex 7 with lithocholic acid (8) calculated with Autodock Vina37 using crystal structure data of both components revealed a probable mode of inclusion (Fig. 5); the steroid skeleton occupies both cavities of the duplex, in contrast to complexes with more flexible singly-bridged dimers.12,13 It should be noted that within the commonly used native CDs the β-CD macrocycle is known to be a better host for steroids than γ-CD while in the duplex series the reverse order is observed, as can be deduced from this and previous31 studies. Solvent accessible surface models illustrated in Fig. 3 suggest that the disulfide moieties – being oriented inside the cavity – occupy some space in the central part of the cavity and may thus cause steric hindrance. Inclusion of sterically bulkier guests may require expulsion of sulfur atoms from the favorable equilibrium positions through rotation about C5–C6 bond (gauche–trans conformation).31 This unfavorable effect is likely to be more pronounced in smaller β-CD duplex thus accounting for higher binding affinity of steroid compounds to larger γ-CD-duplex 7.
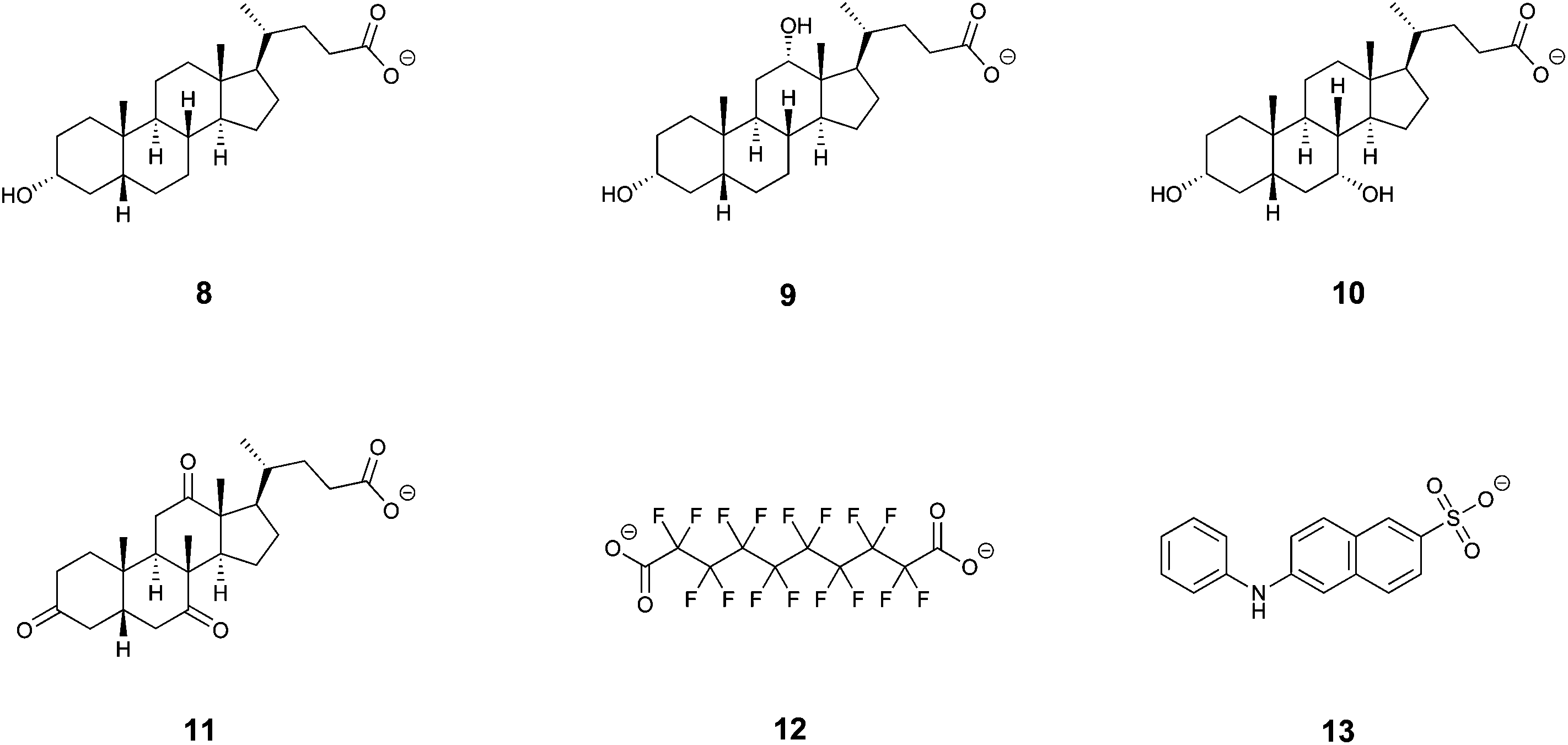 | ||
| Fig. 4 Structure formulas of compounds 8–13 used for the calorimetric study. | ||
 | ||
| Fig. 5 A docking model of a complex of duplex 7 and lithocholic acid (8); (a) side view; (b and c) top down views of both rims of the complex. | ||
| Entry | Guest compound | Binding stoichiometry (ligand:7) |
K ± σK (M−1) | ΔH° ± σH (kcal mol−1) | TΔS° ± σTΔS (kcal mol−1) |
|---|---|---|---|---|---|
| 1 | Lithocholic acid (8) | 1:1 |
1.90 ± 0.43 × 108 | −11.02 ± 0.08 | 0.27 ± 0.19 |
| 2 | Deoxycholic acid (9) | 1:1 |
5.31 ± 2.16 × 107 | −3.57 ± 0.06 | 6.97 ± 0.27 |
| 3 | Chenodeoxycholic acid (10) | 1:1 |
1.05 ± 0.60 × 108 | −5.08 ± 0.08 | 5.86 ± 0.38 |
| 4 | Dehydrocholic acid (11) | 1:1 |
8.06 ± 1.83 × 107 | −6.76 ± 0.05 | 4.03 ± 0.16 |
| 5 | Hexadecafluorodecane-1,10-dioic acid (12) | 1:1 |
3.25 ± 0.43 × 105 | −2.37 ± 0.06 | 5.15 ± 0.13 |
| 6 | 6-(p-Toluidino)-2-naphthalensulfonic acid (13) | 2:1 |
5.78 ± 0.79 × 106 | −13.62 ± 0.15 | −4.38 ± 0.13 |
| 1.06 ± 1.50 × 107 | −10.91 ± 0.13 | −1.32 ± 0.83 |
Hexadecafluorodecane-1,10-dioic acid forms a 1:1 complex with duplex 7 with Ka 3.3 × 105 M−1, that is more than two orders of magnitude lower that the analogous complex with β-CD duplex.31 Moreover, while the complex with β-CD duplex was found to be strongly enthalpy-driven (ΔH° = −12.5 kcal mol−1, TΔS° = −1.7 kcal mol−1), in the larger duplex 7 the entropy contribution overwhelms the enthalpy term and becomes the driving force (ΔH° = −2.4 kcal mol−1, TΔS° = 5.2 kcal mol−1), indicating that a larger space allows larger rotational and translational freedom of the guest molecule.
Out of the complexes showing higher apparent stoichiometry, a convincing fit for the two “subsequent binding sites” model was observed with the sodium salt of 6-(p-toluidino)-2-naphthalensulfonic acid (13), providing two binding constants of similar magnitude (5.78 × 106 and 1.06 × 107 M−1). This is the only complex out of the series showing a negative value for the TΔS° term indicating that rotational and translational freedom of both guest molecules is restricted. It is assumed that each molecule occupies one cavity with the sulfonic group being oriented outside the cavity to aqueous media. The phenyl rings are likely to overlap in part inside the cavity interacting through π–π stacking as deduced from known complexes of planar aromatic compounds in native γ-CD.38
Conclusions
The synthetic procedures described in this paper allow preparation of both C6I,C6IV and C6I,C6V bis-O-debenzylated γ-CD isomers. The latter, being symmetrical, was subsequently converted to dithiol and used for the synthesis of the γ-CD duplex connected with two disulfide bonds. The oxidative dimerization proceeded well in 0.1–1 mM concentration range at pH 9 affording γ-CD duplex in high yield. In contrast to our preceding studies on α- or β-CD homologues the formation of a monomeric product was not observed, probably due to unfavorable relatively large deformation of γ-CD macrocycles required for intramolecular disulfide bond formation.Due to the large solvent accessible volume of the cavity of the γ-CD duplex (740 Å3), the molecule is ideally suited for complexation of relatively large organic molecules such as steroids. Bile acids examined in this study show binding affinities in the range of 5.3 × 107 M−1–1.9 × 108 M−1, i.e. two to three orders of magnitude larger than the native β-CD which has been known to be the best host for steroid structures among native CDs. The ITC titrations revealed that the cavity of γ-CD duplex is able to accommodate multiple smaller molecules or to form higher aggregates with them. Altogether with smaller α-CD or β-CD homologues25,31 we have developed a system of host molecules that enables complexation of the broad range of organic molecules in aqueous media with high binding affinities (Ka ∼ 107–109 M−1). We presume that they could be used in various indicator displacement assays at low analyte concentrations or for drug delivery purposes.
Acknowledgements
This work was financially supported by the Institute of Organic Chemistry and Biochemistry AS CR v.v.i. (Z40550506), COST (Action CM1005) and by Ministry of Education, Youth and Sport of the Czech Republic (LD12019). M. D. is indebted to the Czech Science Foundation for the support of the project 14-03276S.References
- H. E. Dodziuk, Cyclodextrins and Their Complexes, Wiley-VCH, Wienheim, 2006 Search PubMed.
- L. Liu and Q.-X. Guo, J. Inclusion Phenom. Macrocyclic Chem., 2002, 42, 1–14 CrossRef CAS.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- T. Kraus, Curr. Org. Chem., 2011, 15, 802–814 CrossRef CAS.
- F. Bellia, D. La Mendola, C. Pedone, E. Rizzarelli, M. Saviano and G. Vecchio, Chem. Soc. Rev., 2009, 38, 2756–2781 RSC.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- E. V. Anslyn, J. Org. Chem., 2007, 72, 687–699 CrossRef CAS PubMed.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- W. M. Nau, G. Ghale, A. Hennig, H. s. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558–11570 CrossRef CAS PubMed.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS PubMed.
- Y. Liu and Y. Chen, Acc. Chem. Res., 2006, 39, 681–691 CrossRef CAS PubMed.
- Y. Liu, Y. W. Yang, E. C. Yang and X. D. Guan, J. Org. Chem., 2004, 69, 6590–6602 CrossRef CAS PubMed.
- P. R. Cabrer, E. Azvarez-Parrilla, F. Meijide, J. A. Seijas, E. R. Nunez and J. V. Tato, Langmuir, 1999, 15, 5489–5495 CrossRef.
- I. Tabushi, Y. Kuroda and K. Shimokawa, J. Am. Chem. Soc., 1979, 101, 1614–1615 CrossRef CAS.
- O. Bistri, K. Mazeau, R. Auzely-Velty and M. Sollogoub, Chem. – Eur. J., 2007, 13, 8847–8857 CrossRef CAS PubMed.
- R. Breslow and S. Chung, J. Am. Chem. Soc., 1990, 112, 9659–9660 CrossRef CAS.
- D. Q. Yuan, S. Immel, K. Koga, M. Yamaguchi and K. Fujita, Chem. – Eur. J., 2003, 9, 3501–3506 CrossRef CAS PubMed.
- D.-Q. Yuan, K. Koga, I. Kouno, T. Fujioka, M. Fukudome and K. Fujita, Chem. Commun., 2007, 828–830 RSC.
- T. Lecourt, J. M. Mallet and P. Sinay, Tetrahedron Lett., 2002, 43, 5533–5536 CrossRef CAS.
- T. Lecourt, J. M. Mallet and P. Sinay, Eur. J. Org. Chem., 2003, 4553–4560 CrossRef CAS.
- O. Bistri, T. Lecourt, J. M. Mallet, M. Sollogoub and P. Sinay, Chem. Biodiversity, 2004, 1, 129–137 CAS.
- T. Lecourt, P. Sinay, C. Chassenieux, M. Rinaudo and R. Auzely-Velty, Macromolecules, 2004, 37, 4635–4642 CrossRef CAS.
- D. X. Dong, D. Baigl, Y. L. Cui, P. Sinay, M. Sollogoub and Y. M. Zhang, Tetrahedron, 2007, 63, 2973–2977 CrossRef CAS.
- O. Bistri-Aslanoff, Y. Bleriot, R. Auzely-Velty and M. Sollogoub, Org. Biomol. Chem., 2010, 8, 3437–3443 CAS.
- L. Kumprecht, M. Buděšínský, J. Vondrášek, J. Vymětal, J. Černý, I. Císařová, J. Brynda, V. Herzig, P. Koutník, J. Závada and T. Kraus, J. Org. Chem., 2009, 74, 1082–1092 CrossRef CAS PubMed.
- L. Krejčí, M. Buděšínský, I. Císařová and T. Kraus, Chem. Commun., 2009, 3557–3559 RSC.
- A. Wang, W. Li, P. Zhang and C.-C. Ling, Org. Lett., 2011, 13, 3572–3575 CrossRef CAS PubMed.
- R. Breslow, N. Greenspoon, T. Guo and R. Zarzycki, J. Am. Chem. Soc., 1989, 111, 8296–8297 CrossRef CAS.
- L. Kumprecht, M. Buděšínský, P. Bouř and T. Kraus, New J. Chem., 2010, 34, 2254–2260 RSC.
- P. Maloň, L. Bednarová, M. Straka, L. Krejčí, L. Kumprecht, T. Kraus, M. Kubanová and V. Baumruk, Chirality, 2010, 22, E47–E55 CrossRef PubMed.
- A. Grishina, S. Stanchev, L. Kumprecht, M. Buděšínský, M. Pojarová, M. Dušek, M. Rumlová, I. Křížová, L. Rulíšek and T. Kraus, Chem. – Eur. J., 2012, 18, 12292–12304 CrossRef CAS PubMed.
- A. J. Pearce and P. Sinay, Angew. Chem., Int. Ed., 2000, 39, 3610–3612 CrossRef CAS.
- T. Lecourt, A. Herault, A. J. Pearce, M. Sollogoub and P. Sinay, Chem. – Eur. J., 2004, 10, 2960–2971 CrossRef CAS PubMed.
- R. A. Laskowski, J. Mol. Graphics, 1995, 13, 323–330 CrossRef CAS PubMed.
- Due to pKa ∼ 5 of the investigated bile acids in aqueous solutions they are nearly completely dissociated under the conditions (pH 7) of ITC experiments. For reference see: A. Fini and A. Roda, J. Lipid Res., 1987, 28, 755–759 CAS.
- Z. W. Yang and R. Breslow, Tetrahedron Lett., 1997, 38, 6171–6172 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2002, 125, 966–972 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1026740. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02464h |
| This journal is © The Royal Society of Chemistry 2015 |