
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
UV-visible and 1H–15N NMR spectroscopic studies of colorimetric thiosemicarbazide anion sensors†
Kristina N.
Farrugia
a,
Damjan
Makuc
bc,
Agnieszka
Podborska
d,
Konrad
Szaciłowski
def,
Janez
Plavec
bc and
David C.
Magri
*a
aDepartment of Chemistry, Faculty of Science, University of Malta, Msida, MSD 2080, Malta. E-mail: david.magri@um.edu.mt; Fax: +(356) 2340 3320; Tel: +(356) 2340 2276
bSlovenian NMR Centre, National Institute of Chemistry, Hajdrihova 19, SI-1000 Ljubljana, Slovenia
cEN→FIST Centre of Excellence, Trg Osvobodilne fronte 13, SI-1000 Ljubljana, Slovenia
dAGH University of Science and Technology, Faculty of Non-Ferrous Metals, al. Mickiewicza 30, 30-059 Kraków, Poland
eJagiellonian University, Faculty of Chemistry, ul. Ingardena 3, 30-060 Kraków, Poland
fAcademic Centre of Materials and Nanotechnology, AGH University of Science and Technology, al. Mickiewicza 30, 30-059 Kraków, Poland
First published on 18th November 2014
Abstract
Four model thiosemicarbazide anion chemosensors containing three N–H bonds, substituted with phenyl and/or 4-nitrophenyl units, were synthesised and studied for their anion binding abilities with hydroxide, fluoride, acetate, dihydrogen phosphate and chloride. The anion binding properties were studied in DMSO and 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO–H2O by UV-visible absorption and 1H/13C/15N NMR spectroscopic techniques and corroborated with DFT studies. Significant changes were observed in the UV-visible absorption spectra with all anions, except for chloride, accompanied by dramatic colour changes visible to the naked eye. These changes were determined to be due to the deprotonation of the central N–H proton and not due to hydrogen bonding based on 1H/15N NMR titration studies with acetate in DMSO-d6–0.5% water. Direct evidence for deprotonation was confirmed by the disappearance of the central thiourea proton and the formation of acetic acid. DFT and charge distribution calculations suggest that for all four compounds the central N–H proton is the most acidic. Hence, the anion chemosensors operate by a deprotonation mechanism of the central N–H proton rather than by hydrogen bonding as is often reported.
:1 DMSO–H2O by UV-visible absorption and 1H/13C/15N NMR spectroscopic techniques and corroborated with DFT studies. Significant changes were observed in the UV-visible absorption spectra with all anions, except for chloride, accompanied by dramatic colour changes visible to the naked eye. These changes were determined to be due to the deprotonation of the central N–H proton and not due to hydrogen bonding based on 1H/15N NMR titration studies with acetate in DMSO-d6–0.5% water. Direct evidence for deprotonation was confirmed by the disappearance of the central thiourea proton and the formation of acetic acid. DFT and charge distribution calculations suggest that for all four compounds the central N–H proton is the most acidic. Hence, the anion chemosensors operate by a deprotonation mechanism of the central N–H proton rather than by hydrogen bonding as is often reported.
Introduction
Molecular chemosensors for the detection of anions have gained significant interest in the last two decades.1,2 The topic has become an established area of research in the field of supramolecular chemistry.3–9 This increased interest is owing to the significant role that anionic species play in biological systems and environmental ecosystems.10,11 The standard paradigm has been the use of receptor units, such as conjugated ureas and thioureas, as potential hydrogen bond donors.12–16Hydrogen bonding between receptors and anions is correlated with the acidity of the receptor protons.1 The introduction of electron-withdrawing substituents, notably on a phenyl substituent, enhances the acidity of the anion binding subunit. However, as the acidity of the receptor protons increases, the likelihood of deprotonation also increases. The result is a dichotomy between the realm of supramolecular chemistry involving hydrogen bonding and the realm of acid–base indicator chemistry involving deprotonation.8,17 The possibility of a competition between hydrogen bonding and deprotonation is now recognized by researchers in the field, which is dependent on many parameters including the basicity of the anion, the stability of the conjugate base, and the solvent in addition to the acidity of the anion receptor.18
Pyrrolylamidothiourea-based anion receptors undergo a color change in DMSO with F−, AcO−, C6H5COO− and H2PO4− resulting in dramatic spectral changes in the UV-vis spectra.19–21 X-ray crystallography was used by the Gale group to confirm that deprotonation occurs at the thiourea N–H proton next to the pyrrole moiety after the addition of only one equivalent of anion. Gunnlaugsson and co-workers have reported naphthalimide thiosemicarbazide anion chemosensors with striking colour changes from yellow/green to red/purple.22 The interactions are considered to result from complexation, with the exception of excess fluoride. However, most recently, Gunnlaugsson has reported a pyridine-based thiosemicarbazide derivative that undergoes deprotonation in acetonitrile.23 The titration profiles were dependent on the basicity of the anion with OH−, F− and AcO− requiring two equivalents, while H2PO4− and SO42− requiring significantly higher molar equivalents to reach titration completion. These observations are thought to result from an initial unstable hydrogen bond complex, followed by the deprotonation of the central thiourea N–H proton as concluded from X-ray crystallography data.22,23
Previous studies by the groups of Jiang24 and Lin25 also include work on thiosemicarbazides. Jiang reported the anion sensing ability in acetonitrile while Lin conducted studies in DMSO and aqueous 95:5 DMSO–H2O. In both solvent systems, a bathochromic shift was observed on addition of basic anions including fluoride, acetate and dihydrogen phosphate with concomitant colour changes. The presence of water did not alter the anion–receptor interaction significantly in DMSO.25 Furthermore, in both studies, the spectral changes were assigned to a 1:1 hydrogen bond complex based on 1H NMR titrations, among other arguments. However, a recent study on thiosemicarbazide anion sensors with electron-withdrawing substituents suggests that the interaction is due to deprotonation as tested with hydroxide.23
In this study we examine the anion recognition of a series of simple model thiosemicarbazide-based chemosensors 1–4 in aqueous DMSO and DMSO to gain further insight into the mechanism of action between hydrogen bonding and deprotonation. The four diarylthiosemicarbazide anion sensors differ in the number and position of the electron-withdrawing 4-nitrophenyl substituent. We examine the anion binding/deprotonation properties of 1–4 using UV-visible absorption spectroscopy and 1H NMR titration experiments with anions of varying basicity including OH−, F−, AcO−, H2PO4− and Cl−. In addition we employ two-dimensional 1H/15N and 1H/13C NMR correlation spectroscopy to probe the chemistry of the three N–H bonds in order to gain a better understanding of the mechanism of action.
Results and discussion
Synthesis
The synthesis of the chemosensors 1–4 is shown in Scheme 1. The compounds were synthesised from commercially available chemicals by reacting phenyl hydrazine 5 or 4-nitrophenyl hydrazine 6 with phenyl isothiocyanate 7 or 4-nitrophenyl isothiocyanate 8 in a one-step reaction by refluxing in dry CH3CN for 2 hours. All four products 1–4 were collected as solids after trituration from CHCl3 in 57–85% yields. The compounds were fully characterised using spectroscopic techniques including 1H, 13C and 15N NMR, IR and HRMS (see the Experimental section for data).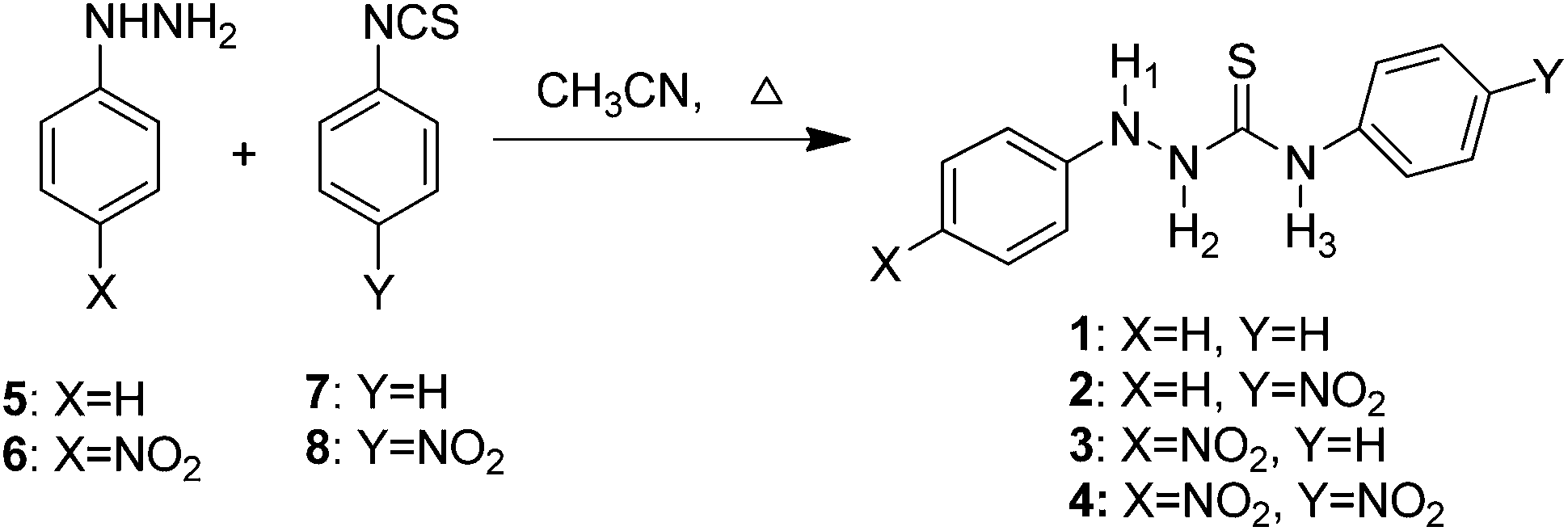 | ||
| Scheme 1 Synthesis of the thiosemicarbazide anion sensors 1–4 in dry CH3CN on refluxing for 2 hours. | ||
UV-visible spectrophotometric studies
UV-visible spectroscopic studies of receptors 1–4 were carried out in competitive media of 9:1 DMSO–H2O with the sodium salts of OH−, F−, AcO−, H2PO4− and Cl−. Compound 1, lacking a nitrophenyl electron-withdrawing substituent, exhibits almost no absorption between 280 and 700 nm. Upon addition of OH−, F−, and AcO−, 1 displays a broad shoulder appearing at ca. 335 nm accompanied by a slight colour change from colourless to faint yellow (Fig. 1a). No significant changes were observed in the UV-visible spectra with either H2PO4− or Cl− indicating that the receptor is not significantly interacting with these anions in the competitive solvent medium (Fig. S1†). Equilibrium constants of 4.2 and 4.4 were determined for 1 in the presence of OH− and F−. However, the broadness of the absorption shoulder prevented the determination of a stability constant for the other anions tested.
 | ||
| Fig. 1 Colour changes observed for 2.7 × 10−5 M 1–4 in 9:1 DMSO–H2O on addition of up to 4 equivalents of OH−, F−, AcO−, Cl− and H2PO4− as sodium salts. | ||
The 4-nitrophenyl substituted molecule 2 exhibits a λmax at 352 nm (logε = 4.09) in 9:1 DMSO–H2O. Upon increasing concentrations of OH−, F−, AcO− and H2PO4−, the absorption band at 352 nm decreases slightly at the expense of new absorbance bands at 308 and 392 nm, the latter tailing out to ca. 700 nm (Fig. 2). The changes are accompanied by two distinguishable isosbestic points at 338 and 362 nm, and a colour change from almost colourless to brown-yellow (Fig. 1b). Further analysis of the spectral changes clearly shows that all four anions result in similar UV-vis absorption profiles requiring two equivalents of anions to reach the plateau (Fig. 4a). No change was observed upon the addition of Cl−.
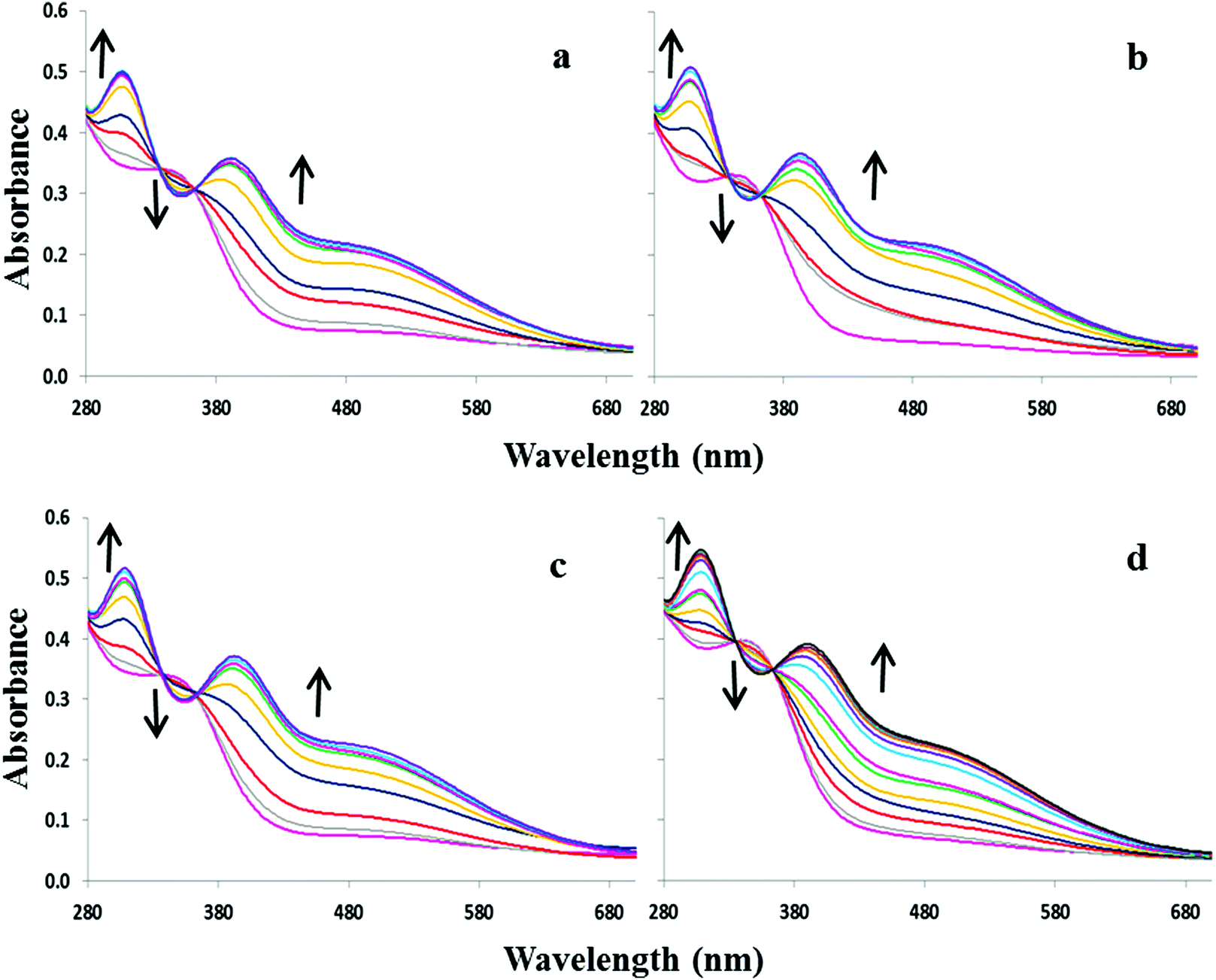 | ||
| Fig. 2 UV-visible absorption spectra of 2.7 × 10−5 M 2 in 9:1 DMSO–H2O upon addition of various anions as sodium salts: (a) OH− (0–4 equivalents), (b) F− (0–4 equivalents), (c) AcO− (0–4 equivalents) and (d) H2PO4− (0–55 equivalents.). | ||
In 9:1 DMSO–H2O compounds 3 and 4 exhibit an intense absorption band at ca. 367 nm (logε = 4.3) and a weaker band at ca. 527 nm (logε = 3.8). The addition of OH−, F−, AcO− and H2PO4− to 3 or 4 resulted in similar spectral changes. The absorbance spectra of 3 with and without subsequent additions of anions are shown in Fig. 3. Compound 3 exhibited a new band at 305 nm and the band at 527 nm increases significantly resulting in isosbestic points at 327 and 418 nm. Similarly for compound 4, titration with the four most basic anions resulted in a significant increase centred at 528 nm with two clear isosbestic points at 296 and 392 nm (Fig. S2†). Distinct colour changes from colourless to intense purple were observed for 3 (Fig. 1c) and color changes from pale pink to dark reddish-pink were observed for 4 (Fig. 1d). UV-vis titration experiments with compounds 3 and 4 in DMSO provided a virtual equivalent set of spectra with a slight red-shift of 5 and 7 nm in the charge transfer band maximum at 532 and 535 nm, respectively, on addition of basic anions (Fig. S3†).
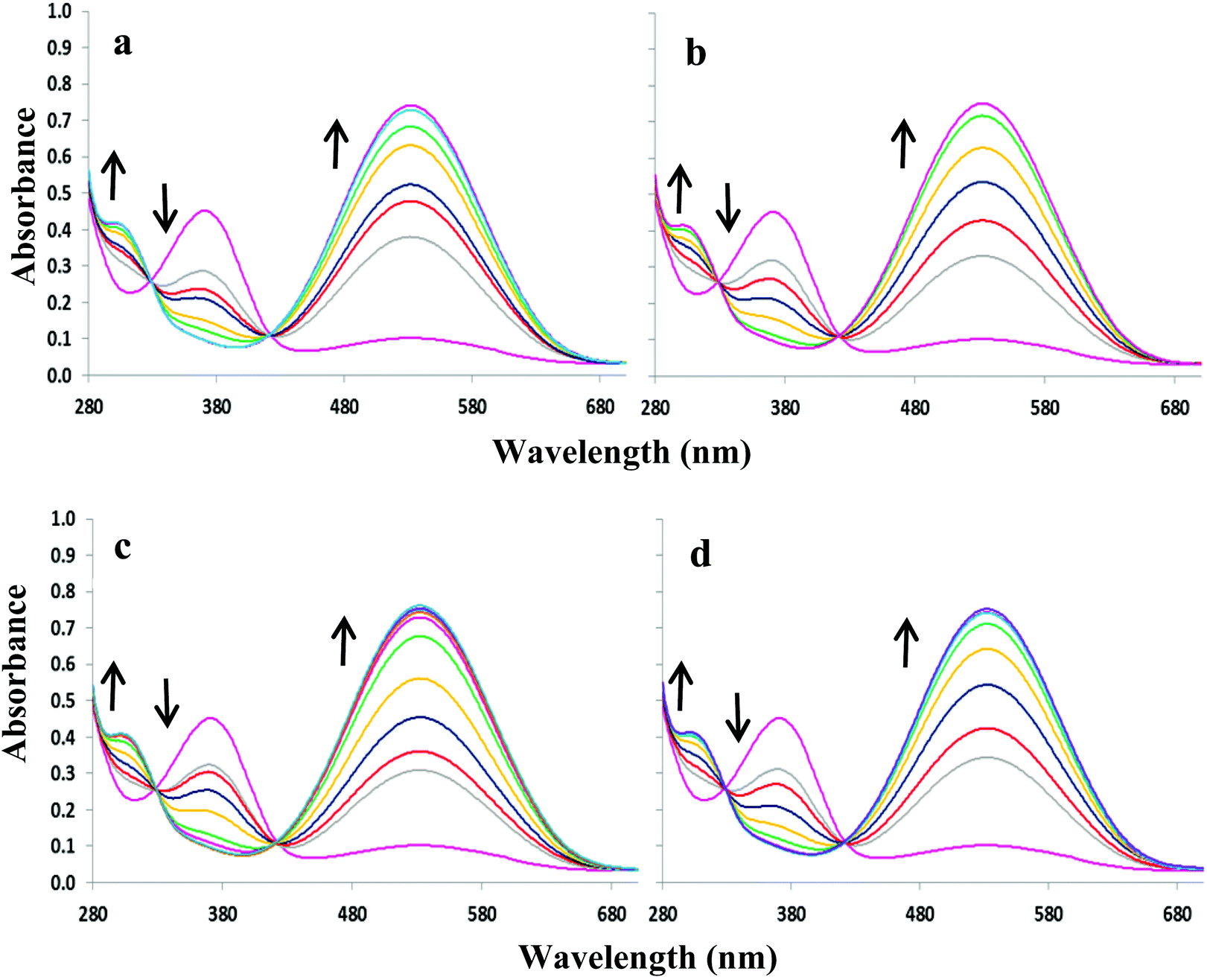 | ||
| Fig. 3 UV-visible absorption spectra of 2.7 × 10−5 M 3 in 9:1 DMSO–H2O upon addition of various anions as sodium salts: (a) OH− (0–4 equivalents), (b) F− (0–4 equivalents), (c) AcO− (0–4 equivalents) and (d) H2PO4− (0–40 equivalents.). | ||
The anion titration profiles in 9:1 DMSO–H2O for compounds 2–4 with OH−, F−, AcO− and H2PO4− are shown in Fig. 4. The addition of 2 equivalents of OH−, F−, AcO− were required to reach the plateau in the case of 2 and 3 (Fig. 4a and 4b, respectively) while with H2PO4− up to 50 equivalents of the anion were necessary. In contrast, titrations with 4 in 9:1 DMSO–H2O (Fig. 4c) and 3 and 4 in DMSO only required the addition of 1 equivalent of OH−, F−, AcO− and H2PO4− (Fig. S4†).
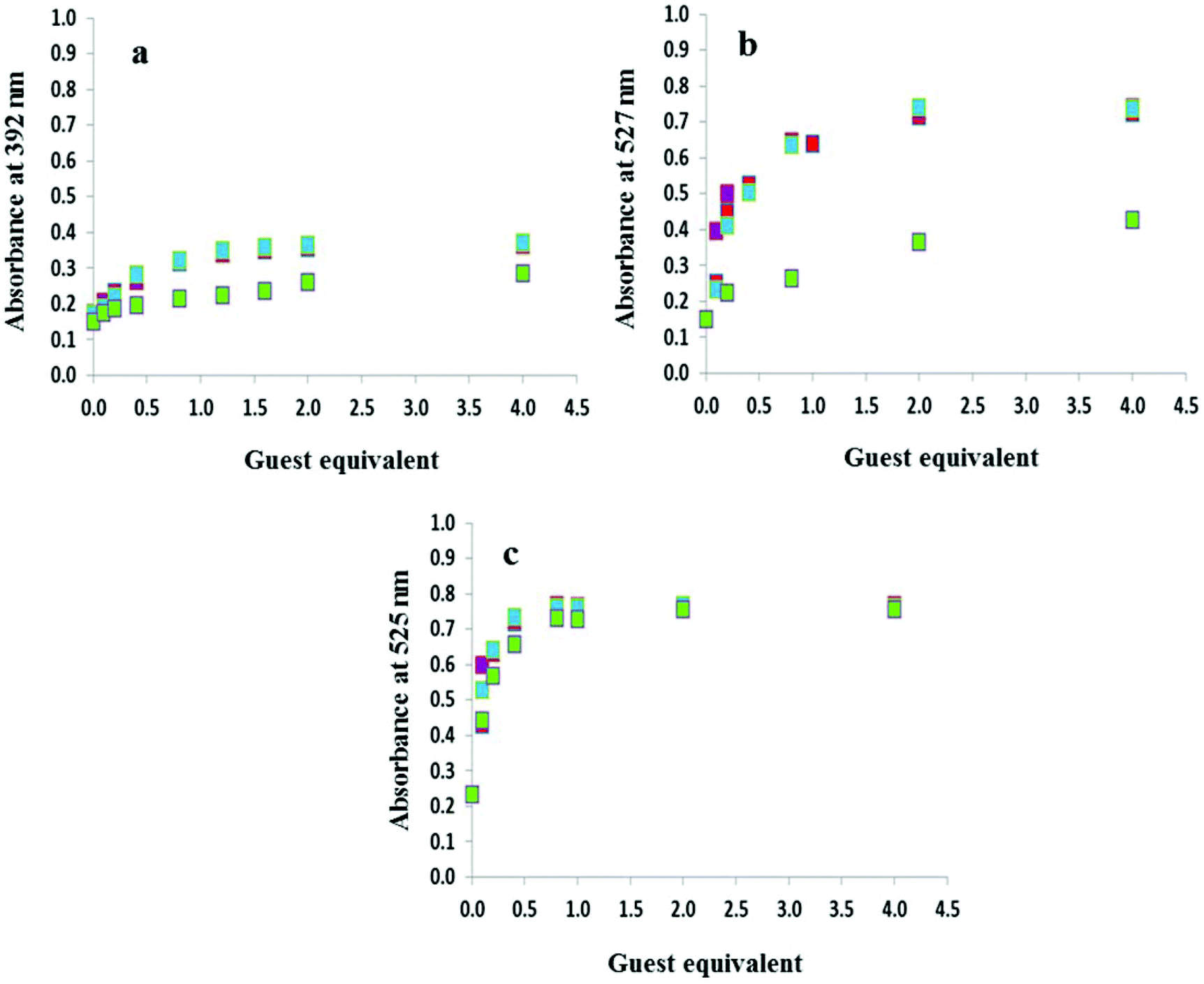 | ||
Fig. 4 Titration profiles in 9:1 DMSO–H2O at a concentration of 2.7 × 10−5 M (a) 2 at 392 nm, (b) 3 at 527 nm and (c) 4 at 525 nm with 0–4 equivalents of anions as sodium salts: OH− ( ), F− ( ), F− ( ), AcO− ( ), AcO− ( ) and H2PO4– ( ) and H2PO4– ( ). ). | ||
Fig. 1 highlights the dramatic colour changes observed for the nitro-containing compounds 2–4 in the presence of basic anions. For all three compounds, the observed colour change is identical for OH−, F−, AcO− and H2PO4−, suggesting the same mechanism of action with this group of anions. No colour change was observed with the weakly basic anion Cl−. The titration profiles confirm that with compound 4 only one equivalent of the anion (hydroxide, fluoride, acetate or dihydrogen phosphate) is necessary to reach the plateau.19 Dissociation constants for OH−, F−, AcO− and H2PO4− with 4 were experimentally identical with a value of logβA− of 5.2. Upon addition of OH−, F− and AcO−, compounds 2 and 3 were near the plateau after one equivalent of the anion, although a second equivalent is required to reach the asymptote. In contrast, at least 40 equivalents of dihydrogen phosphate are required to arrive at the end point. Dissociation constants logβA− of 5.0 were determined for both 2 and 3 in the presence of F−, AcO− and OH− while with H2PO4− the values were an order of magnitude lower at 4.0 and 4.2, respectively. A summary of the dissociation constants is provided in Table 1.
βA−) of compounds 1–4a
| Anions | 9:1 DMSO–H2Ob |
100% DMSOc | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 3 | 4 | |
|
a Determined by the UV-visible absorption titration technique. Estimated errors are given as the range calculated from two titration experiments.
b Sodium salts were used.
c Tetrabutylammonium salts were used.
d The band is a shoulder and does not allow for accurate identification and determination of logβA− (NI: not identified). No anion–receptor interaction is observed.
|
||||||
| OH− | 4.2 ± 0.1 | 5.0 ± 0.1 | 5.0 ± 0.2 | 5.3 ± 0.2 | 4.9 ± 0.1 | 5.4 ± 0.2 |
| F− | 4.4 ± 0.2 | 5.0 ± 0.2 | 5.0 ± 0.3 | 5.1 ± 0.3 | 5.1 ± 0.1 | 5.3 ± 0.1 |
| AcO− | NId | 5.1 ± 0.2 | 5.0 ± 0.1 | 5.4 ± 0.1 | 5.1 ± 0.3 | 5.3 ± 0.1 |
| H2PO4− | NId | 4.0 ± 0.1 | 4.2 ± 0.1 | 5.2 ± 0.2 | 5.1 ± 0.1 | 5.4 ± 0.2 |
It is worth noting that the addition of a large excess of OH− results in distinct colour changes with 3. After four equivalents of hydroxide, the solution is purple with a maximum at 527 nm. However, on continuous addition of excess hydroxide (up to 80 equivalents) the band decreases and gradually shifts to 443 nm, while the absorption band at 305 nm increases in intensity. Accompanying these changes is a shift in the isosbestic point from 418 to 431 nm and a colour change from purple to orange (Fig. S5†). These colour changes are attributed to two sequential deprotonation reactions, a phenomenon which was reported for naphthalimide-based thiosemicarbazides on addition of F−.22 However, in our case, addition of excess fluoride resulted only in an increase in the baseline absorption at ca. 360 nm with no colour change. In contrast, no colour or UV-vis spectroscopic change is observed on addition of 80 equivalents of acetate and dihydrogen phosphate.
1H NMR titration studies
The 1H, 13C and 15N resonances of 1–4 were assigned based on the analysis of 1D proton and carbon spectra as well as 1H–13C and 1H–15N correlations based on 2D HSQC and HMBC spectral analyses. The 1H NMR spectra are given in Fig. S6–S9† and the 1H–15N gHSQC and gHMBC spectra are given in Fig. S10–17.†26 Particular attention was paid to the absolute assignment of the three N–H nitrogen and hydrogen atoms by 15N NMR spectroscopy.27,28 The aryl protons fall within a chemical shift range of 6.7–7.5 ppm for 1 and within 6.7 and 8.2 ppm for 2–4. The aromatic N–H proton, H1, is observed in the range of 8.1–9.3 ppm while the two thiourea protons, H2 and H3, are observed to be further downfield at 9.7–10.3 ppm and 9.8–10.4 ppm, respectively. The chemical shifts are consistently in the order of σH3 > σH2 > σH1. The assignment of these three characteristic proton resonances was assisted using 1H–15N gHSQC and gHMBC NMR correlation experiments. 15N chemical shifts for the three thiourea N–H nitrogen atoms were determined indirectly by 2D correlation studies. The aromatic N–H nitrogen atom, N1, was observed between 96 and 106 ppm, while the two thiourea N–H nitrogen atoms, N2 and N3, were observed in the regions of 131–139 ppm and 124–127 ppm, respectively. The chemical shift of the nitro groups was consistently observed at 370 ppm. 13C NMR spectra are indicative of the thiourea carbonyl, which appears at 181 ppm while the remaining aromatic carbon signals are clustered between 112 and 154 ppm. A summary of the diagnostic 1H, 13C and 15N NMR chemical shifts is given in Table 2.
| Compound | 1H | 13C | 15Na | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | Ar–H | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S S |
Ar–C | N1 | N2 | N3 | NO2 | |
| a 15N NMR δ values were determined by 1H–15N correlations from gHMBC and gHSQC spectra. b Compound 1 lacks a NO2 functionality. c Accounts for two superimposed 15N resonances. | ||||||||||
| 1 | 8.07 | 9.71 | 9.83 | 6.71–7.52 | 181.2 | 113.1–148.0 | 96 | 134 | 124 | —b |
| 2 | 8.15 | 10.11 | 10.33 | 6.74–8.24 | 180.9 | 113.3–147.7 | 97 | 139 | 125 | 370 |
| 3 | 9.20 | 9.89 | 9.94 | 6.82–8.15 | 181.5 | 111.6–154.1 | 106 | 131 | 126 | 371 |
| 4 | 9.28 | 10.29 | 10.37 | 6.84–8.21 | 181.2 | 111.9–153.8 | 106 | 135 | 127 | 370c |
1H NMR titrations with 1.0 × 10−2 M of 3 and 4 in the presence of AcO−, added as the TBA salt, were performed in DMSO-d6–0.5% water and the anion–receptor interactions of the thiosemicarbazides protons were closely monitored. At higher concentrations, colour changes were immediate upon addition of only 0.1 equivalents of the anion. (Fig. 5 and Fig. S18† show the changes observed in the 1H NMR spectra of 4 and 3 upon addition of TBAOAc.) The two thiourea N–H protons H2 and H3 appear at 10.29 and 10.37 ppm, respectively, while the aromatic N–H1 proton appears at 9.28 ppm. As can be seen from Fig. 5, the main resonance shifts were observed between 0 and 1 equivalent of added AcO−. Negligible chemical shift changes are observed for the aromatic C–H protons, whereas considerable changes occur for the N–H protons. After the addition of 1 equivalent of added AcO−, only two N–H proton signals were observed corresponding to H1 and H3.
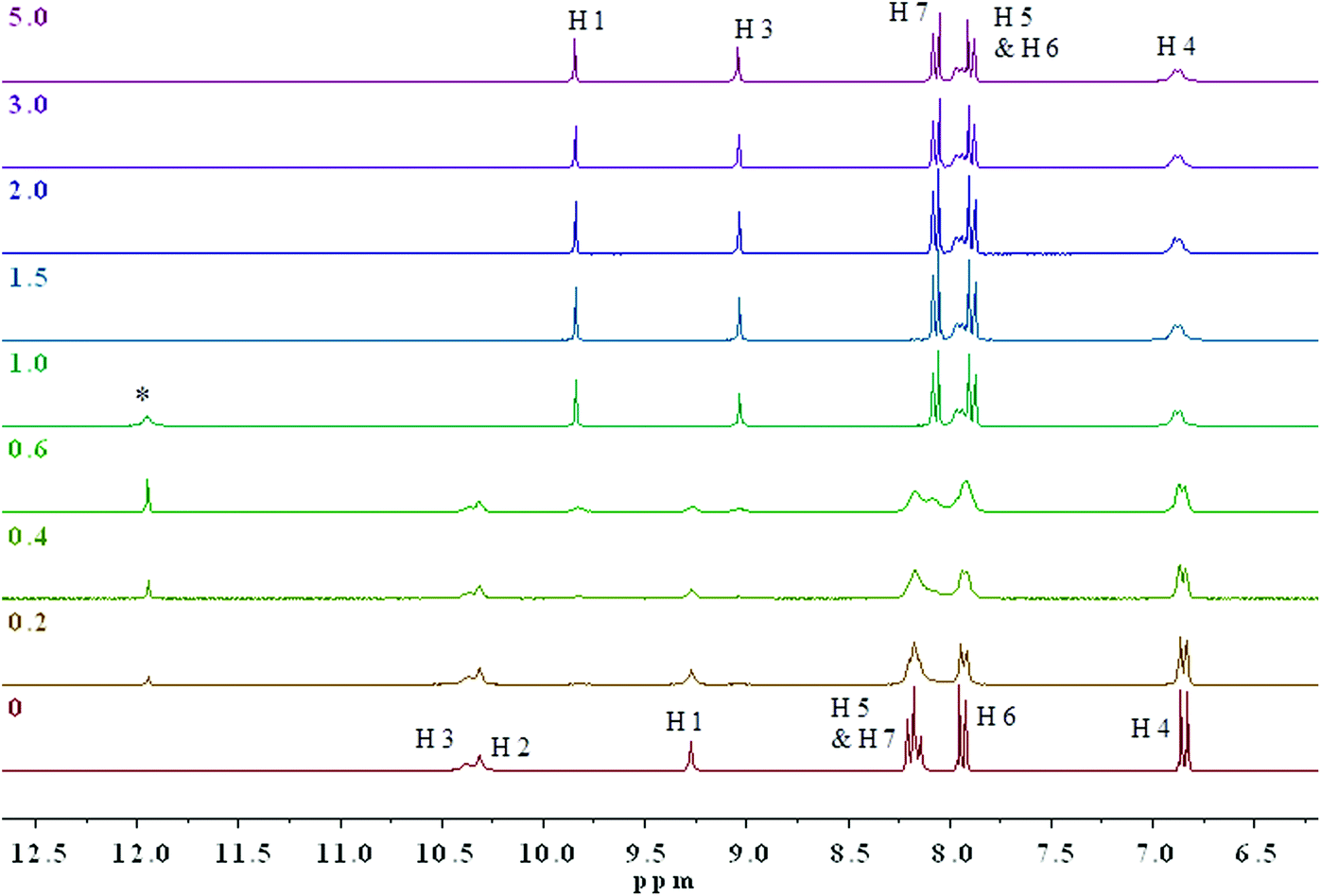 | ||
| Fig. 5 Stacked 1H NMR spectra of 0.01 M 4 upon addition of AcO− in DMSO-d6–0.5% water at 298 K. Numbers on the left correspond to the equivalence of anions added. The assignments of the 1H resonances are shown for the receptor before and after the addition of increasing amount of TBAAcO. The asterisk * marks the resonance due to protonation of acetate. | ||
The combination of 2D 1H–15N/1H–13C gHSQC and gHMBC experiments allowed for the unequivocal assignment of the N–H protons in 3 and 4 in the presence of acetate (Fig. S19–S26†). It was determined that the central thiourea N–H proton H2 disappeared, while the remaining thiourea N–H proton H3 and the aromatic N–H proton H1 are shielded (ΔδH3 = 1.34 ppm) and deshielded (ΔδH1 = 0.56 ppm) to δ 9.03 and 9.84 ppm, respectively (Fig. S19†). These changes are indicative of deprotonation of the thiourea unit by AcO− supporting the conclusion that a deprotonation mechanism occurs. Noticeable changes in the 15N NMR chemical shifts upon addition of the acetate to 3 and 4 are shown in Fig. S18 and S19,† respectively. More specifically, the aromatic N–H1 nitrogen atom was strongly deshielded from δ 106 to 158 ppm (ΔδN1 = 52 ppm), while the thiourea N–H3 nitrogen was only slightly deshielded from δ 127 to 131 ppm (ΔδN3 = 4 ppm) in 4. 15N chemical shift changes have been reported in several previous NMR spectroscopic studies on anion–receptor interactions.26 Typically, the nitrogen nuclei are deshielded by up to 10 ppm when the respective NH donor group is involved in hydrogen-bond interactions in anion–receptor complexes. However, a strongly deshielded aromatic N–H1 nitrogen atom is a characteristic feature resulting from deprotonation of an unsaturated system.29
In general, the anion selectivity trend in DMSO and 9:1 DMSO–H2O was determined to be OH− ≈ F− ≈ AcO− > H2PO4−. In the case of 4, no selectivity for H2PO4− was observed. Our findings differ from those in the literature.24,25 The Lin group determined from UV-visible absorption changes that the selectivity of 3 in DMSO and 95:5 DMSO–H2O is in the order AcO− > F− ≈ H2PO4−. Similarly, the studies by the Jiang group in acetonitrile by UV-visible spectroscopy determined the selectivity for 3 and 4 in the order of AcO− > F− > H2PO4−.22 In both studies hydrogen bonding was proposed as the anion sensing mechanism. However, in both studies no titrations with hydroxide were reported.
Together with the resonances in the 1H NMR spectra, which are confidently assigned to deprotonation of the indicators, a broad signal at 11.95 ppm is observed consistent with the acidic proton of acetic acid resulting from the addition of up to 1.0 equivalent of AcO−. Beyond the addition of 1 equivalent of acetate, no further chemical shift changes were observed. The identity of the signal at 11.95 ppm was confirmed by conducting a separate 1H NMR experiment with an authentic sample of 10 mM glacial acetic acid in DMSO-d6. To the best of our knowledge, this is the first time that direct evidence for deprotonation by NMR with AcO− has been reported with a thiosemicarbazide. Our findings provide convincing evidence in favour of a deprotonation mechanism rather than a complexation mechanism.
DFT studies
The frontier molecular orbitals for the chemosensors 1–4 are shown in Fig. 6. With the exception of 3, the HOMO is mostly delocalized over the arylhydrazine and the thiourea portions of the molecules and the LUMO is mostly delocalized on the other aryl ring. The addition of the nitro group in 2 does not result in a significant change in the spatial distribution of the frontier molecular orbitals. The similarity in the frontier molecular orbitals of 1 and 4 indicates that the overall effect of two nitro substituents on both phenyl rings is a net cancellation of the substituent effect at both terminals of the molecules. The frontier molecular orbitals of 3 are different from the other three molecules. The nitro group on the hydrazine side substantially lowers the energy difference and thus changes the distribution of the energy levels. The DFT calculations clearly illustrate the internal charge transfer (ICT) character within the molecular systems.30,31 The interchange of HOMO and LUMO in the case of 2 also results in a much lower transition energy and higher oscillator strength in this case. Furthermore, charge distribution calculations indicate that the N–H2 hydrogen atom is the most acidic in all cases. Therefore, there is excellent agreement between the experimental and calculated spectra for the deprotonation mechanism as directly observed by 1H and 15N NMR spectroscopy.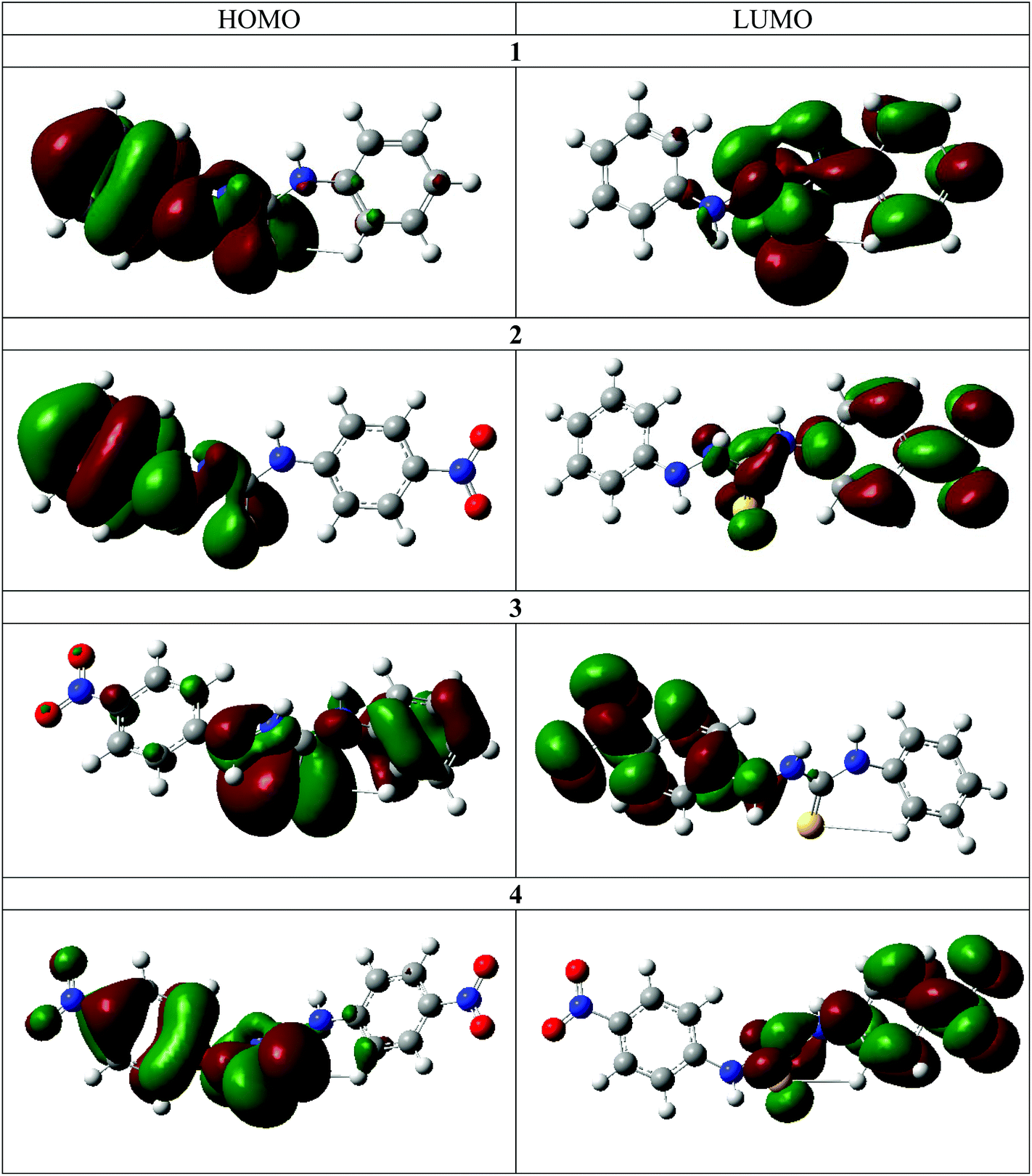 | ||
| Fig. 6 Depiction of the calculated HOMOs and LUMOs for compounds 1–4. | ||
The DFT calculations also support the 2D NMR correlation studies. In all cases the N–H2 proton is the most acidic, as in the neutral molecules it bears the highest positive Mulliken charge (Table 3).32 Furthermore, an increase of the N–H proton acidity upon introduction of NO2 functionalities is clearly evident. In all studied cases, the protons closest to the thiocarbonyl moiety are the most sensitive, which may be a result of a push–pull interaction between the electron donor and electron acceptor (Fig. S27†).
| Compound | N–H1 | N–H2 | N–H3 |
|---|---|---|---|
| 1 | 0.207 | 0.278 | 0.261 |
| 2 | 0.192 | 0.318 | 0.270 |
| 3 | 0.202 | 0.307 | 0.272 |
| 4 | 0.207 | 0.314 | 0.273 |
Conclusions
In conclusion, we have synthesised and studied a series of model thiosemicarbazide-based colorimetric sensors for anions 1–4 in DMSO and 9:1 DMSO–H2O with anions of varying basicity including OH−, F−, AcO− and H2PO4−. The receptors 1–4 possess acidic properties suitable for recognising anionic species through directional hydrogen bonds or anion-induced deprotonation of the thiosemicarbazide functionality. UV-visible absorption spectroscopy revealed that upon addition of these more basic anions, a significant red shift is observed in the ICT absorption band, which is accompanied by dramatic colour changes visible to the naked eye. 1H/15N NMR studies revealed a significant chemical shift in the 15N NMR spectrum with formation of acetic acid at 11.95 ppm in the proton NMR spectra on titration with the acetate. These results suggest that the mechanism of action of 3 and 4 in DMSO-d6–0.5% water with AcO− involves deprotonation of the thiosemicarbazide moiety, in particular at the central thiourea N–H proton, in agreement with the DFT calculations. To the best of our knowledge, this is the first time that 1H/15N NMR titration spectra have been used to gain insight into the chemistry of AcO− with thiosemicarbazides. It is worth emphasising that 2D 1H–13C/15N gHSQC and gHMBC NMR spectroscopy was essential for correctly assigning the chemical shifts of the three N–H proton units in addition to providing insight into a deprotonation mechanism.
Experimental
Chemicals
4-Nitrophenyl isothiocyanate (Sigma-Aldrich), phenyl isothiocyanate (Sigma-Aldrich), 4-nitrophenylhydrazine (BDH) and phenylhydrazine (Scharlau) were used as received. Dimethylsulfoxide-d6 (99.8 atom% D) from Roth was used as received. Acetonitrile (HPLC grade, Sigma-Aldrich) was distilled over calcium hydride and stored in a glass bottle over 3 Å molecular sieves in a desiccator. Other chemicals were used as received without further purification.Instrumentation
The compounds were characterised using 1H and 13C NMR spectra recorded on a Bruker AM250 NMR spectrometer equipped with a 1H/13C dual probe at a frequency of 250.10 MHz and 62.90 MHz, respectively. The acquisition data were processed on a Bruker Aspect 3000 computer using WIN NMR software. The NMR spectra are reported in parts per million (ppm) relative to the dimethylsulfoxide-d6 peak at 2.50 ppm and 39.52 ppm for 1H and 13C NMR, respectively. Infrared (IR) spectra were recorded on a Shimadzu IRAffinity-1 spectrophotometer calibrated versus polystyrene at 1601 cm−1. Solid samples were dispersed in KBr and recorded as clear discs. Ultra-violet absorption spectra were recorded at room temperature using a Jasco V-650 spectrophotometer with 1.0 cm quartz cuvettes. Electrospray time-of-flight (ES-TOF) mass spectra were obtained on a Waters LC Premier instrument. Chemical ionization (CI) used ammonia as the proton source. Melting points were measured on a Gallenkamp melting point apparatus.1H NMR titration experiments and 2D NMR experiments including 1H–15N heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) were performed on a DD2 Agilent Technology NMR spectrometer at a frequency of 297.80 MHz, 74.89 MHz and 30.18 MHz for 1H, 13C and 15N NMR, respectively. Chemical shifts are referenced to the residual solvent signal of DMSO-d6 as noted above while 15N chemical shifts were referenced relative to ammonia (δ 0.00 ppm).
Syntheses
Compounds 1–4 were prepared according to literature procedures with slight modification.20 Due to incomplete or unreported literature data, the compounds were fully characterised. The general procedure was as follows: in a 100 mL round-bottom flask one equivalent of isothiocyanate and one equivalent of hydrazine were dissolved in ca. 20 mL of dry CH3CN and refluxed with magnetic stirring for ca. 2 hours. The reaction was monitored via TLC with silica gel (60F 254, Sigma-Aldrich) on aluminum using 1:1 hexane–ethyl acetate as the eluent and observed under 254 nm UV light. On completion the solution was removed using a rotary evaporator resulting in a crude residue, which was purified by trituration with chloroform, and collected by suction filtration.22
S), 148.0, 139.2, 128.9, 127.9, 125.0, 124.7, 119.8, 113.1; 15N NMR (30 MHz, DMSO-d6, ppm): 134 (1N, thiourea-N2H), 124 (1N, thiourea–N3H), 96 (1N, Ar–N1H); IR (KBr, cm−1): 3252, 3205, 3148, 1599, 1568, 1505, 1481, 1333, 1281, 1209, 1111, 891, 847, 748, 689, 650; HRMS (ESI-TOF): Calculated C13H14N3S [M − H]− 242.0752, Found 242.0762.
S), 147.7, 145.7, 143.1, 128.9, 123.9, 123.5, 120.1, 113.3; 15N (30 MHz, DMSO-d6, ppm): 371 (1N, Ar–NO2), 139 (1N, thiourea–N2H), 125 (1N, thiourea–N3H), 97 (1N, Ar–N1H); IR (KBr, cm−1): 3250, 3203, 3190, 3145, 1600, 1568, 1540, 1330, 1280, 1209, 1111, 891, 846, 748, 688, 648; HRMS (ES-CI-TOF): Calculated C13H14N4SO2 [M + H]+ 289.0759, Found 289.0751.
S), 154.1, 139.0, 138.9, 127.9, 125.7, 125.5, 125.1, 111.6; 15N (30 MHz, DMSO-d6, ppm): 371 (1N, Ar–NO2), 131 (1N, thiourea–N2H), 126 (1N, thiourea–N3H), 105.6 (1N, Ar–N1H); IR (KBr, cm−1): 3319, 3163, 3148, 3084, 1609, 1595, 1549, 1489, 1447, 1339, 1248, 1231, 1178, 1111, 1028, 931, 910, 843, 775, 752, 731, 696, 623; HRMS (ESI-TOF): Calculated C13H14N4SO2 [M + H]+: 289.0759, Found 289.0764.
S), 153.8, 145.6, 143.3, 139.3, 125.8, 124.6, 123.7, 111.9; 15N (30 MHz, DMSO-d6, ppm): 370 (2N, Ar–NO2), 135 (1N, thiourea–N2H), 127 (1N, thiourea–N3H), 106 (1N, Ar–N1H); IR (KBr, cm−1): 3308, 3281, 3144, 3020, 1595, 1553, 1497, 1412, 1331, 1265, 1223, 1111, 934, 856, 843, 748, 707, 634; HRMS (ESI-TOF) Calculated C13H12N5SO4 [M + H]+: 334.0610, Found: 334.0602.
UV-visible absorption spectrometric titrations
UV-visible absorption titrations were performed in 9:1 DMSO–H2O at room temperature with sodium salts. Freshly prepared solutions of 3 × 10−5 M sensor were used. Aliquots of aqueous stock solutions of anions were added (OH−, F−, AcO−, H2PO4− and Cl−) diluted with distilled water. Experiments in DMSO were performed at room temperature with tetrabutylammonium (TBA) salts. The solutions were prepared with 3.4 × 10−5 M of sensor in DMSO with varying volumes of anions.
The absorption data were fitted to the equation −log[A−] = logβA− + log[(Amax − A)/(A − Amin)] where log[A−] is the logarithm molar anion concentration at that point, logβA− is the dissociation constant, Amax is the maximum absorbance at the selected wavelength, Amin is the minimum absorbance at the given wavelength and A is the observed absorbance at that specific wavelength.33 Plotting log[(Amax − A)/(A − Amin)] versus the −log[A−], the logβA− was derived from the slope of the resulting plot. Titrations were repeated at least twice until the results were reproducible.
1H NMR titration experiments
1H NMR titration experiments with anion sensors 3 and 4 were performed in DMSO-d6/0.5% water at 298 K. To 0.6 mL of 10 μM sensor solution, increasing concentrations of TBAAcO salt solution were progressively added and subsequently measured by 1H NMR spectroscopy. Measurements were performed in the same NMR tube on addition of 0.0, 0.2, 0.4, 0.6, 1.0, 1.5, 2.0, 3.0 and 5.0 molar anion equivalents. Individual resonances in the 1H NMR titration profile were assigned on the basis of the chemical shifts, signal integrations, multiplicity, and via1H–13C/15N gHSQC and gHMBC correlation studies. Stacked NMR spectral plots were produced using MestReNova software version 6.Theoretical modeling was performed with Gaussian 09 Rev. D.01 (Gaussian, Inc.).34 The final geometry was obtained by using DFT with the B3LYP (the Becke three-parameter-Lee–Yang–Parr) functional and the 6-311++G(d,p) basis set. Molecular orbitals and surfaces were plotted using GaussView software. Electronic transitions were calculated by using time-dependent DFT with the B3LYP functional and the 6-311++G(d,p) basis set and the polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM) with DMSO as the solvent.
Acknowledgements
We acknowledge the financial support from the University of Malta, the Strategic Educational Pathways Scholarship (STEPS) part-financed by the European Social Fund (ESF) under Operational Programme II, and the European Cooperation in Science and Technology (COST Action CM1005 “Supramolecular Chemistry in Water”). Our appreciation is extended to Prof. Robert M. Borg for assistance with the acquisition of the NMR data. We also gratefully acknowledge the financial support of the Slovenian Research Agency, Ministry of Higher Education, Science and Technology of Republic Slovenia (program no. P1-0242), ENFIST Centre of Excellence. DFT calculations were performed at the Academic Computer Centre CYFRONET AGH within computational grant no. MEiN/SGI3700/UJ/085/2006.References
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed.
- (a) P. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC; (b) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480 RSC; (c) P. A. Gale, Chem. Soc. Rev., 2010, 39, 3746 RSC; (d) C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520 RSC; (e) P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151 RSC; (f) P. A. Gale, Acc. Chem. Res., 2006, 39, 465 CrossRef CAS PubMed.
- (a) L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2013, 42, 3489 RSC; (b) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed.
- (a) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Rev., 2010, 39, 3936 RSC; (b) T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and R. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094 CrossRef CAS PubMed.
- (a) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648 RSC; (b) S. Kubik, Chem. Soc. Rev., 2009, 38, 585 RSC.
- (a) C. Suksai and T. Tuntulani, Chem. Rev., 2003, 32, 192 RSC; (b) M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809 RSC.
- V. Amendola, M. Bonizzoni, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, F. Sancenón and A. Taglietti, Coord. Chem. Rev., 2006, 250, 3094 CrossRef PubMed.
- (a) D.-G. Cho and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1647 RSC; (b) A.-F. Li, J.-H. Wang, F. Wang and Y.-B. Jiang, Chem. Soc. Rev., 2010, 39, 3729 RSC.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem. – Eur. J., 2010, 16, 4368 CrossRef CAS PubMed.
- M. A. Tetilla, M. C. Aragoni, M. Arca, C. Caltagirone, C. Bazzicalupi, A. Bencini, A. Garau, F. Isaia, A. Laguna, V. Lippolis and V. Meli, Chem. Commun., 2011, 47, 3805 RSC.
- (a) V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889 RSC; (b) P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem. – Asian J., 2010, 5, 555 CrossRef CAS PubMed; (c) D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717 CrossRef PubMed; (d) M. Boiochhi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem. – Eur. J., 2005, 11, 3097 CrossRef PubMed; (e) R. M. Duke and T. Gunnlaugsson, Tetrahedron Lett., 2010, 51, 5402 CrossRef CAS PubMed; (f) H. D. Paduka Ali, P. E. Kruger and T. Gunnlaugsson, New J. Chem., 2008, 32, 1153 RSC.
- T. Gunnlaugsson, A. P. Davis, G. M. Hussey, J. Tierney and M. Glynn, Org. Biomol. Chem., 2004, 2, 1856 CAS.
- A. Misra, M. Shahis and P. Dwivedi, Talanta, 2009, 80, 532 CrossRef CAS PubMed.
- X. He, S. Hu, K. Liu, Y. Guo, J. Xu and S. Sho, Org. Lett., 2006, 8, 333 CrossRef CAS PubMed.
- M. Duke and T. Gunnlaugsson, Tetrahedron Lett., 2011, 52, 1503 CrossRef PubMed.
- S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Org. Biomol. Chem., 2003, 1, 741 CAS.
- H. Miyaji and J. L. Sessler, Angew. Chem., Int. Ed., 2001, 40, 154 CrossRef CAS.
- (a) M. Boiocchi, L. Del Boca, D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS PubMed; (b) D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495 RSC; (c) M. Bonizzoni, L. Fabbrizzi, A. Taglietti and F. Tiengo, Eur. J. Org. Chem., 2006, 3567 CrossRef CAS; (d) V. Amendola, D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS PubMed.
- (a) L. S. Evans, P. A. Gale, M. E. Light and R. Quesada, New J. Chem., 2006, 30, 1019 RSC; (b) L. S. Evans, P. A. Gale, M. E. Light and R. Quesada, Chem. Commun., 2006, 965 RSC.
- (a) F.-Y. Wu, Z. Li, L. Guo, X. Wang, M.-H. Lin, Y.-F. Zhao and Y.-B. Jiang, Org. Biomol. Chem., 2006, 4, 624 RSC; (b) L. Nie, Z. Li, J. Han, X. Zhang, R. Yang, W.-X. Liu, F.-Y. Wu, J.-W. Xie, Y.-F. Zhao and Y.-B. Jiang, J. Org. Chem., 2004, 69, 6449 CrossRef PubMed.
- W.-X. Liu, R. Yang, A.-F. Li, A. Li, Y.-F. Gao, X.-X. Luo, Y.-B. Ruan and Y.-B. Jiang, Org. Biomol. Chem., 2009, 7, 4021 CAS.
- T. Gunnlaugsson, P. E. Kruger, P. Jensen, J. Tierney, H. D. Paduka Ali and G. M. Hussey, J. Org. Chem., 2005, 70, 10875 CrossRef CAS PubMed.
- (a) K. Pandurangan, J. A. Kitchen and T. Gunnlaugsson, Tetrahedron Lett., 2013, 54, 2770 CrossRef CAS PubMed; (b) K. Pandurangan, J. A. Kitchen, T. McCabe and T. Gunnlaugsson, CrystEngComm, 2013, 15, 1421 RSC.
- Z. Li, F. Y. Wu, L. Guo, A. F. Li and Y. B. Jiang, J. Phys. Chem. B, 2008, 112, 7071 CrossRef CAS PubMed.
- J. Shao, H. Lin and H.-K. Lin, Talanta, 2008, 75, 1015–1020 CrossRef CAS PubMed.
- (a) D. Makuc, M. Lenarčič, G. W. Bates, P. A. Gale and J. Plavec, Org. Biomol. Chem., 2009, 7, 3505 RSC; (b) D. Makuc, Triyanti, M. Albrecht, J. Plavec, K. Rissanen, A. Valkonen and C. A. Schalley, Eur. J. Org. Chem., 2009, 4854 CrossRef CAS; (c) D. Makuc, M. Albrecht and J. Plavec, J. Supramol. Chem., 2010, 22, 603 CrossRef CAS; (d) D. Makuc, J. R. Hiscock, M. E. Light, P. A. Gale and J. Plavez, Beilstein J. Org. Chem., 2011, 7, 1205 CrossRef CAS PubMed.
- G. J. Martin, M. L. Martin and J.-P. Gouesnard, 15N-NMR Spectroscopy, Springer, Berlin, Heidelberg, 1981 Search PubMed.
- J. Mason, L. F. Larkworthy and E. A. Moore, Chem. Rev., 2002, 102, 913 CrossRef CAS PubMed.
- M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1981, 11B, 1–502 Search PubMed.
- L. Piela, Ideas of Quantum Chemistry, Elsevier, Amsterdam, Netherlands, 2006 Search PubMed.
- G. Y. Li, G. J. Zhao, Y. H. Liu, K. L. Han and G. Z. He, J. Comput. Chem., 2010, 31, 1759 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, A. J. Patty and G. L. Spence, J. Chem. Soc., Perkin Trans. 2, 1993, 1611–1615 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 1H–13C, 1H–15N NMR gHSQC and gHMBC spectra for 1–4, UV-visible titration spectra for 1 and 4 in 9:1 DMSO–water, UV-visible titration spectrum profiles of 3 and 4 in DMSO, addition of an excess of anions with 3 in 9:1 DMSO–water, 1H NMR titration spectra of 3, 1H–15N gHSQC spectra of 3 & 4 with excess acetate, 1H–13C gHSQC and 1H–13C gHMBC NMR spectra of 3 and 4 upon addition of 2 equivalents of AcO− in DMSO-d6–0.5% H2O and charge distribution calculations for 1–4. See DOI: 10.1039/c4ob02091j |
| This journal is © The Royal Society of Chemistry 2015 |