
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl-bis-(scorpiand)-aza receptors differentiate between nucleotide monophosphates by a combination of aromatic, hydrogen bond and electrostatic interactions†
Jorge
González-García
a,
Sanja
Tomić
b,
Alberto
Lopera
a,
Lluís
Guijarro
a,
Ivo
Piantanida
*c and
Enrique
García-España
*a
aICMOL, Departamentos de Química Inorgánica y Orgánica, Facultad de Química, Universidad de Valencia, C/Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain. E-mail: enrique.garcia-es@ uv.es
bLaboratory for Chemical and Biological Crystallography, Division of Physical Chemistry, Ruđer Bošković Institute, HR 10002 Zagreb, Croatia
cLaboratory for Study of Interactions of Biomacromolecules, Division of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Zagreb, Croatia. E-mail: pianta@irb.hr
First published on 26th November 2014
Abstract
Bis-polyaza pyridinophane scorpiands bind nucleotides in aqueous medium with 10–100 micromolar affinity, predominantly by electrostatic interactions between nucleotide phosphates and protonated aliphatic amines and assisted by aromatic stacking interactions. The pyridine-scorpiand receptor showed rare selectivity toward CMP with respect to other nucleotides, whereby two orders of magnitude affinity difference between CMP and UMP was the most appealing. The phenanthroline-scorpiand receptor revealed at pH 5 strong selectivity toward AMP with respect to other NMPs, based on the protonation of adenine heterocyclic N1. The results stress that the efficient recognition of small biomolecules within scorpiand-like receptors relies mostly on the electrostatic and H-bonding interactions despite the competitive interactions in the bulk solvent, thus supporting further optimisation of this versatile artificial moiety.
Introduction
Nucleotides are among the most targeted anionic substrates due to the key roles that they play in biology such as nucleic acid synthesis, transport across membranes, and energy and electron-transfer events. Other relevant functions of nucleotides are related to cell-signalling processes.1 In this respect, the extracellular release of nucleotides serves as a signal during inflammation through the activation of nucleotide receptors P1 and P2.2 Transmembrane protein channels have been implicated in the release of adenosine nucleotides from the intra- to the extracellular space in apoptotic cells.3 In addition, several studies have proved the release of uridine nucleotides during cystic fibrosis.4 Therefore, the development of new abiotic receptors that are able to selectively bind nucleotides in aqueous solution is a relevant goal in chemical and biomedical research.5The interaction among naturally occurring nucleobases and abiotic receptors occurs mainly through columbic interactions, hydrogen bonding and π–π stacking. Very good examples of the involvement of different forces in nucleotide binding are provided by the crystal structures of adducts formed between protonated terpyridinophane or phenanthroline macrocycles with 5′-thymidine triphosphate (5′-TTP).6
Since the dominant driving force in nucleotide binding is charge–charge interaction, a great deal of the studies reported so far describe the selection of the more charged nucleotides with respect to the less charged ones.7–10 Reversed selectivity of ADP over ATP in water was recently reported to occur through the synergistic action of the different binding groups included in a tris(2-aminoethyl)amine receptor containing a pyrimidine group.11
A case of selectivity for AMP over ADP and ATP as a result of strong electrostatic contacts supported by π–π interactions was recently reported in a guanidinium-polypeptide-based polytopic receptor.12
However, achieving base-pair discrimination is a challenging target in biomedical and supramolecular chemistry. Very often base-pair discrimination relies on different hydrogen bonding patterns between the partners which limit its effectivity in water due to competitive hydrogen bonding with the solvent.13 To overcome this difficulty, several years ago sapphyrin and calixpyrrole derivatives appended with nucleobases were designed so that the interaction with the complementary nucleotide was enhanced by hydrogen bonding.14 Favourable discrimination of AMP over the other nucleotide monophosphates (NMPs) by hydrogen bonding was reported using a polytopic receptor with a bis(oxazolin-2-yl)pyridine scaffold.15
Several bis-intercaland-type macrocycles constituted another example of nucleobase discrimination.16 In this work, particularly interesting is the selectivity toward certain nucleobases ascribed to the interaction of the bases with the polyamine-linkers connecting the intercalating subunits.17
On the other hand, Kimura and co-workers reported for the first time, the ability of the Zn2+ complex of cyclen to interact selectively with thymine and uracil through metal ion coordinative bonding to the deprotonated imide group and hydrogen-bonding formation between the amine groups and the pyrimidine carbonyl groups.18 These findings led to development of cyclen derivatives, either with aromatic groups appended in their structure or with more than one cyclen unit.19
Recently, some of us have designed a series of scorpiand-like receptors with different appended motifs in the tail able to discriminate certain nucleobases due to a predominant hydrogen bonding or π–π stacking interaction.20
A further development of the scorpiand-like receptors just mentioned are the double bis-scorpiand receptors PYPOD or PHENPOD, built by connecting two macrocyclic units containing pyridine moieties with pyridine or phenanthroline linkers, respectively (Scheme 1).21 These compounds, particularly PYPOD which have shown selectivity of RNA over DNA,22 are examined in this work for their capability to achieve base-pair discrimination in the recognition of NMPs. The selectivity is analysed in terms of how the functionalities present in the receptors complement AMP, GMP, UMP or CMP.
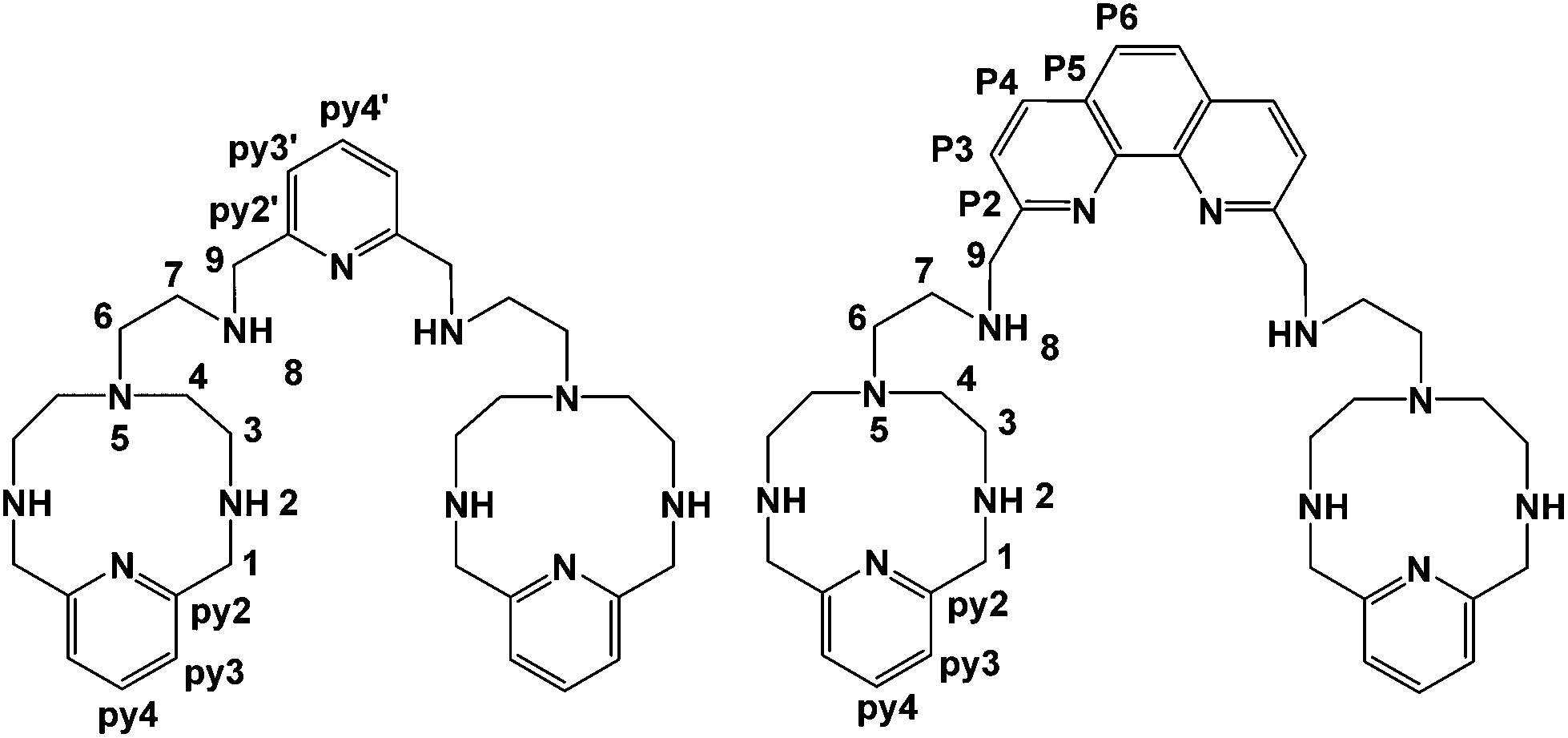 | ||
| Scheme 1 Studied PYPOD (left) and PHENPOD (right). | ||
Results and discussion
Acid–base properties of the nucleotide monophosphates
Before performing any speciation study, it was necessary to calculate the protonation constants of the different NMPs under the same experimental conditions used in the work (Table S1,† NaCl 0.15 M at 298.0 K).From potentiometric analysis, we obtained the protonation constants of the phosphate groups (between 6.1 and 6.3, depending on the nucleotide, see Table S1†) and the stepwise protonation constants of the deprotonated imide nitrogen in the heterocyclic base of GMP and UMP.23 Moreover, the protonation constants of nitrogen N1 in the aromatic ring of AMP and CMP were also determined.24 To confirm these values and get structural information about the protonation sites of the nucleotides, pH dependent 1H and 31P NMR spectra (ESI Chart 1 for labelling and Fig. S1–S5†) were registered in deuterated water. The protonation constants were obtained by treatment of the 1H and 31P NMR chemical shifts at different pD values with the HyperNMR software (Table S1†).25 The values obtained were in reasonable agreement with those calculated by pH-metric analysis.
Interaction of PHENPOD and PYPOD with nucleotide monophosphates
Since both the substrates and the receptors participate in overlapped proton transfer processes, translating the cumulative stability constants into representative stepwise constants is not always straightforward. To do this, the basicity constants of both the substrate and the receptor have to be taken into account and it should be assumed that the interaction will not greatly affect the pH range of existence of the protonated species of nucleotides and receptors. In this way, the stepwise constants (Table S3†) have been calculated for PHENPOD–NMP and PYPOD–NMP systems. Nevertheless, the most unambiguous way to compare the relative stabilities of the different systems and to establish selectivity ratios at different pH values is to use effective constants (Keff). The effective constants are calculated at each pH value as the quotient between the overall amount of complexed species and the overall amounts of free receptors and substrates, independent of the protonation degree (Fig. 1).27
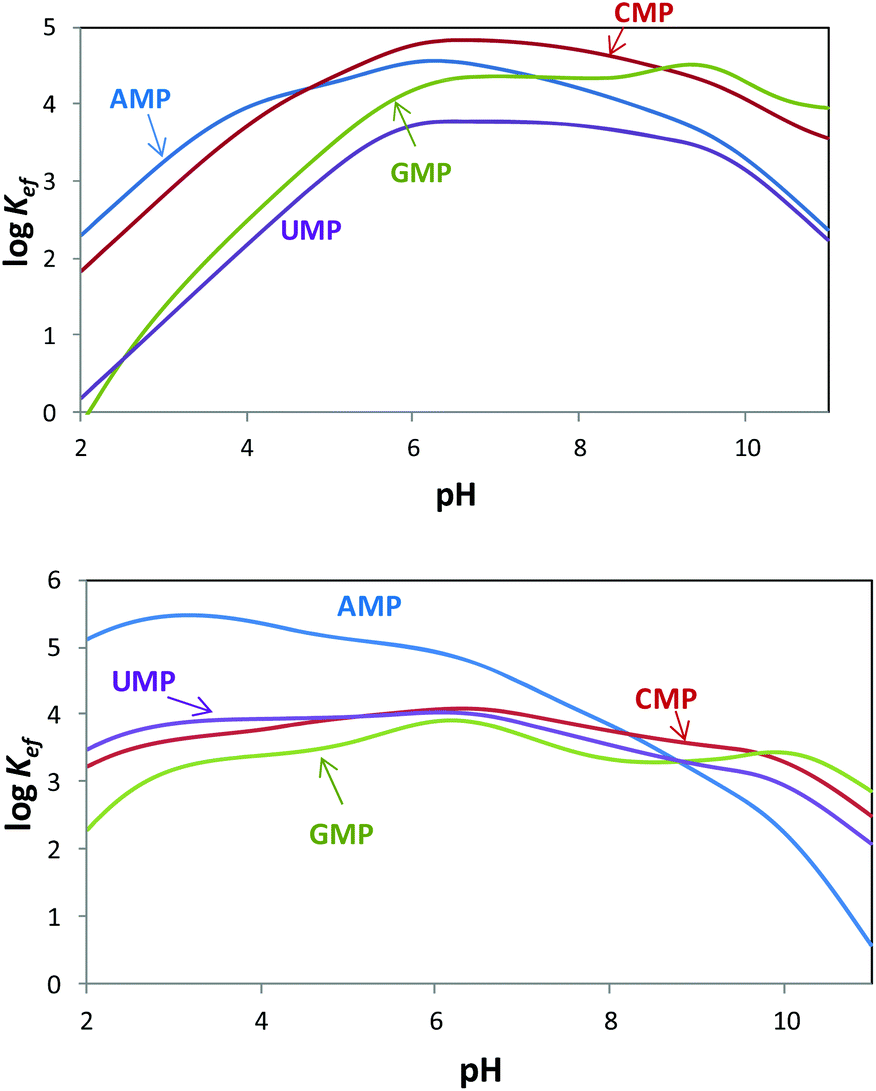 | ||
Fig. 1 Plot of the pH dependence of effective constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Keff/M−1) for the systems PYPOD (up) or PHENPOD (down) with AMP, GMP, CMP and UMP. Keff/M−1) for the systems PYPOD (up) or PHENPOD (down) with AMP, GMP, CMP and UMP. | ||
The effective constants for all the PYPOD–NMP systems show bell-shaped profiles with maximum values of the effective constants in the 6–8 pH range. At higher pH values the positive charge in the receptor would not be high enough to achieve a strong interaction. On the other hand, at pH values below 6 the nucleotide starts to protonate by reducing its negative charge. For the PHENPOD–NMP systems, in particular for AMP, the logKeff-pH profile is different. Although the profile is still bell-shaped the stability observed at acidic pH values is significantly higher, which can likely be attributed to π–π stacking and to the hydrophobicity afforded by the larger condensed aromatic phenanthroline ring. While in the case of PYPOD the situation is not so apparent, PHENPOD clearly recognises AMP over the other three mononucleotides below pH = 8.
In this regard, while the fluorescence emission of PYPOD is too low to perform fluorimetric titrations at any pH value, the intrinsic fluorescence emission of PHENPOD allowed us to carry out titrations with the nucleotides at acidic pH values (Fig. 2). The addition of the nucleotides to a solution of PHENPOD at pH = 5 leads to a decrease in fluorescence which was particularly noticeable for AMP (Fig. 2). Such quenching processes can be attributed to hydrogen bonding and charge interactions between the anionic phosphate moiety and the polyammonium chain of the protonated receptor, which could lead to an intra-complex proton transfer from an ammonium group to a phosphate group making an amine lone pair available for the quenching process.6b The effective constants calculated by processing the fluorimetric data with the HypSpec program that are collected in Table 1 are in reasonable agreement with the values derived from the pH-metric titrations and confirm the selective recognition of AMP by PHENPOD.
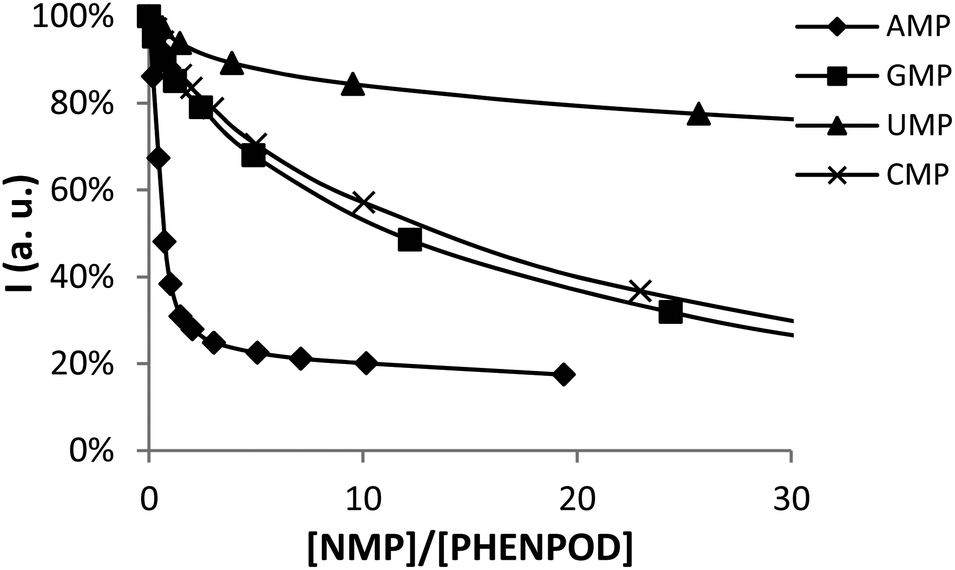 | ||
| Fig. 2 Plot of the fluorescence intensity normalized vs. the molar ratio [NMP]/[PHENPOD]. | ||
| pH = 5.0 | 7.0 | |||
|---|---|---|---|---|
| PYPOD | PHENPOD | PYPOD | PHENPOD | |
| a Calculated from fluorimetric titrations. Values in the brackets inside are standard deviations of the last significant figure. | ||||
| AMP | 4.28 | 5.11 (5.63 (6))a | 4.45 | 4.44 |
| GMP | 3.51 | 3.60 (3.52 (2))a | 4.35 | 3.67 |
| CMP | 4.37 | 3.96 (3.46 (2))a | 4.82 | 3.99 |
| UMP | 3.16 | 3.95 (3.59 (6))a | 3.76 | 3.84 |
The stoichiometries of the PHENPOD–NMP systems were further confirmed by fluorescence emission techniques using Job plots (continuous variation experiments) at pH 5.28 For this purpose, ΔAf = Af (free) − Af (bound) was plotted against the ratio of concentrations R = [L]/([L] + [NMP]), in which Af is the area of the fluorescence emission peaks of PHENPOD, free or bound to the nucleotide, at a constant total ([L] + [NMP]) concentration29 (Fig. 3 and Fig. S8†). In all cases, a maximum is observed at 0.5 supporting a 1:1 stoichiometry. Similar conclusions were derived from Job plots obtained with 1H NMR data (Fig. S11 and S12†).
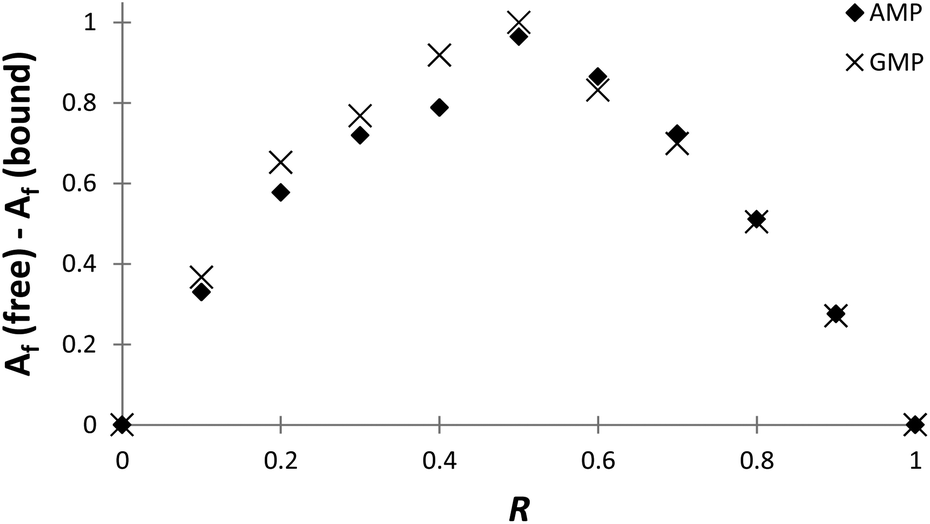 | ||
Fig. 3 Job plot for complex formation between PHENPOD and AMP ( ) or GMP ( ) or GMP ( ). Spectra were recorded at 369 nm in a pH 5.0 solution (0.05 M cacodylate buffer) at a total concentration of [PHENPOD] + [NMP] = 5 × 10−5 M. ). Spectra were recorded at 369 nm in a pH 5.0 solution (0.05 M cacodylate buffer) at a total concentration of [PHENPOD] + [NMP] = 5 × 10−5 M. | ||
Regarding the pyrimidine nucleotides, the plot of the effective constants reveals an unexpectedly high selectivity of PYPOD for CMP over UMP throughout the complete pH range, Keff for CMP being an order of magnitude higher than for UMP (see Fig. 1). This selectivity could originate from different hydrogen bonding patterns of CMP and UMP. Furthermore, since the PYPOD–CMP complex is, under physiological conditions (pH = 6–8), about 3–5 times more stable than the complexes with the purine-nucleotides (AMP and GMP), contribution of aromatic stacking between PYPOD and the nucleobase is not a dominant binding interaction controlling the overall stability of the PYPOD/NMP complex formed.
The selectivity for CMP completely disappears when the central pyridine unit (PYPOD) is substituted by phenanthroline (PHENPOD). In the binding mode of PHENPOD with purines (AMP, GMP), the phenanthroline moiety plays a crucial role interacting with the base by aromatic stacking as confirmed by the NMR experiments (see below). Therefore, the adducts formed in the PHENPOD:purine systems adopt a forced conformation, reducing the impact of electrostatic interactions in comparison with pyridine nucleotides. The high selectivity of PHENPOD for AMP under acidic conditions (pH < 5.5) most likely involves the protonation of adenine at the N1 position. Indeed, it has been observed that adenine heterocycles self-stack more efficiently when 50% of the adenine moieties are protonated at N1.30 The same analogy could be used to explain the preferred stacking between the N1-protonated adenine with the neutral phenanthroline ring in comparison with the non-protonated guanine. Moreover, the data of purine/pyrimidine self-stacking for nucleosides show the trend A > G > T > C, reflecting the decreasing aromaticity and hydrophobic character of their nucleobase residues supporting the stacking preference of adenine for PHENPOD.30
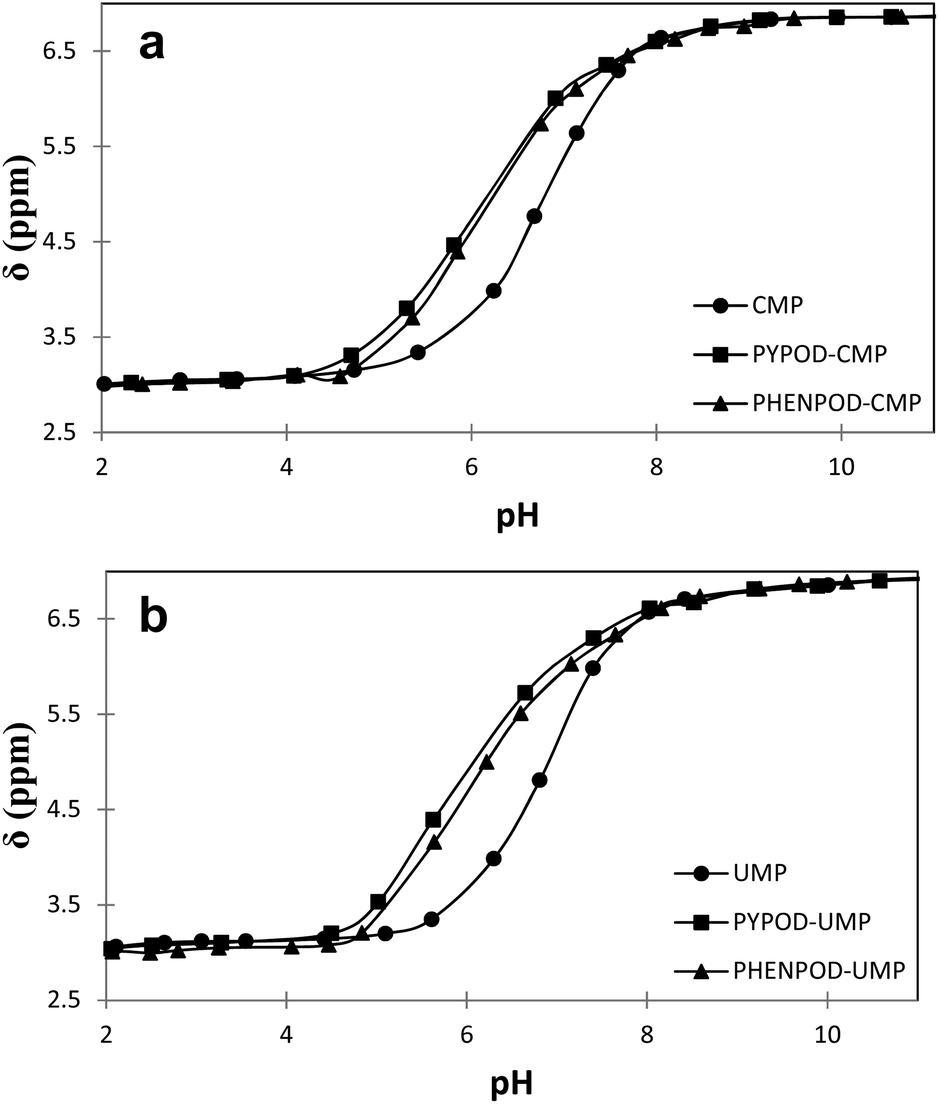 | ||
| Fig. 4 Plot of 31P NMR δ (ppm) vs. pH for free mononucleotides (●) CMP (a) and UMP (b) and for the complexes formed with PYPOD (■) and PHENPOD (▴). | ||
Furthermore, the 1H NMR shifts of the aromatic signals of solutions of the nucleotides and receptors in 1:1 molar ratio (Fig. 5a and b) revealed more pronounced shifts for the PHENPOD–NMP systems than for the PYPOD–NMP systems, suggesting a stronger aromatic stacking between the nucleobases and the phenanthroline. The anomeric proton signal (HR1) of the nucleotide was also significantly shifted upfield upon complexation with PYPOD/PHENPOD (Fig. 5c).
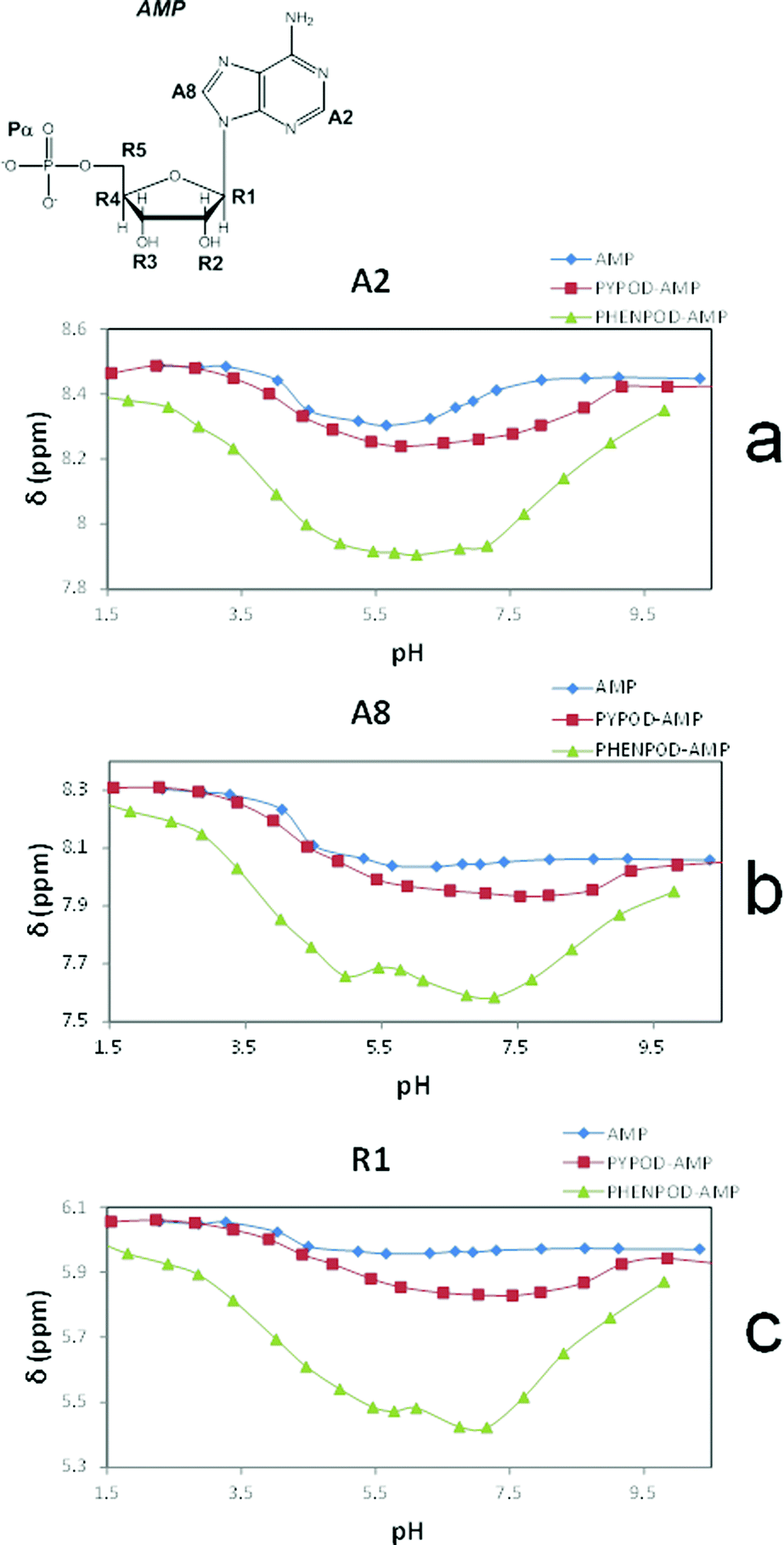 | ||
| Fig. 5 Plot of 1H NMR δ (ppm) vs. pH of: AMP complexes with PYPOD and PHENPOD: aromatic protons (a) HA2 and (b) HA8, anomeric signal (c) HR1. | ||
An important fact is that the 1H NMR shift differed in the acidic pH range, which varied considerably from pD ∼ 1 only for PHENPOD–NMP systems, while no variation was observed for PYPOD–NMP systems until pD 2–3 depending on the system. This difference could be attributed to the stronger interactions exerted by PHENPOD based on a more effective aromatic stacking.
The 1H NMR titrations of PHENPOD and PYPOD with the studied nucleotides at pD ∼ 7.0 resulted in shifts of the proton signals, whereby the shift direction (upfield or downfield) as well as the intensity of the shifts varied significantly from case to case (examples Fig. 6, complete overview see ESI Table S4†). Although the large values of the stability constants prevented their determination by NMR titrations, and NOE experiments did not provide any relevant information, a qualitative analysis of the proton shifts can help to derive several general conclusions. For instance, the PHENPOD-AMP titrations (Fig. 6 UP) revealed strong shifts of the aromatic protons of both the ligand and the nucleobase, pointing toward pronounced aromatic stacking interactions – (a similar situation was observed also for other nucleotide complexes with PHENPOD), thus supporting the significant impact of the large phenanthroline ring in the binding. At variance to that, the PYPOD-nucleotide aromatic proton signal shifts (Fig. 6 DOWN) were smaller and more importantly, most of them shifted in the opposite direction than toward the PHENPOD systems, thus suggesting a different combination of interactions and structural changes, whereby aromatic stacking (as expected) played only a minor role.

However, the PYPOD–NMP systems under neutral conditions were much more appropriate and interesting for modelling due to the selectivity found for CMP over UMP. Also, a close inspection of the hydrogen-bonding patterns revealed distinctive differences, which are summarized in Scheme 2. Such differences were particularly taken into account for the PYPOD–UMP system, which showed the lowest binding constant. Moreover, the small proton NMR shifts of the aromatic protons in the PYPOD–UMP system suggested that aromatic stacking interactions between the central pyridine of PYPOD and uracil could be neglected.
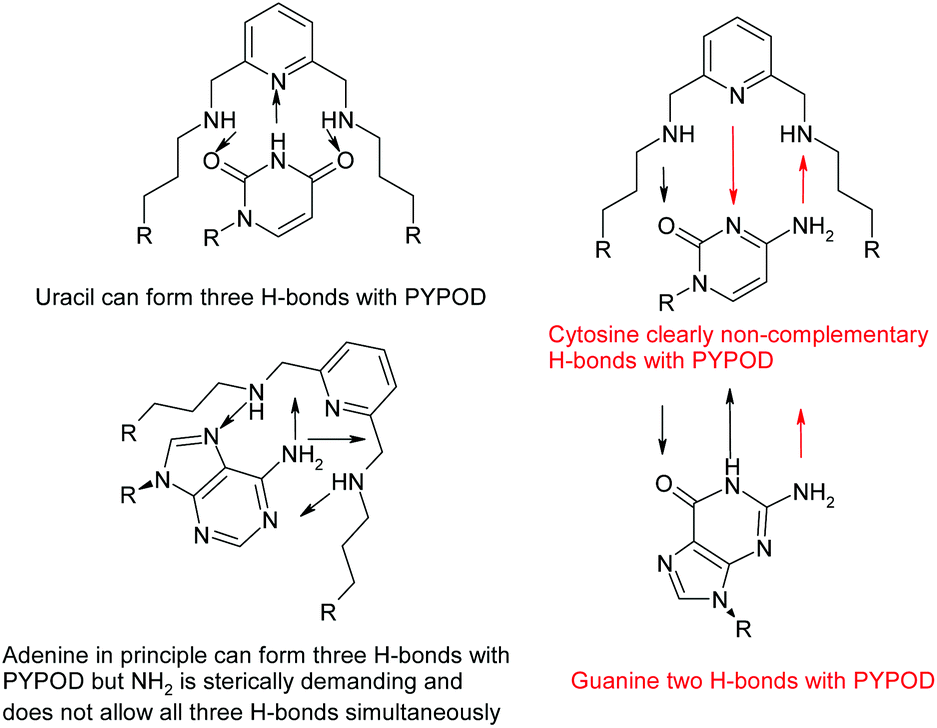 | ||
| Scheme 2 Possibilities of H-bonding of nucleobases inside the PYPOD. | ||
To start with, a PYPOD structure with four positive charges at the secondary ammonium nitrogens (according to the PYPOD protonation constants)21 was constructed and minimised in aqueous medium (Fig. S13†). This PYPOD structure was used to construct the complexes with AMP, CMP and UMP. The molecular dynamics simulations revealed different features between the various complexes. The PYPOD–AMP complex appeared in several conformations and the two most abundant were characterised by aromatic stacking between adenine and pyridine in either (a) approximately perpendicular or (b) approximately coplanar relation (Fig. 7a and 7b). Furthermore, the multiple electrostatic interactions between positively charged aliphatic nitrogens and negatively charged nucleotide phosphates (mostly located within the self-folded structure of the complex) additionally stabilised the system.
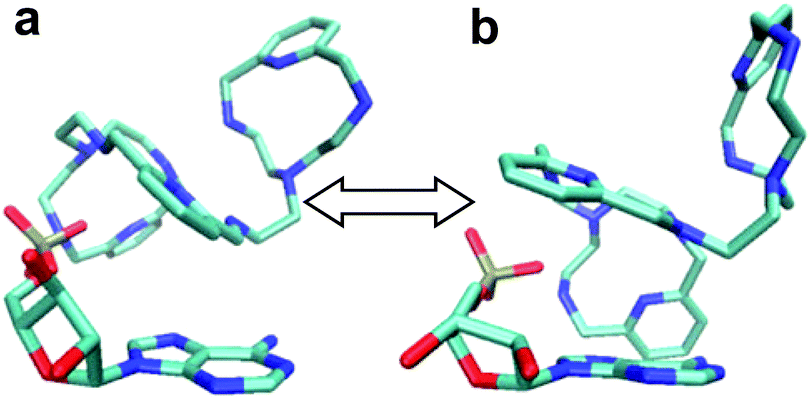 | ||
| Fig. 7 Results of molecular dynamics simulations of the PYPOD–AMP complex: note the central pyridine–adenine orientation (a) approximately perpendicular; (b) approximately coplanar relation. | ||
The PYPOD–CMP complex conformation obtained after 8 ns of distance constrained molecular dynamics simulation (for details see the Experimental section and ESI†) is shown in Fig. 8. The representative conformation was characterised by approximately coplanar stacking as well as more intensive electrostatic interactions in respect of the PYPOD–AMP complex. This is due to the more flexible self-folded structure of the complex caused by the smaller sized nucleobases, namely the larger adenine moiety which requires more space to accommodate in stacking with pyridine and consequently allows for less adjustment of the electrostatically interacting groups (phosphates vs. protonated amines). The minor impact of nucleobase aromatic stacking interactions on the overall stability of the complex is nicely demonstrated by similar binding constants of PYPOD with CMP and AMP despite the large difference in the nucleobase aromatic surface (cytosine has only half of the aromatic surface compared to, e.g., adenine, and therefore usually exhibits an order of magnitude with a lower aromatic stacking affinity).
 | ||
| Fig. 8 Results of molecular dynamics simulations of PYPOD-CMP. | ||
Intriguingly, the PYPOD–UMP complex unfolded during the molecular dynamics simulations, giving rise to a large number of quite different structures. Therefore, a novel starting complex structure was formed, taking into account a unique possibility of H-bond patterns (Scheme 2), and submitted to the molecular dynamics. As none of the obtained major conformations (Fig. S14, ESI†) showed significantly lower energy than the others, they could be discussed simultaneously. Essential for binding was the varying set of electrostatic interactions between the positively charged PYPOD side chains and the negatively charged UMP-phosphate; however H-bonding interactions of uracil also played an important role. Nevertheless, such multi-conformation sets resulted in relatively low overall stability in respect of the PYPOD–UMP complex.
Conclusions
The bis-polyazapyridinophane scorpiands PYPOD and PHENPOD efficiently bound the nucleotides in water by dominant electrostatic interactions between the nucleotide phosphates and the protonated aliphatic amines. Moreover, aromatic stacking interactions between the aryl groups of PHENPOD/PYPOD and the nucleobases also contribute to the binding. A similar synergistic effect of electrostatic and aromatic stacking interactions was recently reported for recognition in the AMP/ADP/ATP series.12Intriguingly, fine regulation of the steric parameters in the scorpiand/nucleotide complex resulted in: (a) selectivity of PYPOD toward CMP in respect of other nucleotides (in particular UMP) based on the adjustment of the aromatic stacking partners with the electrostatic interaction partners as well as with the size and hydrophobicity of the PYPOD binding pocket; (b) selectivity of PHENPOD toward AMP under weakly acidic conditions based on the protonation of adenine heterocyclic N1. Both selectivities brought unparalleled features in the field of aqueous small molecule receptors for nucleotides.
Many small receptors recognised the purine (GMP and/or AMP) nucleotides by combined aromatic stacking interactions with electrostatic/H-bonding contribution within the receptor pocket. Considering the pyrimidine nucleobases, mostly UMP/TMP selective receptors are reported, the selective interactions based on Zn2+-coordination or complementary base recognition. The very recently reported unique example of TMP recognition (even in respect of the UMP) relied on a peculiar combination of binding interactions, including highly flexible aromatic macrocycle appended with a complementary base (adenine).31 However, to the best of our knowledge, there is no low-molecular-weight receptor exhibiting preference toward CMP in respect of the other nucleotides. Therefore, particularly intriguingly, a two orders of magnitude higher affinity of PYPOD toward CMP in comparison with the other pyrimidine nucleotide (UMP) is observed here. There are several applications of such selectivity; for instance interfering with the action of sialyl transferases can be envisioned by the action of PYPOD selectively blocking the formation of CMP-sialic acid (enzyme donor substrate), in that way impairing the regulation of the sialylation of glycans on the cell surface.32 Affinity of PYPOD toward AMP and GMP was slightly lower than toward CMP, pointing out the dominant impact of electrostatic interaction.
Furthermore, several AMP selective receptors are known,33 but none of them has an on/off switchable mechanism so easily controlled as shown here for PHENPOD, which by pH change can ratiometrically switch on/off selectivity toward AMP in respect of other nucleotides (Fig. S9,† right: in mixture of all NMPs at pH 5 PHENPOD binds 72% of AMP, while at physiological pH 7.4 PHENPOD binds 40% of AMP).
Obtained selectivities are still not applicable for the efficient extraction of CMP or AMP from the mixture of nucleotides (including also di- and triphosphates, as well as other forms), however structure–activity relations are useful for further optimisation of PHENPOD and PYPOD as lead compounds. The design and study of new generations of analogues with delicately varied properties of aliphatic linkers (e.g. length, number and position of protonable amino groups) as well as variation of aryl-bridgesare in progress.
Experimental
All the reagents were supplied by commercial suppliers (Sigma-Aldrich) and analysed before using. The synthesis and characterization of PYPOD and PHENPOD have been described previously.21EMF measurements
The potentiometric titrations were carried out at 298.1 ± 0.1 K using NaCl 0.15 M as a supporting electrolyte. The experimental procedure (burette, potentiometer, cell, stirrer, microcomputer, etc.) has been fully described elsewhere.34 The acquisition of the EMF data was performed with the computer program PASAT.35 The reference electrode was an Ag/AgCl electrode in saturated KCl solution. The glass electrode was calibrated as a hydrogen-ion concentration probe by titration of previously standardized amounts of HCl with CO2-free NaOH solutions and the equivalent point determined by the Gran's method,36 which gives the standard potential, E°′, and the ionic product of water (pKw = 13.73(1)).The computer program HYPERQUAD was used to calculate the protonation and stability constants.26 The HYSS37 program was used to obtain the distribution diagrams The pH range investigated was 2.5–11.0. In the binary L–A systems concentrations of the anion and of the receptors ranged from 1 × 10−3 M to 5 × 10−3 M.
NMR measurements
The 1H and 13C spectra were recorded on a Bruker AV 500 spectrometer at 500 MHz for 1H and 125.43 MHz for 13C. The NMR experiments involving 31P were recorded on a Bruker AV 500 spectrometer equipped with a switchable probe. The chemical shifts were recorded in ppm. All spectra were recorded at room temperature and the concentration of L1·6HCl, L2·6HCl, AMP, GMP, UMP and CMP was 2 × 10−3 M in D2O. The pD was adjusted using a concentrated solution of DCl or NaOD in D2O.Spectroscopic measurements
The emission fluorescence spectra were obtained on the Varian Eclipse fluorimeter in aqueous buffer solution (pH = 5, sodium cacodylate buffer, I = 0.05 M), all in quartz cuvettes (1 cm).Molecular modelling
Initial structures of the ligand-mononucleotide complexes PYPOD-AMP, PYPOD-CMP and PYPOD-UMP were prepared using the program VMD1.8,38 taking into account to the assumed electrostatic stabilizations shown in Scheme 2. The systems were parameterized by ANTECHAMBER and xLeaps, the modules available within the AMBER Tools, using the general AMBER force field GAFF.39 The complexes were placed in the centre of the octahedral box filled with TIP3P type water molecules. A water buffer of 10 Å was used and Cl− ions were added to neutralize the systems. The solvated complexes were geometry optimized in 2 cycles using the steepest descent and conjugate gradient methods with the solute molecules constrained by the harmonic potential. After optimization, systems were equilibrated for 2 ns in two steps: during the first step of 50 ps the system was heated from 0 to 300 K under NVT conditions. In the next step the water density was adjusted (NPT conditions). The equilibrated systems were subjected to the 4 ns of productive, unconstrained MD simulations at constant temperature (300 K) and pressure (1 atm) with the time step of 2 fs (SHAKE algorithm was used to restrain the motion of hydrogens). The simulations were performed with thesander program, available within the AMBER11 package,40 using periodic boundary conditions, wherein the electrostatic interactions were calculated using the particle-mesh Ewald method.41 The temperature and pressure was regulated using Langevin dynamics (with collision frequency of 1 ps-1) and the Berendsen barostat,42 respectively. In the direct space the pairwise interactions were calculated within the cut off distance of 11 Å. Since the complexes tended to fall apart (disintegrated), in further simulation of PYPOD–CMP and PYPOD–UMP complexes we used the weak distance constraints for the several PYPOD–UMP pairs of atoms (rup = 3.55 Å, kr = 10 kcal mol−1 Å−2).In total the complexes PYPOD–AMP, PYPOD–CMP and PYPOD–UMP were simulated for 14, 10 and 6 ns, respectively. The trajectories were visualized using the VMD 1.8 program.38
Acknowledgements
Financial support was received from Ministerio de Economia y Competitividad, FEDER and Generalitat Valenciana (Projects CTQ2009-14288-CO4-0, CONSOLIDER INGENIO 2010 CSD2010-00065 and PROMETEO 2011/008) and Ministry of Science, Education and Sport of Croatia (098-0982914-2918, 098-1191344-2860) are gratefully acknowledged. I. P. and E. G.-E. are grateful to COST Action CM1005 for their support in networking and mutual cooperation.Notes and references
- (a) E. Gendaszewska-Darmach and M. Kucharska, Purigenic Signalling, 2011, 7, 193 CrossRef CAS PubMed; (b) B. S. Khakh and G. Burnstock, Sci. Am., 2009, 301, 84 CrossRef CAS PubMed; (c) W. G. Junger, Nat. Rev. Immunol., 2011, 11, 201 CrossRef CAS PubMed.
- M. Idzko, D. Ferrari and H. Eltschig, Nature, 2014, 509, 310 CrossRef CAS PubMed.
- F. B. Chekeni, M. R. Elliott, J. K. Sandilos, S. F. Walk, J. M. Kinchen, E. R. Lazarowski, A. J. Armstrong, S. Penuela, D. W. Laird, G. S. Salvesen, B. E. Isakson, D. A. Bayliss and K. S. Ravichandran, Nature, 2010, 467, 863 CrossRef CAS PubMed.
- E. R. Lazarowski, Purigenic Signalling, 2012, 8, 359 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Supramolecular Chemistry. Concepts and Perspectives, Wiley-VCH, Weinheim, 1995 Search PubMed; (b) J. L. Atwood, Comprehensive and Supramolecular Chemistry, Pergamon, Oxford, 1996 Search PubMed; (c) E. García-España, R. Belda, J. González, J. Pitarch and A. Bianchi, in Receptor for Nucleotides. Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley & Sons, 2012 Search PubMed; (d) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 6603 CrossRef CAS PubMed.
- (a) C. Bazzicalupi, A. Bencini, A. Bianchi, E. Faggi, C. Giorgi, S. Santarelli and B. Valtancoli, J. Am. Chem. Soc., 2008, 130, 2440 CrossRef CAS PubMed; (b) C. Bazzicalupi, A. Bencini, S. Biagini, E. Faggi, S. Meini, C. Giorgi, A. Spepi and B. Valtancoli, J. Org. Chem., 2009, 74, 7349 CrossRef CAS PubMed.
- I. Tabushi, Y. Kobuke and J.-I. Imuta, J. Am. Chem. Soc., 1981, 103, 6152 CrossRef CAS.
- (a) B. Dietrich, M. W. Hosseini and J.-M. Lehn, J. Am. Chem. Soc., 1981, 103, 1282 CrossRef CAS; (b) E. Kimura, M. Kodama and T. Yatsunami, J. Am. Chem. Soc., 1982, 104, 3182 CrossRef CAS.
- (a) A.-S. Delépine, R. Tripier, M. Le Baccon and H. Handel, Eur. J. Org. Chem., 2010, 5380 CrossRef; (b) A.-S. Delepine, R. Tripier and H. Handel, Org. Biomol. Chem., 2008, 6, 1743 RSC.
- (a) G. Malojčić, I. Piantanida, M. Marinić, M. Žinić, M. Marjanović, M. Kralj, K. Pavelić and H.-J. Schneider, Org. Biomol. Chem., 2005, 3, 4373 RSC; (b) A. Sornosa-Ten, M. T. Albelda, J. C. Frias, E. Garcia-Espana, J. M. Llinares, A. Budimir and I. Piantanida, Org. Biomol. Chem., 2010, 8, 2567 RSC.
- (a) P. Arranz-Mascaros, C. Bazzicalupi, A. Bianchi, C. Giorgi, M. D. Gutierrez-Valero, R. Lopez-Garzon, M. L. Godino-Salido and B. Valtancoli, Chem. Commun., 2011, 47, 2814 RSC; (b) P. Arranz-Mascaros, C. Bazzicalupi, A. Bianchi, C. Giorgi, M. L. Godino-Salido, M. D. Gutierrez-Valero, R. Lopez-Garzon and B. Valtancoli, New J. Chem., 2011, 35, 1883 RSC.
- H. Y. Kuchelmeister and C. Schmuck, Chem. – Eur. J., 2011, 17, 5311 CrossRef CAS PubMed.
- A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- (a) V. Kral and J. L. Sessler, Tetrahedron, 1995, 51(2), 539 CrossRef CAS; (b) J. L. Sessler, V. Kral, T. V. Shishkanova and P. A. Gale, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4848 CrossRef CAS PubMed.
- Y. Hisamatsu, K. Hasada, F. Amano, Y. Tsubota, Y. Wasada-Tsutsui, N. Shirai, S. Ikeda and K. Odashima, Chem. – Eur. J., 2006, 12, 7733 CrossRef CAS PubMed.
- (a) L.-M. Tumir, M. Grabar, S. Tomić and I. Piantanida, Tetrahedron, 2010, 66, 2501 CrossRef CAS PubMed; (b) L.-M. Tumir, I. Piantanida, P. Novak and M. Žinić, J. Phys. Org. Chem., 2002, 15, 599 CrossRef CAS; (c) I. Piantanida, B. S. Palm, P. Čudić, M. Žinić and H.-J. Schneider, Tetrahedron Lett., 2001, 42, 6779 CrossRef CAS.
- (a) M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, Supramol. Chem., 1995, 5, 139 Search PubMed; (b) O. Baudoin, F. Gonnet, M.-P. Teulade-Fichou, J.-P. Vigneron, J.-C. Tabet and J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2762 CrossRef CAS.
- (a) M. Shionoya, T. Ikeda, E. Kimura and M. Shiro, J. Am. Chem. Soc., 1994, 116, 3848 CrossRef CAS; (b) S. Aoki and E. Kimura, J. Am. Chem. Soc., 2000, 122, 4542 CrossRef CAS.
- (a) F. Schmidt, S. Stadlbauer and B. Konig, Dalton Trans., 2010, 39, 7250 RSC; (b) S. Aoki and E. Kimura, J. Am. Chem. Soc., 2000, 122, 4542 CrossRef CAS.
- M. Inclán, M. T. Albelda, E. Carbonell, S. Blasco, A. Bauz, A. Frontera and E. García-España, Chem. – Eur. J., 2014, 20, 3730 CrossRef PubMed.
- J. González, J. M. Llinares, R. Belda, J. Pitarch, C. Soriano, R. Tejero, B. Verdejo and E. García-España, Org. Biomol. Chem., 2010, 8, 2367 Search PubMed.
- J. González-García, L. Uzelac, M. Kralj, J. M. Llinares, E. García-España and I. Piantanida, Org. Biomol. Chem., 2013, 11, 2154 Search PubMed.
- (a) J. A. Aguilar, E. García-España, J. A. Guerrero, S. V. Luis, J. M. Llinares, J. F. Miravet, J. A. Ramirez and C. Soriano, J. Chem. Soc., Chem. Commun., 1995, 2237 RSC; (b) G. Kampf, L. E. Kapinos, R. Griesser, B. Lippert and H. Sigel, J. Chem. Soc., Perkin Trans. 2, 2002, 1320 RSC; (c) R. B. Martin and Y. H. Mariam, Met. Ions Biol. Syst., 1979, 8, 57 CAS; (d) H. Sigel, Pure Appl. Chem., 2004, 76, 1869 CAS.
- (a) H. A. Arab, Z. M. Anwar and M. Sokar, J. Chem. Eng. Data, 2004, 49, 256 CrossRef; (b) Y. Kinjo, R. Tribolet, N. A. Corfù and H. Siegel, Inorg. Chem., 1989, 28, 1480 CrossRef CAS.
- (a) C. Frassineti, S. Ghelli, P. Gans, S. Sabatini, M. S. Moruzziy and A. Vacca, Anal. Biochem., 1995, 231, 374 CrossRef CAS PubMed; (b) C. Frassineti, L. Alderighi, P. Gans, A. Sabatini, A. Vacca and S. Ghelli, Anal. Bioanal. Chem., 2003, 376, 1041 CrossRef CAS PubMed; (c) L. Alderighi, A. Bianchi, L. Biondi, L. Calabi, M. D. Miranda, P. Gans, S. Ghelli, P. Losi, L. Paleari, A. Sabatini and A. Vacca, J. Chem. Soc., Perkin Trans. 2, 1999, 2741 RSC; (d) C. Frassineti, L. Alderighi, P. Gans, A. Sabatini, A. Vacca and S. Ghelli, Anal. Bioanal. Chem., 2003, 376, 1041 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- M. T. Albelda, J. Aguilar, S. Alves, R. Aucejo, P. Diaz, C. Lodeiro, J. C. Lima, E. García-España, F. Pina and C. Soriano, Helv. Chim. Acta, 2003, 86, 3118 CrossRef CAS.
- (a) P. Job, Comptes Rendus, 1925, 180, 928 CAS; (b) P. MacCarthy, Anal. Chem., 1978, 50, 2165 CrossRef CAS.
- (a) H. Y. Kuchelmeister and C. Schmuck, Chem. – Eur. J., 2011, 17, 5311 CrossRef CAS PubMed; (b) O. Baudoin, F. Gonnet, M.-P. Teulade-Fichou, J.-P. Vigneron, J.-C. Tabet and J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2762 CrossRef CAS.
- H. Sigel and R. Griesser, Chem. Soc. Rev., 2005, 34, 875 RSC.
- M. Radić Stojković, M. Škugor, S. Tomić, M. Grabar, V. Smrečki, Ł. Dudek, J. Grolik, J. Eilmes and I. Piantanida, Org. Biomol. Chem., 2013, 11, 4077 Search PubMed.
- A. Maggioni, M. von Itzstein, I. B. R. Guzman, A. Ashikov, A. S. Stephens, T. Haselhorst and J. Tiralongo, ChemBioChem, 2013, 14, 1936 CrossRef CAS PubMed.
- X.-F. Shang, H. Su, H. Lin and H.-K. Lin, Inorg. Chem. Commun., 2010, 13, 999 CrossRef CAS PubMed.
- E. García-España, M. J. Ballester, F. Lloret, J. M. Moratal, J. Faus and A. Bianchi, J. Chem. Soc., Dalton Trans., 1988, 101 RSC.
- M. Fontanelli and M. Micheloni, Proceedings of the I Spanish-Italian Congress on Thermodinamics of Metal Complexes, Peñíscola, Castellón, 1990. Program for the automatic control of the microburette and the acquisition of the electromotive force readings Search PubMed.
- (a) G. Gran, Analyst, 1952, 77, 661 RSC; (b) F. J. Rossotti and H. Rossotti, J. Chem. Educ., 1965, 42, 375 CrossRef CAS.
- P. Gans , Programs to determine the distribution of Species in multiequilibria systems from the stability constants and mass balance equations.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. C. Luo, W. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 11, University of California, San Francisco, 2010 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1992, 98, 10089 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81(8), 3684 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic fluorimetric data and potentiometric titrations, detailed experimental conditions, NMR analyses and molecular modelling. See DOI: 10.1039/c4ob02084g |
| This journal is © The Royal Society of Chemistry 2015 |