
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the amino acid composition of cyclic peptides on their self-assembly in lipid bilayers†
Maarten
Danial
a,
Sébastien
Perrier‡
*a and
Katrina A.
Jolliffe
*b
aKey Centre for Polymers & Colloids, The University of Sydney, School of Chemistry, Building F11, Sydney NSW 2006, Australia. E-mail: S.Perrier@warwick.ac.uk
bThe University of Sydney, School of Chemistry, Building F11, Sydney NSW 2006, Australia. E-mail: Kate.Jolliffe@sydney.edu.au
First published on 16th December 2014
Abstract
The effect of amino acid composition on the formation of transmembrane channels in lipid bilayers upon self-assembly of alt-(L,D)-α-cyclic octapeptides has been investigated. Cyclic peptides comprising D-leucine, alternating with different combinations of L-azidolysine, L-lysine(Alloc), L-lysine and L-tryptophan were synthesized and the size of pores formed via self-assembly of these molecules in lipid bilayers was elucidated using large unilamellar vesicle fluorescence assays and dynamic light scattering. Pore formation was examined in large unilamellar vesicles made up of egg yolk phosphatidylcholine or Escherichia coli total lipid extract. From these analyses, we have established that cyclic peptides with charged side chains form large pores while those with neutral side chains form unimeric pores. Furthermore, the cyclic peptides that consist of non-symmetric amino acid configurations possess a higher membrane activity than the cyclic peptides with a symmetric amino acid configuration. In addition, we have found that peptide amphiphilicity plays a vital role in selective partitioning between bilayers that consist of egg yolk phosphatidylcholine and those comprised of E. coli total lipid extract. These results suggest that selective transbilayer channel formation via self-assembly may be a viable alternative for many applications that currently use more expensive, multistep synthesis methods.
Introduction
Protein channels represent one class of structures that maintain cell vitality through sustaining equilibria of ions, water, pH and solutes in organelles and cells. Several prominent examples of protein channels include the aquaporins,1,2 sodium and potassium channels,3 chloride channels,4 and pathogen-derived α-hemolysin.5 These channels provide a direct route of access in which ions and water are transported efficiently across otherwise impermeable phospholipid bilayer membranes.6 The plethora of protein channels in nature also provides inspiration for the design of drug delivery routes,7 sequencing applications,8 or in sensing technologies7,9,10 providing direct access across the phospholipid bilayer membrane. While synthesis of these protein channels is feasible through biotechnological processes, adaptability and modulation of properties such as channel cavity size and ion selectivity to suit different applications poses a formidable challenge. The self-assembly process to build hierarchical, supramolecular structures via a bottom-up approach, however, provides an alternative route that permits access to application-tailored artificial channels. In addition, the self-assembly process also avoids low-yielding, multistep synthesis procedures to create functional structures thereby lowering the cost, increasing the ease of production and ultimately making self-assembled transmembrane channels desirable for use in a range of the aforementioned applications.Artificial transmembrane channels have been created via self-assembly from crown-ethers,11 peptides,12,13 oligoamides,14 nucleotides,15,16 cyclodextrins,17,18 and aromatic macrocycles.19–21 Features of these and other artificial transmembrane channels including ion transport selectivity as well as specificity have been reviewed recently.22–24 Additionally, some remarkable examples of solute-specific transport using self-assembled structures have also been reported. Liu et al. reported a light-responsive ion channel based on a benzocrown ether bound to an azobenzene group with an dodecyl aliphatic tail.25 Upon adoption of the cis conformation with 365 nm UV light, the benzocrown ethers stacked to form a 2.8 Å channel, which had an increased affinity with the lipid bilayer compared to the trans-ion channel, thereby showing that stimuli responsive ion-specific channels can be created. In another example, Langecker et al. constructed a 2 nm artificial channel through the molecular self-assembly of a double stranded DNA stem with cholesterol-modified single stranded DNA to form a cap and mediate lipid attachment.15 Single molecule translocation experiments showed that these artificial channels were able to discriminate single DNA molecules.
Cyclic peptides that self-assemble into nanotubular structures have also offered a versatile route to create artificial channels.26,27 This is in part due to employing high yielding solid phase peptide synthesis methods, which provides access to large amounts of material. Furthermore, through modifying the length of the peptide, precise molecular control over channel diameter can be achieved making it a viable route to create channels with different diameters. Cyclic peptides that contain alternating α-D,L amino acids have therefore been an attractive template to form well-defined nanotubes and channels with cavity diameters ranging from 2 to 13 Å.13,28–32 Another appealing feature of these self-assembling cyclic peptides is that the alternating absolute configuration projects the amino acid side chains out of the plane of the cyclic peptide and enables chemical modification of the peptide side chains without causing significant interference with the extensive hydrogen bonding perpendicular to the plane of the cyclic peptide.
Functionalization of the cyclic peptide side chains with (macro)molecules and the effect upon the properties of nanotubes33–38 and artificial channels39–41 is an exciting recent development of these systems. Although, the effect of the amino acid configuration and type of peptide side chains upon artificial channel formation has been studied,13,27–30,39,42–44 only a selection of studies have investigated the lipid bilayer partitioning of self-assembling cyclic peptides that contain non-natural β- or γ-amino acids.45,46 To the best of our knowledge, no previous study conducted has described the effects on lipid bilayer partitioning of octameric cyclic peptides that contain non-natural α-L-amino acids.
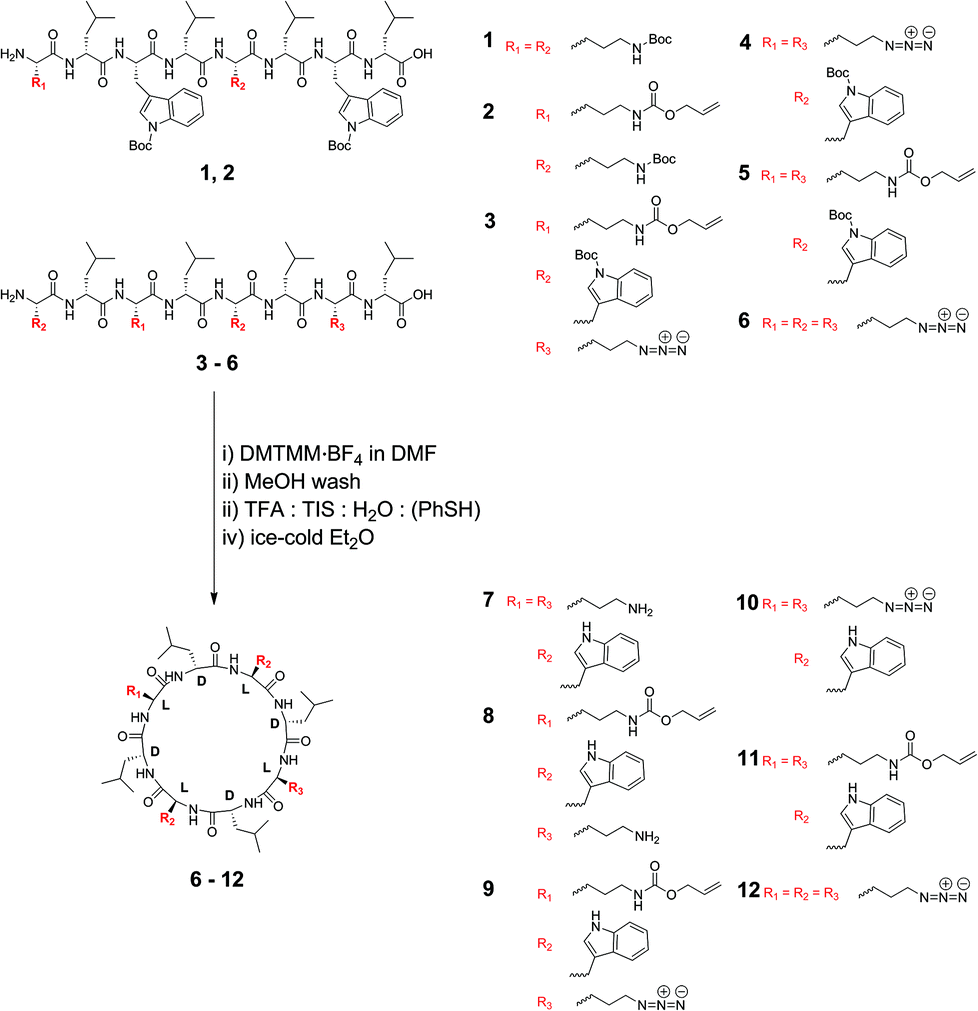
In this paper we investigate the effect upon lipid bilayer channel formation of six different cyclic peptides that are comprised of natural and non-natural α-L amino acids or have amino acid configurations that have not previously been examined. The type of pore formed was elucidated via dynamic light scattering and two independent, pore-size-discriminating large unilamellar vesicle assays, which either had calcein or 5(6)-carboxyfluorescein entrapped. Furthermore, the concentration-dependent pore-formation is examined for two types of LUVs, consisting of Egg yolk phosphatidylcholine (EggPC) and Escherichia coli total lipid extract, respectively.
Results and discussion
Synthesis
Cyclic peptides that consist of alternating L- and D-α-amino acids have previously been shown to adopt a low-energy, flat ring conformation that self-assembles into nanotubes in solution47,48 as well as in lipid bilayers.13,30 Although studies have explored variations in the type of cyclic peptide on phospholipid channel formation, only a limited number of variations in amino acid constituents have been investigated (for a recent review refer to Montenegro et al.27). For instance, Ghadiri et al. noted that octameric cyclic peptides that consisted only of L-glutamine and D-leucine did not have the appropriate surface characteristics to partition into lipid bilayers.13 Furthermore, additional studies predominantly explored application of cyclic peptides that contain α-D,L-amino acids,13,30,39,43,44 β-amino acids45 or γ-amino acids.46 We chose to use cyclic peptides comprising eight α-amino acids as opposed to analogs with four, six or ten residues or β-/γ-amino acids for several reasons. Firstly, cyclic peptides with eight amino acid residues can easily be synthesized to high purity from commercially available or readily accessible starting materials without requiring reverse phase purification protocols. Secondly, cyclic peptides comprising six amino acids or less possess small cavity sizes (<0.6 nm) through which limited solute transport may be observed. Thirdly, based on experiments using N-alkylated cyclic peptides, the tetrameric, hexameric and decameric analogues exhibit lower association constants than the octameric systems, suggesting stronger self-assembly into nanotubes for the cyclic octapeptides.26 In this work, we therefore investigated the ability to form lipid bilayer pores by self-assembly of octameric cyclic peptides. These cyclic peptides consisted of natural and non-natural amino acids, with the number of leucine residues held constant, while the alternating side chains were varied to allow an investigation of side chain functionality on membrane partitioning. Cyclic peptides (7–11) included two L-tryptophan residues, which had previously been shown to be required for bilayer partitioning,13 while cyclic peptide 12, which excluded tryptophan, was synthesized to establish whether the L-azidolysine side chain was capable of promoting phospholipid bilayer partitioning and channel formation in the absence of Trp. A combination of lysine-derived charged (amine, -NH3+), polarizable (azido, -N3), and allyloxycarbonyl (-Alloc) side chains were employed to establish the role of these different functionalities on membrane partitioning, without varying the distance of the functional group from the peptide core or significantly altering the size of the side chains. Cyclic peptides 8 and 9 were unsymmetrically substituted by positioning different side chains on opposite sides of the peptide core to establish whether this resulted in changes to membrane partitioning. In addition and for the first time, the role of these amino acid side chains on channel formation in phospholipid bilayers made from zwitterionic EggPC or negatively charged E. coli total lipid extract was investigated.Synthesis of the required linear peptide precursors (1–6) was achieved through solid phase peptide synthesis49 followed by mild, selective cleavage from a chlorotrityl chloride resin using 20% hexafluoroisopropanol in dichloromethane (Scheme 1) without deprotection of the Boc groups. Although the sequence of amino acids within the linear peptide chain was varied to terminate with L-lysine (1 and 2), with L-tryptophan (3–5) or with L-azidolysine (6) at the N-terminus, this difference did not influence the overall linear peptide synthesis yields or purity. Subsequently, cyclization of the Boc-protected linear peptide precursors was achieved under high dilution via addition of a 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTMM·BF4) reagent (Scheme 1). The DMTMM·BF4 reagent has been shown to be an effective coupling agent that can improve cyclization yields50 and can be produced at high synthesis scales.51
 | ||
| Scheme 1 Synthesis of cyclic peptides used in this study. | ||
The structure of the cyclic peptides was verified by MALDI-FTICR mass spectrometry as well as NMR spectroscopy. Specifically, the disappearance of the signals attributable to the N-terminal α-CH at ∼4.4 ppm and the C-terminal α-CH at ∼4.7 ppm, respectively confirmed cyclization for the peptides terminating with lysine derivatives 1, 2 and 6. Similarly, cyclization of the peptides 3–5, which have N-terminal tryptophan(Boc) was confirmed by the disappearance of the signals attributable to the N-terminal α-CH and C-terminal α-CH at ∼4.1 ppm and ∼4.7 ppm, respectively. As shown previously,41 cyclization of the linear peptides can also be determined using distortionless enhancement by polarization transfer-edited heteronuclear single quantum coherence (HSQC) NMR spectroscopy (Fig. S1–S6†). In addition, the tendency of the cyclic peptides to aggregate, in contrast to the linear peptide precursors, was confirmed by dynamic light scattering.
Characterization of pore type
The structure of the phospholipid bilayer channel formed was characterized using pore-size-discriminating large unilamellar vesicle (LUV) assays along with dynamic light scattering (DLS) analysis. As shown in Fig. 1, the main modes of transbilayer channel formation are via unimeric pores, barrel staves and bilayer (carpet-like) disruption.52 To assess which transbilayer channels are formed, LUVs with either calcein or 5(6)-carboxyfluorescein entrapped were prepared. The formation of barrel staves or lipid bilayer disruption as depicted in Fig. 1b and c was assayed with the calcein-entrapped LUVs. Calcein serves as a reporter fluorescent dye (average diameter ∼ 1.5 nm), which is self-quenched at high concentrations and upon dilution (<8 μM) results in a fluorescence band maximum at 505 nm (ESI Fig. S7†). Molecular confinement of calcein within the lumen in the LUVs leads to self-quenching while release via barrel staves or lipid bilayer disruption causes a reduction in calcein self-quenching. In addition to this assay, dynamic light scattering was performed to exclude the possibility of LUV lysis as a result of lipid bilayer disruption within the time-frame of the LUV assay, thereby corroborating the formation of barrel staves. The presence of smaller pores, e.g. unimeric pores (channel diameter < 0.8 nm) as depicted in Fig. 1a, was revealed with LUVs that contained the pH responsive fluorescent dye 5(6)-carboxyfluorescein, which has a weak fluorescence emission in acidic pH and a strong fluorescence emission at pH > 7. The formation of unimeric pores upon the addition of the cyclic peptides was established using LUV assays with a pH gradient across the phospholipid bilayer, in which the internal LUV environment consisted of a phosphate buffer at pH 5.8 and the external environment consisted of a phosphate buffer at pH 7.4. Collapse of the transbilayer pH gradient due to the formation of transbilayer channels causes an efflux of protons and thus neutralizes the internal LUV environment and results in an increase in 5(6)-carboxyfluorescein fluorescence emission.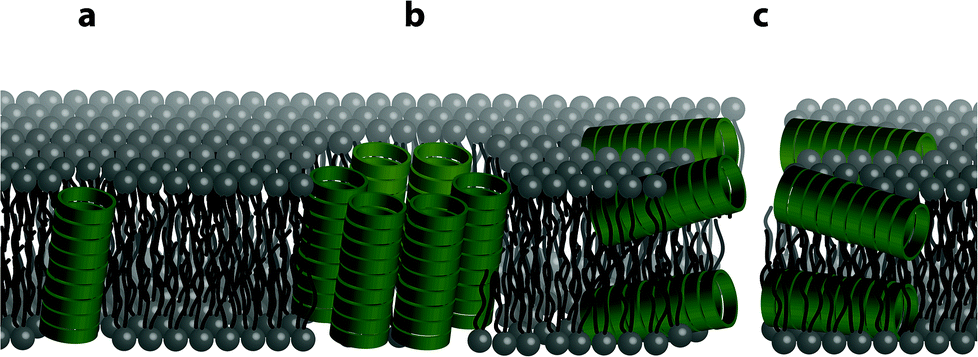 | ||
| Fig. 1 Illustration showing the different possible modes of bilayer channel formation. Formation of cyclic peptide channels in a lipid bilayer can occur as (a) a unimeric pore, (b) a barrel stave or (c) through a bilayer (carpet-like) disruption. | ||
Dynamic light scattering experiments of the LUVs were employed to assess the integrity of the lipid bilayer upon the addition of the cyclic peptides. Upon the formation of unimeric pores and barrel staves, LUV integrity should remain stable unless a high osmotic difference between the internal and external environment causes breakdown of the LUV lipid bilayer. Thus in all experiments, no osmotic potentials across lipid bilayers were introduced. Conversely, lipid bilayer disruption and breakdown of the LUVs into smaller aggregates should be evidenced by DLS by a distribution shift to smaller hydrodynamic radius. As a control, triton X-100 was added to LUVs in order to assess the changes in the hydrodynamic radius distribution upon LUV lysis and hence loss of lipid bilayer integrity.
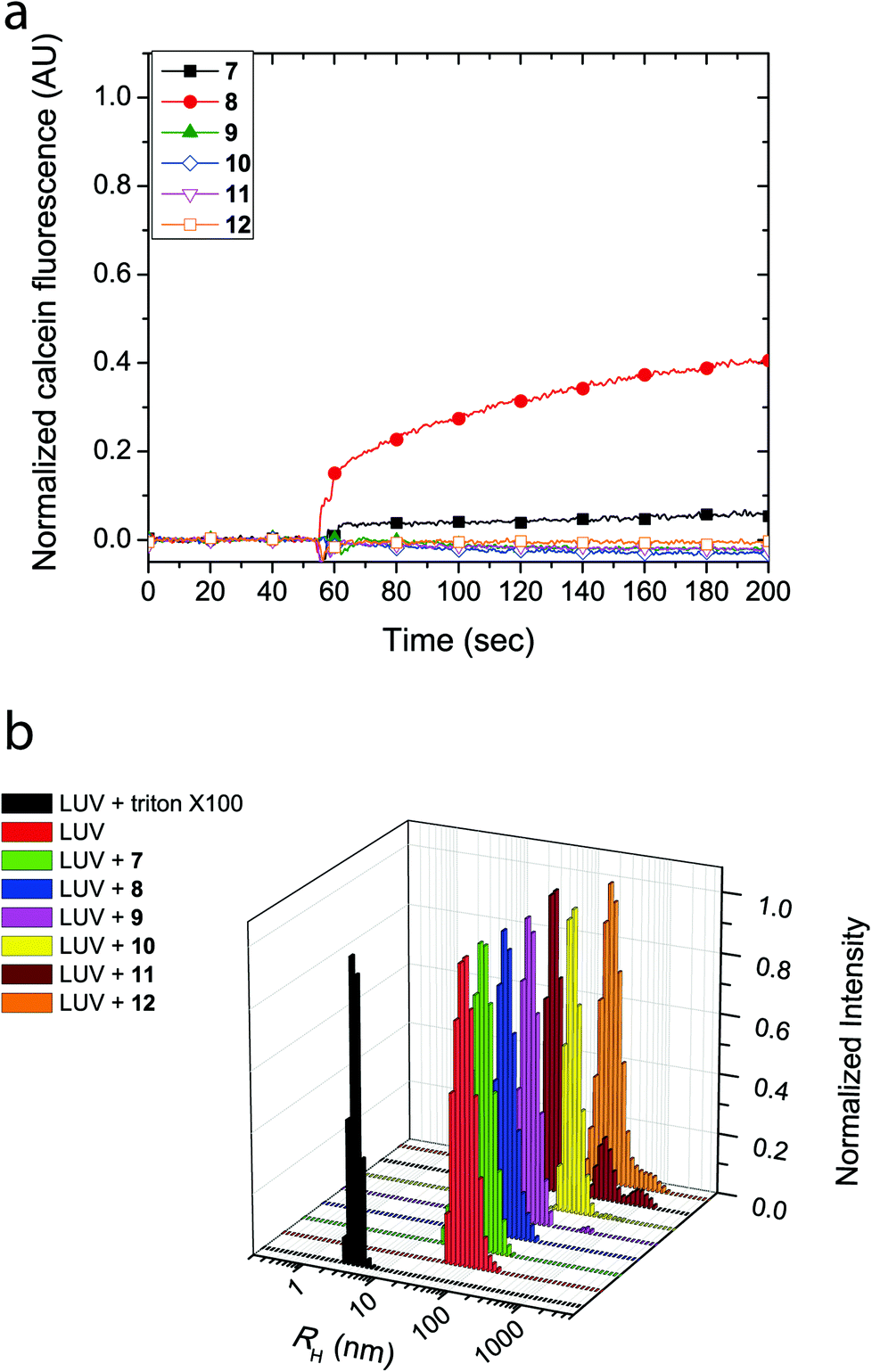
The cyclic peptides were examined for their lipid bilayer partitioning and their propensity to form barrel staves or bilayer disruption using calcein-entrapped LUVs made from egg yolk phosphatidylcholine (EggPC) or made from E. coli total lipid extract. As presented in Fig. 2a, the addition of cyclic peptides 7 and 8, which contain the positively charged lysine residues, to LUVs prepared from EggPC resulted in immediate calcein release and fluorescence increases of 80% and 65%, respectively, after 200 s. In contrast, little to no change in fluorescence was observed upon the addition of cyclic peptides 9–12 to EggPC LUVs containing calcein. Furthermore as shown in Fig. 2b, hydrodynamic radius (RH) distributions of the LUV solutions centered at ∼55 nm obtained from dynamic light scattering indicated that, within the time-frame of the experiment, LUV lysis did not result upon the addition of cyclic peptides 7–12. The size distribution profile upon the addition of cyclic peptide 10, however, revealed an additional species that was attributed to the formation of large aggregates (RH ∼ 200–300 nm) caused by limited solubility of this cyclic peptide in aqueous buffer (Fig. 2b). Further corroboration for the formation of barrel staves as opposed to bilayer disruption due to the addition of cyclic peptides was achieved via concentration dependent measurements (vide infra) as well as comparison to LUV lysis through the addition triton X-100, which leads to a hydrodynamic radius centered at 3–4 nm (Fig. 2b). Taken together, these results strongly suggest that the cause for calcein dye leakage from the LUVs upon the addition of cyclic peptides 7 and 8, both of which have positively charged side chains, results from the formation of barrel staves rather than via bilayer disruption.
 | ||
| Fig. 2 (a) Calcein dye leakage experiments with EggPC LUVs upon addition of cyclic peptides (final concentration = 19 μM) at approximately 60 seconds. The fraction of calcein fluorescence measured is relative to 100% LUV lysis in the presence of triton X-100. (b) Dynamic light scattering (CONTIN) hydrodynamic radius (RH) distributions obtained with EggPC LUVs upon the addition of cyclic peptides or triton X-100. | ||
Similarly, LUVs prepared from E. coli total lipid extract also showed release of calcein upon addition of cyclic peptides 7 and 8, albeit only leading to a fluorescence increase of 6% and 40%, respectively (Fig. 3a). Notably, the asymmetric amphiphilic cyclic peptide 8, which contains L-Lysine and L-Lysine(Alloc) residues on opposite sides of the cyclic peptide ring caused a higher calcein dye release from the LUV than the symmetric cyclic peptide 7, which contains two L-Lysine residues. As shown in Fig. 3b, dynamic light scattering confirmed that the LUVs remained intact in the presence of the cyclic peptides suggesting that bilayer disruption and LUV lysis was not the cause of calcein release. Further evidence that rules out the formation of carpet-like bilayer disruptions was evident from concentration dependent experiments with 5(6)-carboxyfluorescein entrapped LUVs (vide infra). Similar to experiments performed with EggPC LUVs, dynamic light scattering revealed large aggregates (RH ∼ 200–300 nm) in mixtures of the LUVs and cyclic peptides 10 and 12. These aggregates were attributed to the limited solubility of cyclic peptides 10 and 12 in aqueous solution but also suggested that minimal phospholipid bilayer partitioning was taking place in the form of unimeric pores.
 | ||
| Fig. 3 (a) Calcein dye leakage experiments with LUVs made from E. coli total lipid extract upon addition of cyclic peptides (final concentration = 19 μM) at approximately 60 seconds. The fraction of calcein fluorescence measured is relative to 100% LUV lysis in the presence of triton X-100. (b) Dynamic light scattering (CONTIN) hydrodynamic radius (RH) distributions obtained with E. coli LUVs upon the addition of cyclic peptides or triton X-100. | ||
The formation of large pores in both calcein-entrapped LUVs composed of EggPC and E. coli total lipid extract was also evident in the assay with 5(6)-carboxyfluorescein entrapped LUVs. As Fig. 4 shows, the highest fluorescence response was due to cyclic peptide 7 and 8 whereby the activity difference between LUVs made from EggPC or E. coli total lipid extract was consistent with observations made in the calcein-entrapped LUV assays (vide supra). In contrast to the experiments with calcein-entrapped LUVs, the addition of cyclic peptides 9–12 to LUVs containing 5(6)-carboxyfluorescein resulted in a 20–40% fluorescence increase in EggPC LUVs and a 10–35% fluorescence increase in LUVs made from E. coli total lipid extract. Notably, a higher degree of unimeric pore formation was evident with cyclic peptide 9 compared to cyclic peptides 10–12. Once again, asymmetry within cyclic peptide 9 with a single L-azidolysine opposite a single L-lysine(Alloc) may promote the lipid bilayer partition activity over symmetric cyclic peptides 10–12, which do not possess two different amino acids opposite each other in the cyclic peptide ring. A slight difference between cyclic peptide induced pore formation in LUVs made from EggPC and E. coli total lipid extract was observed (Fig. 4). The lower degree of pore formation of cyclic peptide 12 in LUVs made from E. coli total lipid extract agrees with a higher degree of aggregation observed by DLS measurements (Fig. 3b). The low degree of pore formation in both types of LUVs upon the addition of cyclic peptide 12, which lacks tryptophan residues, was consistent with previous studies in which L-tryptophan was replaced with L-glutamine.13
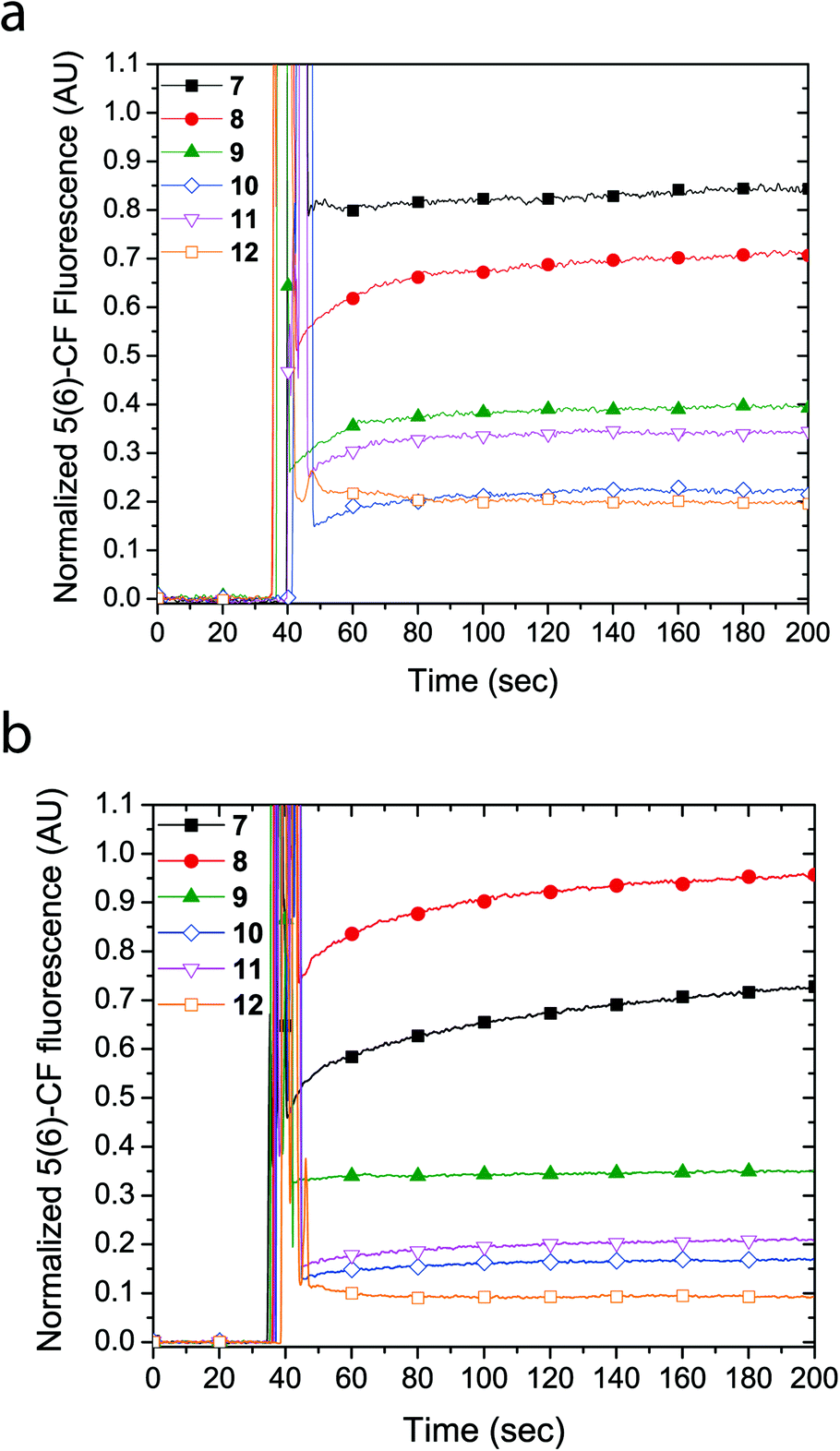 | ||
| Fig. 4 Proton efflux measured using 5(6)-carboxyfluorescein entrapped in LUVs made from (a) EggPC or (b) E. coli total lipid extract. The cyclic peptide was added to the LUV mixture ∼ 40 seconds (final concentration = 19 μM). The fluorescence signal is normalized against 100% LUV lysis upon the addition of triton X-100. | ||
Concentration dependence on pore formation
A concentration titration of the cyclic peptides was performed on LUVs made from EggPC and E. coli total lipid extract to establish the concentration-dependent pore-formation activity as well as support previous experiments that provide information for barrel stave formation over carpet-like bilayer disruption. As Fig. 5 shows, an increase in concentration of the cyclic peptide corresponded to a non-linear increase in fluorescence emission upon incubation with 5(6)-carboxyfluorescein-entrapped LUVs. While these trends reveal that the concentration of all cyclic peptides required to reach 50% proton efflux varies, the concentration-dependent fluorescence response also indicated that no ‘leaky’ phospholipid membranes were formed upon the addition of cyclic peptide and thus affirms the absence of phospholipid bilayer disruption. Comparison of titration results revealed a larger fluorescence transition observed with LUVs made from EggPC than with LUVs made from E. coli total lipid extract (Fig. 5). Although the source of these variations between LUVs composed of EggPC and LUVs composed of E. coli total lipid extract is still unclear, these results may have implications for antibacterial applications.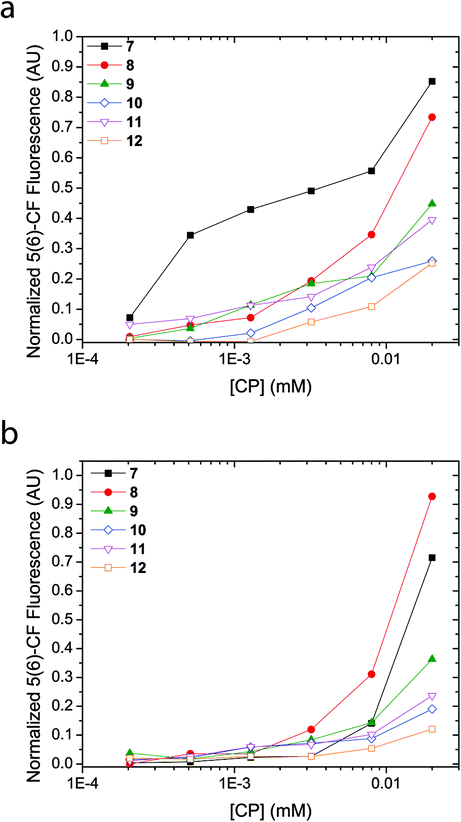 | ||
| Fig. 5 5(6)-carboxyfluorescein fluorescence response as a function of cyclic peptide concentration. A non-linear concentration dependent fluorescence response indicates unimeric pore formation in 5(6)-carboxyfluorescein-entrapped LUVs made from (a) EggPC and (b) E. coli total lipid extract. | ||
Another purpose of the titration series presented in Fig. 5 was to perform a Hill analysis,53 which ultimately reveals the concentration of cyclic peptide required to reach 50% proton efflux (EC50) as well as the Hill coefficient (n). However, the results obtained via the Hill analysis were not reliable since the majority of the cyclic peptides (9–12) did not reach 50% proton efflux within the concentration range assayed. Applying a non-linear regression modeling procedure on the data presented in Fig. 5 led to large errors so EC50 values and Hill coefficients could not be determined. While addition of data points at higher concentrations of cyclic peptide would remedy the error, attempts to increase the concentration of cyclic peptide in the assay while keeping the concentration of DMSO at 2 vol.% were unsuccessful and resulted in precipitation. Precipitation in the LUV assay does not provide an accurate measure for channel activity determined via the 5(6)-carboxyfluorescein-entrapped LUV assay because the fraction of active cyclic peptides in solution is unknown. Alternatively, an elevated concentration of cyclic peptides could also be achieved by addition of a larger aliquot to the solution containing LUVs. However, the increased DMSO content would result in a compromised phospholipid bilayer stability and may cause contents of the LUV to leak out in to the external medium thereby influencing the result obtained by fluorescence spectroscopy. Since determination of the EC50 and Hill coefficient cannot be attained reliably in this contribution, information on the cooperativity, stoichiometry and stability of the transbilayer supramolecular assembly will be subject of future work.
Although membrane activity of peptides and proteins is still poorly understood, the differences observed in pore formation by cyclic peptides 7–12 (barrel stave formation by 7 and 8 and unimeric pore formation by 9–12) can be attributed to differences in how the different functional groups interact with the lipid bilayer, together with the differences in charge and intrinsic lipid curvature profiles of the EggPC and E. coli total lipid extract lipid components. It is expected that while the positively charged side chains of 7 and 8 might interact with the lipid headgroups, they would not interact with the hydrophobic membrane interior. This may explain the barrel-stave pores formed by these peptides in which the positively charged side chains can be effectively screened from the membrane lipids by projection into the interior of the macropores and/or towards the neighbouring ‘staves’. In contrast, unimeric pores are observed for compounds 9–12, which bear side chains more likely to partition into the interior of the membrane.
Significant differences between pore formation by 7 and 8 were also observed in vesicles with differing lipid compositions. These may be a result of the differences in intrinsic curvature between EggPC and E. coli total lipid extract. The intrinsic curvature of a bilayer is induced by the size ratio of the lipid head group to the lipid acyl tail domain and ultimately establishes whether phospholipid bilayers are energetically stable. In phospholipid bilayers, cyclic peptides 7 and 8 could form barrel staves as illustrated in Fig. 6a and b, respectively. EggPC consists of zwitterionic phosphatidylcholine lipids (99.5%), e.g. 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and a small proportion of sphingomyelin (0.5%),54 which has been determined to possess near-zero intrinsic curvature.55 A near-zero intrinsic curvature implies that a low energy barrier has to be overcome if the bilayer folds around the barrel stave without exposing the hydrophobic acyl domain to the cyclic peptide amine side chains (Fig. 6a). As a result of near-zero intrinsic curvature of EggPC bilayers, we propose that the positively charged L-lysine amine side chains of cyclic peptide 7 intercalate with the zwitterionic lipid headgroups of EggPC (Fig. 6a). Conversely, the addition of amphiphilic cyclic peptide 8 to LUVs made from EggPC suggests barrel stave formation as illustrated in Fig. 6b. This proposed structure has been suggested to be stabilized by van der Waals interactions between the acyl tail domain of the phospholipid bilayer. In this system, the L-lysine(Alloc) side chain could interact with the lipid tails leading to the projection of positively charged L-lysine side chains into the barrel stave cavity (Fig. 6b).
 | ||
| Fig. 6 Two proposed modes of barrel staves upon interaction of (a) cyclic peptide 7 with phospholipid bilayers made from EggPC, which possesses near-zero intrinsic lipid curvature and (b) cyclic peptide 8 with phospholipid bilayers made from E. coli total lipid extract, which has strong negative intrinsic lipid curvature. | ||
In contrast to EggPC, the composition of E. coli total lipid extract consists of zwitterionic phosphatidylethanolamine (57.5%), negatively charged phosphatidylglycerol (15.1%) and negatively charged cardiolipin (9.8%).54 This lipid composition leads to a bilayer with strong negative intrinsic curvature55 and would explain the low calcein release observed upon addition of cyclic peptide 7 to the LUVs as the scenario illustrated in Fig. 6a in which bilayer folding takes place to accommodate interactions with positively charged L-lysine side chains of cyclic peptide 7 is energetically unfavorable. Barrel stave formation upon addition of amphiphilic cyclic peptide 8 to bilayers made from E. coli total lipid extract is proposed in Fig. 6b. In this proposal, the L-lysine(Alloc) side chain on cyclic peptide 8 intercalates with the phospholipids via van der Waals interactions.
Conclusions
In this paper the propensity of six structurally different cyclic peptides to self-assemble into transbilayer pores in phospholipid bilayers made from egg yolk phosphatidylcholine and from E. coli total lipid extract was investigated. The type of transbilayer pore formed by each cyclic peptide was elucidated via pore-size discriminating LUV assays and dynamic light scattering of the LUVs in the presence of the cyclic peptide. The results obtained from the calcein-entrapped LUV assay indicate that cyclic peptides with amine sidechains (7 and 8), which would be expected to be protonated under the assay conditions (pKa ∼ 10.5), had the propensity to form larger pores than the non-charged cyclic peptides (9–12) in LUV bilayers. The LUV assays also revealed that symmetric cyclic peptide 7 with two amino side-chains had low activity upon incubation with LUVs made from E. coli total lipid extract while having the highest activity of all peptides investigated with LUVs made from egg yolk phosphatidylcholine. Conversely, asymmetric cyclic peptide 8 with only one amino side chain demonstrated higher pore forming activity in LUVs made from E. coli total lipid extract than observed in LUVs made from egg yolk phosphatidylcholine. This result demonstrates that the amphiphilicity of the cyclic peptide plays a pivotal role in partitioning of these molecules within bilayers made from E. coli total lipid extract in contrast to bilayers made from egg yolk phosphatidylcholine. Furthermore, the 5(6)-carboxyfluorescein-entrapped LUV assays also demonstrated that incorporation of L-tryptophan and L-lysine(Alloc) residues in the cyclic peptide promote pore formation. Together, these results indicate that new combinations of amino acids within a self-assembling cyclic peptide can lead to the emergence of novel biomembrane–active materials.Experimental
Materials
All reagents and solvents were used as received from commercial suppliers unless specifically mentioned. The cyclization coupling agent 4-(4,6-dimethoxy-(1,3,5)triazin-2-yl)-4-methyl-morpholinium tetrafluoroborate (DMTMM·BF4) was prepared as described by S. A. Raw.51 The Fmoc-protected L-azidolysine was prepared as described by Chapman et al.37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5 v/v/v, 3 mL) for 4 h under an atmosphere of nitrogen. The reaction mixture was afterwards concentrated under reduced pressure and then triturated with cold diethylether to give 9 as an off-white solid in quantitative yield. HRMS (MALDI-FTICR) m/z [M + Na]+ calculated: 1213.6857, found: 1213.6837. See Fig. S3† for HSQC NMR.
:5:5:5 v/v/v/v, 3 mL) for 3 h under an atmosphere of nitrogen. The reaction mixture was afterwards concentrated under reduced pressure and then triturated with cold diethylether to give 11 as an off-white solid in quantitative yield. HRMS (MALDI-FTICR) m/z [M + Na]+ calculated: 1271.7163, found: 1271.7147. See Fig. S5† for HSQC NMR.
:5:5 v/v/v, 3 mL) for 4 h under an atmosphere of nitrogen. The reaction mixture was afterwards concentrated under reduced pressure and then triturated with cold diethylether to give 9 as an off-white solid in quantitative yield. HRMS (MALDI-FTICR) m/z [M + Na]+ calculated: 1213.6857, found: 1213.6837. See Fig. S3† for HSQC NMR.
:5:5:5 v/v/v/v, 3 mL) for 3 h under an atmosphere of nitrogen. The reaction mixture was afterwards concentrated under reduced pressure and then triturated with cold diethylether to give 11 as an off-white solid in quantitative yield. HRMS (MALDI-FTICR) m/z [M + Na]+ calculated: 1271.7163, found: 1271.7147. See Fig. S5† for HSQC NMR.
Large unilamellar vesicle assays
Calcein- or 5(6)-carboxyfluorescein-entrapped LUVs consisting of either egg yolk phosphatidylcholine or E. coli total lipid extract were prepared via extrusion method as described in Danial et al.41 Briefly, egg yolk phosphatidylcholine (10 mg) or E. coli total lipid extract (10 mg) was dissolved in chloroform (50 mL) after which slow removal of the solvent via rotary evaporation yielded a lipid film. The lipid film was then hydrated with a solution containing either calcein (40 mM) in sodium phosphate buffer (10 mM, pH 7.4) or 5(6)-carboxyfluorescein (30 μM) in sodium phosphate buffer (10 mM, pH 5.8) for 1–2 hours and placed in a sonic bath for 30 seconds. The suspension of lipid vesicles is then subjected to five freeze–thaw cycles followed by extrusion (29 times) through a 100 nm pore-size polycarbonate membrane using the Avanti® mini-extruder kit. The large unilamellar vesicles (LUVs) were then purified from unencapsulated fluorescent dye using a Sephadex G-200 column (25 cm × 4.0 cm) using phosphate buffer (10 mM, pH 7.4) in the case of calcein-entrapped LUVs and using phosphate buffer (10 mM, pH 5.8) in the case of 5(6)-carboxyfluorescein-entrapped LUVs. Elution of the calcein-entrapped LUVs was monitored visually through the appearance of a light yellow band from the column. Elution of the 5(6)-carboxyfluorescein-entrapped LUVs from the column was monitored using scattering of laser light observed by holding a hand-held laser pointer at the column nozzle. Both calcein- and 5(6)-carboxyfluorescein-entrapped LUVs were diluted in sodium phosphate buffer (10 mM, pH 7.4) to yield LUV solutions with a final lipid concentration 0.1 mgmL−1. The size of the LUVs was verified using dynamic light scattering. All solutions containing LUVs were used in fluorescence experiments on the same day as preparation.
Time-dependent fluorescence was performed using a Perkin-Elmer LS50B luminescence spectrometer at 20 °C. Calcein or 5(6)-carboxyfluorescein-entrapped large unilamellar vesicles (2 mL, 0.1 mgmL−1, 0.13 mM) were aliquoted into a disposable PMMA fluorescence cuvette. For the calcein-entrapped LUVs, the excitation wavelength was set at 493 nm and the emission wavelength was set at 505 nm, with excitation slit set at 2.5 nm and emission slit set at 10.0 nm. For the 5(6)-carboxyfluorescein-entrapped LUVs, the excitation wavelength was set at 493 nm and the emission wavelength was set at 510 nm, with the excitation slit set at 10.0 nm and the emission slit set at 15.0 nm. The emission of the baseline at 505 nm was monitored for ∼50 seconds followed by the addition of the cyclic peptide (40 μL of 1 mM stock solution in DMSO). In a separate experiment, triton X-100 (40 μL, 10% w/v in 10 mM phosphate buffer, pH 7.4) was added to the cuvette containing LUVs to establish the maximum (calcein and 5(6)-carboxyfluorescein) fluorescence, fT that can be attained in the assay. The change in 5(6)-carboxyfluorescein emission, Δf (in percent) was determined using the following equation:
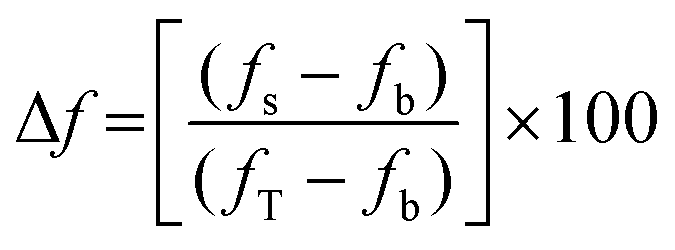 | (1) |
Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed on a Malvern ZetaSizer Nano S equipped with a 633 nm laser and a detector at 173° backscattering angle operating at 20 °C. Calcein-entrapped LUVs (2 mL, 0.1 mgmL−1, 0.13 mM) made from egg yolk phosphatidylcholine or E. coli total lipid extract were aliquoted into a disposable PMMA fluorescence cuvette to which the cyclic peptide (40 μL of 1 mM stock solution in DMSO) was added. The samples were allowed to equilibrate for 10 minutes before the DLS measurement was initiated. Particle (CONTIN) size distributions were analyzed using the intensity distribution using the ‘general purpose-normal resolution’ mode in the Zetasizer software (v 6.32).
Acknowledgements
The Australian Research Council Discovery (K.A.J. and S. P.) and Future Fellowship (S. P.) Programmes are acknowledged for funding. Ms. Carmen My-Nhi Tran (The University of Sydney) is acknowledged for performing preliminary fluorescence experiments and Dr Philip G. Young (The University of Sydney) is acknowledged for synthesizing several cyclic peptides. Dr Nick Proschogo (The University of Sydney) is thanked for performing MALDI-FTICR and electrospray ionization–mass spectrometry on the peptides presented in this paper. Dr Ming Liang Koh (The University of Sydney) is thanked for proofreading this manuscript.Notes and references
- K. Murata, K. Mitsuoka, T. Hiral, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599–605 CrossRef CAS PubMed.
- L. S. King, D. Kozono and P. Agre, Nat. Rev. Mol. Cell Biol., 2004, 5, 687–698 CrossRef CAS PubMed.
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1866 CrossRef CAS.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CAS.
- S. Majd, E. C. Yusko, Y. N. Billeh, M. X. Macrae, J. Yang and M. Mayer, Curr. Opin. Biotechnol., 2010, 21, 439–476 CrossRef CAS PubMed.
- A. De La Escosura-Muñiz and A. Merkoçi, ACS Nano, 2012, 6, 7556–7583 CrossRef PubMed.
- B. A. Cornell, V. L. B. Braach-Maksvytis, L. G. King, P. D. J. Osman, B. Raguse, L. Wieczorek and R. J. Pace, Nature, 1997, 387, 580–583 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- V. E. Carmichael, P. J. Dutton, T. M. Fyles, T. D. James, J. A. Swan and M. Zojaji, J. Am. Chem. Soc., 1989, 111, 767–769 CrossRef CAS.
- R. R. Ketchem, W. Hu and T. A. Cross, Science, 1993, 261, 1457–1460 CAS.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. Yuan, A. J. Clements, S. V. Harding, G. Szabo, Z. Shao and B. Gong, J. Am. Chem. Soc., 2008, 130, 15784–15785 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS PubMed.
- N. Madhavan, E. C. Robert and M. S. Gin, Angew. Chem., Int. Ed., 2005, 44, 7584–7587 CrossRef CAS PubMed.
- N. Badi, L. Auvray and P. Guégan, Adv. Mater., 2009, 21, 4054–4057 CrossRef CAS.
- X. Zhou, G. Liu, K. Yamato, Y. Shen, R. Cheng, X. Wei, W. Bai, Y. Gao, H. Li, Y. Liu, F. Liu, D. M. Czajkowsky, J. Wang, M. J. Dabney, Z. Cai, J. Hu, F. V. Bright, L. He, X. C. Zeng, Z. Shao and B. Gong, Nat. Commun., 2012, 3 Search PubMed.
- L. Chen, W. Si, L. Zhang, G. Tang, Z. T. Li and J. L. Hou, J. Am. Chem. Soc., 2013, 135, 2152–2155 CrossRef CAS PubMed.
- W. Si, L. Chen, X. B. Hu, G. Tang, Z. Chen, J. L. Hou and Z. T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS PubMed.
- T. M. Fyles, Chem. Soc. Rev., 2007, 36, 335–347 RSC.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed.
- T. Liu, C. Bao, H. Wang, Y. Lin, H. Jia and L. Zhu, Chem. Commun., 2013, 49, 10311–10313 RSC.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- J. Montenegro, M. R. Ghadiri and J. R. Granja, Acc. Chem. Res., 2013, 46, 2955–2965 CrossRef CAS PubMed.
- T. Suga, S. Osada and H. Kodama, Bioorg. Med. Chem., 2012, 20, 42–46 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- J. R. Granja and M. R. Ghadiri, J. Am. Chem. Soc., 1994, 116, 10785–10786 CrossRef CAS.
- N. Khazanovich, J. R. Granja, D. E. McRee, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1994, 116, 6011–6012 CrossRef CAS.
- J. Taira, S. Osada, R. Hayashi, T. Ueda, M. Jelokhani-Niaraki, H. Aoyagi and H. Kodama, Bull. Chem. Soc. Jpn., 2010, 83, 683–688 CrossRef CAS.
- J. Couet, J. D. Jeyaprakash, S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297–3301 CrossRef CAS PubMed.
- W. S. Horne, N. Ashkenasy and M. R. Ghadiri, Chem. – Eur. J., 2005, 11, 1137–1144 CrossRef CAS PubMed.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831–7838 CrossRef CAS.
- C. Reiriz, R. J. Brea, R. Arranz, J. L. Carrascosa, A. Garibotti, B. Manning, J. M. Valpuesta, R. Eritja, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2009, 131, 11335–11337 CrossRef CAS PubMed.
- R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2011, 2, 1956–1963 RSC.
- R. Chapman, K. A. Jolliffe and S. Perrier, Aust. J. Chem., 2010, 63, 1169–1172 CrossRef CAS.
- L. Motiei, S. Rahimipour, D. A. Thayer, C.-H. Wong and M. R. Ghadiri, Chem. Commun., 2009, 3693–3695 RSC.
- M. Danial, C. M.-N. Tran, P. G. Young, S. Perrier and K. A. Jolliffe, Nat. Commun., 2013, 4, 2780 Search PubMed.
- M. Danial, C. M.-N. Tran, K. A. Jolliffe and S. Perrier, J. Am. Chem. Soc., 2014, 136, 8018–8026 CrossRef CAS PubMed.
- J. T. Fletcher, J. A. Finlay, M. E. Callow, J. A. Callow and M. R. Ghadiri, Chem. – Eur. J., 2007, 13, 4008–4013 CrossRef CAS PubMed.
- J. Sánchez-Quesada, H. S. Kim and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 2503–2506 CrossRef.
- D. Wang, L. Guo, J. Zhang, R. W. Roeske, L. R. Jones, Z. Chen and C. Pritchard, J. Pept. Res., 2001, 57, 301–306 CrossRef CAS.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651–656 CrossRef CAS.
- R. García-Fandiño, M. Amorín, L. Castedo and J. R. Granja, Chem. Sci., 2012, 3, 3280–3285 RSC.
- P. De Santis, S. Morosetti and R. Rizzo, Macromolecules, 1974, 7, 52–58 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- S. L. Mellor, D. A. Wellings, J.-A. Fehrentz, M. Paris, J. Martinez, N. J. Ede, A. M. Bray, D. J. Evans and G. B. Bloomberg, in Fmoc Solid Phase Peptide Synthesis, Oxford Univ. Press, New York, NY, USA, 2000 Search PubMed.
- S. J. Butler and K. A. Jolliffe, Org. Biomol. Chem., 2011, 9, 3471–3483 CAS.
- S. A. Raw, Tetrahedron Lett., 2009, 50, 946–948 CrossRef CAS PubMed.
- Y. Shai, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 55–70 CrossRef CAS.
- A. V. Hill, Biochem. J., 1913, 7, 471–480 CAS.
- Phospholipid compositions of Egg yolk phosphatidylcholine and E. coli total lipid extract were derived from Avanti Polar lipids, Inc. http://www.avantilipids.com.
- B. Kollmitzer, P. Heftberger, M. Rappolt and G. Pabst, Soft Matter, 2013, 9, 10877–10884 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the linear peptide synthesis and additional fluorescence and NMR data are included. See DOI: 10.1039/c4ob02041c |
| ‡ Current affiliation: The University of Warwick, Department of Chemistry, Coventry CV4 7AL, UK; Faculty of Pharmacy and Pharmaceutical Sciences, Monash University, VIC 3052, Australia. |
| This journal is © The Royal Society of Chemistry 2015 |