
Modulation of human IAPP fibrillation: cosolutes, crowders and chaperones
Mimi
Gao
a,
Kathrin
Estel
b,
Janine
Seeliger
b,
Ralf P.
Friedrich
c,
Susanne
Dogan
b,
Erich E.
Wanker
c,
Roland
Winter
b and
Simon
Ebbinghaus
*a
aDepartment of Physical Chemistry II, Ruhr-University Bochum, 44780 Bochum, Germany. E-mail: simon.ebbinghaus@rub.de
bPhysical Chemistry I – Biophysical Chemistry, Department of Chemistry and Chemical Biology, TU Dortmund, Otto-Hahn-Str. 6, 44227 Dortmund, Germany
cMax Delbrueck Center for Molecular Medicine, Robert-Roessle-Str. 10, 13125 Berlin, Germany
First published on 11th November 2014
Abstract
The cellular environment determines the structure and function of proteins. Marginal changes of the environment can severely affect the energy landscape of protein folding. However, despite the important role of chaperones on protein folding, less is known about chaperonal modulation of protein aggregation and fibrillation considering different classes of chaperones. We find that the pharmacological chaperone O4, the chemical chaperone proline as well as the protein chaperone serum amyloid P component (SAP) are inhibitors of the type 2 diabetes mellitus-related aggregation process of islet amyloid polypeptide (IAPP). By applying biophysical methods such as thioflavin T fluorescence spectroscopy, fluorescence anisotropy, total reflection Fourier-transform infrared spectroscopy, circular dichroism spectroscopy and atomic force microscopy we analyse and compare their inhibition mechanism. We demonstrate that the fibrillation reaction of human IAPP is strongly inhibited by formation of globular, amorphous assemblies by both, the pharmacological and the protein chaperones. We studied the inhibition mechanism under cell-like conditions by using the artificial crowding agents Ficoll 70 and sucrose. Under such conditions the suppressive effect of proline was decreased, whereas the pharmacological chaperone remains active.
Introduction
Since the abnormal accumulation of insoluble fibrillar protein aggregates in tissues and organs, either intracellularly or extracellularly located, has been linked with protein misfolding diseases, such as Alzheimer's disease, Parkinson's disease, Huntington's disease and type 2 diabetes mellitus, much effort has been devoted to unravel their pathogenetic mechanism and to find therapeutic interventions. Proteins, involved in those diseases, are generally intrinsically disordered and can fold into amyloidogenic (fibrillar) aggregates. Such amyloids are highly structured stable protein assemblies that have been shown to be cytotoxic.1,2 However, recent studies suggest that amyloids can even be protective to the cells and that transient soluble oligomers are the primary cytotoxic species.2–4The two competing pathways between folding and aggregation may be rationalized by a double funnel-shaped energy landscape.5 The early steps of aggregation involve the formation of partially folded intermediates that are prone to form aggregated species.6 Such intermediates are formed by perturbing the native conformational ensemble through physical-chemical parameters such as temperature, pressure and pH,7–9 but also by adding cosolutes to the solution10–15 or with the aid of chaperones, either molecular16–18 or pharmacological.19–22
Although such effects have been studied extensively, the interpretation of their impact on the folding and aggregation landscape often remains ambiguous. Here we present a comprehensive kinetic and thermodynamic study to directly compare the effects of different classes of aggregation modulators and cosolutes on protein aggregation and fibrillation of a disease-related amyloidogenic polypeptide: (i) osmolytes or chemical chaperones, (ii) pharmacological chaperones, and (iii) protein chaperones.
Osmolytes or chemical chaperones
Osmolytes or chemical chaperones are cellular occurring small organic molecules that function as responders to equilibrate cellular osmotic pressure and thus to maintain protein stability and functionality.23,24 Typical osmolytes are carbohydrates (e.g. trehalose), polyoles (e.g. glycerine, sorbitol, inositol), amino acids (e.g. glycine, proline, taurine), and methylamines (e.g. trimethylamine N-oxide (TMAO), betaine).25 They preferentially interact with the solvent water in comparison to the protein surface. This preferential hydration effect (also referred to as the osmophobic effect) stabilizes proteins enthalpically by shifting the folding equilibrium towards more compact (native-like) structures with eventually less solvent accessible surface area (SASA).26–28 However, it is often unclear how the increased monomer stability translates to aggregation as osmolytes can either promote or inhibit aggregation.11,14 Different mechanisms have been discussed on how osmolytes could modulate the aggregation pathway. Aggregation-prone folding intermediates could be ameliorated by osmolytes inhibiting aggregation.11,15,29 On the other hand, fibrillar structures are compact with low SASA. Thus, osmolytes could enhance fibrillation by stabilizing these structures.13,14,30,31Macromolecular cosolutes
Macromolecular cosolutes such as dextran, Ficoll and polyethylene glycol (PEG), are often used to mimic the densely crowded environment of a living cell which is filled up to a volume of 30–40%32 by differently sized biomolecules. Macromolecular crowding restricts the accessible volume by the excluded volume effect (an effective repulsion) and thus entropically stabilizes proteins by favouring the more compact native structure of the protein.33,34 However, for artificial macromolecular crowders like dextran, enthalpic stabilization may play an important role as well. We recently showed that the stabilizing effect is mediated by a preferential hydration mechanism, rather than an excluded volume effect.35 In contrast to osmolytes, macromolecular crowders generally promote aggregation. It was shown that aggregation of α-synuclein36,37 and apolipoprotein C-II38 is accelerated by artificial crowders. The effect is commonly interpreted by the stabilization of more compact fibrillar structures along the aggregation pathway.39Pharmacological chaperones
Pharmacological chaperones, also known as low-concentration working small molecules, become attractive in the light of therapeutic applications, as high and non-physiological concentrations of osmolytes are required to efficiently modulate protein aggregation. A group of polyphenols including EGCG20 and resveratrol22 has been discovered as potent inhibitors of amyloid formation. In contrast to chemical chaperones, pharmacological chaperones inhibit aggregation and subsequent fibrillation by direct interaction. Due to their planar structure and hydrophobic nature it was suggested that they either bind to partially folded regions within the monomer, lead to formation of nontoxic off-pathway oligomers, or destabilize the fibrillar states upon binding to mature fibrils essentially via hydrophobic interactions.40 Recently, an orcein-related small molecule, O4, has been shown to reduce cytotoxicity of Aβ involved in Alzheimer's disease by stimulating its aggregation.19Heat shock proteins or other non-amyloidogenic proteins
Heat shock proteins or other non-amyloidogenic proteins are a third class of chaperones which often coexist as secondary components in amyloid deposits. Such secondary components include glycosaminoglycans (e.g. heparin, heparin sulfate or chondroitin sulfate), proteoglycans (e.g. perlecan), plasma glycoproteins (e.g. the serum amyloid P component [SAP]), apolipoprotein E and collagen. The function of these non-amyloidogenic secondary amyloid components, concerning their influence on the aggregation and the pathogenity of the species evolved, is not completely understood yet. However, an accelerating effect on peptide aggregation and fibril formation could be demonstrated for different glycosaminoglycans, such as heparin.41–45 In contrast, the plasma protein SAP has been shown to have an inhibitory effect on protein aggregation, e.g. of transthyretin and Aβ.46,47 A possible reason for this inhibitory effect could be the chaperone activity of SAP, which was uncovered through a partial back folding of a denatured lactate dehydrogenase into its active conformation.48 The plasma glycoprotein SAP belongs to the pentraxin protein family, whose quaternary structure consists of five non-covalently bound β-sheet-rich subunits. However, it is ambiguous until now if the protein is present as a pentamer or combines to a decameric structure.49–51 Each subunit of SAP contains two calcium binding sites and exhibits a specific calcium dependent ligand binding to, e.g., oligosaccharides, glycosaminoglycans or phosphoethanolamino groups.49,52 This binding might also be responsible for the colocalisation of SAP in many forms of amyloid fibrils.49,53,54The effect of potential chaperones on the aggregation kinetics and thermodynamics has generally been investigated in diluted buffer solutions, neglecting the highly crowded milieu in cells.55,56 Here, we present a comprehensive study of different chaperones and cosolute effects on protein aggregation and fibrillation. Using different biophysical methods such as thioflavin T (ThT) fluorescence spectroscopy, attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy, circular dichroism (CD) spectroscopy, atomic force microscopy (AFM), and a cytotoxicity assay, we study the chaperone effects on human islet amyloid polypeptide (human IAPP) fibrillation. Human IAPP is a 37 amino acid residues short polypeptide, coproduced with insulin in β-cells. Its amyloidogenesis is involved in the development of type 2 diabetes mellitus.2In vitro studies showed that human IAPP fibrillation is membrane-mediated57 and thus the human IAPP-mediated cytotoxicity is proposed to be initiated by intracellular aggregation and presumably caused by disrupting membrane integrity of different cellular compartments.58,59 Mimicking the cellular environment by different chaperones and cell-like cosolutes, we focus on the mechanism and the efficiency of the pharmacological chaperone O4, the chemical chaperone proline and the natural protein chaperone SAP. We find that the effect of small-molecule and macromolecular crowders on human IAPP aggregation and fibrillation is largely determined by enthalpic effects rather than their excluded volume contribution, and thus significantly depends on the chemical nature of the crowding agent. Moreover, the protein's chemical makeup itself also determines whether the cosolute effect is accelerative or suppressive. Through the action of pharmacological and natural protein chaperones, the fibrillation reaction can be largely suppressed via formation of non-toxic off-pathway oligomeric species. Our results illustrate the importance of understanding different cosolute and crowding effects to understand protein aggregation in vivo. This is crucial for the development of pharmacological chaperones.
Experimental section
Sample preparation
Ficoll 70, dopamine and proline were obtained from Sigma-Aldrich. O4 was purchased from TimTec and freshly dissolved in DMSO to 20 mM stock solutions. Serum amyloid P component (SAP), human and rat IAPP (islet amyloid polypeptide or amylin) were purchased from Merck Millipore and Bachem Holding, respectively. For Atto565-labelled human IAPP, the C-terminal Y37K was chemically ligated to the Atto dye via NHS ester chemistry and was performed by PSL Heidelberg. Before use, IAPP peptides were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) yielding a concentration of 0.5 mg mL−1 and incubated for at least 30 min to ensure an aggregate- and seed-free sample.Protein aggregation and disaggregation
In order to study the disaggregation ability of O4, synthetic human IAPP, produced by the laboratory of Dr Volkmer-Engert (Institute for Medical Immunology, Charité, Berlin, Germany), was dissolved in HFIP overnight, sonicated for 30 min, and lyophilized. Monomeric human IAPP dilutions for ThT binding assays, CD spectroscopy and dot blotting were prepared by dissolving the lyophilized peptide in H2O and sonification for 30 min. Protein concentration was measured at 280 nm in an OD Eppendorf UVette with an Eppendorf BioPhotomenter and adjusted to 60 μM with distilled water. Aggregation reaction was started by mixing a 30 μM human IAPP peptide stock solution with 2× Tris-buffer (40 mM Tris/HCl, pH 7.4) to a final concentration of 30 μM, and incubation at 37 °C and 300 rpm for indicated time periods. All concentrations of fibrillar human IAPP species refer to the equivalent concentration of human IAPP monomers. Influence of O4 and controls on human IAPP aggregation were tested by diluting 20 mM DMSO stock solutions with 2× Tris-buffer to a concentration of 60 μM prior to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 incubation with the 60 μM human IAPP peptide stock solution. Disaggregation reactions were performed with human IAPP pre-aggregated for 48 h and equimolar amounts of O4 and controls diluted in Tris-buffer.
:1 incubation with the 60 μM human IAPP peptide stock solution. Disaggregation reactions were performed with human IAPP pre-aggregated for 48 h and equimolar amounts of O4 and controls diluted in Tris-buffer.
Thioflavin (ThT) fluorescence spectroscopy
Appropriate amounts of peptide (10 μM or 50 μM) were lyophilized overnight and dissolved in ThT-containing DPBS (10 μM ThT, 1.5 mM KH2PO4, 8.1 mM Na2HPO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4) or Tris/Ca-buffer (10 μM ThT, 10 mM Tris-HCl, 140 mM NaCl, 2 mM CaCl2, pH 7.4) in the absence or presence of corresponding amounts of the tested small molecules or proteins. In a 96-well plate, the ThT fluorescence intensity was measured at 25 °C every 10 min after shaking for 10 s at an emission wavelength of 482 nm (excitation at 440 nm) using a plate reader (InfiniteM200, Tecan). The ThT data were background corrected by subtraction of the ThT fluorescence intensity of the corresponding buffer and the lowest intensity value during the lag phase was set to zero (offset correction). For normalization, the intensity values were divided by the maximum intensity of measurement. For studying fibril disaggregation induced by O4, preformed human IAPP aggregates (30 μM) were mixed with equimolar amounts of O4 or with the DMSO control solution together with 40 μM ThT in phosphate buffer (100 mM sodium phosphate, 10 mM NaCl, pH 7.4) and measured at 37 °C.Fluorescence anisotropy
Lyophilized C-terminally Atto565-labelled human IAPP and label-free human IAPP were separately dissolved in Tris/Ca-buffer (10 mM Tris-HCl, 140 mM NaCl, 2 mM CaCl2, pH 7.4) to a concentration of 10 μM each and rapidly mixed to yield a 1:9 ratio. The measurements were performed within the lag time of human IAPP aggregation on a K2TM multifrequency phase fluorometer by ISS (Champaign, IL, USA) and analysed by the software Vinci made by ISS (Champaign, IL, USA). The anisotropy was measured with an excitation wavelength of 550 nm and emission wavelength of 585 nm. Lyophilized, in Tris/Ca-buffer dissolved SAP was added stepwise.
Circular dichroism (CD) spectroscopy
Preformed human IAPP aggregates (30 μM) were mixed with equimolar O4 dilutions. After 24 h incubation CD spectra were recorded with a 1 mm light path on a J-720 (Jasco, Tokyo, Japan) spectrometer. Solutions containing corresponding amounts of O4 but no human IAPP were incubated in parallel; reference CD spectra were recorded and subtracted from the CD signals to isolate the human IAPP-specific changes in CD spectra.Attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy
ATR-FTIR spectra were recorded using a Nicolet 6700 infrared spectrometer equipped with a liquid nitrogen cooled MCT (HgCdTe) detector and an ATR out-of compartment accessory consisting of a liquid jacked ATR flow-through cell (Thermo Scientific) with a trapezoidal Si-crystal (80 × 10 × 4 mm3, angle of incidence: 45°, Resultec) as the internal reflection element (IRE). For each FTIR spectrum, 128 scans with a spectral resolution of 2 cm−1 were taken. The ATR flow-cell was tempered to 25 °C. For each measurement, a buffer spectrum was collected before a solution of 10 μM IAPP dissolved in DPBS (pH 7.4 in D2O) in presence or absence of small molecules was injected. Spectra were recorded every 5 min for 20 h. Data analysis was carried out using the GRAMS software (Thermo Electron). After buffer subtraction, the spectra were baseline corrected between 1710 and 1585 cm−1.Atomic force microscopy (AFM)
50 μM lyophilized human IAPP were dissolved in 10 mM NaH2PO4 (pH 7.4), 10 μM ThT and in presence or absence of corresponding amounts of the small-molecule additives. Due to weak adsorption of IAPP onto mica in the presence of salts, human IAPP was matured in the absence of salts. For analysing the influence of SAP on human IAPP, 50 μM lyophilized human IAPP were dissolved in Tris/Ca-buffer (10 mM Tris-HCl, 2 mM CaCl2, 10 μM ThT, pH 7.4) in the presence and absence of 50 μM SAP. After performing a ThT assay for 20 h, aliquots were taken from the 96 well plate, diluted to 25 μM with water and deposited on freshly cleaved mica. The samples were dried with a stream of nitrogen, rinsed with water, again dried with a stream of nitrogen and freeze-dried overnight. AFM measurements were performed in the tapping mode on a MultiMode scanning probe microscope equipped with a NanoScope IIIa controller (Digital Instruments) using an E-Scanner (scan size 15 μm × 15 μm) and a MMMC cantilever holder (both Veeco Instruments) equipped with a silicon SPM sensor (PPP-NCHR, NanoAndMore). The dried samples were scanned in air with drive frequencies around 240 kHz and drive amplitudes between 60 and 150 mV. Images were scanned at room temperature with rates between 1.0 and 1.5 Hz. Image analysis and processing were done using the software Nanoscope 5 (Vecco Instruments). For height determinations, more than 100 human IAPP fibrils were analysed at each condition.For O4-induced human IAPP disaggregation studies, preformed human IAPP aggregates were incubated with O4 at equimolar concentration for 24 h. 15 μM IAPP samples of 10 μL were adsorbed for 10 min on the freshly cleaved mica (Nanoworld), glued to a microscope slide, washed with freshly filtered deionized water (4 × 30 μL) and dried overnight. AFM images were recorded on a Nanowizard II/Zeiss Axiovert setup (JPK) using intermittent contact mode and FEBS cantilevers (Veeco).
Dot blot assay
Dot blot assays to detect human IAPP amyloid oligomers were performed as described previously.60 Briefly, fibrillar human IAPP aggregates were incubated for 24 h with O4 and 10 μL aliquots of 15 μM human IAPP disaggregation reactions were spotted onto nitrocellulose membranes. Membranes were blocked for 1 h with 10% non-fat milk in PBS-T (137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, 10 mM Na2HPO4, 0.05% Tween 20). After washing membranes were incubated with the aggregate-specific conformational anti-IAPP antibody 91 in PBS-T containing 3% BSA and developed using horseradish peroxidase conjugated secondary antibodies (Promega, Germany). Dot blots were quantified using TotalLab TL120 from Nonlinear Dynamics.Quantification of SDS and NP40 stable amyloid aggregates
In filter retardation assays (FRAs) insoluble aggregates were monitored by adding 40 μL of human IAPP disaggregation reactions to an equal volume of 4% sodium dodecyl sulfate (SDS) and 100 mM dithiothreitol (DTT) and boiled at 98 °C for 5 min. For the quantification of native NP40 stable aggregates 40 μL of human IAPP aggregation and disaggregation reactions were mixed to an equal volume of 1% NP40 (Calbiochem). 40 μL of the SDS or NP40 treated samples were filtered through a cellulose acetate membrane with 0.2 μm pores (OE66, Schleicher and Schuell, Germany). Membranes were blocked in PBS-T containing 3% skim milk. Aggregates retained on the filter membrane were detected using the self-made IAPP 91 antibody and secondary antibodies conjugated to horseradish peroxidase (Promega, Germany).Cell viability assay
INS-1E cells61 were seeded at 105 cells mL−1 into a 96 well plate and grown for 24 h before incubated with human IAPP in the presence or absence of the small-molecule additives. After lyophilizing, 10 μM human IAPP with the appropriate amount of the small-molecule compounds were dissolved in cell culture medium (RPMI 1640, supplemented with 2 mM glutamine, 5% FCS, 10 mM HEPES, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin) and exposed to the INS-1E cells. In case of testing the cytotoxicity of human IAPP fibrils, 50 μM human IAPP were dissolved in DPBS, pH 7.4 and incubated for 24 h before 5× diluted with cell culture medium and exposed to the cells. After 24 h of incubation at 37 °C under 5% CO2, the solutions were removed, the cells were washed once with fresh medium and incubated for another 24 h with a WST-1 solution (2.5% Cell Proliferation Reagent WST-1 (Roche), 7.5% PBS, 90% cell culture medium). The absorbance was measured at 450 nm with reference at 630 nm. Percentage cell viability was calculated based on the absorbance measured relative to that of cells exposed to the small molecules alone, without human IAPP.Results and discussion
The pharmacological chaperone O4 acts as a thermodynamic inhibitor on human IAPP fibrillation
The in vitro amyloid formation of human IAPP can be interpreted by a nucleation-dependent polymerization mechanism.3,10,62 The stochastic formation of aggregation nuclei defines the length of the lag phase and precedes fibril growth. Thus, the aggregation kinetics exhibits a sigmoidal-shaped profile.63 We used thioflavin (ThT) as an extrinsic probe to measure the human IAPP aggregation kinetics (Fig. 1b). The excitation maximum shifts from 385 to 450 nm and the fluorescence maximum at 482 nm is enhanced upon non-covalent binding to amyloid cross-β structures.64 Here, the effect of addition of the ocrein-related small molecule O4 (Fig. 1a) on the fibrillation reaction was investigated in a dose-dependent manner (Fig. 1b). At 5-fold molar excess of O4, the ThT fluorescence intensity remained unchanged during the observed time period, suggesting that the fibrillation reaction was almost completely inhibited. At lower O4 concentrations (2 and 10 μM), the final ThT fluorescence intensity was reduced, the aggregation kinetics remained unchanged, however.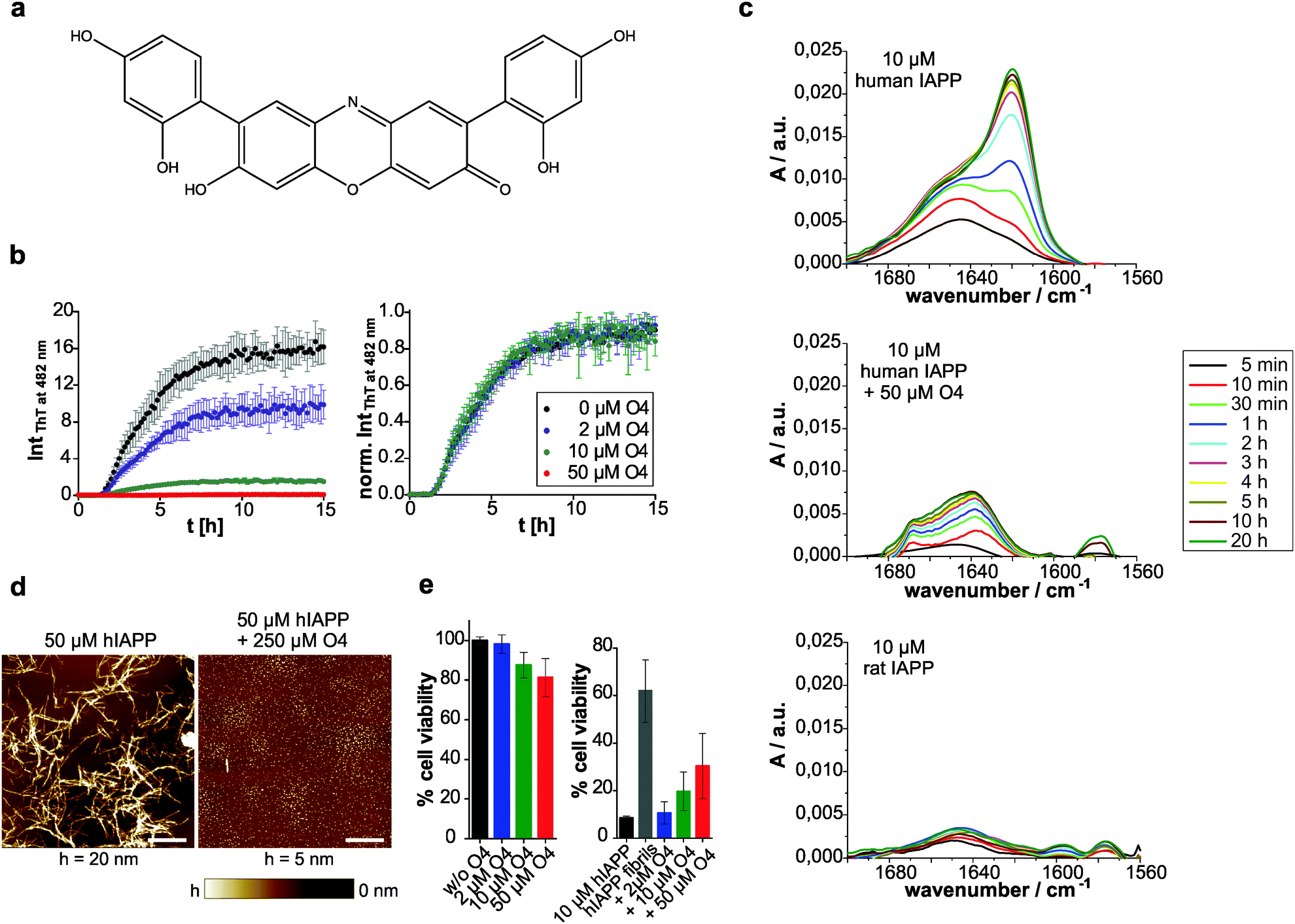 | ||
| Fig. 1 Effect of O4 on human IAPP fibrillation in diluted buffer. (a) Chemical structure of O4. (b) Dose-dependent effect of O4 on the aggregation time course of 10 μM human IAPP in DPBS, pH 7.4, monitored by ThT fluorescence spectroscopy with absolute (left) and normalized (right) ThT fluorescence intensities. n ≥ 4, error bars indicate mean ± s.d. (c) ATR-FTIR spectra of the time-dependent shift of the amide-I′ band of 10 μM human IAPP without and with O4. For comparison, the time-dependent secondary structure of non-amyloidogenic ratIAPP is shown. (d) Morphology of amyloidogenic human IAPP (hIAPP) in the absence and presence of O4 after by AFM. Scale bar, 1 μm. (e) Pancreatic INS-1E cells were exposed to 10 μM human IAPP in the presence and absence of O4 for 24 h. O4 is fractionally toxic to the INS-1E cells (left), but cytotoxicity of human IAPP is partially reduced by O4 treatment (right). n = 2–4, error bars indicate mean ± s.d. | ||
Further, we investigated the structural transition of human IAPP by time-resolved attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy. The secondary structural changes of human IAPP during aggregation were examined by analyzing the amide-I′ band region (Fig. 1c). In the absence of O4, the peak maximum of the amide-I′ band underwent a time-dependent shift from 1645 cm−1 towards 1621 cm−1, consistent with the temporal structural change from initially disordered and partially α-helical structures to the formation of intermolecular β-sheets during fibril formation of human IAPP.10 The time-dependent IR absorbance increase of the amide-I′ band was due to the adsorption of human IAPP on the IRE surface upon aggregation. In contrast, at 5-time molar excess of O4, this time-dependent structural transition of human IAPP was not observed, indicating that O4 stabilized the disordered and partially α-helical structures and thus inhibited the fibril formation of human IAPP. Furthermore, the adsorption of the human IAPP/O4 adducts to the IRE surface failed to appear, indicating stabilization of soluble human IAPP species. These results are consistent with results obtained from the time-resolved secondary structure of the non-amyloidogenic ratIAPP2 (Fig. 1c). Thus, we show that O4 inhibits human IAPP aggregation by acting on the pre-fibrillar stage. However, it is unclear whether non-fibrillar assemblies, such as off-pathway oligomers, form at a later stage. To address this issue, we used atomic force microscopy (AFM) in order to identify whether the aggregation pathway of human IAPP was changed in the presence of O4. We show that in the presence of O4 small, globular species of human IAPP (1.2 ± 0.4 nm) were formed and the formation of fibrillar structures (8.8 ± 2.5 nm) was suppressed (Fig. 1d). However, individual short mature human IAPP fibrils as shown in Fig. 1d were still observed.
To test whether the O4-redirected aggregation pathway and/or the O4-stabilized human IAPP species were toxic, we performed a WST-1 cell viability assay using the pancreatic β-cell line INS-1E (Fig. 1e). O4 itself featured concentration-dependent but fractional toxicity for the cells. In the presence of human IAPP, the percentage cell viability was calculated based on the absorbance measured relative to that of cells exposed to culture media containing O4 but without peptide. Mature fibrils of human IAPP have been reported to be largely non-toxic65 and our data show that around 60% cells remained alive upon 24 h incubation with preformed human IAPP fibrils, whereas only around 8% of the INS-1E cells survived the aggregation process of human IAPP when monomeric human IAPP was applied. When O4 was added to monomeric human IAPP before addition to the cells, we found a dose-dependent increase of cell viability, indicating that the O4-stabilized human IAPP species featured less toxicity. Hence, our data support the hypothesis that O4 stabilizes off-pathway species of human IAPP, thereby reducing its cytotoxicity. With the unchanged aggregation kinetics at low concentrations of O4 we demonstrate that O4 does not affect the original human IAPP aggregation pathway from a monomeric disordered structure via formation of nuclei and oligomers to fibrillation, but favours a competing off-pathway stabilization of non-fibrillar, globular assemblies of human IAPP with increasing O4 concentration. Therefore, O4 seems to act as a thermodynamic aggregation inhibitor. Interestingly, although a 25% identity and 50% similarity exists between human IAPP and Aβ in their primary structure,66 O4 has been shown to be a potent stimulator of Aβ aggregation by stabilizing β-sheet-rich fibrils, acting as a molecular clip via hydrophobic interactions.19 These different observations imply that O4's mode of action on protein aggregation is highly dependent on the exact chemical properties of the primary structure. It has been demonstrated that a certain order of the hydrophobic amino acid residues within the Aβ peptide is required to bind to O4.19 Thus, the direct and hydrophobic interaction between O4 and human IAPP might require certain conformations of the peptide that can be easily adopted in the non-fibrillar state due to its pronounced conformational dynamics as revealed by recent molecular dynamics simulation.67 In conclusion, our data show that O4 is able to thermodynamically stabilize non-fibrillar, small globular species of human IAPP by preventing the formation of ordered fibrils and thus the formation of toxic on-pathway oligomers.
O4 disassembles preformed β-sheet-rich human IAPP fibrils
We then investigated whether O4 has the ability to influence the stability and structure of preformed human IAPP aggregates. Hence, we aggregated 60 μM human IAPP for 3 days to generate mature human IAPP fibrils and subsequently incubated these aggregates for another two days with O4 at an equimolar concentration. The composition of the resulting human IAPP aggregate species was subsequently characterized using dot blot and filter retardation assays (FRA) (Fig. 2a–c). We found that O4 treatment significantly reduces the binding of the fibril-specific anti-IAPP antibody AB91 to β-sheet-rich human IAPP fibrils, suggesting that compound treatment alters the abundance or structure of insoluble aggregates (Fig. 2a). In agreement with these findings, we found a reduction of human IAPP aggregates in presence of O4, when samples were analysed using native NP40 (Fig. 2b) or denaturating SDS filter assays (Fig. 2c). This indicates that the compound can destabilize normally very stable preformed human IAPP aggregates. To further characterize the effect of O4 on preformed human IAPP aggregates, we performed a quantitative, time-resolved analysis of human IAPP disaggregation using a ThT binding assay. We found that compared to the control, ThT binding to O4 treatment aggregates was dramatically reduced in cell-free assays (Fig. 2d), supporting the hypothesis that O4 treatment alters the structure and stability of preformed human IAPP aggregates. This view was confirmed by CD spectroscopy analysis, indicating that the β-sheet content of O4 treated aggregates compared to untreated control samples was reduced (Fig. 2e). Finally, we analysed the effect of O4 on preformed human IAPP fibrils by AFM, indicating that compound treatment decreases the abundance of typical β-sheet-rich amyloid aggregates (Fig. 2f). Together these biochemical and biophysical studies indicate that O4 does not only interfere with spontaneous human IAPP polymerization (Fig. 1b–d) but also can modulate the characteristic structural properties of preformed β-sheet-rich amyloid fibrils in vitro.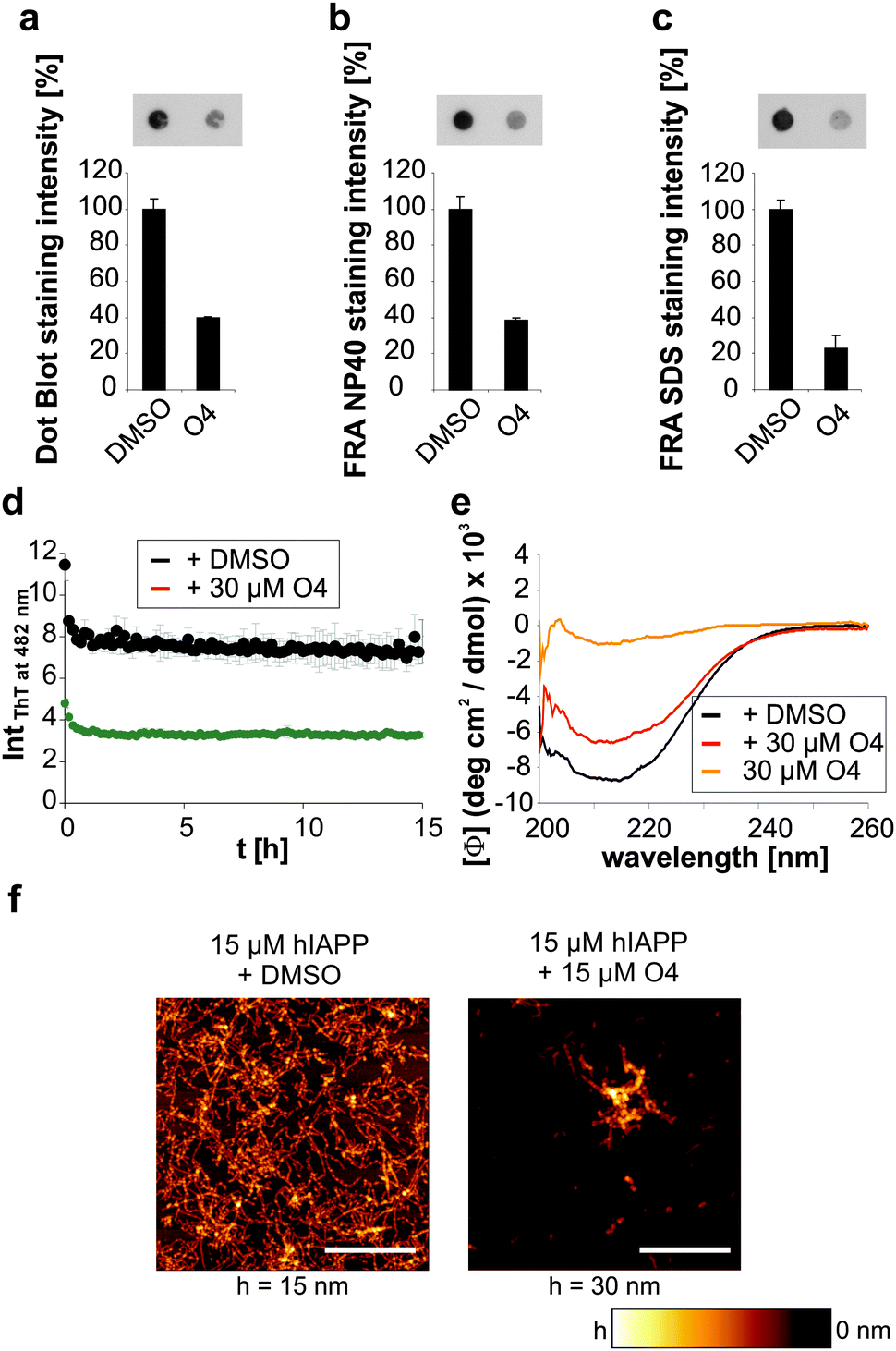 | ||
| Fig. 2 Influence of O4 IAPP disaggregation. Effect of equimolar amounts of O4 (30 μM) on human IAPP disaggregation monitored by (a) dot blot, (b) NP40 and (c) SDS FRAs using the human IAPP antibody 91. n = 4, error bars indicate mean ± s.d. Images show representative signal intensities of the samples. (d) Effect of O4 on human IAPP disaggregation monitored by ThT binding. n = 4, error bars indicate mean ± s.d. (e) Effect of O4 on secondary structure of human IAPP during disaggregation analysed by CD spectroscopy. (f) AFM images after 24 h incubation of preformed human IAPP (hIAPP) aggregates with O4 at equimolar concentration. Scale bar, 1 μm. | ||
Macromolecular crowding effects modulate IAPP fibrillation and O4 inhibition
We then compared the effects of the macromolecular crowder Ficoll 70 and sucrose on human IAPP aggregation. We found that both have approximately the same suppressive effect on fibril formation of human IAPP, indicated by the reduced final ThT fluorescence intensities compared to those obtained in pure buffer solution (Fig. 1b and 3). The lag time remained unchanged and the apparent growth rate constant was reduced by half with respect to DPBS (Table 1), revealing a marked decrease of the fibril elongation rate. The macromolecular crowder Ficoll 70 and its monomeric equivalent sucrose modulate the kinetics in the same way. Thus, similar to what we previously found regarding their effect on protein stability, the cosolute effect is dominated by the common chemical nature of the crowding agent, rather than by its macromolecular characteristics.35 In other words, the steric excluded volume effect does not have a major effect on human IAPP aggregation. Additionally, recent studies have demonstrated that Ficoll 70 is not fully inert, but features a pronounced binding behaviour to monomeric IAPP peptides.68 As sucrose is known to be excluded from the protein surface,69,70 the observed suppressive effect of sucrose on human IAPP fibril formation is rather caused by an enthalpic preferential hydration mechanism.27 The macromolecular crowder Ficoll 70 modulated human IAPP aggregation via the same avenue. Our results are in line with previous independent studies on Aβ and α-synuclein aggregation. Sucrose (2 g L−1) enhances and accelerates the aggregation reaction of Aβ.31 Similarly, Ficoll 70 (150 g L−1) accelerates α-synuclein aggregation.37 However, in both studies Ficoll 70 and sucrose accelerated the aggregation. Thus, the enthalpic effect that depends on the chemical nature of the crowding agent and on the protein's chemical make-up itself determines whether the cosolute effect is accelerative or suppressive.| Lag time [h] | App. growth rate constant [h−1] | |
|---|---|---|
| DPBS, pH 7.4 | 1.1 ± 0.1 | 4.3 ± 0.5 |
| 300 g L−1 sucrose | 1.3 ± 0.1 | 1.4 ± 0.2 |
| 300 g L−1 Ficoll 70 | 1.3 ± 0.3 | 1.4 ± 0.4 |
Next, we tested if this cosolute effect interferes with pre-fibrillar species that the pharmacological chaperone O4 interacts with, and thus changes its inhibition efficiency. We found similar dose-dependent inhibitory effects of O4 in the presence of sucrose and Ficoll 70 with respect to the diluted scenario (Fig. 3). Under macromolecular crowding conditions, the fibril elongation step of human IAPP was slightly retarded by addition of equimolar O4 (Fig. 3b). Moreover, Fig. 3b shows that the inhibitory effect of equimolar O4 was less effective in the presence of Ficoll 70 compared to DPBS or sucrose (Table 2). Thus, macromolecular crowding does modulate the O4 aggregation inhibition. One reason could be the increased formation of O4 aggregates21 in the crowded solution which decreases the effective concentration of O4. Such aggregates would be expected to be inactive for modulating the fibrillation reaction of human IAPP.20 These results illustrate the importance of considering the in-cell crowding effect for the development of pharmacological chaperones.
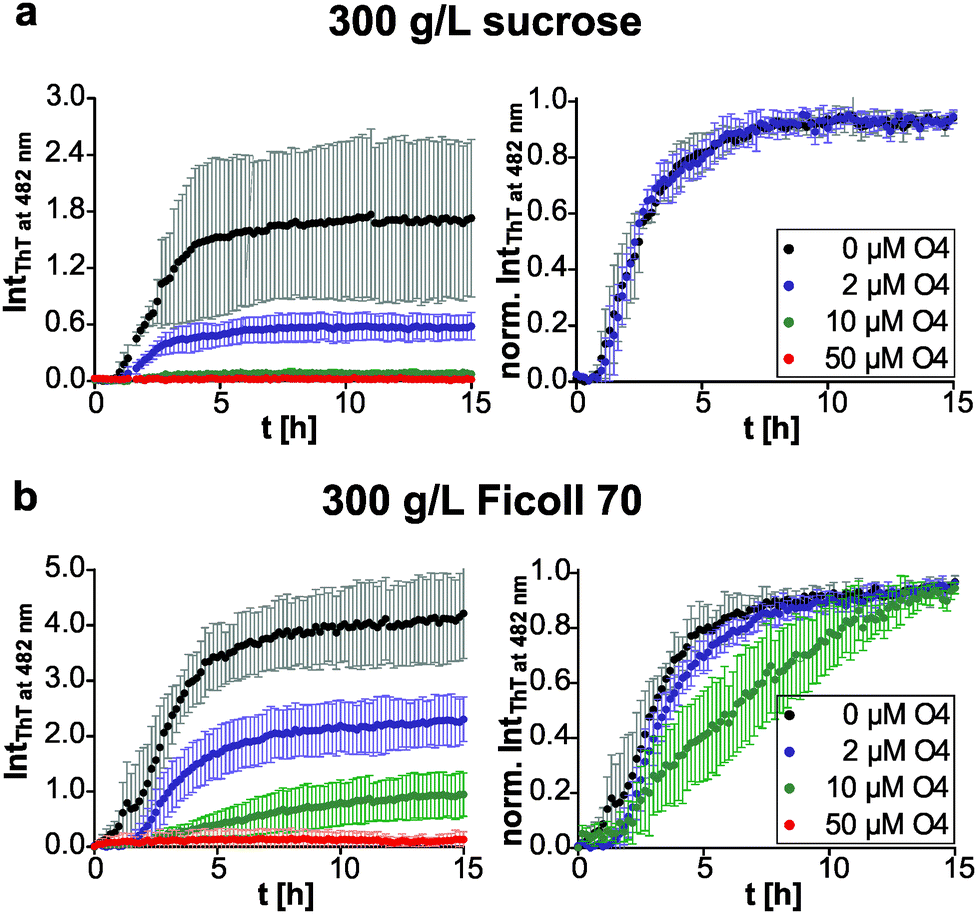 | ||
| Fig. 3 Effect of O4 on human IAPP fibril formation under crowding conditions monitored by ThT fluorescence spectroscopy. Dose-dependent effect of O4 on the aggregation time course of 10 μM human IAPP in (a) 300 g L−1 sucrose and (b) its polymer Ficoll 70. The left panel shows the time-dependent ThT fluorescence intensity using absolute values, whereas those are normalized to the maximum value of each measurement for better comparison of the underlying kinetics and shown in the right panel. n ≥ 4, error bars indicate mean ± s.d. | ||
| % fibril formation compared to 0 μM O4 | |||
|---|---|---|---|
| DPBS pH 7.4 | 300 g L−1 sucrose | 300 g L−1 Ficoll 70 | |
| 2 μM O4 | 58.3 ± 1.9 | 33.6 ± 17.9 | 56.9 ± 14.2 |
| 10 μM O4 | 9.7 ± 1.5 | 4.6 ± 2.9 | 23.7 ± 10.7 |
| 50 μM O4 | 0.6 ± 0.2 | 0.6 ± 0.5 | 3.4 ± 5.0 |
Chemical chaperone proline redirects human IAPP to form amorphous assemblies
We further tested the natural amino acid proline, a common osmolyte in plants,72 but also known to be cryoprotective for membranes.73 By using an equimolar mixture of human IAPP and proline, we found no significant change in the aggregation kinetics using the ThT fluorescence assay (Fig. 4a). Since proline can be upregulated up to >0.4 M in E. coli cells under stress conditions,74 proline was tested in the concentration range between 0.1 and 1 M. The indirect working mechanism of proline via preferential hydration of proteins requires high amounts of the osmolyte and thus starting from 0.5 M proline we found a weak concentration-dependent retardation of the elongation phase only, but a dose-dependent decrease of the amount of human IAPP fibrils formed (Fig. 4a). Next, we tested whether proline redirects the aggregation pathway of human IAPP. To this end, we used AFM to show that in the presence of proline fibrillar human IAPP species were formed, though unfavoured (Fig. 4b). As a higher human IAPP concentration was needed for sample preparation, the concentrations of both, peptide and cosolute, were increased to 50 μM and 5 M, respectively. Proline caused a shortening of the fibrils, whose heights were shifted from 4.2–15.7 nm to 2.0–8.3 nm. More interestingly, globular, amorphous aggregates were formed apart from the fibrillar assemblies. This suggests that proline disfavours fibril formation of human IAPP and diverts the amyloidogenesis into an alternative aggregation pathway where shorter and smaller, fibrillar and non-fibrillar species are formed. | ||
| Fig. 4 Fibrillation of human IAPP in the presence of proline. (a) Time-dependent ThT fluorescence intensities showing the aggregation time courses of 10 μM human IAPP when proline is added to DPBS, pH 7.4, with absolute (left) and normalized (right) data. n ≥ 4, error bars indicate mean ± s.d. (b) Morphological differences of human IAPP (hIAPP) aggregates upon incubation with proline. AFM images of human IAPP assemblies in the absence and presence of proline. Scale bar, 1 μm. | ||
This is consistent with the hypothesis of proline working as an osmophobic cosolute. The osmolyte does not interact directly, but is excluded from the protein's surface leading to increased number and strength of hydrogen bonds for the native vs. denative states.70 Such preferential exclusion might cause human IAPP to form non-fibrillar aggregates in order to minimize exposed surface area. However, the osmolyte effect cannot be generalized for any intrinsically disordered protein (IDP). Previously, we showed that TMAO and betaine did not affect the morphology of human IAPP fibrils formed, but did modulate the aggregation kinetics.10
Further, we investigated the osmophobic effect of proline on the fibril formation of human IAPP under macromolecular crowding conditions.56 Again, we used Ficoll 70 as a polymer crowding agent with its monomer sucrose as control. Interestingly, in the presence of sucrose the additional effect of proline was suppressed (Fig. 5a). At similar concentrations the osmolyte effect of sucrose (0.88 M) dominated. Based on the transfer model,27 sucrose is the more effective protecting osmolyte compared to proline. That is consistent with transfer free energy values of peptide backbone units from 1 M osmolyte to water (sucrose: −259.4 J mol−1; proline: −200.8 J mol−1).75 In contrast to O4, the effect of proline on human IAPP fibrillation was abolished under macromolecular crowding conditions. We could not observe any significant decrease of the fibril formation or retardation of the fibril elongation by proline (Fig. 5b). In conclusion, our comparative investigation reveals that the osmolytic effect of proline and the effect of Ficoll 70 as macromolecular crowder are non-additive on the human IAPP fibrillation reaction due to their chemical nature rather than to geometrical (steric) effects.
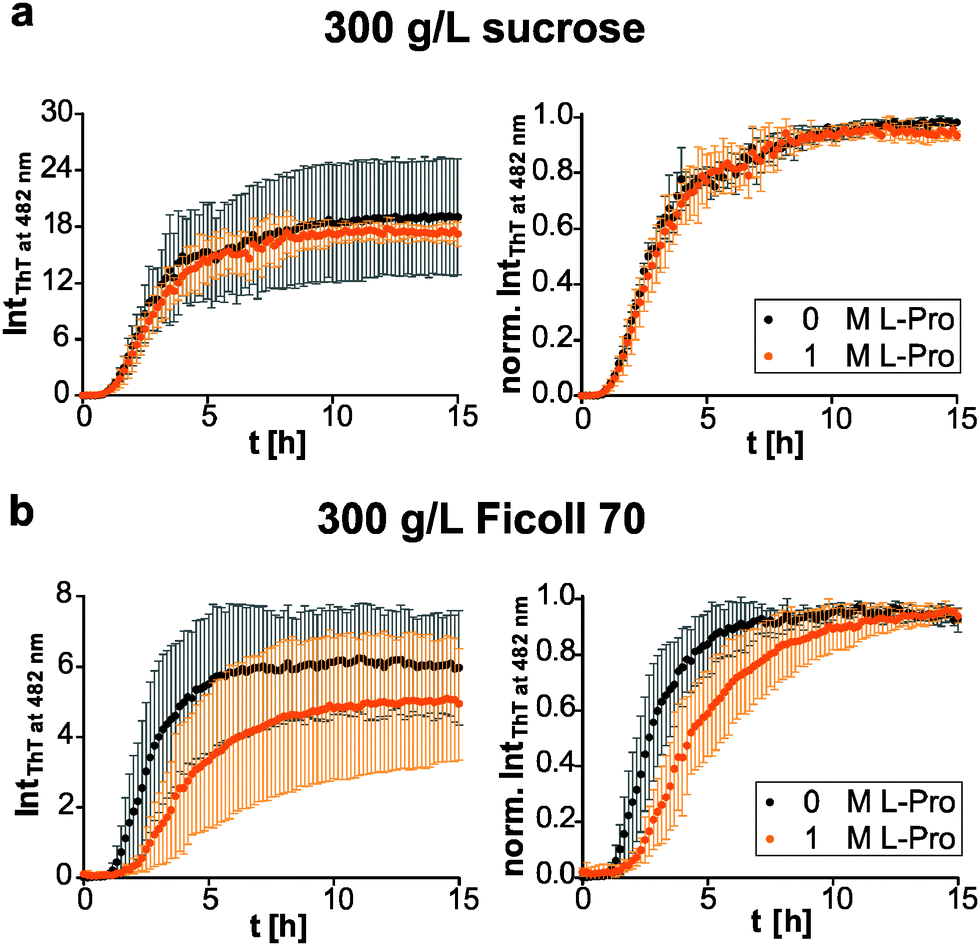 | ||
| Fig. 5 Osmolyte effect under crowding conditions. Aggregation time course of 10 μM human IAPP in the presence of 1 M proline in (a) 300 g L−1 sucrose and (b) Ficoll 70 (left). In addition, the ThT fluorescence intensities are normalized to the maximum value of each measurement for better comparison of the underlying kinetics (right). n ≥ 4, error bars indicate mean ± s.d. | ||
Potential protein chaperone serum amyloid P component suppresses IAPP fibril formation
The plasma glycoprotein SAP is a secondary amyloid component commonly found in amyloid deposits. To study the influence of SAP on the aggregation process and fibril formation of human IAPP, we performed SAP concentration-dependent ThT fluorescence spectroscopic measurements and compared the results with those on human IAPP as well as pure SAP as control (Fig. 6a). In contrast to the sigmoidal ThT data for mere human IAPP, the ThT assay of the pure SAP solution shows no increase of fluorescence intensity and therefore no unspecific binding of ThT to SAP. Combined incubation of human IAPP and SAP led to a SAP concentration-dependent extension of human IAPP lag-phases as well as a deceleration of elongation times. Moreover, a reduced ThT fluorescence intensity in the stationary phase of human IAPP aggregation was observed for a 2-fold excess of SAP. It can be concluded that due to the interaction of human IAPP and SAP a retardation or even partial inhibition of human IAPP nucleation and fibril formation takes place. The loss of the typical sigmoidal aggregation profil of human IAPP aggregation points towards a change of the aggregation mechanism, being different from the characteristic nucleation dependent polymerization reaction of human IAPP on its own. Such findings could be explained by the chaperone activity of SAP. SAP seems to be able to bind early oligomeric human IAPP species, stabilize them or even refold them into the native conformation of human IAPP. Such effect would explain both, the extended nucleation phase as well as the retarded elongation of human IAPP fibril formation.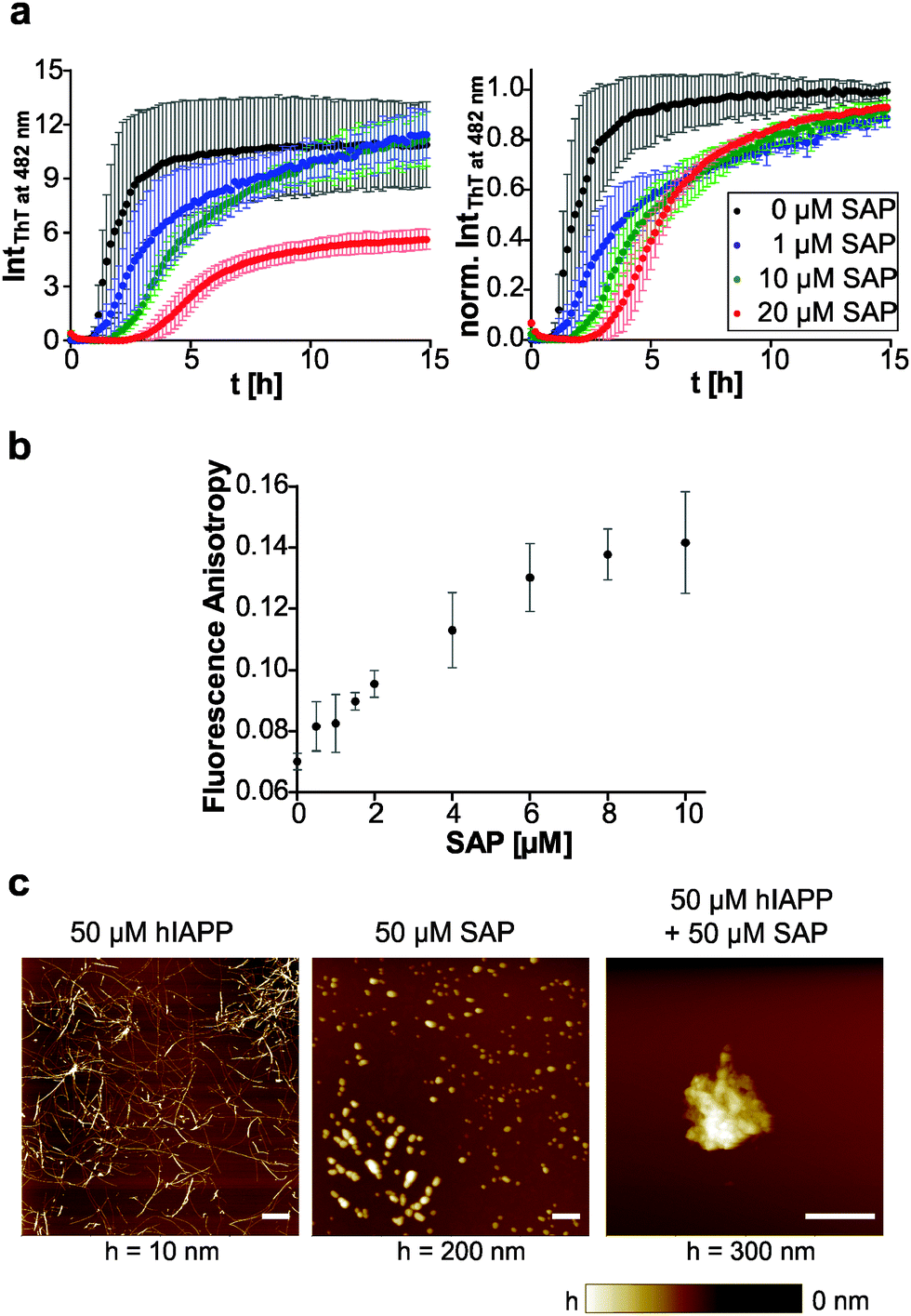 | ||
| Fig. 6 Fibrillation of human IAPP in the presence of SAP. (a) Time-dependent ThT fluorescence intensities showing the aggregation time courses of 10 μM human IAPP when SAP is added, absolute (left) and normalized (right) data. n ≥ 4, error bars indicate mean ± s.d. (b) Fluorescence anisotropy measurement of fluorescence-labelled (Atto565) 10 μM human IAPP upon step-wise addition of SAP. (c) Morphological differences of human IAPP assemblies in the absence and presence of SAP as determined by AFM. Scale bar, 1 μm. | ||
To further investigate the binding of SAP to early IAPP aggregate species, we performed fluorescence-anisotropy measurements within the lag time of the human IAPP aggregation process using a mixture of fluorescence-labelled and label-free human IAPP upon increasing the SAP concentration. As shown in Fig. 6b, the anisotropy values increase with rising SAP concentration, clearly indicating binding of SAP to early oligomeric species of human IAPP. Furthermore, a binding ratio of 1:1 may be inferred as the anisotropy value is saturated at an equimolar ratio of human IAPP and SAP. In case of the peptide Aβ, SAP has been reported to bind to both the fibrillar and the monomeric state in a calcium-dependent reaction.47 However, the binding motif and the nature of interaction are largely unknown.
Additional AFM experiments were conducted to reveal morphological changes of human IAPP in the presence of SAP during the aggregation process. Fig. 6c displays mere human IAPP fibrils on the mica surface with their characteristic mean heights of 3.3 ± 2.4 nm. SAP itself is detected as globular species on mica with mean heights of 50.6 ± 23.0 nm. By incubating an equimolar mixture of human IAPP and SAP, larger aggregates with a total height of ∼170 nm were observed on mica. Additionally, single globular SAP species could still be found. However, no IAPP fibrils could be detected anymore. This again indicates binding of SAP to human IAPP and formation of combined human IAPP/SAP aggregates which seem to prevent human IAPP from fibril formation.
Conclusions
In summary, we have shown that the pharmacological chaperone O4 is an efficient inhibitor of human IAPP aggregation. O4 does not modulate the aggregation mechanism but stabilizes human IAPP at an early non-fibrillar stage. Thereby, O4 redirects the aggregation pathway towards off-pathway globular species, reducing the human IAPP cytotoxicity. Further, O4 can interact with human IAPP aggregates at a mature stage and disassemble fibrils into smaller less stable structures. The preferential exclusion effect of proline leads to similar modulation effects, but less efficient. In contrast, we show that SAP modifies the human IAPP aggregation kinetics. The protein chaperone directly interacts with early non-fibrillar species. However, the products are also off-pathway globular aggregates. Crowding agents modulate the human IAPP aggregation even in the absence of chaperones. The comparison of Ficoll 70 to its monomeric equivalent sucrose shows that these crowders modulate the aggregation kinetics via an osmolyte-like mechanism rather than a macromolecular crowding effect. This mechanism interferes with the inhibitory effect of O4 and proline suggesting that cosolute effects in the crowded cell need to be considered when studying the mechanism of chaperone interaction. The sensitivity of IAPP fibrillation to cosolute effects shows that age-related changes of cellular environment could play a crucial role in the onset of type 2 diabetes mellitus.Acknowledgements
We thank the group of Dr Pierre Maechler (Geneva University Hospital, Switzerland) for providing the INS-1E cell line. We acknowledge funding from the Ministry of Innovation, Science and Research of the State of North Rhine-Westphalia (Rückkehrerprogramm), the Cluster of Excellence RESOLV (EXC 1069) funded by the German Research Foundation (DFG) and the grants from BMBF (NGFN-Plus 01GS08132).Notes and references
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384 CrossRef CAS PubMed.
- K. Weise, D. Radovan, A. Gohlke, N. Opitz and R. Winter, ChemBioChem, 2010, 11, 1280 CrossRef CAS PubMed.
- F. U. Hartl and M. Hayer-Hartl, Nat. Struct. Mol. Biol., 2009, 16, 574 CAS.
- V. N. Uversky, Front. Biosci., 2009, 14, 5188 CrossRef CAS.
- R. Soong, J. R. Brender, P. M. Macdonald and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 7079 CrossRef CAS PubMed.
- D. Radovan, V. Smirnovas and R. Winter, Biochemistry, 2008, 47, 6352 CrossRef CAS PubMed.
- Y. Li, W. Xu, Y. Mu and J. Zhang, J. Chem. Phys., 2013, 139, 55102 CrossRef PubMed.
- J. Seeliger, K. Estel, N. Erwin and R. Winter, Phys. Chem. Chem. Phys., 2013, 15, 8902 RSC.
- Z. Ignatova and L. M. Gierasch, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13357 CrossRef CAS PubMed.
- T. Borwankar, C. Röthlein, G. Zhang, A. Techen, C. Dosche and Z. Ignatova, Biochemistry, 2011, 50, 2048 CrossRef CAS PubMed.
- F. Scaramozzino, D. W. Peterson, P. Farmer, J. T. Gerig, D. J. Graves and J. Lew, Biochemistry, 2006, 45, 3684 CrossRef CAS PubMed.
- V. N. Uversky, J. Li and A. L. Fink, FEBS Lett., 2001, 509, 31 CrossRef CAS.
- A. Arora, C. Ha and C. B. Park, FEBS Lett., 2004, 564, 121 CrossRef CAS.
- J. R. Peinado, F. Sami, N. Rajpurohit and I. Lindberg, FEBS Lett., 2013, 587, 3406 CrossRef CAS PubMed.
- P. J. Muchowski, G. Schaffar, A. Sittler, E. E. Wanker, M. K. Hayer-Hartl and F. U. Hartl, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7841 CrossRef CAS PubMed.
- F. Seyffer, E. Kummer, Y. Oguchi, J. Winkler, M. Kumar, R. Zahn, V. Sourjik, B. Bukau and A. Mogk, Nat. Struct. Mol. Biol., 2012, 19, 1347 CAS.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93 CrossRef CAS PubMed.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558 CAS.
- B. Y. Feng, B. H. Toyama, H. Wille, D. W. Colby, S. R. Collins, B. C. H. May, S. B. Prusiner, J. Weissman and B. K. Shoichet, Nat. Chem. Biol., 2008, 4, 197 CrossRef CAS PubMed.
- R. Mishra, D. Sellin, D. Radovan, A. Gohlke and R. Winter, ChemBioChem, 2009, 10, 445 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Annu. Rev. Phys. Chem., 2013, 64, 273 CrossRef CAS PubMed.
- P. H. Yancey, J. Exp. Biol., 2005, 208, 2819 CrossRef CAS PubMed.
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Science, 1982, 217, 1214 CAS.
- D. W. Bolen and I. V. Baskakov, J. Mol. Biol., 2001, 310, 955 CrossRef CAS PubMed.
- S. N. Timasheff, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 67 CrossRef CAS PubMed.
- R. Gilman-Politi and D. Harries, J. Chem. Theory Comput., 2011, 7, 3816 CrossRef CAS.
- Y. S. Kim, S. P. Cape, E. Chi, R. Raffen, P. Wilkins-Stevens, F. J. Stevens, M. C. Manning, T. W. Randolph, A. Solomon and J. F. Carpenter, J. Biol. Chem., 2000, 276, 1626 CrossRef PubMed.
- D. S. Yang, C. M. Yip, T. H. J. Huang, A. Chakrabartty and P. E. Fraser, J. Biol. Chem., 1999, 274, 32970 CrossRef CAS PubMed.
- J. Fung, A. A. Darabie and J. McLaurin, Biochem. Biophys. Res. Commun., 2005, 328, 1067 CrossRef CAS PubMed.
- S. B. Zimmerman and S. O. Trach, J. Mol. Biol., 1991, 222, 599 CrossRef CAS.
- H.-X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375 CrossRef CAS PubMed.
- A. P. Minton, J. Biol. Chem., 2001, 276, 10577 CrossRef CAS PubMed.
- M. Senske, L. Törk, B. Born, M. Havenith, C. Herrmann and S. Ebbinghaus, J. Am. Chem. Soc., 2014, 136, 9036 CrossRef CAS PubMed.
- M. D. Shtilerman, T. T. Ding and P. T. Lansbury, Biochemistry, 2002, 41, 3855 CrossRef CAS PubMed.
- V. N. Uversky, E. M. Cooper, K. S. Bower, J. Li and A. L. Fink, FEBS Lett., 2002, 515, 99 CrossRef CAS.
- D. M. Hatters, A. P. Minton and G. J. Howlett, J. Biol. Chem., 2002, 277, 7824 CrossRef CAS PubMed.
- R. J. Ellis and A. P. Minton, Biol. Chem., 2006, 387, 485 CrossRef CAS PubMed.
- F. L. Palhano, J. Lee, N. P. Grimster and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 7503 CrossRef CAS PubMed.
- S. Jha, S. Patil, J. Gibson, C. E. Nelson, N. N. Alder and A. T. Alexandrescu, J. Biol. Chem., 2011, 286, 22894 CrossRef CAS PubMed.
- A. Abedini, S. M. Tracz, J.-H. Cho and D. P. Raleigh, Biochemistry, 2006, 45, 9228 CrossRef CAS PubMed.
- G. M. Castillo, J. A. Cummings, W. Yang, M. E. Judge, M. J. Sheardown, K. Rimvall, J. B. Hansen and A. D. Snow, Diabetes, 1998, 47, 612 CrossRef CAS.
- J. A. Cohlberg, J. Li, V. N. Uversky and A. L. Fink, Biochemistry, 2002, 41, 1502 CrossRef CAS PubMed.
- B. Klajnert, M. Cortijo-Arellano, M. Bryszewska and J. Cladera, Biochem. Biophys. Res. Commun., 2006, 339, 577 CrossRef CAS PubMed.
- K. Andersson, M. Pokrzywa, I. Dacklin and E. Lundgren, PLoS One, 2013, 8, e55766 CAS.
- S. Janciauskiene, P. García de Frutos, E. Carlemalm, B. Dahlbäck and S. Eriksson, J. Biol. Chem., 1995, 270, 26041 CrossRef CAS PubMed.
- A. R. Coker, A. Purvis, D. Baker, M. B. Pepys and S. P. Wood, FEBS Lett., 2000, 473, 199 CrossRef CAS.
- J. Emsley, H. White, B. P. O'Hara, G. Oliva, N. Srinivasan, I. J. Tickle, T. L. Blundell, M. B. Pepys and S. P. Wood, Nature, 1994, 367, 338 CrossRef CAS PubMed.
- I. J. Sørensen, O. Andersen, E. H. Nielsen and S. E. Svehag, Scand. J. Immunol., 1995, 41, 263 CrossRef PubMed.
- J. A. Aquilina and C. V. Robinson, Biochem. J., 2003, 375, 323 CrossRef CAS PubMed.
- H. Hamazaki, J. Biol. Chem., 1987, 262, 1456 CAS.
- M. Pepys, T. Rademacher, S. Amatayakul-Chantler, P. Williams, G. Noble, W. Hutchinson, P. Hawkins, S. Nelson, J. Gallimore and J. Herbert, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5602 CrossRef CAS.
- M. B. Pepys, R. F. Dyck, F. C. de Beer, M. Skinner and A. S. Cohen, Clin. Exp. Immunol., 1979, 38, 284 CAS.
- L. A. Munishkina, A. L. Fink and V. N. Uversky, Curr. Alzheimer Res., 2009, 6, 252 CrossRef CAS.
- Y.-Q. Fan, J. Lee, S. Oh, H.-J. Liu, C. Li, Y.-S. Luan, J.-M. Yang, H.-M. Zhou, Z.-R. Lü and Y.-L. Wang, Int. J. Biol. Macromol., 2012, 51, 845 CrossRef CAS PubMed.
- J. Seeliger, K. Weise, N. Opitz and R. Winter, J. Mol. Biol., 2012, 421, 348 CrossRef CAS PubMed.
- T. Gurlo, S. Ryazantsev, C.-J. Huang, M. W. Yeh, H. A. Reber, O. J. Hines, T. D. O'Brien, C. G. Glabe and P. C. Butler, Am. J. Pathol., 2010, 176, 861 CrossRef CAS PubMed.
- M. Magzoub and A. D. Miranker, FASEB J., 2012, 26, 1228 CrossRef CAS PubMed.
- Q.-L. Ma, G. P. Lim, M. E. Harris-White, F. Yang, S. S. Ambegaokar, O. J. Ubeda, C. G. Glabe, B. Teter, S. A. Frautschy and G. M. Cole, J. Neurosci. Res., 2006, 83, 374 CrossRef CAS PubMed.
- A. Merglen, S. Theander, B. Rubi, G. Chaffard, C. B. Wollheim and P. Maechler, Endocrinology, 2004, 145, 667 CrossRef CAS PubMed.
- J. W. Kelly, Nat. Struct. Biol., 2000, 7, 824 CrossRef CAS PubMed.
- T. Eichner and S. E. Radford, Mol. Cell, 2011, 43, 8 CrossRef CAS PubMed.
- R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, R. K. Grover, R. Roy and S. Singh, J. Struct. Biol., 2005, 151, 229 CrossRef CAS PubMed.
- J. Seeliger and R. Winter, Subcell. Biochem., 2012, 65, 185 CAS.
- J. Seeliger, F. Evers, C. Jeworrek, S. Kapoor, K. Weise, E. Andreetto, M. Tolan, A. Kapurniotu and R. Winter, Angew. Chem., Int. Ed., 2012, 51, 679 CrossRef CAS PubMed.
- M. Andrews and R. Winter, Biophys. Chem., 2011, 156, 43 CrossRef CAS PubMed.
- J. Seeliger, A. Werkmüller and R. Winter, PLoS One, 2013, 8, e69652 CAS.
- P. Attri, P. Venkatesu and M.-J. Lee, J. Phys. Chem. B, 2010, 114, 1471 CrossRef CAS PubMed.
- R. Politi and D. Harries, Chem. Commun., 2010, 46, 6449 RSC.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036 CrossRef CAS PubMed.
- Y. Yoshiba, T. Kiyosue, K. Nakashima, K. Yamaguchi-Shinozaki and K. Shinozaki, Plant Cell Physiol., 1997, 38, 1095 CrossRef CAS.
- A. S. Rudolph and J. H. Crowe, Cryobiology, 1985, 22, 367 CrossRef CAS.
- U. Dinnbier, E. Limpinsel, R. Schmid and E. P. Bakker, Arch. Microbiol., 1988, 150, 348 CrossRef CAS.
- M. Auton and D. W. Bolen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15065 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |