
From NdFeB magnets towards the rare-earth oxides: a recycling process consuming only oxalic acid
Tom Vander Hoogerstraetea,
Bart Blanpainb,
Tom Van Gervenc and
Koen Binnemans*a
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F – P.O. Box 2404, B-3001 Heverlee, Belgium. E-mail: Koen.Binnemans@chem.kuleuven.be
bKU Leuven, Department of Materials Engineering (MTM), Kasteelpark Arenberg 44 – P.O. Box 2450, B-3001 Heverlee, Belgium
cKU Leuven, Department of Chemical Engineering, De Croylaan 46 – P.O. Box 2423, 3001 Leuven, B-3001 Heverlee, Belgium
First published on 14th November 2014
Abstract
A chemical process which consumes a minimum amount of chemicals to recover rare-earth metals from NdFeB magnets was developed. The recovery of rare-earth elements from end-of-life consumer products has gained increasing interest during the last few years. Examples of valuable rare earths are neodymium and dysprosium because they are important constituents of strong permanent magnets used in several large or growing application fields (e.g. hard disk drives, wind turbines, electric vehicles, magnetic separators, etc.). In this paper, the rare-earth elements were selectively dissolved from a crushed and roasted NdFeB magnet with a minimum amount of acid, further purified with solvent extraction and precipitated as pure oxalate salts. The whole procedure includes seven steps: (1) crushing and milling of the magnet into coarse powder, (2) roasting to transform the metals into the corresponding oxides, (3) the selective leaching of the rare-earth elements with acids (HCl, HNO3) to leave iron behind in the precipitate, (4) extracting remaining transition metals (Co, Cu, Mn) into the ionic liquid trihexyl(tetradecyl)phosphonium chloride, (5) precipitating the rare earths by the addition of oxalic acid, (6) removing the precipitate by filtration and (7) calcining the rare-earth oxalates to rare-earth oxides which can be used as part of the feedstock for the production process of new magnets. The magnet dissolution process from the oxides utilized four molar equivalents less acid to dissolve all rare earths in comparison with a dissolution process from the non-roasted magnet. Moreover, the less valuable element iron is already removed from the magnet during the dissolution process. The remaining transition metals are extracted into the ionic liquid which can be reused after a stripping process. Hydrochloric acid, the side product of the rare-earth oxalate precipitation process, can be reused in the next selective leaching process. In this way, a recycling process consuming only air, water, oxalic acid and electricity is developed to recover the rare earths from NdFeB magnets in very high purity.
Introduction
In 2011, many industrialized countries were facing a rare-earth crisis. For the first time in history, the demand for these elements was higher than the supply and the rare-earth metal prices dramatically increased, some of them by a factor of 20.1 This issue was due to three different reasons. First, China had a monopoly on the rare-earth market at that time and supplied approximately 97% of the global production of rare-earth oxides in terms of volume. Secondly, China set a strong export quota on rare earths. Thirdly, Europe has only a few primary rare-earth ore deposits.2,3 In 2010, the European Commission reported that the rare-earth elements had the highest supply risk of all elements in the near future.4,5 The U.S. Department of Energy also wrote in a similar report that the rare-earth elements neodymium, dysprosium, terbium, europium and yttrium had the highest supply risk for the United States in the near and medium future.6Three possibilities exist for maintaining the rare-earth supply. First, the old rare-earth mines can be reopened but this will take some time.2 Secondly, rare earths can be substituted by other less expensive rare earths or d-block metals, but this often results in products which are less effective and usually requires a new product design.2 Thirdly, recycling of rare earths from scrap produced throughout the production process or from end-of-life consumer products has also gained considerable interest. The third option has several advantages over primary ore mining:1 (1) a diminishing dependency on other rare-earth mining countries such as China, (2) a smaller amount of chemicals, air and water pollution and a lower energy consumption, (3) production of the rare earths without concerns about radioactive trace elements (Th and U) present in ores and for which the regulations are very strict, (4) helping the rare-earth market to stay in balance (mitigating the so-called “balance problem”).7
Neodymium–iron–boron (NdFeB) magnets are commercially available since the middle of the 1980s.8–10 They followed the ferrite and SmCo magnets and have a much higher magnetic strength than magnets from previous generations at temperatures close to room temperature. However, ferrite magnets still represent about 85% of the market due to their lower price. On the other hand, NdFeB magnets are gaining ground due to the increasing importance of miniaturisation in many applications. The smaller and lighter rare-earth permanent magnets can be used in several applications without losing magnetic strength.
The NdFeB magnet production, expressed in volumes, represented about 20% share of the rare earth market in 2012.11 However, in terms of value, the magnets take more than half of the market share (51%). There is a lack of recent numbers, but it is expected that the current numbers are even higher due to the growing demand for permanent magnets in the last few years.12,13 Moreover, it was reported that China produced 20 ktons of NdFeB scrap during the production of NdFeB magnets in 2010.14
There is literature available dealing with NdFeB magnet recycling.15–21 These include hydrogen decrepitation and direct reuse,22–24 selective precipitation,25–31 liquid metal extractions,26,32–36 glass slag methods,37 solvent extraction,38 direct melting,39,40 or gas phase extractions.41–46 However, large scale industrial recycling plants for the rare-earth recovery from end-of-life NdFeB magnets or scrap are scarce and mainly magnet producers are recycling their production scrap.16,47 It was reported that the company Hitachi Metals is recycling over 90% of their powder and solid NdFeB scrap (NEOMAX© trademark).16,47–50 The Japanese company Santoku Corporation has started a recycling route for NdFeB magnet from which more details are known.51 First, the magnets are grinded into particles smaller than 75 μm by using a jaw crasher. In a second step, they oxidize the powder in a NaOH solution at elevated temperatures. Third, the rare earths are leached selectively by a pH controlled HCl solution and separated from other elements by solvent extraction with 2-ethylhexyl phosphonic mono-2-ethylhexyl ester (PC-88A) extractant. One of their patents is also describing a recycling process for the recovery of iron and boron.52 They use a thermite reaction in which FeB and a slag are formed and separated.
The number of studies on the selective leaching of the rare earths from roasted NdFeB magnets is limited. In a leaching process, metals are selectively dissolved in the leachate leaving the undesired metals behind in the residue. Bounds reported in 1994 that Rhône-Poulenc (now part of Solvay) developed a selective leaching process to recover rare earths.53 However, no details are given about the process, and no magnet recycling activities are currently executed at their plant in La Rochelle (France). Koyama et al. have reported a selective leaching process as well.16,54–56 They used a very diluted solution of 0.02 M HCl and an autoclave at 180 °C to leach 99% of the rare earths and only 0.5% of iron after 2 h. A higher concentration of acid (0.1 M HCl) gave no selectivity because iron dissolved as well into the solution. Lower concentrations (0.01 or 0.001 M HCl) and different conditions (24 h at 60 °C) gave no full dissolution of the rare earths and gave surprisingly also extraction percentages of 14.0 and 1.2% of iron, respectively. There are some recent published patents on rare-earth recycling including a roasting and selective leaching step. However, these patents are not, or at best only vaguely, describing the chemical background and have not been under a scientific referee process.57–60
In this paper, the selective leaching process of rare earths from NdFeB magnets is described in detail with emphasis on the mechanism of the dissolution process. A NdFeB magnet with a known composition was first crushed and milled into smaller particles, and a roasting process was applied and studied. Afterwards, the rare earths were selectively leached by the inorganic acids HCl and HNO3. The difference between a leaching process with HCl from the roasted and a non-roasted magnet in terms of mechanism, acid consumption and purity of the leachate is explained. Remaining transition metals in the leachate, such as cobalt, copper and manganese, were extracted from chloride media towards the undiluted ionic liquid trihexyl(tetradecyl)phosphonium chloride. The rare earths were precipitated as oxalate salts by addition of oxalic acid. This results in a metal-free solution, highly concentrated in HCl, which is a side product of the rare-earth oxalate formation, and can be reused in a new leaching experiment. The oxalate salts can be calcined to obtain rare-earth oxides, which can be used as direct starting products for NdFeB magnet manufacturing. The whole process is developed in such a way that a very low amount of chemicals is consumed (only air, water and oxalic acid), large volumes of waste streams are avoided (only water, carbon dioxide and the leaching residue) and the aqueous leaching phase and the ionic liquid phase can be reused.
Experimental
Materials and methods
The demagnetised NdFeB magnet (Fig. 1A) was obtained from the University of Birmingham (UK), where its composition was measured with ICP (Table 1). The sum of all concentrations (95.72 wt%) was slightly lower than the theoretical value (100 wt%). This is due to experimental errors during the analysis. | ||
| Fig. 1 (A) The NdFeB magnet before crushing and milling, (B) NdFeB powder (<400 μm) before roasting, (C) roasting and powder fire at high temperatures and (D) the leachate colored pink-violet by neodymium (right) and the remaining orange/brown iron(III) hydroxide precipitate (left). | ||
| Fe | 58.16 |
| Nd | 25.95 |
| Co | 4.22 |
| Dy | 4.21 |
| B | 1.00 |
| Nb | 0.83 |
| O | 0.41 |
| Al | 0.34 |
| Pr | 0.34 |
| C | 0.07 |
| Si | 0.06 |
| Mn | 0.05 |
| Cu | 0.04 |
| Ni | 0.02 |
| N | 0.02 |
| Total | 95.72 |
A silicon solution in isopropanol was obtained from SERVA Electrophoresis GmbH (Heidelberg, Germany). A gallium standard (1000 mg L−1) was purchased from Merck (Overijse, Belgium). The leaching acids HNO3 (65 wt%, 14.3 M) and HCl (37 wt%, 12.0 M) were purchased from Chemlab NV (Zedelgem, Belgium) and VWR (Leuven, Belgium) respectively. CaCl2·4H2O (99.995%) was obtained from Merck (Darmstadt, Germany) and NH4NO3 (99.9%) and NH4Cl (99.9%) from VWR (Leuven, Belgium). Trihexyl(tetradecyl)phosphonium chloride (>97%) (Cyphos® IL 101) was purchased from Cytec (Toulouse, France) and H2C2O4·2H2O (>99.5%) from J. T. Baker. All chemicals were used as received, without further purification. A Heraeus Megafuge 1.0 centrifuge was used to remove the leachate from the remaining solid particles after the leaching or solvent extraction experiments. A TGA Q500 (TA instruments) and an alumina crucible were used for thermogravimetric analysis (TGA) of the NdFeB powder. The sample was heated at 20 °C min−1 in dry air (60 mL min−1). A LECO oxygen analyser was used to determine the oxygen content of the powders after grinding or roasting. ICP-MS (Agilent 7500ce) was used to determine the composition of the magnet. The pH was measured by a Sevencompact pH/ion meter S220 and a Slimtrode (Hamilton) electrode. A Retsch ZM100 centrifugal mill (Gemini BV) was used for grinding of the magnet and a muffle furnace for roasting. Metal concentrations were determined with a benchtop total reflection X-ray fluorescence (TXRF) spectrometer (Picofox S2, Bruker) which has an accuracy of 8.4% and 6.5% and a precision of 2.7% and 2.5% for the rare earths and for the transition metals respectively. Pretreatment of the TXRF sample carriers was performed with silicone oil in isopropanol.
Grinding and roasting
The magnet was first crushed between two aluminium metal plates by applying a force of 10 tons with a hydraulic press. NdFeB magnets are brittle and break quite easily into smaller pieces. A sieve of 400 μm was used, (Fig. 1B) and part of the undersize particles were milled with a centrifugal mill to obtain particles <80 μm. Note that we refer to <80 and <400 μm powder as a powder in which the cross section is maximum 80 μm and 400 μm, respectively. The roasting temperature and uptake of oxygen as a function of time and temperature were studied with TGA. The magnet powder was first heated at 20 °C min−1 up to 950 °C and kept at this temperature for another 480 min. The sample chamber was purged with dry air (60 mL min−1) and the initial sample mass in the alumina crucible was 15.988 mg.The influence of the temperature on the mass gain of the NdFeB magnet was studied as well on larger scales. A powder sample (5 g) was placed into a ceramic crucible and heated to the desired temperature without blowing extra air or oxygen into the muffle furnace. The difference and increase in mass was calculated from the mass before and after the roasting experiments. The mass gain (%) after the roasting process is defined as:
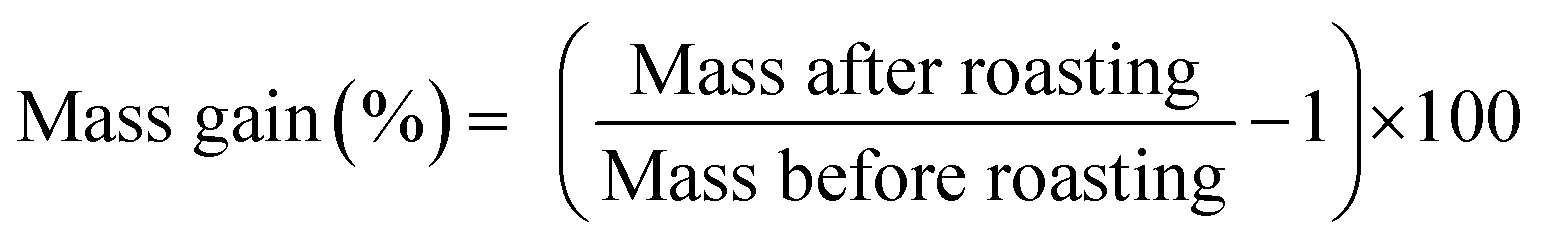 | (1) |
Roasting as a function of time was studied at a temperature of 650 °C. This temperature was chosen to test if also lower temperatures but longer heating times could be used for the roasting process. The sample was removed from the furnace after certain time intervals and allowed to cool down to room temperature. Afterwards, it was weighed and then placed again in the furnace. The <400 μm powder used in all leaching experiments was kept for 15 h at 950 °C to obtain full conversion of the metal into the oxides.
Leaching
Leaching experiments were performed in 4 mL vials. A sample of the <400 μm powder (0.1 g) was mixed with different amounts of acid and further diluted with water until the total volume of the leaching phase was 1 mL. A stirring bar was added and the vials were closed with a sealed lid. The mixtures were stirred for 15 h in an oil bath of 80 °C. Afterwards, the vials were centrifuged for 10 min at 5300 rpm and the leachate was removed from the leaching residue. We will refer to the leaching residue as the sum of non-reacted iron(III) oxide and precipitated iron(III) hydroxide. The leachate was centrifuged a second time, as it was often impossible to avoid that part of the iron(III) hydroxide precipitate was entrained into the leachate causing errors during the determination of the metal content in the leachate. The metal content and the pH of the aqueous phase were measured after the second centrifugation step. 2 mL of a 37 wt% (12.0 M) HCl solution was added to the remaining brown precipitate and stirred again for 1 h at 80 °C in an oil bath to dissolve the leaching residue. All liquid phases of the sample were mixed and diluted to a final volume of 25 mL. The leaching experiments with HNO3 and the mixtures HCl/7.5 M CaCl2 and HNO3/7.5 M NH4NO3 were performed in the same way as the leaching experiments with HCl. The precipitate obtained after a leaching process with HNO3 was dissolved as well in HCl, as full dissolution of the leaching residue in HNO3 was slower and incomplete. The percentage extraction (%E) is defined as:
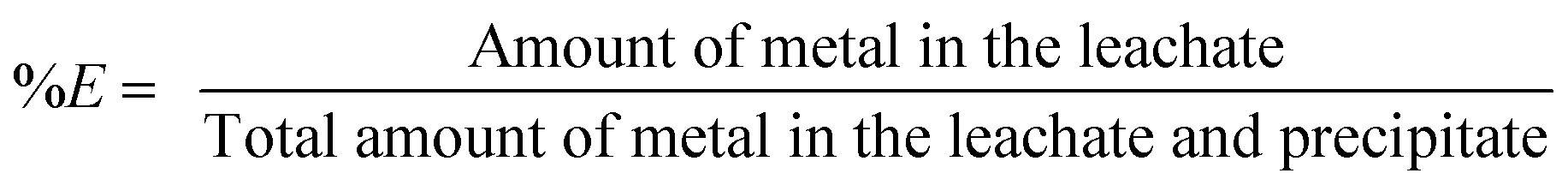 | (2) |
The relative metal concentrations in the leachate as a function of time were studied by dissolving 0.1 g of roasted NdFeB magnet powder in 1 mL of diluted HCl with various acid concentrations. The HCl concentration was chosen in this way that the molar ratio between HCl and the rare earths molecules n(HCl)/n(Ln), was kept constant at a value of 3.5. The sample was heated and stirred at 80 °C and cooled down after each time interval. An aliquot of the leachate (1 μL, 0.1 vol%) was removed and directly, without dilution or addition of a standard, measured on its relative metal content with TXRF. Afterwards, the vial was placed back in the oil bath to continue the leaching experiment. Only the four main metals present in the magnet (Fe, Nd, Dy and Co) were considered and the relative mass percentages in the leachate were calculated as:
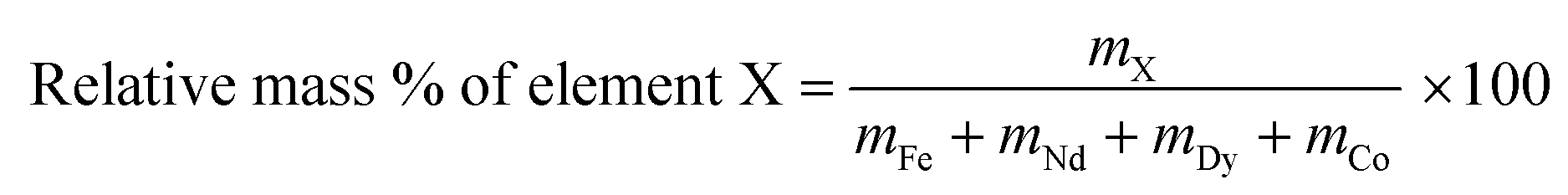 | (3) |
The experiments with increasing metal concentration on the aqueous phase were performed by mixing different amounts of NdFeB magnet oxide powder with 1 mL of diluted HCl. The molar ratio between HCl and the rare-earth molecules (n(HCl)/n(Ln)) was kept constant at a value of 3.5.
The metal contents of all leachates were measured with TXRF. An aliquot of 50 or 100 μL of the aqueous phase was mixed with a 1000 mg L−1 gallium standard and diluted to 1 mL with Milli-Q water. The concentration of gallium was chosen in the range of the lowest metal concentrations present in the leachate. The sample carriers were pretreated with 20 μL of silicone oil dissolved in isopropanol and dried for 5 min in an furnace at 60 °C. Afterwards, an aliquot of 5 μL of the sample was added on the sample carrier and dried for another 10 min. The carriers were measured for 500 s or for 1000 s when the iron concentration was very low. Longer measuring times were necessary at low iron concentrations because the Kα line of iron overlaps partially with the Lα line of dysprosium.
Solvent extraction
The solvent extraction experiments were performed with 1 mL of a mixture containing 25.0 g L−1 dysprosium and 3.0 g L−1 cobalt. A higher concentration of cobalt, in comparison with the concentrations found after the leaching process, was chosen to obtain a peak in the TXRF spectrum with an intensity that is high enough to come out of the higher background spectrum due to the very high rare-earth concentrations in the solution. Dysprosium was chosen instead of neodymium to avoid peak overlap between the L lines of neodymium and the K line of cobalt in the TXRF spectrum. Trihexyl(tetradecyl)phosphonium chloride has been chosen as ionic liquid because it is hydrophobic, non-fluorinated and it can be used in undiluted form. The advantages of using this ionic liquid are describe elsewhere.61–63 The aqueous phase contained 3.5 M NH4Cl and the HCl concentration was ranged between 0 and 1 M HCl. The organic phase consists of 1 mL of trihexyl(tetradecyl)phosphonium chloride which was presaturated with water containing the same NH4Cl and HCl concentration as used in the extraction experiments. The volume ratio was one and the mixtures were shaken for 1 h at 1800 rpm at 60 °C. Afterwards, they were centrifuged for 5 min at 5300 rpm and the metal ion concentration was measured with TXRF. The separation factor αCo,Dy is defined as:
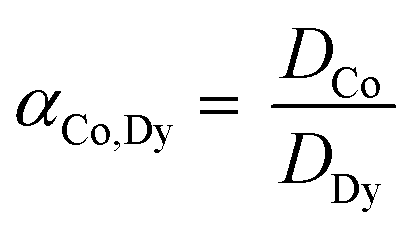 | (4) |
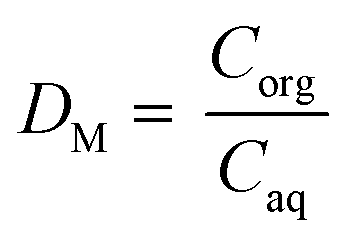 | (5) |
Precipitation
Different amounts of oxalic acid were added to 10 mL of a solution containing 3.5 M of NH4Cl and 26.7 g L−1 neodymium. The mixtures were stirred for 2 h at 800 rpm at room temperature. Afterwards, they were centrifuged and the remaining metal content in the aqueous phase was measured with TXRF. The percentage precipitation (%P) of neodymium was calculated by the following formula:
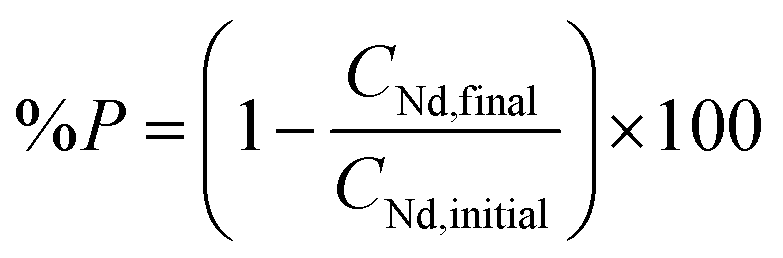 | (6) |
Complete process
A sample of roasted NdFeB magnet powder (3.1 g) was mixed with 30 mL of an aqueous phase containing 1.35 mL of 37 wt% (12.0 M) HCl and 3.5 M NH4Cl (n(HCl)/n(Ln) = 3.5). The mixture was stirred intensively for three days at 100 °C in a sealed vial in order to obtain a pH above two. Afterwards, part of the precipitate was filtered off on a double paper filter. The remaining precipitate, still present in the leachate after the first filtration step, was filtered on a syringe filter with a pore size of 45 μm. The iron free aqueous phase was brought into contact with 5 mL of the ionic liquid trihexyl(tetradecyl)phosphonium chloride presaturated with water. The extraction was performed by shaking at 60 °C for 1 h at 1800 rpm. Afterwards, the mixture was centrifuged for 5 min at 2000 rpm and the aqueous phase was separated from the organic phase with a syringe. Oxalic acid was slowly added to this solution under constant stirring until the pH of the solution was not significantly changing anymore (0.623 g). The rare-earth oxalates were filtered on a Buchner filter.Results and discussion
Crushing and roasting
First, the oxygen content of the NdFeB magnet particles with a size smaller than <400 μm (after crushing and sieving) and <80 μm (after milling) were measured. Important to mention is that a powder fire and a temperature increase were observed after milling the <400 μm powder to particles of <80 μm. When the mill was opened, fresh air came into contact with the hot and oxygen-sensitive magnet powder and the powder took fire. The experiment was therefore not repeated because the high temperature could lead to deformation of the mill. Knowing the oxygen content is important as it has a significant effect on the calculated total mass gain. The maximum uptake of oxygen and maximum mass gain of the magnet can be calculated based on the composition of the magnet (Table 1). At complete oxidation of the magnet, the mass of the magnet should increase by 35.6 wt%. At this point, the oxygen content in the magnet is 26.3 wt%. The oxygen content of the fine powder (<400 μm) before roasting was 0.56 wt%, which is slightly higher than the 0.41 wt% of oxygen present in the magnet before size reduction (Table 1). This means that thermal oxidation of the magnet in the presence of air is slow and there is no need to be concerned about a possible powder fire (Fig. 1C). The situation was different for the <80 μm magnet particles, where a powder fire was observed after milling the <400 μm powder with the centrifugal mill. This is partly due to the higher surface area of the particles, but the main reason is probably the increasing temperature during milling which increases the oxidation rate. The powder fire itself contributed as well to an increase in temperature. As a consequence, the mass gain of the <80 μm powder after milling increased to 5.95 wt%.A small amount of the <400 μm NdFeB powder (15.99 mg) was first analysed thermogravimetrically with TGA. Heating to 1000 °C at 10 °C min−1 and immediately cooling down did not fully oxidize the sample. This was confirmed in three ways. First, the slope of the TGA curve was still steep at 1000 °C suggesting that the maximum uptake of oxygen was not yet reached. Secondly, hydrogen gas was released when the remaining non-oxidized particles were brought into contact with an acid. Thirdly, the maximum theoretical uptake of oxygen, based on the composition from Table 1, was not yet reached. Therefore, a new TGA experiment was performed in which the sample was heated at 20 °C min−1 to 950 °C and held there for 6 h (Fig. 2). A temperature of 950 °C was selected because this was the maximum temperature of the muffle furnace used for the roasting experiments.
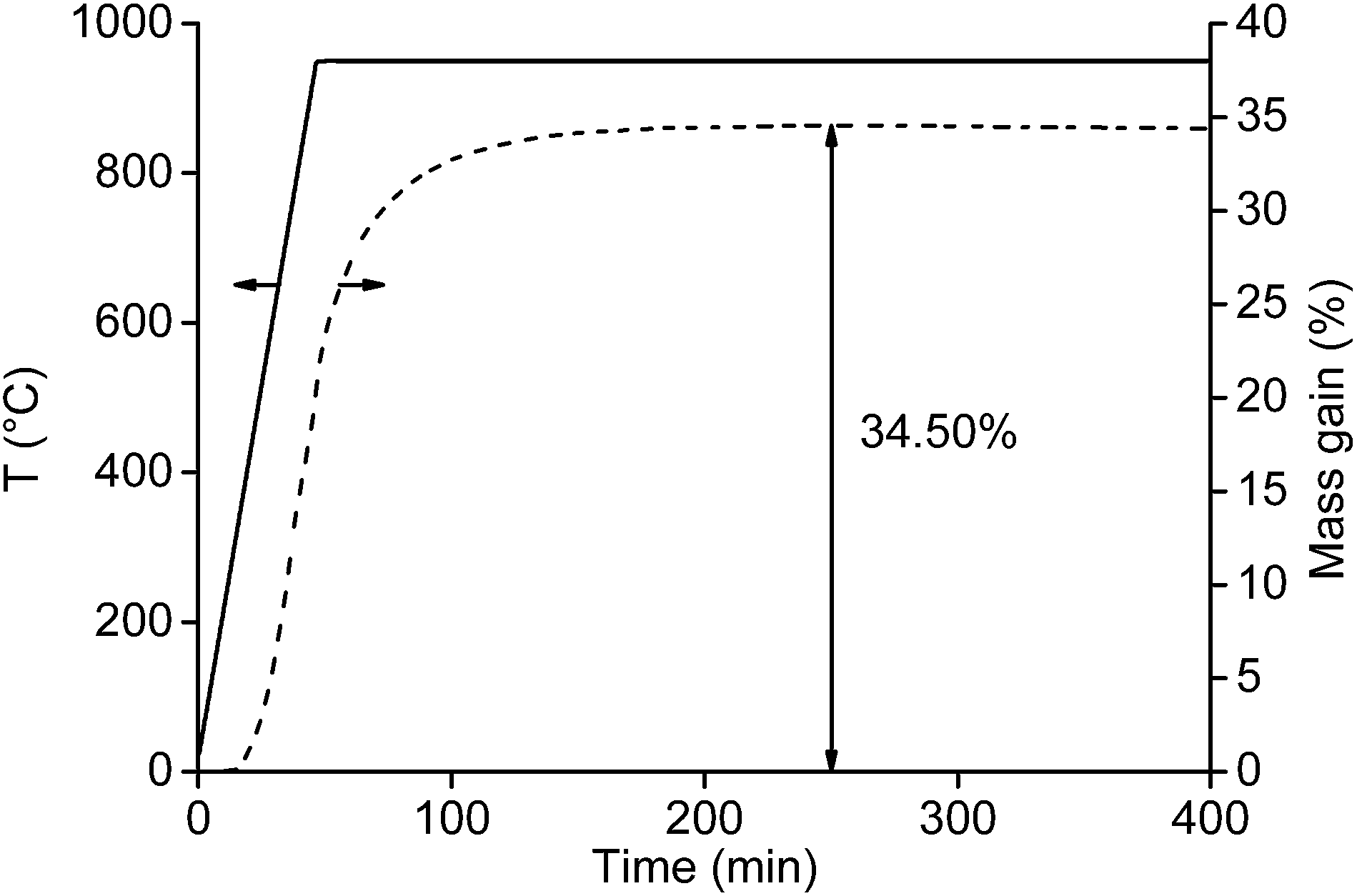 | ||
| Fig. 2 Thermogravimetric analysis of the magnet powder <400 μm. The NdFeB magnet powder was first heated to 950 °C (heating rate: 20 °C min−1) and kept at this temperature for another 6 h. The temperature of the furnace is given by the solid line and the mass gain (%) by the dashed line. | ||
The maximum mass was not yet reached after 45 min when the furnace obtained its highest temperature (950 °C). Additional 200 min at 950 °C were required to obtain a constant mass of the sample. At this point, the maximum mass gain was 34.5%, which is slightly lower than the theoretical value calculated from the composition given in Table 1 (35.6%).
Afterwards, the roasting with higher amounts of NdFeB powder was carried out in order to have enough material for the leaching experiments. The mass gain (%) as a function of the temperature was studied by keeping 5 g of NdFeB powder for 1 h at a specific temperature (Fig. 3). It was observed that mass gain was low until a temperature of 300 °C, which is similar to what has been observed in the TGA experiment (Fig. 2). Afterwards, a significant increase in oxidation occurred and full oxidation was obtained at 800 °C for the <80 μm powders and at 950 °C for the <400 μm powder. The powder was also analysed on its oxygen content with the oxygen analyser. Oxygen concentrations of 23.6 and 24.2 wt% were obtained at 950 °C for the <400 μm powder and at 900 °C for the <80 μm powder, respectively. This is slightly lower than the oxygen concentrations which can be calculated based on the initial composition of the magnet (26.3%). The difference could be due to experimental errors. However, it is also possible that the samples used in the oxygen determination and TGA experiments were not of perfectly identical composition. In principle, the size of the particles smaller than <400 μm powder ranges from 0 to <400 μm. This has its consequences when sampling the powder from the vial. The larger particles will be on top of the smaller particles, leading to a slightly inhomogeneous distribution of the powder in the vial. This is also reflected by the presence of small outliers present in the curve presenting the mass gain as a function of the temperature for the <400 μm powder.
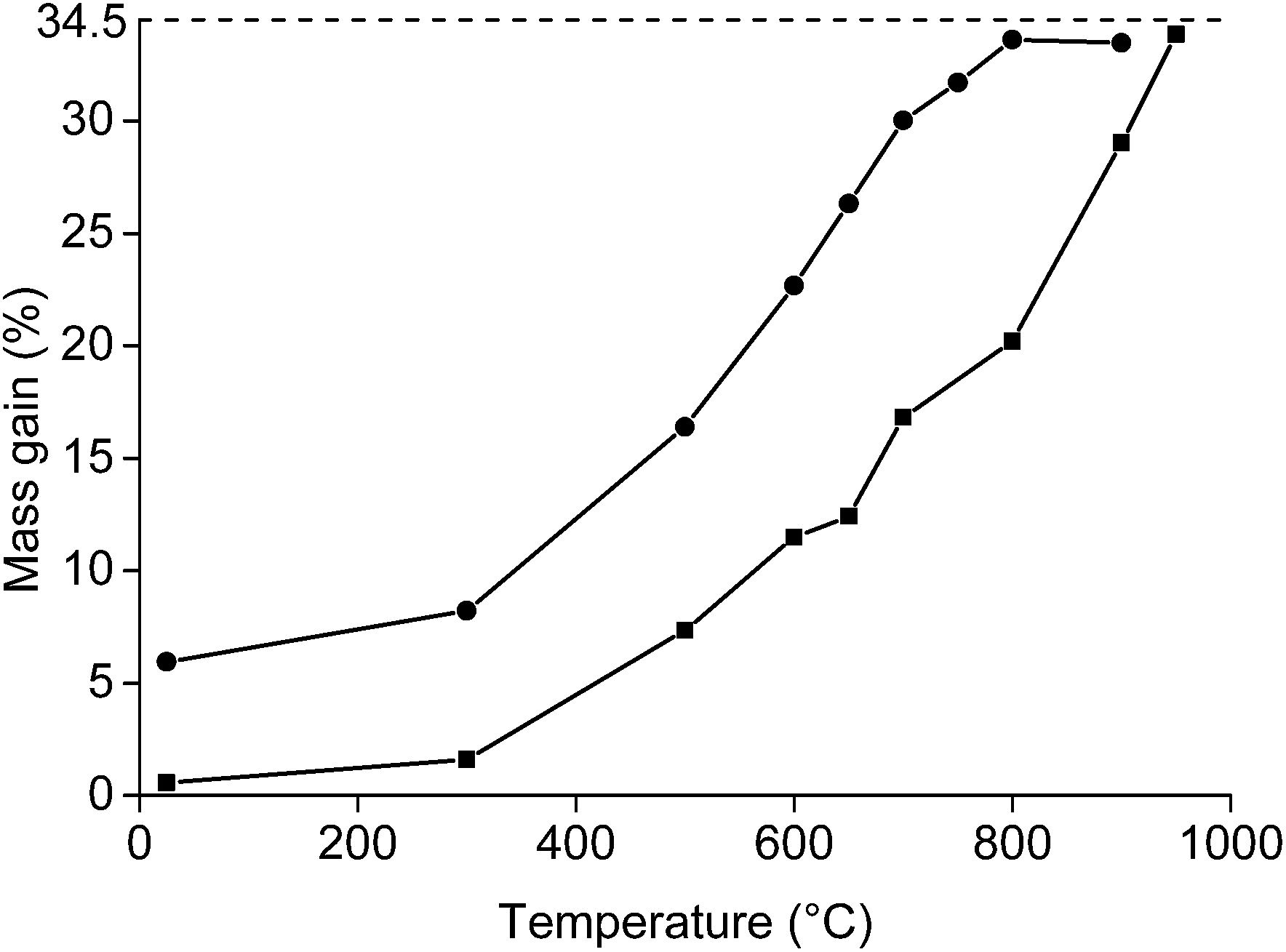 | ||
| Fig. 3 Mass gain (%) of the <400 μm (■) and <80 μm (●) NdFeB magnet powder as a function of the temperature at which it was held for 1 h. The dashed line is drawn at 34.5%, which is the maximum mass gain determined with TGA. | ||
Roasting as a function of the time was studied as well (Fig. 4). The powder was placed in a muffle furnace at 650 °C. A sharp increase was observed in the oxidation during the first minutes of the roasting process. Afterwards, the oxidation rates decreased. A mass gain of 20.8% was obtained for the <400 μm powder if it was heated for 20 h at 650 °C. The mass gain of the <80 μm powder was 33.2% when it was placed in the furnace for 23 h. This value is very similar to the degree of oxidation when it was placed for 1 h in the furnace at 800 °C (33.6%). A further increase in mass over time at constant temperature was also observed in the TGA measurement.
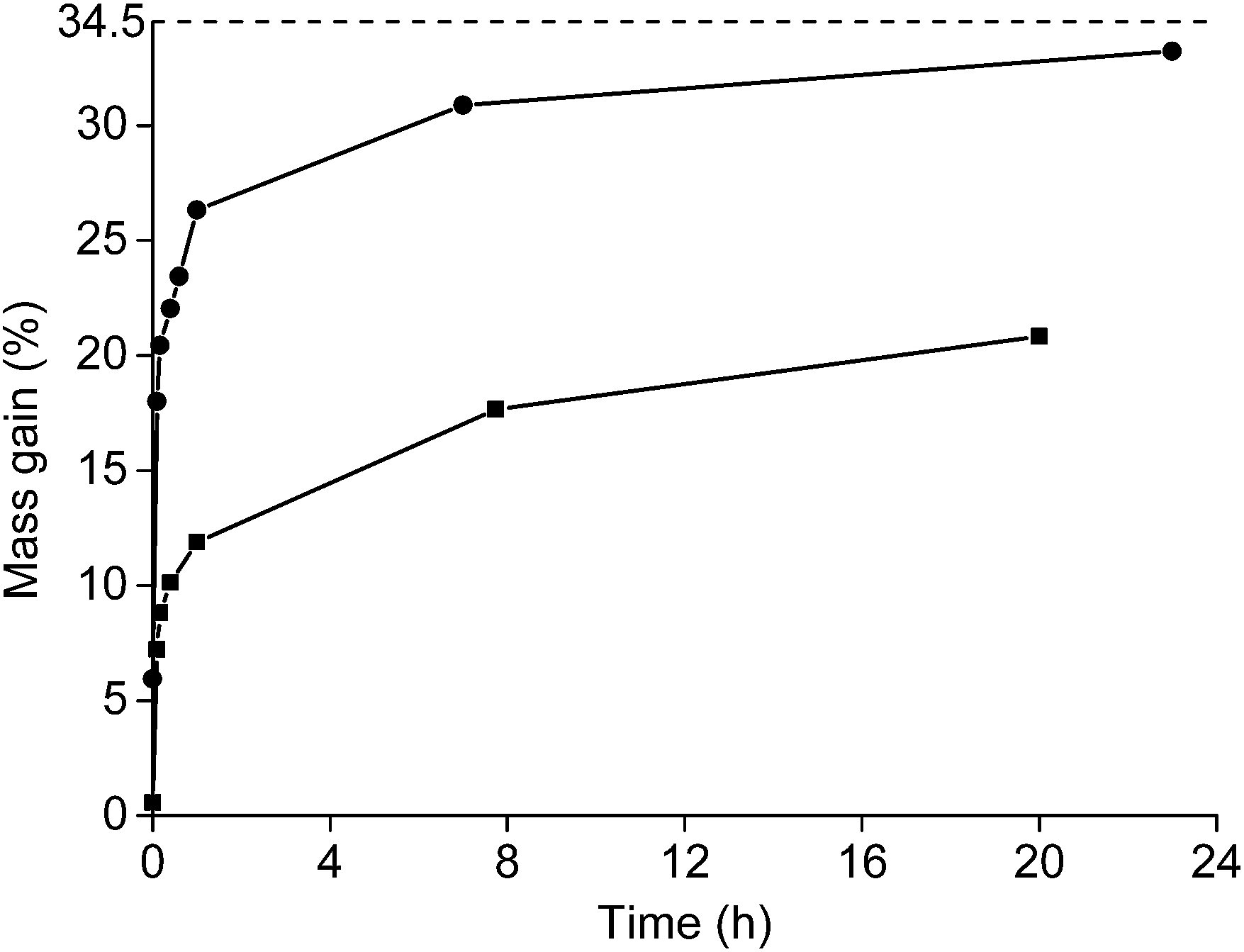 | ||
| Fig. 4 Mass gain (%) of the <400 μm (■) and <80 μm (●) NdFeB magnet powder as a function of the roasting time at a temperature of 650 °C. The dashed line is drawn at 34.5%, which is the maximum mass gain determined with TGA. | ||
Leaching
The acid HCl was studied first as leaching agent (Fig. 5). Different amounts of acid were mixed with 0.1 g of the <400 μm powder and stirred for 15 h at 80 °C in closed vials of 4 mL. Afterwards, the concentration in the leachate was measured and the remaining precipitate was dissolved in 37 wt% acid and analysed as well on its remaining metal content. It was chosen to plot the number of moles of acid added over the total moles of rare earths (Ln = Nd + Pr + Dy) present in the roasted magnet (n(HCl)/n(Ln)) on the X-axis instead of plotting the amount of HCl or initial HCl concentration. In this way, it was possible to correct for small differences in the weighed powder. Moreover, it is also easier to use the optimal values towards other experiments such as the scale-up experiment. The leaching percentages of the four main metals present in the magnet (Fe, Nd, Dy, Co) were reported as a function of the ratio n(HCl)/n(Ln). It was observed that the rare earths were leached much faster than the transition metals. A linear increase in extraction percentage was observed for neodymium and dysprosium going to a maximum around 3.5. This value was slightly higher than the expected value of 3.0, which is the chloride-metal ratio found in rare earth chlorides. This is probably due to the fact that other elements co-leached (e.g. Co) and maybe because the composition of the magnet in Table 1 is not fully correct. At an n(HCl)/n(Ln) ratio of 3.5, only 16% of cobalt and no iron was leached to the solution. The leaching percentages for neodymium and dysprosium were above 90% for all higher n(HCl)/n(Ln) ratios. A second important data point is found at an n(HCl)/n(Ln) ratio of 10. At this point, the rare earths and cobalt are almost fully extracted into the leachate, whereas iron was dissolved for only 25% (Fig. 5).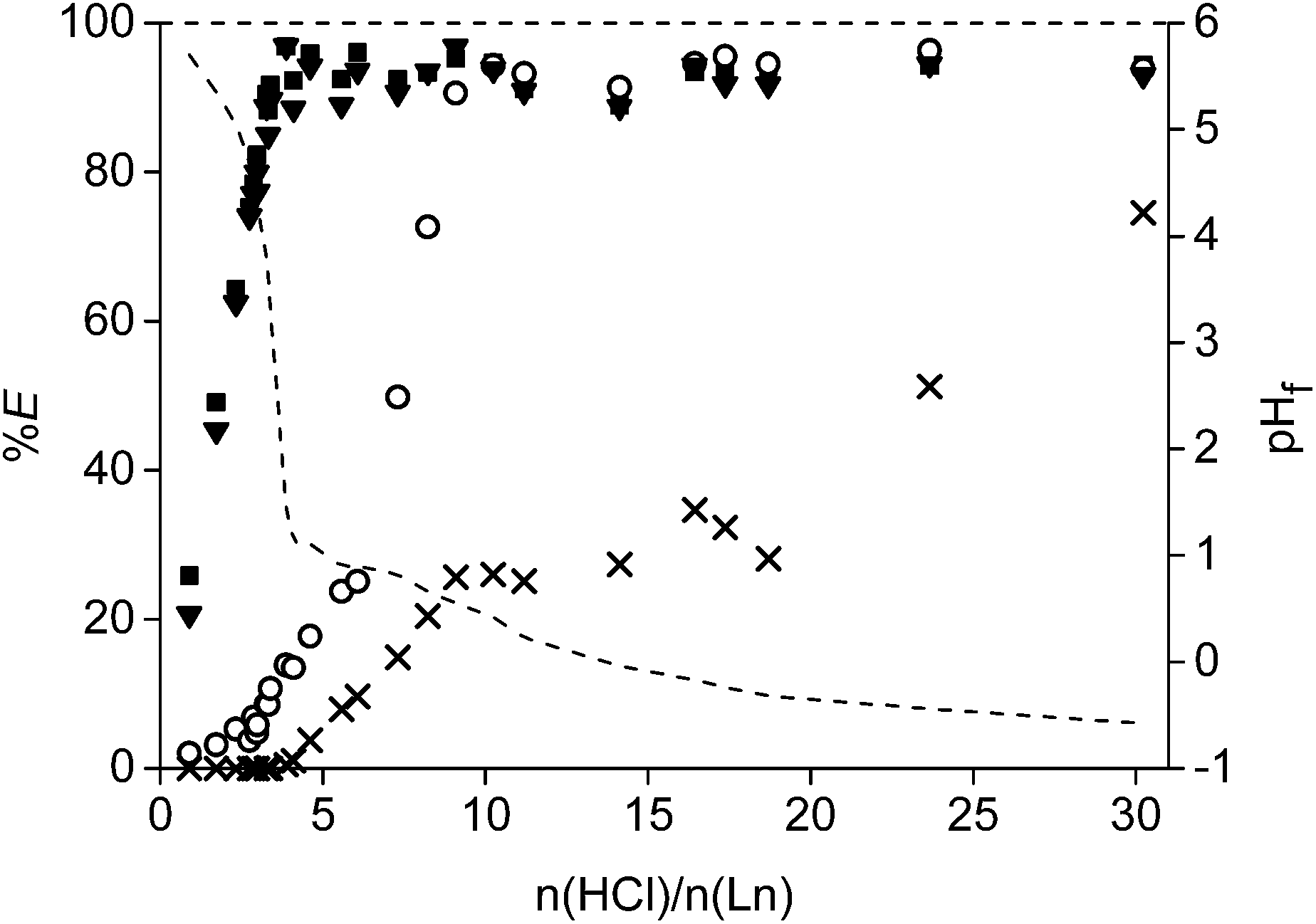 | ||
| Fig. 5 Extraction percentages (%E) of neodymium (■), dysprosium (▼), cobalt (○) and iron (×) as a function of the molar ratio of HCl over the rare earths present in the roasted NdFeB magnet powder (15 h at 80 °C) The dashed line shows the measured final pH values (pHf) after the leaching experiments. Relative standard deviation < 6.9%. | ||
The actual leaching percentages, especially for the rare earths, are definitely higher that the measured values and probably close to 100%. The fact that we reach only 90% is due to the relatively small volume (1 mL) of leaching solution in comparison to the amount of NdFeB magnet (0.1 g). A small volume was chosen in order to get a concentrated leachate which is beneficial for industrial solvent extraction processes which can follow on the leaching process. Therefore, it is impossible to remove all dissolved rare earths quantitatively from the wet iron(III) hydroxide precipitate. This will cause errors when dissolving the remaining precipitate and measuring its rare-earth metal ion concentration. Washing of the precipitate in order to remove the remaining rare earths was considered but it was difficult to avoid removal of part of iron present as some kind of suspension. The formation of a poorly formed precipitate or suspension is probably due to the formation of different iron(III) hydroxide species at the different pH levels obtained during the consumption of the acid in the leaching process. More technical equipment, such as a solid bowl centrifuge, are necessary to quantitatively remove all the dissolved rare earths from the iron precipitate.64
Leaching of the other elements than Fe, Co, Dy and Nd was not studied into detail due to overlapping peaks in the TXRF spectrum or because the elements gave X-ray signals which are too weak to be measured (e.g. light elements). However, elements present in the magnet such as niobium (0.83 wt%) and silicon (0.06 wt%) are probably not leached because their halide salts are not stable in aqueous environment. Boron oxide (B2O3, 1.00 wt%), formed during the roasting process from boron, has a moderate solubility in water and can be leached as boric acid.65 However, part of the boron could be present in the leaching residue as well when working with a solution with a pH higher than 2.25 The purity of the rare earths (Nd + Dy + Pr) after the leaching procedure increased from 30.5 wt% in the magnet to a minimum of 90% in the leachate at an n(HCl)/n(Ln) ratio of 3.5. All elements initially present in the magnet were included in this calculation, except iron and part of the cobalt which were left behind in the precipitate. The exact purity is definitely higher that the reported one as also elements such as niobium and silicon were included. The purity drops at higher n(HCl)/n(Ln) ratio because of the increasing concentration of cobalt and iron in the leachate.
An identical leaching process with HNO3 was performed as well and gave very similar results (Fig. 6). In addition, leaching with HCl and HNO3 in the presence of a concentrated chloride or nitrate matrix (3.75 M of CaCl2 or 7.5 M of NH4NO3 respectively) was tested as well to prove that the leachate could be used directly into interesting and cheap solvent extraction systems to further purify and separate the rare earths.62,63 The idea is that the leachate, containing high amounts of CaCl2 or NH4NO3, can be reused (after removal of the rare earths) as diluent for the acid used in a new NdFeB leaching experiment. In this way, a closed aqueous loop is generated and an aqueous waste stream with a high salt concentration is avoided. No large differences were observed for the mixtures in comparison with the pure acids. However, it seems to be more difficult to obtain a pH higher than 2 when using these salts.
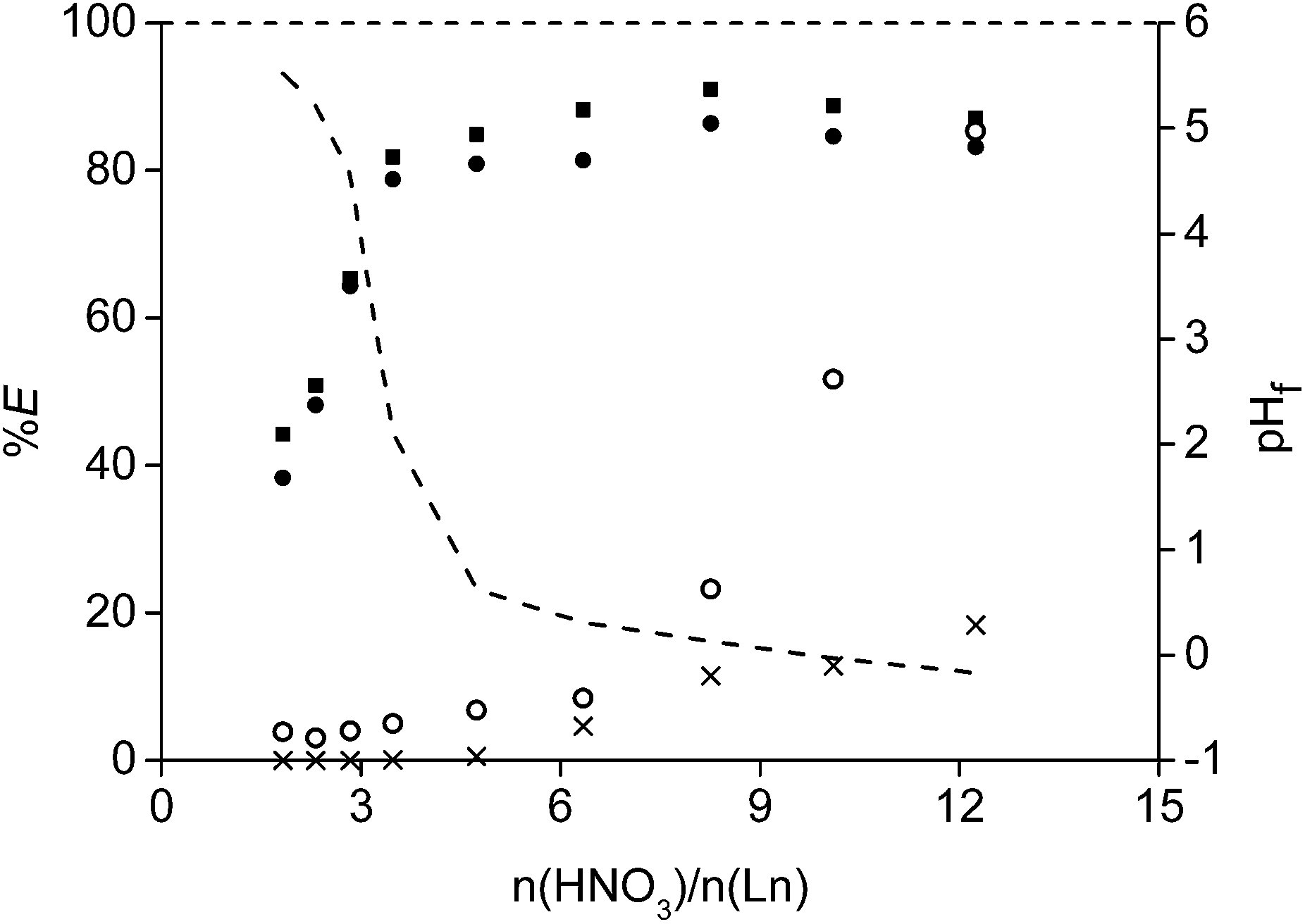 | ||
| Fig. 6 Percentage extraction (%E) of neodymium (■), dysprosium (▼), cobalt (○) and iron (×) as a function of the molar ratio of HNO3 over the rare earths present in the roasted NdFeB magnet powder. The dashed line shows the measured final pH values (pHf) after the leaching experiments. Relative standard deviation < 6.9%. | ||
A leaching process with HCl was performed as well on the non-roasted magnet powder in order to compare the results with a leaching process from a roasted magnet (Fig. 6 and 7). No metal selectivity could be obtained over the whole HCl concentration range. Moreover, 15 equivalents of acid were necessary to obtain full dissolution of the rare earths and cobalt. This requires about four times more acid to leach and dissolved all rare earths and cobalt in comparison with a leaching process from the roasted magnets. In addition, cobalt and the rare earths were enriched when leaching was performed from the oxides because only 30% of iron went into solution at a n(HCl)/n(Ln) ratio of 10. Such a selective dissolution process was not observed in the case of leaching from the non-roasted magnet.
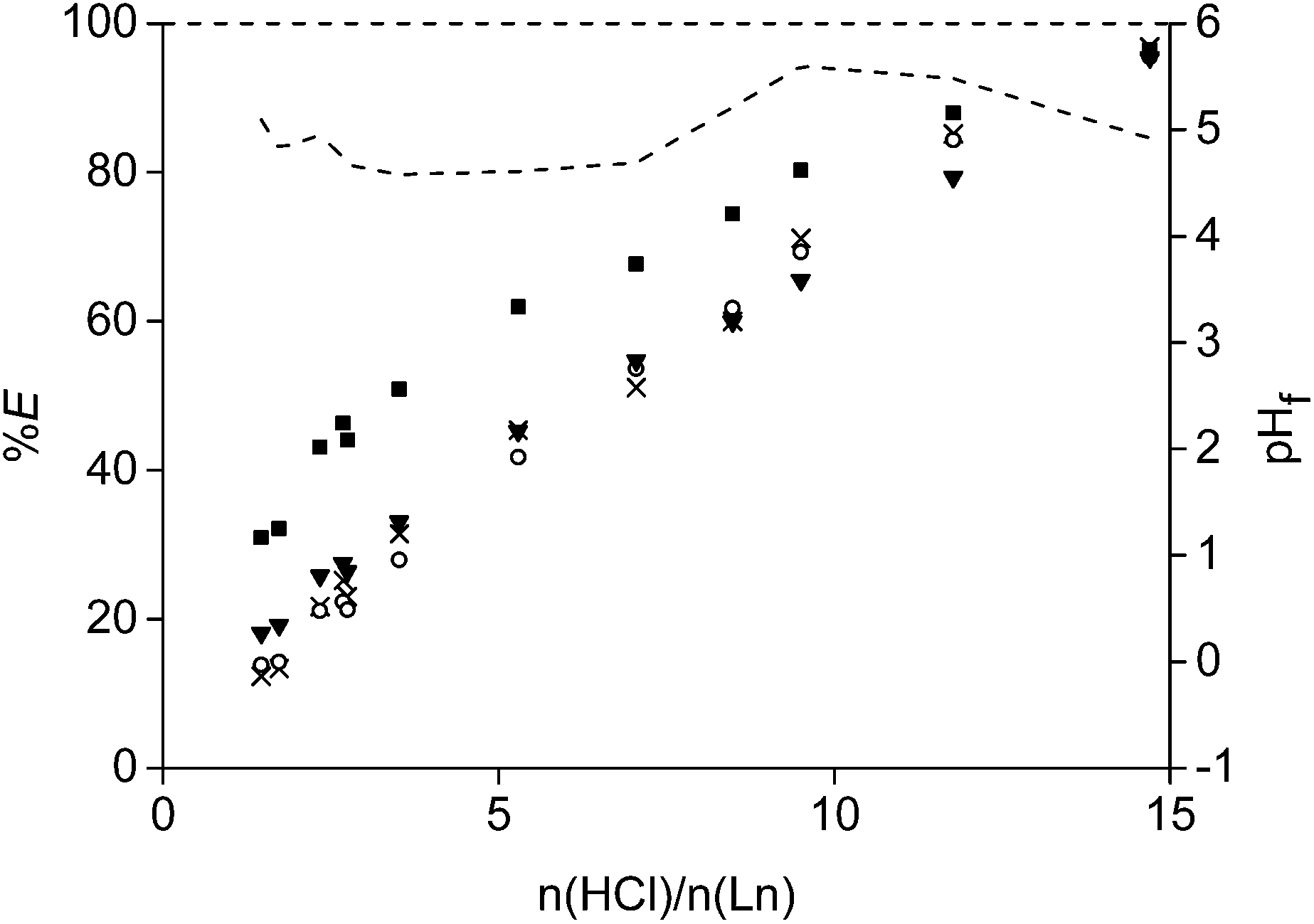 | ||
| Fig. 7 Percentage extraction (%E) of neodymium (■), dysprosium (▼), cobalt (○) and iron (×) as a function of the initial HCl leaching concentration for NdFeB magnet powder which was not roasted. The dashed line presents the measured final pH values (pHf) after the leaching experiments. Relative standard deviation < 4.2%. | ||
The speed of the leaching experiments was studied by mixing 1 mL of diluted HCl with 0.1 gram of roasted magnet in a vial of 4 mL with a stirring bar of 1.5 cm and at a temperature of 80 °C. The n(HCl)/n(Ln) ratio was 3.5 and a very small volume (0.001 mL) of leachate was removed after several time intervals to measure the actual metal ion concentrations with TXRF (Fig. 8). Only the main metals were measured and the ratios were calculated as given in eqn (3).
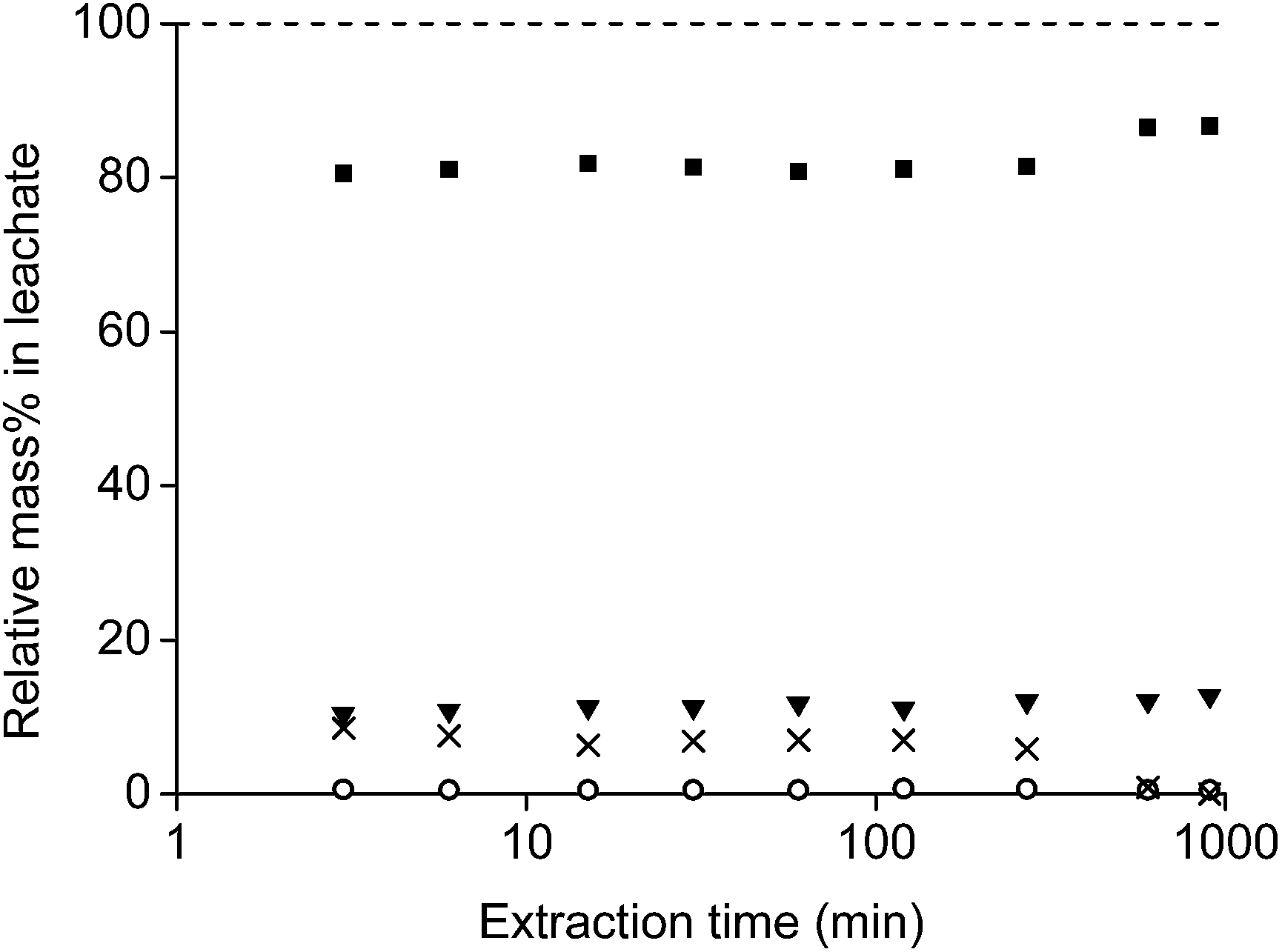 | ||
| Fig. 8 Relative mass percentages of neodymium (■), dysprosium (▼), cobalt (○) and iron (×) in the leachate as a function of extraction time for 0.1 gram or roasted NdFeB magnet in 1 mL of a diluted HCl solution with a n(HCl)/n(Ln) ratio of 3.5. | ||
The relative concentrations of the four main elements (Nd, Dy, Fe and Co) in the leachate were stable for 120 min (2 h). At this point, the concentration of HCl in the solution was significantly lower than in the initial stage and the relative iron concentration started to decrease. After 900 min (15 h), the pH was just above 2 and all iron was precipitated. One should take into account that the leaching time and kinetics depends on several parameters such as the stirring speed, size of the stirring bar, temperature, the applied acid, powder size and amount, etc. This was also observed by Koyama et al.16,54–56 0.02 M HCl and an autoclave at 180 °C leached 99% of the rare earths and only 0.5% of iron after 2 h. A higher concentration (0.1 M HCl) gave no selectivity because also iron went into the solution. Lower concentrations (0.01 or 0.001 M HCl) and different conditions (24 h at 60 °C) gave no full leaching of the rare earths and gave surprisingly also dissolutions percentages of 14.0 and 1.2% iron, respectively.
It was concluded that under the conditions applied in this work, 15 h of stirring is sufficient to obtain a leachate in equilibrium with the remaining precipitate. However, it is possible that the leachate is not yet in equilibrium with the leaching residue. Longer stirring times could probably further reduce the iron content and increase the purity of the rare earths in the leachate as will be shown later.
The differences between leaching from the NdFeB oxides or from the non-roasted magnets can be explained with some basic iron chemistry. Iron(III) oxide is formed as most stable iron compound during the roasting process at 950 °C in air and at atmospheric pressure.66,67 Addition of the acid (e.g. HCl) gives a similar dissolution reaction for the rare-earth oxides (Ln2O3) as for iron(III) oxide (Fe2O3). Important to mention is that probably mixed metal oxides (e.g. NdFeO3) are formed, instead of pure Ln2O3 or Fe2O3 which are used in the simplified eqn (7) and (8):68
| Ln2O3 + 6HClaq → 2LnCl3 + 3H2O | (7) |
| Fe2O3 + 6HClaq → 2FeCl3 + 3H2O | (8) |
FeCl3 is not stable above a pH of 2 and undergoes hydrolysis in water:
| 2FeCl3 + 6H2O → 2Fe(OH)3,s + 6HClaq | (9) |
A detailed study of the formed iron(III) hydroxide species is outside the scope of this paper and is extensively described elsewhere,67 It is important to mention here that, depending on the pH values, also mixed oxide/hydroxide can be formed. The acid, which is released during the reaction written in eqn (9), can undergo again a reaction as given by eqn (7) or (8). The rare-earth chlorides are stable at higher acidic pH values than iron(III). In this way, only rare earths dissolve in the aqueous phase as long as there is no excess of acid compared to the amount of rare earths present in the leachate and precipitate. This is why iron starts to leach when the final pH decreases below 2 (in this case around n(HCl)/n(Ln) > 3.5) (Fig. 5).
Different reactions occur when leaching was performed on the non-roasted NdFeB magnet powder. The redox half reactions and their standard reduction potential (E0) for the rare earths neodymium, dysprosium, and for the acidic protons are:69
| 2H+ + 2e− ⇌ H2 E0 = 0 V | (10) |
| Nd3+ + 3e− ⇌ Nd E0 = −2.323 V | (11) |
| Dy3+ + 3e− ⇌ Dy E0 = −2.295 V | (12) |
Giving a global redox reaction:
| 6HClaq + 2Ln → 3H2,g + 2LnCl3 | (13) |
A similar reaction proceeds for iron. Here, iron(II) is formed instead of iron(III) because it has a lower standard reduction potential:
| Fe2+ + 2e− ⇌ Fe E0 = −0.447 V | (14) |
| Fe3+ + 3e− ⇌ Fe E0 = −0.037 V | (15) |
Half reactions (9) and (13) give the global redox reaction:
| Fe + 2HClaq → FeCl2 + H2,g | (16) |
Iron(II) chloride is more stable against hydrolysis than iron(III) chloride. Therefore, it will also go into solution, even at a final pH value of 5 (Fig. 6). The electrochemical reaction from iron(II) to iron(III) in water is spontaneous but slow and difficult when no oxidant (e.g. oxygen gas) is present in solution. This is why no selectivity could be obtained when leaching was performed from the non-roasted magnet powder. However, the same full dissolution process with HNO3 from the non-roasted magnet powder was not possible as it reacts electrochemically with Fe(II) to Fe(III) with the formation of H2O and NOx gas. The overall reaction can be described as:70
| 3Fe2+ + 4H+ + NO3− ⇌ 3Fe3+ + 2H2O + NO | (17) |
Metal concentration
It was tested whether higher amounts of NdFeB oxide powder could be used as well with 1 mL of aqueous leaching phase. Therefore, the amount of oxide powder was increased but the n(HCl)/n(Ln) ratios were kept constant, once at a ratio of 3.5, and once at a ratio of 10. The amount of remaining aqueous phase in the precipitate, after removal of the leachate, also increased when the sample amount increased. This was due the higher mass of the remaining precipitate. This made calculation of the leaching percentages very difficult and incorrect. Therefore, only the metal concentrations found in the leachate are reported.The concentration of neodymium, dysprosium, and cobalt increased almost proportionally with increasing NdFeB magnet oxide amounts at a n(HCl)/n(Ln) ratio of 3.5 (Fig. 9 and 10). The metal concentration in the aqueous phase after a selective rare-earth leaching process of 0.8 g of magnet oxide powder with 1 mL of 4.3 M HCl were 175 g L−1 neodymium, 27 g L−1 dysprosium and 7 g L−1 cobalt. No iron was leached up to this point. The pH decreased from 3.30 to 2.76 while increasing the sample amount. The pH of the leachate after addition of 1 mL of diluted HCl to 1.2 g of metal oxide powder was 1.41 and significantly lower than the other pH values obtained for lower amounts. It was already shown in Fig. 5 that differences in pH, especially around a value of 2, can have a large influence on iron leaching from the magnet oxide powder. 1.7 g L−1 iron was dissolved in the leachate as a consequence of the lower pH. This is probably due to the higher amount of NdFeB oxide powder and in agreement with our previous experiments where we found that leaching in the presence of higher salt concentrations (CaCl2 or NH4NO3) was kinetically slower. Longer stirring and leaching times could probably solve this problem. It is noteworthy that there is still metal selectivity in the leaching process with higher sample amounts (at n(HCl)/n(Ln) ratios of 3.5). This could be beneficial for industrial applications where smaller equipment volumes and highly concentrated metal solutions are a financial and practical advantage, respectively (process intensification).71
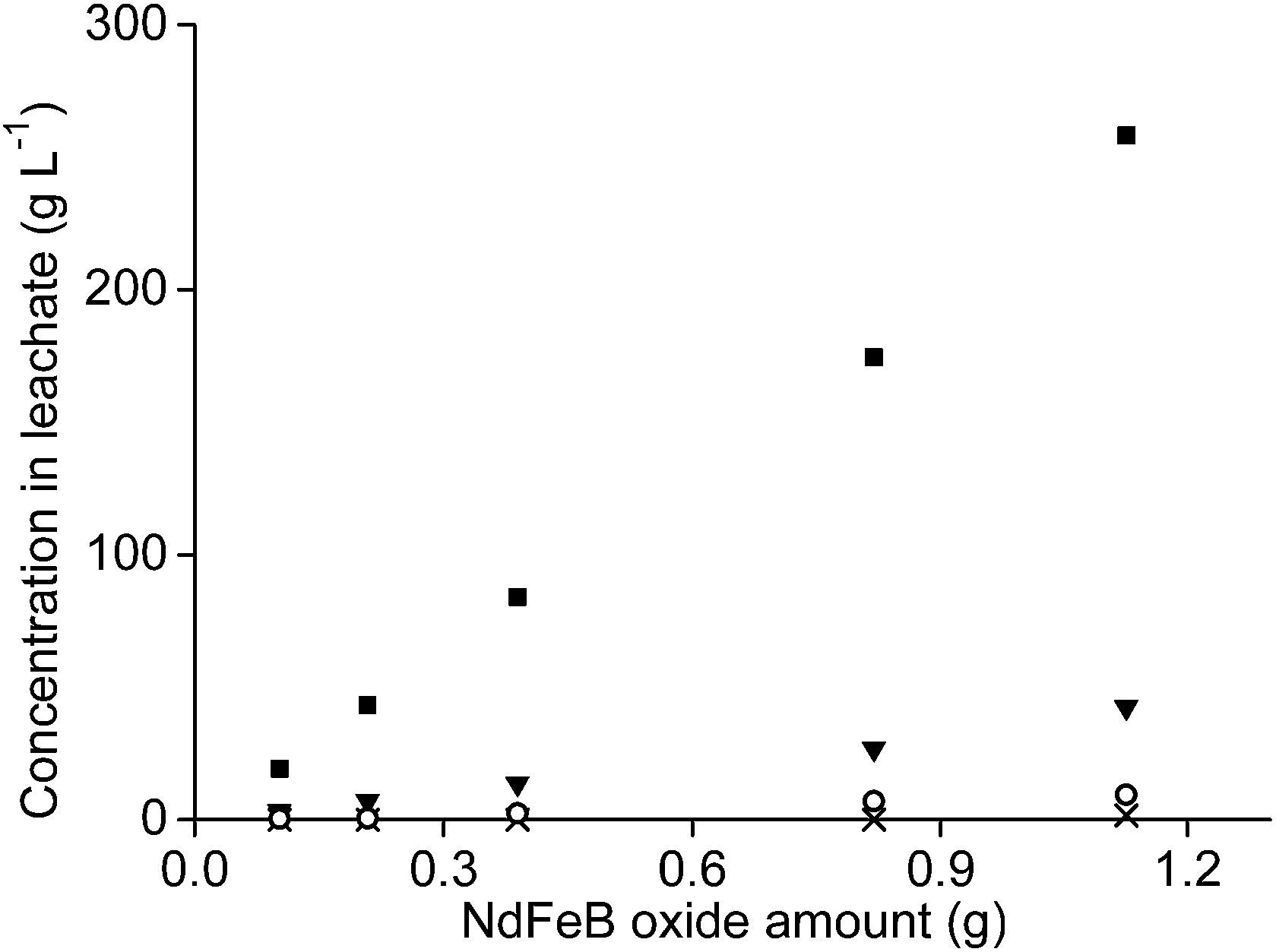 | ||
| Fig. 9 Concentration of neodymium (■), dysprosium (▼), cobalt (○) and iron (×) in the leachate as a function of the amount of magnet oxide powder involved in leaching with 1 mL of aqueous phase. The n(HCl)/n(Ln) ratio was 3.5 and the pH after leaching between 3.3 and 2.76. The data point with the highest concentrations had a pH of 1.41. Standard deviation < 7.6%. | ||
 | ||
| Fig. 10 Color of the leachate when the amount of NdFeB magnet oxide powder, dissolved in 1 mL of diluted HCl and at constants n(HCl)/n(Ln) ratio of 3.5, was increased from left to right. The purple colour is characteristic for neodymium and the orange colour is due to a small amount of dissolved iron. | ||
The metal concentrations of neodymium, dysprosium and cobalt also increased linearly with increasing sample amount when the leaching was performed at an n(HCl)/n(Ln) ratio of 10. Here, the pH decreased from 0.42 to −0.52 when the NdFeB oxide powder amount was increased. The decreasing pH is probably a kinetic effect. The concentration of iron in the leachate did not increase proportionally with increasing sample amount but the slope was slightly increasing and that relatively more iron was leached than the other elements at lower pH values (Fig. 11).
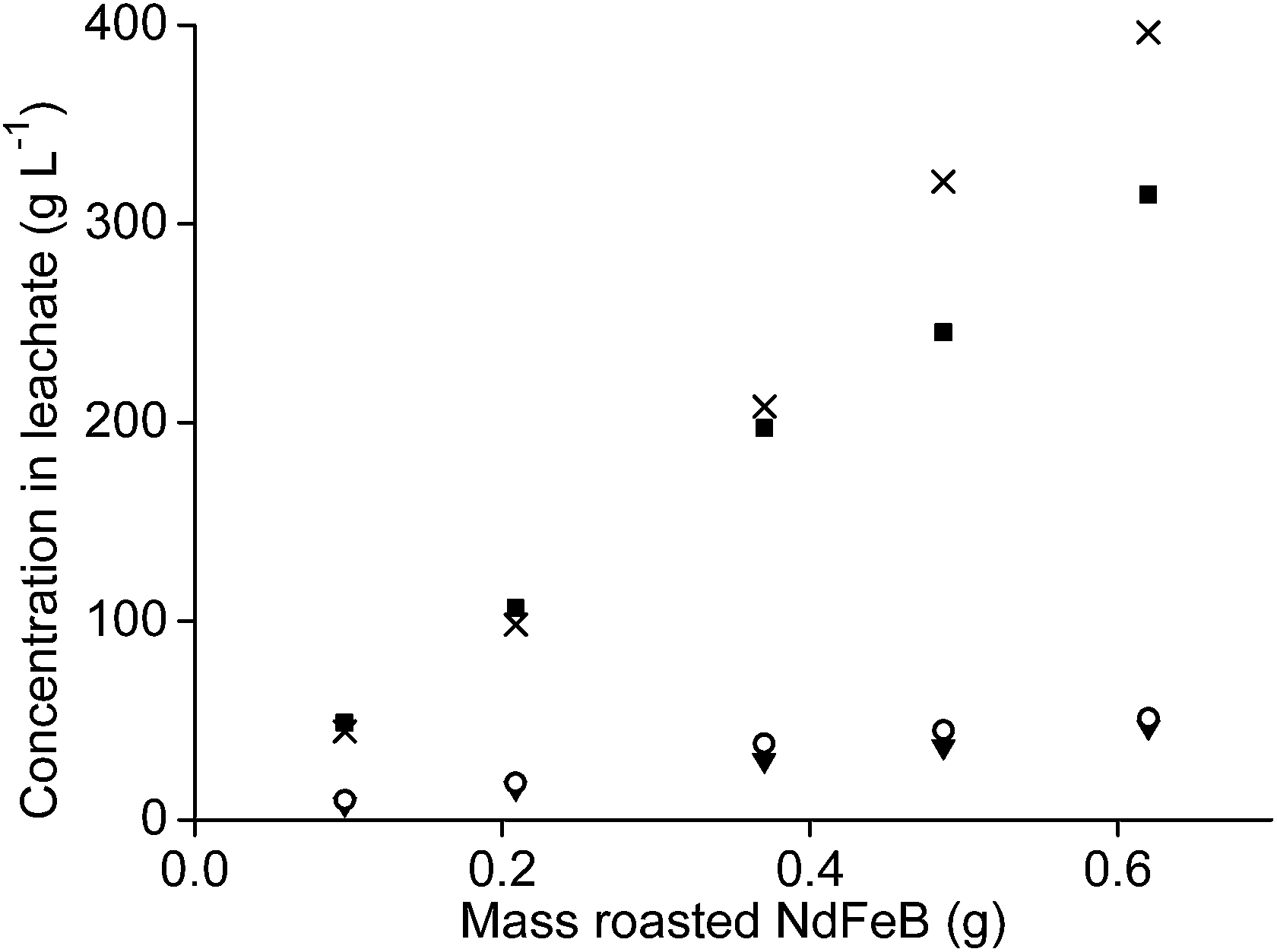 | ||
| Fig. 11 Concentration of neodymium chloride (■), dysprosium chloride (▼), cobalt chloride (○), and iron(II,III) chloride (×) as a function of the amount of magnet oxide powder involved in leaching with 1 mL of aqueous phase. The n(HCl)/n(Ln) value was 10 and the pH after the extraction between 0.42 and −0.56. Relative standard deviation < 4.2%. | ||
Solvent extraction
The 1 mL aliquot of aqueous phase obtained after the leaching process of 0.1 g of NdFeB oxide powder, contained 23 g L−1 of rare earths and about 0.3 g L−1 of cobalt at a n(HCl)/n(Ln) ratio of 3.5. A solvent extraction process using the undiluted ionic liquid trihexyl(tetradecyl)phosphonium chloride (Cyphos® IL 101) was chosen to further purify the aqueous phase. This ionic liquid is an excellent extractant for iron(III), copper(II), zinc(II), manganese(II) and cobalt(II), whereas other metals such as nickel or rare earths are not extracted from concentrated aqueous chloride solutions.62 The acid HCl can be used as salting-out agent with very high separation factors. Unfortunately, further processing of the rare earths (e.g. precipitation with oxalic acid) is very difficult from a highly acidic solution and would ask for a large amount of a base or an excess of the precipitating reagent. Therefore, we selected 3.5 M NH4Cl as salting-out agent. A value of 3.5 was chosen because higher amounts of rare earths start to extract at higher NH4Cl concentrations and an extra scrubbing step of the ionic liquid phase would be necessary to avoid losses of rare earths. On the other hand, the addition of a small amount of HCl can increase the extraction of cobalt and decrease the extraction of rare earths.62 As a consequence, also the separation factor αCo,Dy would increase with increasing acidity of the aqueous phase. A separation study between 3.0 g L−1 cobalt and 25 g L−1 dysprosium in the presence of 3.5 M NH4Cl and different amounts of HCl in the aqueous phase was performed and the separation factors are represented in Fig. 12. The concentration of cobalt was made 10-fold higher than that found in the leachate and dysprosium instead of neodymium was chosen as rare earth. In this way, the overlapping peaks in the TXRF spectrum between cobalt (very low concentration) and neodymium (very high concentration) were avoided. The separation factor between cobalt and dysprosium was about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with an extraction percentage of 0.5% for dysprosium and 99.1% for cobalt when no acid is present. The separation factor increased towards 130000 at 1 M of HCl.
000 with an extraction percentage of 0.5% for dysprosium and 99.1% for cobalt when no acid is present. The separation factor increased towards 130000 at 1 M of HCl.
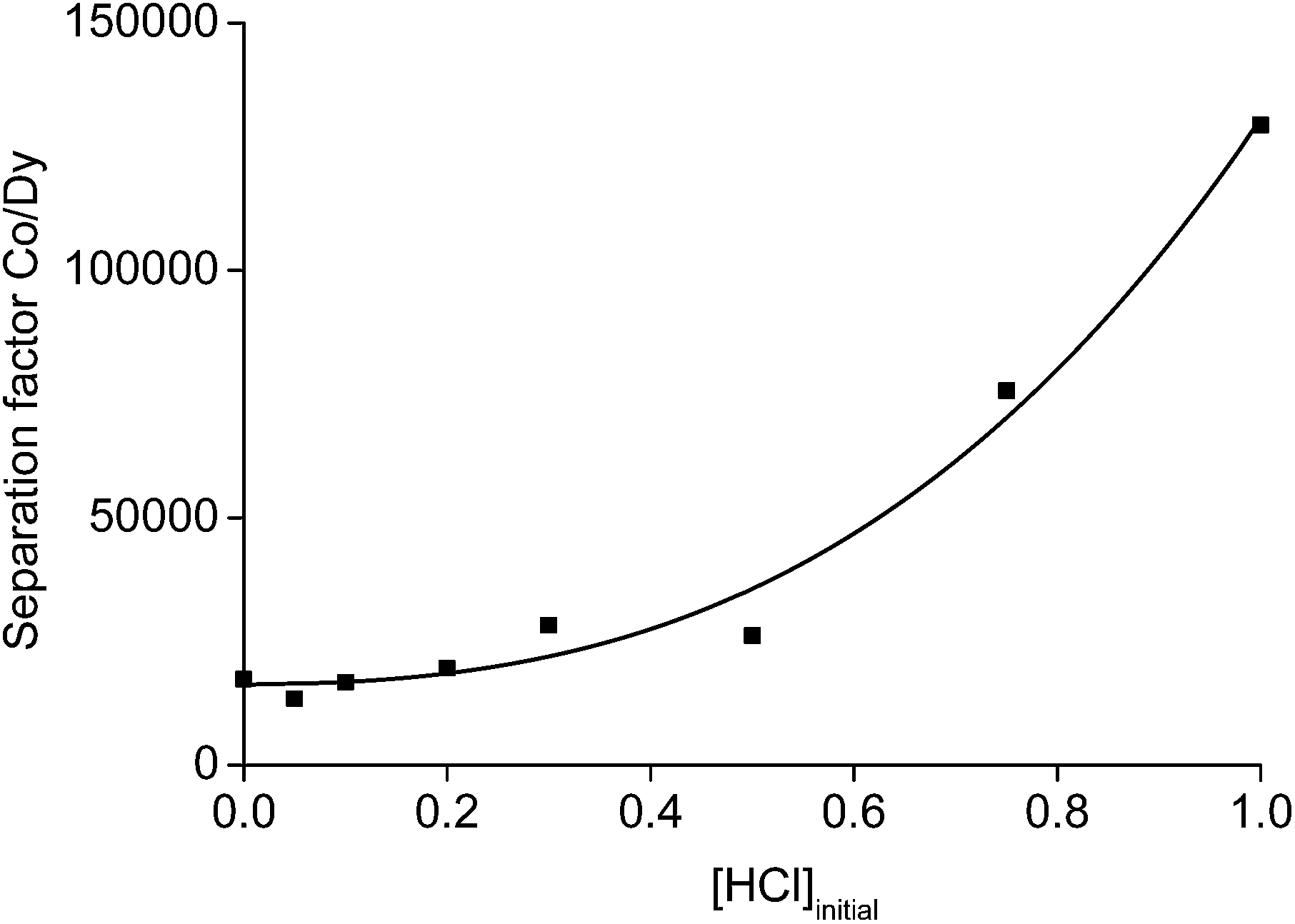 | ||
| Fig. 12 The separation factors αCo,Dy between cobalt and dysprosium as a function of the initial HCl concentration of the aqueous phase. The aqueous phase contains 3.0 g L−1 cobalt, 25 g L−1 Dy and 3.5 NH4Cl. The organic phase was trihexyl(tetradecyl)phosphonium chloride mixed with the aqueous phase in a volume ratio of 1. | ||
Precipitation
The aqueous phase is now pure in rare-earth metals, but they have to be removed from the aqueous phase. The starting materials of NdFeB magnets are rare-earth oxides, so the end product of the recycling loop should be rare-earth oxides too. Oxalic acid is an organic acid which forms very strong water insoluble complexes with rare earths. The following precipitation reaction occurs when oxalic acid is added to the aqueous phase:| 2LnCl3 + 3H2C2O4,s → Ln2(C2O4)3,s↓ + 6HClaq | (18) |
Each rare-earth ion liberates three acidic H+ atoms to the solution (eqn (18)). The acidic aqueous phase generated by the precipitation process can be reused for new leaching and solvent extraction experiments. However, oxalic acid should remain in the aqueous phase because this would decrease the leaching percentage of the rare earths due to the formation of rare-earth oxalate salts in a following leaching experiment. Moreover, the acid which would be generated would decrease the pH and would bring iron(III) into solution. This can only be avoided if eqn (18) is complete and no oxalic acid is present in the aqueous phase after the precipitation reaction. It was observed that the reaction was complete when the initial aqueous phase contained no HCl. The amount of neodymium precipitated from a solution containing 3.5 M NH4Cl increased linear towards 100% at a value n(oxalic acid)/n(Nd) of 1.5 (Fig. 13). This suggests that all oxalic acid is consumed as long as rare-earth ions are still present in the solution. Unfortunately, this is not the case when working at, for instance, initial HCl concentrations of 0.5 M. Under these conditions, 100% precipitation of the rare earths occurred only at a value n(oxalic acid)/n(Nd) of 2 which means that there is still a significant amount of oxalic acid dissolved in the aqueous phase which is a problem towards the reuse of the aqueous phase in a new leaching experiment. No precipitation of ammonium oxalate was observed for similar mixtures without metals, which proves that the precipitate is pure in rare-earth metals. The precipitate can be washed to remove traces of NH4Cl, but can be calcined immediately as well, because NH4Cl decomposes to HCl and NH3 at the temperature necessary to calcinate the rare-earth oxalates.
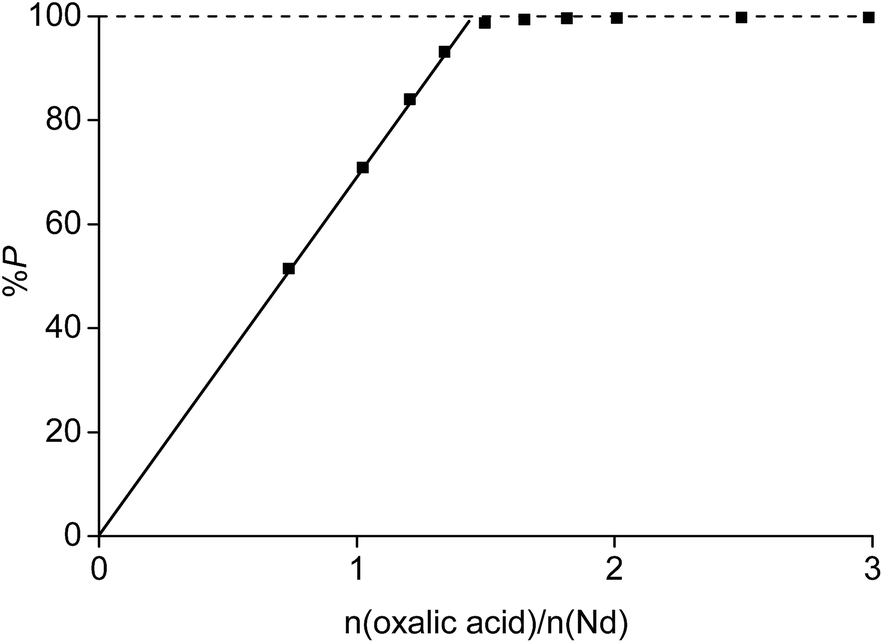 | ||
| Fig. 13 Percentage precipitation (%P) as a function of the molar ratio of oxalic acid added to the solution over neodymium present in the aqueous phase n(oxalic acid)/n(Nd). The aqueous phase contained of 26.7 g L−1 neodymium and 3.5 M NH4Cl. | ||
Overall process
The roasting, leaching, solvent extraction and the oxalate precipitation process were optimised. A complete recycling process can now be applied to NdFeB magnets. A flow chart of the process can be found in Fig. 14 and the details in the experimental section.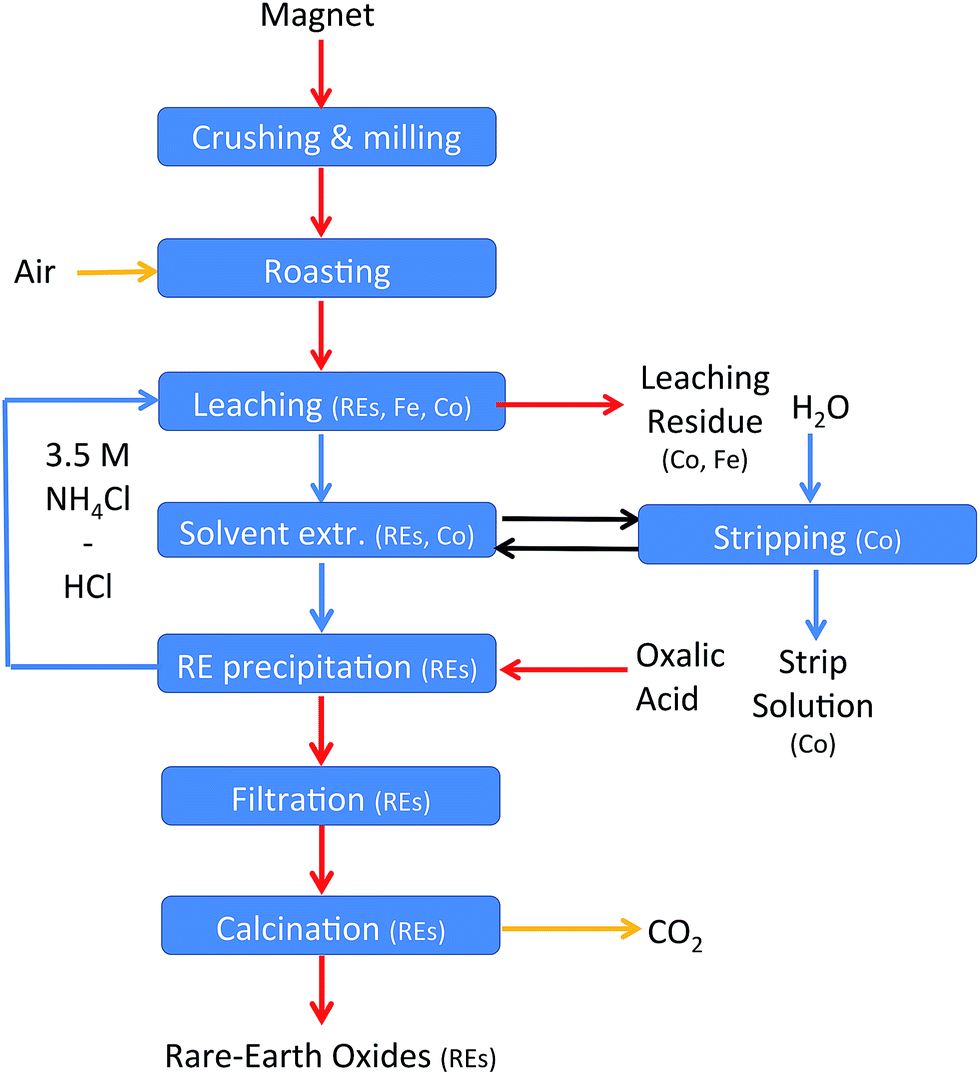 | ||
| Fig. 14 Flow chart of the recycling scheme. Yellow arrows: gas steams, blue arrows: aqueous streams, black arrows: ionic liquid steam, and red arrows: solid streams. | ||
The recycling process for NdFeB magnets includes seven steps. The rare-earth metals were leached out of the magnet oxide powder after the crushing and roasting step and iron was left behind. A sample of roasted NdFeB magnet powder (3.1 g) was therefore mixed with 30 mL of an aqueous phase containing 1.35 mL of 37 wt% (12.0 M) HCl and 3.5 M NH4Cl (n(HCl)/n(Ln) = 3.5). The temperature was increased up to 100 °C instead of the previously used 80 °C because a temperature of 80 °C was not high enough to obtain a pH above 2 after five days of stirring. The slower kinetics towards equilibrium of the process is probably due to the high salt concentration (3.5 M NH4Cl) and the higher volume of the leachate. A pH value of 2.5 was obtained after five days of stirring at 100 °C in 3.5 M NH4Cl. The leaching residue was removed by filtration. Unfortunately, about 15% of the aqueous phase was lost in this process due to water absorption in the filter pores and in the leaching residue. Specialized equipment, such as a solid bowl centrifuge, can significantly reduce these losses.64 Afterwards, the leachate (about 25 mL) was brought into contact with 5 mL of the ionic liquid trihexyl(tetradecyl)phosphonium chloride to remove the remaining transition metals from the rare earths. The organic phase contained mainly cobalt (3.3 g L−1) but also some traces of the other transition metals copper (0.02 g L−1) and manganese (0.03 g L−1) but no nickel. The extraction of these metals is in agreement with earlier studies performed in our laboratory.62 About 0.13 g L−1 neodymium and 0.10 g L−1 dysprosium were lost in the organic phase. The next step involved a precipitation step with oxalic acid. HCl is released in this step (eqn (18)) and the end of the reaction can be followed by measuring the pH. The pH was stable (pH = 0.15) after the addition of 0.623 g of oxalic acid. It is better to avoid the addition of an excess of oxalic acid in this step to prevent precipitation of the rare earths during the following leaching step. The metal concentrations in the aqueous phase after the precipitation process were 2.4 g L−1 of neodymium and 0.4 g L−1 of dysprosium. The aqueous phase, concentrated in HCl, can be reused as leaching solution for new NdFeB oxide powder. In this way, also losses of the rare earths in the oxalate precipitation step are avoided because the remaining rare earths are brought back into the process.
The aqueous stream containing HCl and NH4Cl is a closed loop. HCl, regenerated during the precipitation process, can be reused in the leaching step. However, part of the aqueous phase is lost during the filtration steps. Therefore, a small amount of acidified water has to be added in order to get an aqueous phase with the same volume and acid concentration as in the first leaching step. A second, rather small loss of HCl is due to cobalt leaching, which is not regenerated by oxalate precipitation and HCl formation. Apart from this, oxalic acid is the only chemical which is consumed during the process. For each rare-earth atom present in the magnet, 1.5 molar equivalents of oxalic acid are necessary in the process. Small amounts of HCl and NH4Cl are lost in the precipitate during the precipitation step. Also oxygen is consumed which is freely available from the air. Water can be used as stripping agent to remove cobalt from the ionic liquid phase.61,72
The process contains three streams of waste. First, the leaching residue, which is concentrated in iron, but also cobalt and niobium are present. Secondly, there is an aqueous waste stream which is generated during the stripping process of cobalt and extracted rare-earths from the ionic liquid. This stream contains mainly cobalt but also neodymium, dysprosium and manganese. The solubility of the ionic liquid into the aqueous phase is expected to be lower than 100 ppm.72 Thirdly, harmless water and carbon dioxide gas are released during the calcination step of the rare-earth oxalates.
The process consumes energy on the lab scale, especially during the roasting step. However, NdFeB magnets are reacting exothermic with oxygen at higher temperature (Fig. 1C). The heat released in this step, in combination with a more appropriate furnace, could significantly reduce energy consumption on an industrial scale. Moreover, fluidized bed roasters can operate in a net energy producing mode.70
Conclusions
A proof-of-principle is given in which the rare-earths metals are recovered from the NdFeB magnet in high purity by using a milling, roasting, leaching, solvent extraction and precipitation scheme. It was shown that roasting of the NdFeB magnet powder, prior to the dissolution process, significantly increases the relative concentrations of the rare earths in the leachate compared to dissolution of a non-roasted NdFeB magnet. Moreover, less acid is consumed and the purity of the rare earths in the leachate increases to values higher than 90%. The difference between a dissolution process starting from a roasted or non-roasted magnet was explained. During the roasting process, iron(III) is formed which is not stable to hydrolysis when the final pH of the leachate is higher than 2. Iron(II), which is more stable at higher pH values, is formed when the non-roasted magnet is dissolved in acid, resulting in a less pure rare-earth leachate. The leachate, free of iron, was then used in a solvent extraction process with the undiluted ionic liquid trihexyl(tetradecyl)phosphonium chloride to remove remaining traces of cobalt. Afterwards, the rare earths were precipitated by the addition of oxalic acid and can be calcined to obtain pure rare-earth oxides which can be used as part of the feedstock for the production of NdFeB magnets. The whole process consumes only oxalic acid, air and water and produces three waste streams: the leaching residue, the aqueous stripping solution and CO2 formed during calcination of the rare-earth oxalates. Further optimization of the process, in order to increase of the yields of each process step, is currently under investigation in our group. Moreover, the process is still energy-consuming; especially the roasting step. Further investigations will focus on a more energy efficient selective dissolution process of the magnet. Also the removal of boron, probably accumulating in the aqueous NH4Cl stream, will be investigated in the near future.Acknowledgements
The authors thank the KU Leuven for financial support (GOA/13/008 and IOF-KP RARE3). The authors would like to thank Pencheng Yang (KU Leuven) and Umicore for the oxygen determination with their LECO oxygen analyser and Wout Veulemans (KU Leuven) for the help with the grinding experiments. We thank Allan Walton from the University of Birmingham (UK) for providing the NdFeB magnet and Dorien Baeten (KU Leuven) for the help with the TGA experiments.Notes and references
- M. Humphries, Report: Rare Earth Elements: The Global Supply Chains, CRS, Report for Congress, R41347, http://www.crs.gov, 2012 Search PubMed.
- D. Schüler, M. Buchert, R. Liu, S. Dittrich and C. Merz, Report: Study on Rare Earths and Their Recycling, Final Report for The Greens/EFA group in the European Parliament, http://www.oeko.de, 2011 Search PubMed.
- N. Charles, J. Tuduri, D. Guyonnet, J. Melleton and O. Pourret, Rare earth elements in Europe and Greenland: a geological potential?, An overview, 12th Meeting of the Society of Geology Applied to Mineral Deposits (SGA), pp. 1698–1701, 2013 Search PubMed.
- European Commission, Report: Critical Raw Materials for the EU, Report of the ad-hoc working group on defining critical raw materials, http://ec.europa.eu/, 2010 Search PubMed.
- M. A. de Boer and K. Lammertsma, ChemSusChem, 2013, 6, 2045–2055 CrossRef CAS PubMed.
- U. S. Department of Energy, Report: 2011 Critical Materials Strategy, http://energy.gov/node/349057, 2011 Search PubMed.
- K. Binnemans, P. T. Jones, K. Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- O. Gutfleisch, M. A. Willard, E. Brück, C. H. Chen, S. G. Sankar and J. P. Liu, Adv. Mater., 2011, 23, 821–842 CrossRef CAS PubMed.
- J. J. Croat, J. F. Herbst, R. W. Lee and F. E. Pinkerton, Appl. Phys. Lett., 1984, 44, 148–149 CrossRef CAS.
- K. H. J. Buschow, IEEE Trans. Magn., 1994, 30, 565–570 CrossRef CAS.
- Great Western Minerals Group LTD, http://wenku.baidu.com/view/016f216825c52cc58bd6be11.html, 2014.
- W. T. Benecki, T. K. Claggett and S. R. Trout, Permanent Magnets 2010–2020: A Comprehensive Overview of the Global Permanent Magnet Industry, W. T. Benecki, LLC, 2010 Search PubMed.
- K. Habib and H. Wenzel, J. Cleaner Prod., 2014, 84, 348–359 CrossRef CAS.
- J. Lin, P. R. China Patent CN102206755, 2013.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- M. Tanaka, T. Oki, K. Koyama, H. Narita and T. Oishi, in Handbook on the Physics and Chemistry of Rare Earths Including Actinides, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, vol. 43, pp. 159–211, 2013 Search PubMed.
- J. H. Rademaker, R. Kleijn and Y. Yang, Environ. Sci. Technol., 2013, 47, 10129–10136 CAS.
- X. Du and T. E. Graedel, J. Ind. Ecol., 2011, 15, 836–843 CrossRef CAS.
- C. D. Anderson, C. G. Anderson and P. R. Taylor, Can. Metall. Q., 2013, 52, 249–256 CrossRef CAS.
- O. Takeda and T. Okabe, Metall. Mater. Trans. E, 2014, 1, 160–173 Search PubMed.
- V. Kumar, M. K. Jha, A. Kumari, R. Panda, J. R. Kumar, and J. Y. Lee, in Rare Metal Technology 2014, ed. N. R. Neelameggham, S. Alam, H. Oosterhof, A. Jha and S. Wang, Wiley, pp. 81–88, 2014 Search PubMed.
- I. R. Harris, J. Speight and A. Walton, WO2012101398, 2012.
- B. Sprecher, Y. Xiao, A. Walton, J. Speight, R. Harris, R. Kleijn, G. Visser and G. J. Kramer, Environ. Sci. Technol., 2014, 48, 3951–3958 CrossRef CAS PubMed.
- K. Lee, K. Yoo, H. S. Yoon, C. J. Kim and K. W. Chung, Geosyst. Eng., 2013, 16, 286–288 CrossRef CAS.
- C. H. Lee, Y. J. Chen, C. H. Liao, S. Popuri, S. L. Tsai and C. E. Hung, Metall. Mater. Trans. A, 2013, 44, 5825–5833 CrossRef CAS.
- T. W. Ellis, F. A. Schmidt and L. L. Jones, in Methods and opportunities in the recycling of rare earth based materials, ed. K. C. Liddell, R. G. Bautista and R. J. Orth, Symposium on Metals and Materials Waste Reduction, Recovery and Remediation, at the 1994 Materials Week, Meeting Location: Rosemont, IL Date: October 03–06, 1994, The Minerals, Metals & Materials Society, Warrendale, Pennsylvania, pp. 199–208, 1994 Search PubMed.
- J. W. Lyman and G. R. Palmer, US5129945 A, 1992.
- T. Itakura, R. Sasai and H. Itoh, J. Alloys Compd., 2006, 408–412, 1382–1385 CrossRef CAS.
- M. Gasgnier, L. Albert, J. Derouet, L. Beaury and P. Caro, C. R. Acad. Sci., Ser. IIb: Mec., Phys., Chim., Astron., 1994, 318, 915–920 CAS.
- K. Ishioka, M. Matsumiya, M. Ishii and S. Kawakami, Hydrometallurgy, 2014, 144–145, 186–194 CrossRef CAS.
- W. Lai, M. Liu, C. Li, H. Suo and M. Yue, Hydrometallurgy, 2014, 150, 27–33 CrossRef CAS.
- Y. Xu, L. S. Chumbley and F. C. Laabs, J. Mater. Res., 2000, 15, 2296–2304 CrossRef CAS.
- H. Okamoto, J. Phase Equilib., 1991, 12, 249–250 CrossRef.
- A. A. Nayeb-Hashemi, J. B. Clark and L. J. Swartzendruber, Bull. Alloy Phase Diagrams, 1985, 6, 235–238 CrossRef CAS.
- O. Takeda, T. H. Okabe and Y. Umetsu, J. Alloys Compd., 2004, 379, 305–313 CrossRef CAS.
- O. Takeda, K. Nakano and Y. Sato, Mater. Trans. JIM, 2014, 55, 334–341 CrossRef CAS.
- T. Saito, H. Sato and T. Motegi, J. Alloys Compd., 2006, 425, 145–147 CrossRef CAS.
- Y. Kikucki, M. Matsumiya and S. Kawakami, Solvent Extr. Res. Dev., Jpn., 2014, 21, 137–145 CrossRef.
- K. Asabe, A. Saguchi, W. Takahashi, R. Suzuki and K. Ono, Mater. Trans. JIM, 2001, 42, 2487–2491 CrossRef CAS.
- A. Saguchi, K. Asabe, W. Takahashi, R. Suzuki and K. Ono, Mater. Trans. JIM, 2002, 43, 256–260 CrossRef CAS.
- G. Y. Adachi, K. Shinozaki, Y. Hirashima and K. I. Machida, J. Less-Common Met., 1991, 169, L1–L4 CrossRef CAS.
- J. Jiang, T. Ozaki, K. I. Machida and G. Y. Adachi, J. Alloys Compd., 1997, 260, 222–235 CrossRef CAS.
- K. Murase, K. I. Machida and G. Y. Adachi, J. Alloys Compd., 1995, 217, 218–225 CrossRef CAS.
- M. Itoh, K. Miura and K. I. Machida, J. Alloys Compd., 2009, 477, 484–487 CrossRef CAS.
- K. Miura, M. Itoh and K. I. Machida, J. Alloys Compd., 2008, 466, 228–232 CrossRef CAS.
- T. Uda, Mater. Trans. JIM, 2002, 43, 55–62 CrossRef CAS.
- T. Akahori, Y. Miyamoto, T. Saeki, M. Okamoto and T. H. Okabe, in Magnesium Technology 2014, ed. M. Alderman, M. V. Manuel, N. Hort and N. R. Neelameggham, John Wiley & Sons, Inc., Hoboken, NJ, USA, pp. 35–38, 2014 Search PubMed.
- T. Akahori and M. Okamoto, WO2013132924, 2013.
- T. Akahori and Y. Hiroshige, in Design for Innovative Value Towards a Sustainable Society, ed. M. Matsumoto, Y. Umeda, K. Masui and S. Fukushige, Springer Netherlands, pp. 525–529, 2012 Search PubMed.
- T. Saeki, T. Akahori, Y. Miyamoto, M. Kyoi, M. Okamoto, T. H. Okabe, Y. Hiroshige and T. Nemoto, in Rare Metal Technology 2014, Wiley, pp. 103–106, 2014 Search PubMed.
- M. Kamiyoshi, H. Miyamoto, N. Nishigaki, Y. Taira and T. Irie, Recycling Process of Rare Earth from Re-Fe-B Sintered Magnets at SANTOKU Corporation, Lecture given at the 8th International Conference on f-Elements, ICFE-8, Udine, Italy, 26–31 August 2012 Search PubMed.
- A. Kaneko and H. Ohrai, WO2007119846, 2007.
- C. O. Bounds, in The recycle of sintered magnet swarf, ed. K. C. Liddell, R. G. Bautista and R. J. Orth, Symposium on Metals and Materials Waste Reduction, Recovery and Remediation, at the 1994 Materials Week Meeting, Location: Rosemont, IL, The Minerals, Metals & Materials Society, Warrendale, Pennsylvania, 1994, pp. 173–186 Search PubMed.
- K. Koyama, A. Kitajima and M. Tanaka, Kidorui, 2009, 54, 36–37 CAS.
- K. Koyama and M. Tanaka, in Proceedings of MMIJ Fall Meeting, The Mining and Materials Processing Institute of Japan, vol. 2, Sapporo, pp. 57–58, 2009 Search PubMed.
- K. Koyama and M. Tanaka, in The Latest Technology Trend and Rescource Strategy of Rare Earths, ed. K. Machida, CMC Press, Tokyo, pp. 127–131, 2011 Search PubMed.
- J. Lin, CN102206755, 2011.
- J. Li, Z. Daming, X. Zhuli and J. Xianping, CN102011020, 2012.
- C. J. Kim, H. S. Yoon and K. W. Chung, KR101147643, 2012.
- K. W. Chung, H. S. Yoon and C. J. Kim, KR101183579, 2012.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- T. Vander Hoogerstraete and K. Binnemans, Green Chem., 2014, 16, 1594–1606 RSC.
- R. Merkl and W. Steiger, Miner. Metall. Process., 2012, 29, 6–12 CAS.
- A. Fortuny, M. T. Coll and A. M. Sastre, Sep. Purif. Technol., 2012, 97, 137–141 CrossRef CAS.
- T. Jiang, Y. B. Zhang, Z. C. Huang, G. H. Li and X. H. Fan, Ironmaking Steelmaking, 2008, 35, 21–26 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley, New York, 2003 Search PubMed.
- H. S. Yoon, C. J. Kim and J. S. Kim, J. Korean Inst. Resour. Recycl., 2004, 13, 43–48 CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis Group, Boca Raton, 2007 Search PubMed.
- I. R. Epstein, K. Kustin and L. J. Warshaw, J. Am. Chem. Soc., 1980, 102, 3751–3758 CrossRef CAS.
- T. Van Gerven and A. Stankiewicz, Ind. Eng. Chem. Res., 2009, 48, 2465–2474 CrossRef CAS.
- S. Wellens, R. Goovaerts, C Möller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
| This journal is © The Royal Society of Chemistry 2014 |