A one-pot two-step efficient metal-free process for the generation of PEO-b-PCL-b-PLA amphiphilic triblock copolymers†
Brieuc Guillerma,
Vincent Lemaurb,
Bruno Ernouldc,
Jérôme Cornilb,
Roberto Lazzaronib,
Jean-François Gohyc,
Philippe Duboisa and
Olivier Coulembier*a
aCenter of Innovation and Research in Materials and Polymers (CIRMAP), Laboratory of Polymeric and Composite Materials, University of Mons (UMONS), Place du Parc, 23 7000 Mons, Belgium. E-mail: olivier.coulembier@umons.ac.be
bCenter of Innovation and Research in Materials and Polymers (CIRMAP), Laboratory for Chemistry of Novel Materials, University of Mons, 23 Place du Parc, B-7000, Mons, Belgium
cInstitute of Condensed Matter and Nanosciences (IMCN), Bio- and Soft Matter (BSMA), Université catholique de Louvain, Place L. Pasteur 1, B-1348, Louvain-la-Neuve, Belgium
First published on 28th January 2014
Abstract
In an effort to reduce hazardous chemicals, a one-pot two-step process with active bases and inactive salts was developed for the synthesis of high molar mass PEO-block-PCL-block-P(L- or D,L-LA) amphiphilic triblock copolymers. A series of poly(ε-caprolactone) (PCL)/poly(L- or D,L-lactide) (P(L- or D,L-LA)) di- and triblock copolymers have been prepared in bulk from metal-free catalytic systems and starting from either 1-pyrenemethanol or poly(ethylene oxide) (PEO) macroinitiator. The controlled generation of such structures was obtained after screening and comparing a wide variety of organic activators. Narrower dispersity characterizing each sample prepared from the PEO macroinitiator were elucidated by theoretical modeling. Finally, the ability of those triblock copolymers to self-associate in water was studied by dynamic light scattering and compared to PEO-b-P(CL-co-LA) copolymers.
Introduction
Nowadays amphiphilic block copolymers attract much attention since they represent a new class of functional structures that are unique building sequences with a number of applications.1 The self-association of amphiphilic block copolymers in water is one of the most interesting properties and results in micelle2,3 or vesicle4 formation. This property is used in many applications such as solubilization of molecules,5 drug delivery,6 nanoreactors7 and membranes.8Over the last few years, a great number of amphiphilic block copolymers have been developed, particularly those composed of aliphatic polyester segments known as biodegradable and biocompatible for most.9–13 If the ring-opening polymerization (ROP) of cyclic esters has been known for a long time, the customary use of organometallic compounds for driving the process has detrimental effects on the performance of the final polymers. Purposely, another pathway to prepare polyesters has been developed twenty years ago and consists in the metal-free ROP technique, also called organocatalysis. Many organic compounds were studied for the synthesis of poly(lactide) (PLA) or poly(ε-caprolactone) (PCL) such as 4-dimethylaminopyridine (DMAP),14–19 phosphines,20 Brönsted acids,21–26 thiourea-amine,27–31 carbenes32–35 and phosphazenes.36–40 These organocatalysts have enabled the preparation of well-defined functional polymeric materials with predictable molecular weights and narrow dispersity values. Since most of those polymerizations were carried out in organic solution, they do not totally fit with a clean chemistry process. Indeed, these processes utilize (preferably renewable) raw materials, reduce waste and avoid the use of toxic and/or hazardous reagents and solvents in the manufacture and application of chemical products. Upon consideration it becomes then quite clear that the association of a metal-free with a solvent-free process represents the goal to achieve as already exemplified for simple organic and polymerization reactions.41,42 Some studies already present results obtained from metal-free polymerizations of cyclic esters in bulk.15,17,20,24–26,39,43–48 Nevertheless, some organocatalysts used in bulk can cause degradation of polyesters,34 transesterification side reactions or co-initiation. Moreover, polymerizations in bulk require high temperatures, implying the risk for organic structures to thermally degrade.17,20,49
In this work, we describe a process for the synthesis of amphiphilic block copolymers composed of a polyester hydrophobic part: PCL and/or poly((L or D,L)lactide) (P((L or D,L)LA)) and a hydrophilic chain based on poly(ethylene oxide) (PEO). In order to fulfil clean chemistry requirements and control the polymerization of each block, we first studied the homopolymerization in bulk of ε-CL and (L or D,L)LA with organocatalysts already applied in solution polymerizations. Then, we investigated the synthesis of PEO-b-PCL and PEO-b-P((L or D,L)LA) amphiphilic diblock copolymers. This was supplemented by analyzing the impact of the PEO macroinitiator on the control of the polymerization of ε-CL with a molecular modeling approach. Calculated distances between the initiator and the catalyst had allowed to establish a relationship between the dispersity of the polymer and the initiator chain length.
Afterwards, a one-pot two-step process for the syntheses of amphiphilic triblock copolymers PEO-b-PCL-b-P((L or D,L)LA) involving a set of active bases and inactive salts was developed. Finally, formations of micellar structures in water from these amphiphilic block copolymers were compared to the one obtained from statistical PEO-b-P(CL-co-(L or D,L)LA) copolymers.
With respect to previous studies in the literature, this is the first report on the controlled synthesis of high molecular weight amphiphilic triblock copolymers composed of ε-CL and (L or D,L)LA and obtained from an organocatalyst in bulk, i.e. without organic solvent.
Experimental section
Materials
1-Pyrenemethanol, trifluoroacetic acid (TFA), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), 4-dimethylaminopyridine (DMAP), triphenylphosphine, molecular iodine, phenol, 4-chlorophenol, methyl tosylate, adenine, N-benzyl-N-methylpyrrolidinium chloride, 1,1,3,3-tetramethylguanidine, 1,3-diphenylguanidine, dicyclohexylcarbodiimide (DCC), fumaric acid, 2-tert-butyl-1,1,3,3-tetramethylguanidine, tert-butylimino-tris(dimethylamino)phosphorane (P1-t-Bu), 2,6-di-tert-butylpyridine, 4-methoxypyridine, picolinic acid, 2-pyridine sulfonic acid, 3-phenylpyridine, isoquinoline, benzoic acid, lauric acid, phenylphosphonic acid, isobutyric acid and trifluoromethanesulfonic acid were purchased from Aldrich and dried in appropriate conditions. L-LA and D,L-LA (Galactic, Belgium) were recrystallized from dried toluene and stored in a glove box. α-Methyl, ω-hydroxyl poly(ethylene oxide) (PEO) (Fluka, Mw ∼ 5000 g mol−1) was dried by three azeotropic distillations of toluene, then dried at 60 °C in vacuo overnight and stored in a glove box. ε-Caprolactone (ε-CL) was distilled in the presence of CaH2 under reduced pressure at 90 °C and stored in a glove box.Analytical techniques
1H NMR spectra were recorded using a Bruker AMX-500 apparatus at r.t. in CDCl3 (4 mg/0.6 mL). 13C NMR spectra were recorded using a Bruker AMX-300 apparatus at r.t. in CDCl3 (50 mg/0.6 mL). MALDI mass spectra were recorded using a Waters QToF Premier mass spectrometer equipped with a nitrogen laser, operating at 337 nm with a maximum output of 500 J m−2 delivered to the sample in 4 ns pulses at 20 Hz repeating rate. Time-of-flight mass analyses were performed in the reflectron mode at a resolution of about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The matrix-trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) was prepared as 10 mg mL−1 solution in acetone. The matrix solutions (1 μL) were applied to a stainless steel target and air dried. Polymer samples were dissolved in dichloromethane to obtain 1 mg mL−1 solutions. 1 μL aliquots of these solutions were applied onto the target area already bearing the matrix crystals, and then air dried. Finally, 1 μL of a solution of NaI (2 mg mL−1 in acetonitrile:water (1:1)) was applied onto the target plate. For the recording of the single-stage MALDI-MS spectra, the quadrupole (rf-only mode) was set to pass ions from m/z 1000 to 10000 and all ions were transmitted into the pusher region of the time-of-flight analyzer where they were mass analyzed with 1 s integration time. Data were acquired in continuum mode until acceptable averaged data were obtained. Number-average molecular weights and molecular weight distributions (Đ = Mw/Mn) values of polymers determined by Size Exclusion Chromatography (SEC) were based on experiments conducted in chloroform at 35 °C or in THF at 30 °C at a flow rate of 1 mL min−1, using an isocratic pump (VE 1122, Viscotek) a set of two PLgel 5 mm MIXED-C ultra high efficiency column and a Shodex SE 61 differential refractive index detector. A volume of 100 μL of sample solution in chloroform or THF (concentration 0.3% w/v) was injected. Polystyrene standards (Polymer Laboratories) with narrow molecular weight distributions were used to generate a calibration curve. Thermogravimetric analyses (TGA) were carried out on a TA Instruments Q500 with a heating rate of 10 °C min−1 under a flow of nitrogen, using Pt crucibles and 10 mg of sample for each analysis.
000. The matrix-trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) was prepared as 10 mg mL−1 solution in acetone. The matrix solutions (1 μL) were applied to a stainless steel target and air dried. Polymer samples were dissolved in dichloromethane to obtain 1 mg mL−1 solutions. 1 μL aliquots of these solutions were applied onto the target area already bearing the matrix crystals, and then air dried. Finally, 1 μL of a solution of NaI (2 mg mL−1 in acetonitrile:water (1:1)) was applied onto the target plate. For the recording of the single-stage MALDI-MS spectra, the quadrupole (rf-only mode) was set to pass ions from m/z 1000 to 10000 and all ions were transmitted into the pusher region of the time-of-flight analyzer where they were mass analyzed with 1 s integration time. Data were acquired in continuum mode until acceptable averaged data were obtained. Number-average molecular weights and molecular weight distributions (Đ = Mw/Mn) values of polymers determined by Size Exclusion Chromatography (SEC) were based on experiments conducted in chloroform at 35 °C or in THF at 30 °C at a flow rate of 1 mL min−1, using an isocratic pump (VE 1122, Viscotek) a set of two PLgel 5 mm MIXED-C ultra high efficiency column and a Shodex SE 61 differential refractive index detector. A volume of 100 μL of sample solution in chloroform or THF (concentration 0.3% w/v) was injected. Polystyrene standards (Polymer Laboratories) with narrow molecular weight distributions were used to generate a calibration curve. Thermogravimetric analyses (TGA) were carried out on a TA Instruments Q500 with a heating rate of 10 °C min−1 under a flow of nitrogen, using Pt crucibles and 10 mg of sample for each analysis.
Dynamic Light Scattering (DLS) measurements were performed on a Malvern CGS-3 equipped with He–Ne laser (λ = 633 nm). Measurements were carried at scattering angles of 60, 90 and 135° (cell diameter: 10 mm) and the temperature was controlled at 25 °C. The experimental autocorrelation function was analyzed using the CONTIN and cumulant methods. The method of the cumulants was generally used to analyze DLS results, while size distribution histograms were obtained by the CONTIN method. Via the cumulant method, the polydispersity index (PDI) of the particles was estimated from the ratio μ2/Γ12 in which Γ1, the relaxation frequency, and μ2 represent the first and second cumulant, respectively. The apparent hydrodynamic radius (RH) was determined from either the cumulant method or the CONTIN algorithm using the Stokes–Einstein equation:
| RH = kBT/(6πηD) |
Methodology for the modeling study
Molecular mechanics (MM) and molecular dynamics (MD) simulations have been performed with the Materials Studio 6.0 package.50 For all calculations, the Dreiding force field51 was used with the atomic charges assigned on the basis of the PCFF force-field.52 A cut-off of 12.5 Å has been set for the description of the non-bonded interactions.Ring-opening polymerization of cyclic esters
1H NMR (CDCl3) δ (ppm): 8.4–8.0 (m, aromatic protons of pyrene), 5.8 (s, pyrene–CH2–O), 4.1–3.9 (t, –CH2–O–CO), 3.65 (t, –CH2–OH), 2.3–2.2 (d, –CO–CH2–), 1.7–1.5 (m, CH2–CH2–CH2–CH2–O–), 1.5–1.3 (m, –CH2–CH2–CH2–CH2–O–).
Mn,NMR = 11500 g mol−1, Mn,SEC = 13400 g mol−1, Đ = 1.8, yield = 71%.
1H NMR (CDCl3) δ (ppm): 8.4–8.0 (m, aromatic protons of pyrene), 5.9 (dd, pyrene–CH2–O), 5.3–5.1 (m, –O–CHCH3–CO–), 4.35 (q, –CHCH3–OH), 1.6–1.5 (d, –O–CHCH3–CO–).
Mn,NMR = 8700 g mol−1, Mn,SEC = 12000 g mol−1, Đ = 1.20, yield = 46%.
1H NMR (CDCl3) δ (ppm): 4.1–3.9 (t, –CH2–O–CO PCL), 3.65 (t, –CH2–OH PCL), 3.6 (t, –O–CH2–CH2–O–PEO), 3.4 (s, –CH2–O–CH3 PEO), 2.3–2.2 (d, –CO–CH2–PCL), 1.7–1.5 (m, CH2–CH2–CH2–CH2–O–PCL), 1.5–1.3 (m, –CH2–CH2–CH2–CH2–O–PCL).
Mn,NMR (PCL) = 3100 g mol−1, Mn,SEC = 13700 g mol−1, Đ = 1.2, yield = 68%.
1H NMR (CDCl3) δ (ppm): 5.3–5.1 (m, –O–CHCH3–CO–PLA), 4.35 (q, –CHCH3–OH PLA), 3.6 (t, –O–CH2–CH2–O–PEO), 3.4 (s, –CH2–O–CH3 PEO) 1.6–1.5 (d, –O–CHCH3–CO–PLA).
Mn,NMR,PLA = 4400 g mol−1, Mn,SEC = 12500 g mol−1, Đ = 1.1, yield = 35%.
1H NMR (CDCl3) δ (ppm): 8.4–8.0 (m, aromatic protons of pyrene), 5.8 (s, pyrene–CH2–O), 5.3–5.1 (m, –O–CHCH3–CO–PLA), 4.1–3.9 (t, –CH2–O–CO PCL), 4.35 (q, –CHCH3–OH PLA), 2.3–2.2 (d, –CO–CH2–PCL), 1.7–1.5 (m, CH2–CH2–CH2–CH2–O–PCL), 1.6–1.5 (d, –O–CHCH3–CO–PLA), 1.5–1.3 (m, –CH2–CH2–CH2–CH2–O–PCL).
Mn,NMR,PLA = 3800 g mol−1, Mn,NMR,PCL = 9600 g mol−1, Mn,SEC = 23700 g mol−1, Đ = 1.5, yield = 49%.
1H NMR (CDCl3) δ (ppm): 5.3–5.1 (m, –O–CHCH3–CO–PLA), 4.1–3.9 (t, –CH2–O–CO PCL), 4.35 (q, –CHCH3–OH PLA), 3.6 (t, –O–CH2–CH2–O–PEO), 3.4 (s, –CH2–O–CH3 PEO), 2.3–2.2 (d, –CO–CH2–PCL), 1.7–1.5 (m, CH2–CH2–CH2–CH2–O– PCL), 1.6–1.5 (d, –O–CHCH3–CO–PLA), 1.5–1.3 (m, –CH2–CH2–CH2–CH2–O–PCL).
Mn,NMR,PLA = 6100 g mol−1, Mn,NMR,PCL = 3500 g mol−1, Mn,SEC = 18500 g mol−1, Đ = 1.1, yield = 58%.
1H NMR (CDCl3) δ (ppm): 5.3–5.1 (m, –O–CHCH3–CO–PLA), 4.1–3.9 (t, –CH2–O–CO PCL), 4.35 (q, –CHCH3–OH PLA), 3.6 (t, –O–CH2–CH2–O–PEO), 3.4 (s, –CH2–O–CH3 PEO), 2.3–2.2 (d, –CO–CH2–PCL), 1.7–1.5 (m, CH2–CH2–CH2–CH2–O–PCL), 1.6–1.5 (d, –O–CHCH3–CO–PLA), 1.5–1.3 (m, –CH2–CH2–CH2–CH2–O–PCL).
Mn,NMR = 15900 g mol−1, Mn,SEC = 20600 g mol−1, Đ = 2.0, yield = 82.2%.
Results and discussion
Synthesis of PCL and P((L and D,L)LA) homopolymers
ROP of L-lactide (L-LA), D,L-lactide (D,L-LA) and ε-caprolactone (ε-CL) was studied with different organocatalysts in bulk. The temperature of polymerizations was adapted regarding the type of monomer used. All polymerizations were run for 30 minutes with 1-pyrenemethanol (PyOH) as the initiator. It has to be noted that the molar masses determined by size exclusion chromatography (SEC) are given without any Mark-Houwink correction.Studies of bulk polymerization of ε-CL with organocatalysts (guanidine: TBD;43 Brønsted acids: triflic, trichloroacetic, maleic, fumaric acids,24 lactic, tartaric, hexanoic, propionic and citric acids;25,26 phosphazene: 2-tert-butylimino-2-diethylamino-1,3-dimethdimethylperhydro-1,3,2-diazaphosphorine (BEMP)39) reveal the difficulty of producing PCL with a high molecular weight in a short reaction time. Herein, three catalysts were used to polymerize ε-CL: molecular iodine (I2), trifluoroacetic acid (TFA) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). The polymerizations were performed at a temperature above the melting temperature of PCL, i.e. 60 °C and for a [ε-CL]0/[PyOH]0 ratio of 100.
All results are summarized in Table 1. As inspired by the state-of-the-art, molecular iodine53 and TFA24 were used as catalysts. In both cases, polymerizations were carried out at 70 °C with an excess of catalyst as compared to the initial feed of initiator (entries 1&2, Table 1). Both resulted in oligomer formation with molecular weights lower than 1600 g mol−1. As compared to the first two catalysts, a very interesting result was obtained with TBD. In that particular case, PCL was synthesized from 0.25 equivalent of TBD with respect to the PyOH content and at 90 °C (entry 3). SEC analysis gave an experimental molar mass value of 13400 g mol−1 whereas the dispersity index (Đ) was 1.8.
To attest the control over the ε-CL ROP in terms of molar mass and end-groups fidelity, the PCL obtained with TBD was characterized by 1H NMR (Fig. 1) spectroscopy and matrix-assisted laser desorption ionization time-of-flight spectrometry (MALDI-ToF) (Fig. S1†). Both techniques attest for the fidelity of the end-groups and allow determining an experimental molar mass of 8000 g mol−1 in perfect agreement with the theoretical one.
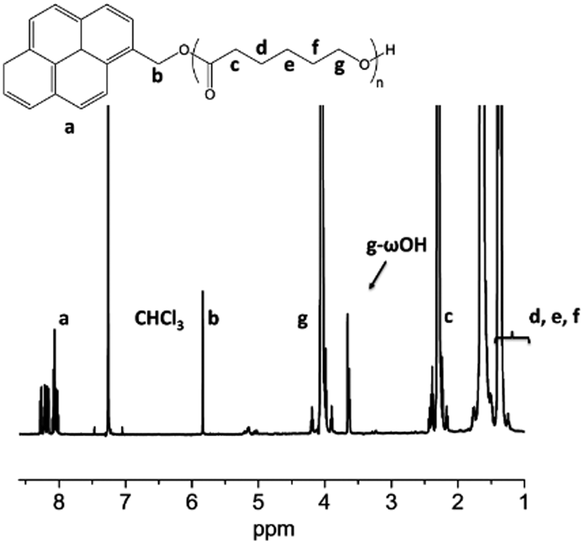 | ||
| Fig. 1 1H NMR spectrum (CDCl3, 21 °C, 500 MHz) of PCL obtained in the presence of TBD and as initiated by 1-pyrenemethanol. | ||
A second screening was performed for the polymerization of L-LA in bulk with 9 different molecules or association of molecules (Table 2). The difficulty associated to the polymerization of L-LA lies in avoiding the racemization of the L-LA unit or the presence of side transesterification reactions. For each catalyst used, the stereospecificity and the absence of side transesterification reactions have been checked by differential scanning calorimetry (DSC) and MALDI-ToF experiments, respectively. All polymerizations were carried out at a temperature of 140 °C above the melting temperature of the L-lactide isomer, i.e., 98 °C and for a [L-LA]0/[PyOH]0 ratio of 100.
| Entry | Catalyst | [Catalyst]0/[PyOH]0 | Mn,SECa (g mol−1) | Đa | Yield (%) |
|---|---|---|---|---|---|
| a SEC in CHCl3 PS standards, 1 mL min−1, T = 30 °C. | |||||
| 1 | 1,1,3,3-Tetramethyl-guanidine | 2/1 | 12200 |
1.4 | 35 |
| 2 | 1,3-Diphenyl-guanidine | 2/1 | 4500 | 1.1 | 20 |
| 3 | 1,2,3-Triphenyl-guanidine | 2/1 | 5800 | 1.1 | 15 |
| 4 | 2-tert-Butyl-1,1,3,3-tetramethyl-guanidine | 2/1 | 3400 | 2.9 | 15 |
| 5 | P1-t-Bu | 2/1 | 7300 | 1.5 | 60 |
| 6 | DMAP | 2/1 | 8600 | 1.4 | 45 |
| 7 | DMAP/DCC | 2/2/1 | 4900 | 1.3 | 39 |
| 8 | DMAP/fumaric acid | 2/0.4/1 | 7300 | 1.3 | 40 |
| 9 | DMAP/TFA | 2/0.4/1 | 12000 |
1.2 | 46 |
All results are summarized in Table 2. Acyclic substituted guanidines (1,1,3,3-tetramethylguanidine, 1,3-diphenylguanidine, 1,2,3-triphenylguanidine and 2-tert-butyl-1,1,3,3-tetramethylguanidine) have first been examined. Polymerizations were carried out with 2 equivalents of the catalyst with respect to the initiator. In the cases of 1,3-diphenylguanidine (entry 2), 1,2,3-triphenylguanidine (entry 3) and 2-tert-butyl-1,1,3,3-tetramethylguanidine (entry 4), the polymers obtained have a molecular weight of max. 5800 g mol−1 with very low yields (between 15 and 20%). The most interesting result was reached with 1,1,3,3-tetramethylguanidine (entry 1) as the catalyst: the polymer has a molecular weight up to 12000 g mol−1 and a Đ of 1.4. If tert-butylimino-tris(dimethylamino)phosphorane (P1-t-Bu) also demonstrated its ability to catalyze the L-LA ROP in bulk (Mn,SEC = 7300 g mol−1, Đ ∼ 1.4), the results obtained from the 4-dimethylaminopyridine (DMAP)-based molecules were even more stimulating (entries 6 to 9). In that series, pristine DMAP and DMAP associated with other molecules such as DCC, fumaric acid and TFA were studied. In all cases, 2 equivalents of DMAP were used relative to the initiator. For the pristine DMAP (entry 6), the PLA synthesized exhibits a molecular weight of 8600 g mol−1 and a Đ of 1.4. However, the MALDI-ToF analysis revealed the presence of transesterification side reactions and side initiation (Fig. S2†). Similar results were obtained with 2 equivalents of DMAP and DCC as catalytic system moreover the molecular weight was too low: 4900 g mol−1 (Fig. S3†). Finally, DMAP associated with fumaric acid and TFA, both characterized by different pKa values (3.03 & 4.44 for fumaric acid and 0.30 for TFA), were studied. In both cases, 2 equivalents of DMAP and 0.4 equivalent of acid were used relative to the initiator. In the case of DMAP/fumaric acid as the catalyst, the polymer exhibits a molecular weight of 7300 g mol−1 and a Đ of 1.3. When TFA was used instead of fumaric acid, a better result was obtained, i.e. a molecular weight of 12000 g mol−1 and a Đ of 1.2. Besides, a molecular weight of 8000 g mol−1 is determined by 1H NMR, in close agreement with the theoretical one (Mn,th = 6700 g mol−1). A very low amount of epimerization is observed by the appearance of a melting transition at 146 °C, as determined by DSC analysis.
MALDI-ToF mass spectrometry was used to confirm the control of the L-LA polymerization with DMAP/TFA as catalyst. The MS spectrum (Fig. 2) shows one main population. The main series is centered at m/z = 7316.3 Da and the interval between two consecutive peaks of the family corresponds to the molecular weight of the lactide (144.13 Da) unit. However, we can notice the presence of small peaks every 72.06 Da corresponding to transesterified PLA chains. However the relative intensity of these peaks is very low, indicating a limited extent of transesterification side reactions. Otherwise the main series corresponds exactly to the expected structure with a sodium cation, i.e., a PLA carrying a pyrene head-group, coming from the initiator and a hydrogen atom at the ω-end-group.
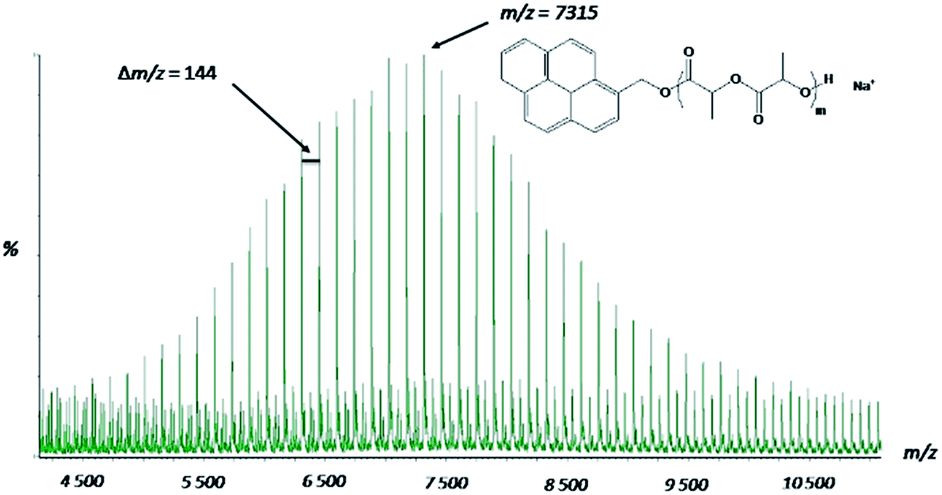 | ||
| Fig. 2 MALDI-Tof spectrum of the PLA obtained in presence of DMAP/TFA (Mn = 7315 g mol−1) (matrix: DCTB and salt: NaI). | ||
In the case of D,L-LA monomer, the polymerization was carried out with the same experimental conditions as for L-LA, i.e. 140 °C, 30 minutes, DMAP/TFA (2/0.4 eq. with respect to the initiator) as the catalyst, and PyOH as the initiator. The polymer exhibits a molar mass of 11500 g mol−1 and a Đ of 1.4 by SEC.
Synthesis of PEO-b-PCL and PEO-b-P((L and D,L)LA) amphiphilic diblock copolymers
The synthesis of the poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) amphiphilic diblock copolymer was first investigated by applying the most efficient ROP conditions obtained for the homopolymerization of ε-CL (Scheme 1). For that purpose, the polymerization was catalyzed by 0.25 equivalent of TBD and initiated by a hydrophilic PEO monomethyl ether macroinitiator with a molar mass of 5000 g mol−1. The reaction was carried out at 90 °C for 30 minutes and with a [ε-CL]0/[PEO]0 ratio of 100. | ||
| Scheme 1 Synthesis of PEO-b-PCL amphiphilic diblock copolymer. | ||
By SEC analysis, the PEO-b-PCL copolymer shows a chromatogram characterized by molecular weight of 13700 g mol−1 (Đ of 1.2) clearly shifted from the PEO macroinitiator SEC trace (Fig. 3). Interestingly, if initiating the ε-CL from a PEO-OH helps to significantly reduce the associated dispersity value (from 1.8 with PyOH to 1.2 in this case), it has also a tremendous impact on the global kinetics of the process. Indeed, by comparing the relative intensities of both PCL and PEO blocks in the 1H NMR spectrum, a PCL molar mass of 3100 g mol−1 is only achieved. As compared to its homopolymerization from PyOH, the ε-CL ROP propagation has been reduced by a factor of 3.
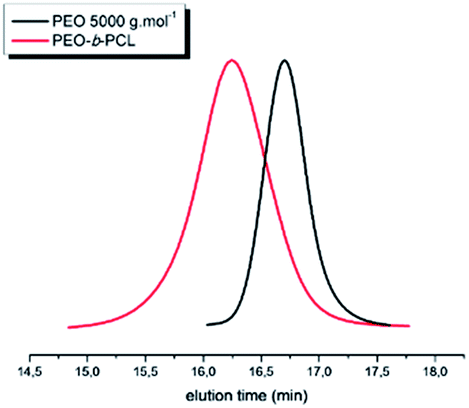 | ||
| Fig. 3 SEC profiles of the PEO-b-PCL diblock copolymer (red line) compared to the PEO macroinitiator (black line) (SEC in THF (+2% TEA), PS standards, 1 mL min−1, T = 35 °C). | ||
In a second stage, the synthesis of PEO-b-P((L or D,L)LA) amphiphilic diblock copolymers was also studied (Scheme 2). The catalyst giving the best result for L-LA homopolymerization was used, i.e. 2 equivalents of DMAP associated to 0.4 equivalent of TFA (with respect to the initiator feed). The PEO macroinitiator was the same as that used for ε-CL ROP and the polymerizations were performed at 140 °C for [L-LA]0/[PEO]0 of 100.
 | ||
| Scheme 2 Synthesis of PEO-b-P((L or D,L)LA) amphiphilic diblock copolymers. | ||
Two PEO-b-P((L and D,L)LA) copolymers were synthesized and regardless of the monomer, similar results were achieved (Table 3). The relative molecular weights determined by SEC were close to 12500 g mol−1 with Đ values around 1.1 (Fig. 4). In perfect agreement with the theoretical molecular masses (Mn,th P(D,L-LA) = 5000 g mol−1 and Mn,th P(L-LA) = 4000 g mol−1), experimental molar masses of 4500 and 4900 g mol−1 were determined by 1H NMR for both P(L-LA) and P(D,L-LA) segments, respectively. All these results indicate a good control for the polymerizations of L- and D,L-LA. Similarly to the ε-CL polymerization, the initiation from a PEO macroinitiator drastically reduces the global kinetics of the process (even at 140 °C) while maintaining very narrow dispersity values.
 | ||
| Fig. 4 SEC profiles of PEO-b-P(D,L-LA) (blue line, on the left) and PEO-b-P(L-LA) (blue line, on the right) diblock copolymers compared to the macroinitiator PEO (black line) (SEC in THF (+2% TEA), PS standards, 1 mL min−1, T = 35 °C). | ||
Impact of PEO on the molecular weight dispersity of the copolymers
A molecular modeling study was further performed to rationalize the decrease in dispersity when going from the PyOH initiator to a PEO-OH initiator. This study was carried out on the polymerization of ε-CL because of the most pronounced difference in the results. The key idea of the simulations is to compare the time that the three components (monomer/initiator/catalyst) spend in close vicinity to allow for the polymerization process. Two different systems were considered according to the optimized ratios of the chemical synthesis. To simulate the polymerization process of PCL with the PyOH initiator in the presence of the TBD catalyst, a unit cell containing 400 ε-CL monomers, four PyOH initiator molecules and one TBD molecule has been built (system 1) by randomly distributing all the molecules in the unit cell. The four PyOH have been replaced by four PEO-OH chains made of 110 monomer units (Mn ∼ 5000 g mol−1) for the second system (system 2). In all simulations, the unit cell has been replicated using periodic boundary conditions to generate an infinite system. The cell parameters of the unit cells have been chosen to ensure a density of the system of 1 g cm−3. In order to reduce the computational efforts, TBD has been positioned in close contact with PyOH or the PEO chain at the starting point of the simulations. In practice, the distance between the nitrogen atom #1 (Table 4) of TBD and the hydrogen atom of the hydroxyl group of the initiator has been fixed at 2 Å. For each Molecular Mechanics (MM) geometry-optimized system, ten 50 ps-long Molecular Dynamics (MD) runs have been performed within the NVT ensemble (constant Number of particles, Volume and Temperature; T = 413 K). The constraint on the distance between the nitrogen atom of TBD and the hydrogen atom of the initiator has then been removed. Finally, the MM-optimized geometry of the last frame of each MD run has been used as the starting point of 3 ns-long MD simulations during which snapshots of the systems have been saved every 10 ps.
|
|||||||
|---|---|---|---|---|---|---|---|
| System | Number of frames during the molecular dynamic runs | Average | Standard deviation | ||||
| 1 | 287 | 22 | 159 | 301 | 7 | 109 | 107 |
| 42 | 78 | 47 | 109 | 37 | |||
| 2 | 2 | 167 | 2 | 22 | 65 | 64 | 57 |
| 95 | 96 | 14 | 132 | 41 | |||

In order to explain the decrease in dispersity when going from the PyOH initiator (system 1) to the PEO-OH initiator (system 2), our simulations focus on the time the three reactants can spend together to initiate the polymerization reaction and by extension to propagate it. To probe this parameter, the distance between the nitrogen atom of TBD and the hydrogen atom of the hydroxyl group of the initiator (dN–H) (Table 4) has been measured during ten independent MD runs (Fig. S4†). In practice, for each of the 301 snapshots recorded during those 3 ns MD runs, the average number of frames for which dN–H is smaller than 5 Å has been reported in Table 4. A larger number implies that the catalyst is staying a longer time close to the initiator, therefore enabling the addition of a larger number of monomer units. Interestingly, Table 4 points out that the number of snapshots is larger for PyOH (109 snapshots versus 64 snapshots for system 1 versus system 2, respectively). More importantly, the shape of the distribution is significantly different: while TBD and PEO-OH always separate during the first half of the MD runs, the catalyst molecule can stay close to PyOH for the whole duration of the simulation. This translates into a larger distribution of snapshots with a distance shorter than 5 Å for PyOH, as reflected by the standard deviation of the two distributions (σ = 107 for system 1 versus 57 for system 2).
The width of these distributions thus suggests that a larger dispersity of polymer chain lengths is expected with the PyOH initiator, which is consistent with the decrease in dispersity when using PEO-OH as the initiator. The fact that TBD is staying longer in close vicinity to PyOH can be understood by the rigid and planar structure of PyOH, which does not allow fast diffusion within the medium, compared to the terminal monomer units of the flexible PEO-OH chain. Note that the close vicinity of TBD and the growing PyOH-based chains should be preserved during the propagation reaction due to the limited diffusion of pyrene. Finally, the different average values in Table 4 also explain the lower rate of the polymerization of ε-CL with PEO-OH as the initiator, compared to PyOH.
Synthesis of PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers
In the third part of the work, the syntheses of amphiphilic triblock copolymers PEO-b-PCL-b-P((L and D,L)LA) were investigated following a one-pot two-step process: bulk polymerization of ε-CL followed by the polymerization of L- or D,L-LA. Since bases like TBD have already been proved to poison PLA through quick transesterification side reactions in bulk leading to racemization and degradation of PLA54 it also represents the only metal-free catalyst able to produce high molecular weight PCL in less than one hour in bulk. Our first interest is to rely on the possibility to quench the catalytic activity of the guanidinium base by formation of a salt totally inert and spectator regarding the DMAP activation of the LA comonomer.In order to find the quenching agent of the guanidinium base, degradation studies of PLA in the presence of different salts of TBD were performed. In a first step, salts of TBD were formed with several Brønsted acids showing different pKa: phenylphosphonic acid (pKa = 1.3), fumaric acid (pKa = 3 and 4.4), benzoic acid (pKa = 4.2), isobutyric acid (pKa = 4.9) and lauric acid (pKa = 7.5). These salts were then placed in the presence of pure P(L-LA) up to 10% in weight. These mixtures and pristine P(L-LA) were then studied by thermogravimetric analysis (TGA) (Fig. 5 and Table 5).
 | ||
| Fig. 5 Degradation curves of pristine P(L-LA) and mixtures composed of P(L-LA) and salts of TBD/acids (10 °C min−1, N2). | ||
| Acid | Tdegradation (°C) | pKa |
|---|---|---|
| Phenylphosphonic acid | 170 | 1.3 |
| Fumaric acid | 190 | 3.0 and 4.4 |
| Benzoic acid | 150 | 4.2 |
| Isobutyric acid | 150 | 4.9 |
| Lauric acid | 150 | 7.5 |
The study showed that the degradation of pristine P(L-LA) (Mn = 10000 g mol−1) was initiated around 350 °C whereas P(L-LA) in the presence of TBD starts to degrade at 150 °C. In the presence of TBD salts, P(L-LA) degradation temperatures ranging from 150 to 190 °C according to the acid are observed. The highest degradation temperature was achieved with the salt of TBD and fumaric acid: P(L-LA) starts to degrade at 190 °C, which is much higher than the temperature at which the polymerization of lactide occurred (140 °C).
Prior to the synthesis of both PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers, PCL-b-P((L and D,L)LA) diblock copolymers were first studied (Scheme 3). For each block, a [M]0/[I]0 ratio of 100 was chosen. The copolymers were obtained in two steps: first polymerization of ε-CL with 0.25 equivalent of TBD as the catalyst and 1-pyrenemethanol as the initiator. After 30 minutes at 90 °C, TBD was quenched by addition of 1.05 equivalents of fumaric acid compared to TBD. Finally, after 10 minutes at 90 °C, the polymerization of (L- or D,L)LA is carried out at 140 °C for 30 minutes, with 2 equivalents of DMAP and 0.4 equivalent of TFA as the catalyst.
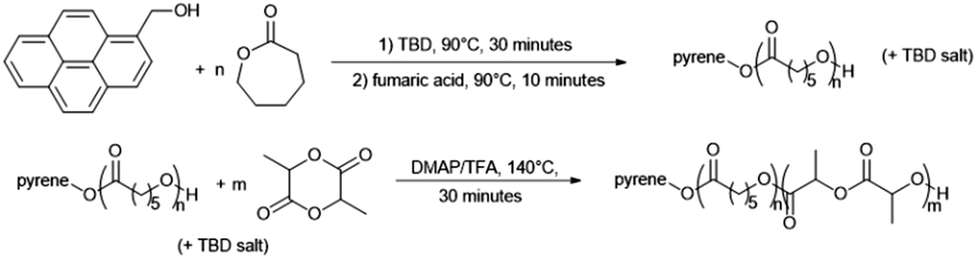 | ||
| Scheme 3 Synthesis of PCL-b-P(L-LA) diblock copolymer. | ||
Table 6 shows the molar masses of PCL and PCL-b-P(L-LA) determined by SEC in THF. An increase of molar masses is observed between PCL (Mn = 14600 g mol−1) and PCL-b-P(L-LA) (Mn = 23700 g mol−1). Moreover Đ decreases during the generation of the second block (Đ = 2.0 for PCL and Đ = 1.5 for the diblock copolymer), suggesting a low amount of transesterification. As attested by 13C NMR analysis (Fig. 6), the presence of a pure diblock topology is proven by the presence of only two signals in the carbonyl region at 169.6 and 173.6 ppm and characterizing both PLA and PCL segments without any statistical co-sequence.55
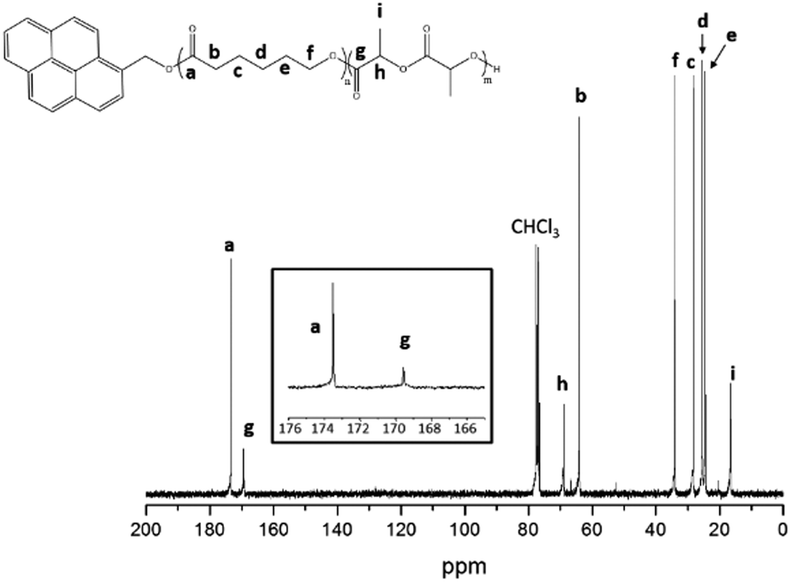 | ||
| Fig. 6 13C NMR spectrum (CDCl3, 21 °C, 300 MHz) of PCL-b-P(L-LA) diblock copolymers totally free from PCL and P(L-LA) homopolymers. | ||
Another proof for the formation of exclusive PCL-b-P(L-LA) diblock copolymer was given by 1H NMR spectroscopy (Fig. 7). The spectrum of the PCL precursor shows signals of α and ω end-groups at 5.80 and 3.65 ppm due to α-pyrene–CH2–O and ω–CH2–OH groups, respectively. The spectrum of the diblock copolymer points to the almost complete disappearance of the ω–CH2–OH triplet at 3.65 ppm in favor of a new quadruplet appearing at 4.35 ppm, which is the signature of the PLA ω-CH–OH end-group only. The reinitiation ratio was calculated and estimated above 99%. Furthermore no polymerization of L-LA occurred in presence of TBD/fumaric acid salt. All these data confirm that this salt is then inert during the L-LA polymerization (at least for the temperature studied here).
 | ||
| Fig. 7 1H NMR spectra (CDCl3, 21 °C, 500 MHz) of PCL (on the left) and PCL-b-P(L-LA) (on the right). | ||
Then, PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers were synthesized. The same experimental conditions as employed for the PCL-b-P(L-LA) diblock copolymers were used for the synthesis of PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers (Scheme 4).
 | ||
| Scheme 4 Synthesis of PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers. | ||
PEO-b-PCL-b-P((L and D,L)LA) amphiphilic triblock copolymers were characterized by SEC. Both copolymers exhibit similar molecular weights of 18000 and 19000 g mol−1 with Đ each equal to 1.4 (Table 7). The shift of the SEC traces for both triblock copolymers to lower elution times in comparison with the PEO macroinitiator and the PEO-b-PCL diblock copolymers clearly indicated the effectiveness of block copolymerization of (L or D,L)LA (Fig. 8).
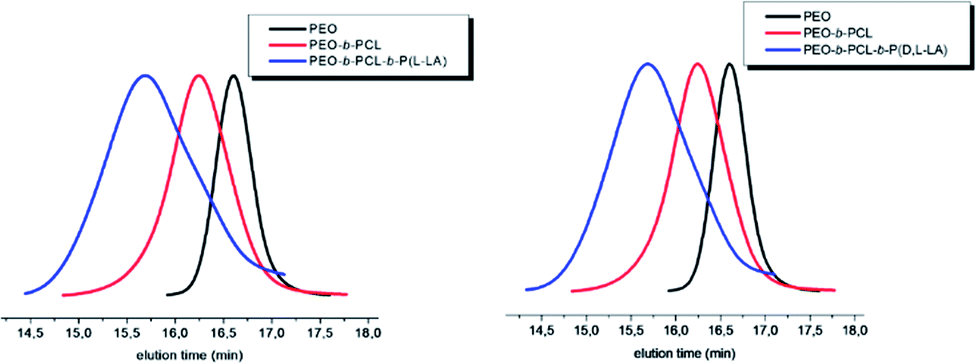 | ||
| Fig. 8 SEC profiles of PEO (black line), PEO-b-PCL diblock copolymers (red line), PEO-b-PCL-b-P(L-LA) (blue line on the right) and PEO-b-PCL-b-P(D,L-LA) (blue line on the left) triblock copolymers (SEC in THF (+2% TEA), PS standards, 1 mL min−1, T = 35 °C). | ||
Self-association of amphiphilic copolymers in water
The ability of block copolymers to self-assemble in selective solvents allows for the formation of various well-defined micellar nanostructures with tunable sizes and morphologies.56,57 In the present study, solutions of amphiphilic triblock copolymers (0.3 mg mL−1) were prepared according to Eisenberg's method.58 The non-selective solvent is acetone which is a fairly good solvent for both PCL, P((L or D,L)LA) and PEO blocks, dialyzed against Milli-Q water.56,57 Thus in water, we expected copolymers to form micelles composed of a polyester micellar core surrounded by a PEO corona. The final concentration of those micelles in water after the dialysis step was set to 0.1 wt%.Dynamic Light Scattering (DLS) experiments were performed on those aqueous solutions to determine the hydrodynamic radius (RH) of the micelles, and obtain information about their shape and their dispersity. For all the copolymers, the RH of the micelles ranged from 27 to 35 nm, whatever the method used (Table 8). Interestingly, the sizes of the micelles were very similar, regardless of the structure of the different amphiphilic copolymers, block or statistical (see ESI for experimental details†). Only a little difference can be observed depending on the molecular weight of the hydrophobic blocks PCL-b-P((L or D,L)LA) or P(CL-co-(L or D,L)LA). Micelles formed by the PEO-b-P(CL-co-D,L-LA) amphiphilic diblock copolymer displayed a RH close to 35 nm with a Mn,hydrophobic part = 10900 g mol−1 whereas the size of the micelles formed by the PEO-b-PCL-b-P(D,L-LA) amphiphilic triblock copolymer was 27.5 nm with a Mn,hydrophobic part = 5500 g mol−1. This result implies that the size of the micelles depends essentially on the molecular weight of the hydrophobic block, in agreement with the literature. Some measurements showed the appearance of small peaks due to very small objects (less than 10 nm). These peaks can be attributed to the presence of unimers. Similar RH values were obtained regardless of the angle of measurements (Table S2†), indicating the formation of spherical micelles.55 Finally, the results confirmed the formation of quite monodisperse micelles, showing a dispersity index around 0.03–0.1 (Fig. S5†).
| Copolymer | CONTIN | Cumulant | |
|---|---|---|---|
| RH,app | RH,app | Dispersity | |
| PEO-b-P(CL-co-L-LA) | 31.5 | 31.5 | 0.04 |
| PEO-b-P(CL-co-D,L-LA) | 35.5 | 35 | 0.10 |
| PEO-b-PCL-b-P(L-LA) | 32 | 32 | 0.07 |
| PEO-b-PCL-b-P(D,L-LA) | 27.5 | 27.5 | 0.04 |
Conclusion
This work reports the controlled synthesis of di- and triblock amphiphilic copolymers by applying a process that avoids the use of organic solvent and any metal catalyst. Whatever the desired topology, the amphiphilic nature of the copolymers is obtained by association of an hydrophilic PEO segment to hydrophobic PCL and/or P(L or D,L)LA blocks. As a first step, a global screening of different organic molecules and salts allowed to highlight that TBD and DMAP/TFA are fully appropriate to polymerize in bulk ε-CL and (L or D,L)LA monomers, respectively. Quite interestingly, and supported by simulation results, it comes out that using a PEO macroinitiator highly improves the control in term of final dispersities. Importantly, in order to access PEO-b-PCL-b-P((L and D,L)LA) triblock copolymers, a one-pot two-step process involving various metal-free active bases and inactive salts has been designed that result in negligible transesterification reactions. Finally, the self-organization of PEO-b-PCL-b-P((L or D,L)LA) or PEO-b-P(CL-co-(L or D,L)LA) amphiphilic triblock copolymers in water was studied. Well-defined spherical micelles were formed with a RH between 27 and 35 nm and a very low dispersity of 0.1.Acknowledgements
This work has been supported by the European Commission and Région Wallonne FEDER program (‘Revêtements fonctionnels’ project – Smartfilm), OPTI2MAT program of excellence, by the Interuniversity Attraction Pole Programme (P7/05) initiated by the Belgian Science Policy Office, the Région Wallonne EMBIOSE project and by FNRS-FRFC. O.C. is Research Associate and J.C. a Research Director of the F.R.S.-FNRS.Notes and references
- P. A. a. B. Lindman, Amphiphilic Block Copolymers: Self-Assembly and Applications, Elsevier, 2000 Search PubMed.
- B. Guillerm, V. Darcos, V. Lapinte, S. Monge, J. Coudane and J.-J. Robin, Chem. Commun., 2012, 48, 2879–2881 RSC.
- B. Guillerm, S. Monge, V. Lapinte and J.-J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1118–1128 CrossRef CAS.
- S. J. Holder, N. A. J. M. Sommerdijk, S. J. Williams, R. J. M. Nolte, R. C. Hiorns and R. G. Jones, Chem. Commun., 1998, 1445–1446 RSC.
- L. M. Bronstein, D. M. Chernyshov, G. I. Timofeeva, L. V. Dubrovina, P. M. Valetsky and A. R. Khokhlov, Langmuir, 1999, 15, 6195–6200 CrossRef CAS.
- R. T. Liggins and H. M. Burt, Adv. Drug Delivery Rev., 2002, 54, 191–202 CrossRef CAS PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- D. Quémener, G. Bonniol, T. N. T. Phan, D. Gigmes, D. Bertin and A. Deratani, Macromolecules, 2010, 43, 5060–5065 CrossRef.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS PubMed.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, W. Van Camp, O. Coulembier, P. Degée, R. Flammang, F. Du Prez and P. Dubois, React. Funct. Polym., 2008, 68, 990–1003 CrossRef CAS.
- S. C. Lee, Y. Chang, J.-S. Yoon, C. Kim, I. C. Kwon, Y.-H. Kim and S. Y. Jeong, Macromolecules, 1999, 32, 1847–1852 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Curr. Opin. Colloid Interface Sci., 2001, 6, 3–10 CrossRef CAS.
- F. Nederberg, E. F. Connor, T. Glausser and J. L. Hedrick, Chem. Commun., 2001, 2066–2067 RSC.
- T. Trimaille, M. Möller and R. Gurny, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4379–4391 CrossRef CAS.
- O. Coulembier and P. Dubois, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1672–1680 CrossRef CAS.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- C. Bonduelle, B. Martín-Vaca, F. P. Cossío and D. Bourissou, Chem. – Eur. J., 2008, 14, 5304–5312 CrossRef CAS PubMed.
- J. Kadota, D. e. Pavlović, J.-P. Desvergne, B. Bibal, F. d. r. Peruch and A. Deffieux, Macromolecules, 2010, 43, 8874–8879 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Möck, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844–851 CrossRef CAS.
- S. p. Gazeau-Bureau, D. Delcroix, B. Martín-Vaca, D. Bourissou, C. Navarro and S. p. Magnet, Macromolecules, 2008, 41, 3782–3784 CrossRef CAS.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316–4320 CrossRef CAS.
- P. V. Persson, J. Schröder, K. Wickholm, E. Hedenström and T. Iversen, Macromolecules, 2004, 37, 5889–5893 CrossRef CAS.
- F. Sanda, H. Sanada, Y. Shibasaki and T. Endo, Macromolecules, 2001, 35, 680–683 CrossRef.
- J. Casas, P. V. Persson, T. Iversen and A. Córdova, Adv. Synth. Catal., 2004, 346, 1087–1089 CrossRef CAS.
- P. V. Persson, J. Casas, T. Iversen and A. Córdova, Macromolecules, 2006, 39, 2819–2822 CrossRef CAS.
- R. Zhu, R. Wang, D. Zhang and C. Liu, Aust. J. Chem., 2009, 62, 157–164 CrossRef CAS.
- A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749–6754 CrossRef CAS PubMed.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- D. J. Coady, A. C. Engler, H. W. Horn, K. M. Bajjuri, K. Fukushima, G. O. Jones, A. Nelson, J. E. Rice and J. L. Hedrick, ACS Macro Lett., 2011, 1, 19–22 CrossRef.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS PubMed.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., 2007, 119, 2681–2684 CrossRef.
- N. E. Kamber, W. Jeong, S. Gonzalez, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2009, 42, 1634–1639 CrossRef CAS.
- O. Coulembier, L. Mespouille, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2006, 39, 4001–4008 CrossRef CAS.
- O. Coulembier, M. K. Kiesewetter, A. Masson, P. Dubois, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 4719–4721 CrossRef CAS PubMed.
- P. Brignou, M. Priebe Gil, O. Casagrande, J.-F. o. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS.
- M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem. – Eur. J., 2010, 16, 13805–13813 CrossRef CAS PubMed.
- L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS PubMed.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- J. De Winter, O. Coulembier, P. Gerbaux and P. Dubois, Macromolecules, 2010, 43, 10291–10296 CrossRef CAS.
- A. Bañón-Caballero, G. Guillena and C. Nájera, Helv. Chim. Acta, 2012, 95, 1831–1841 CrossRef.
- C. J. Hawker, J. L. Hedrick, E. Malmstrom, M. Trollsas, R. M. Waymouth and U. M. Stehling, Polym. Prepr., 1997, 38, 412–413 CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2007, 40, 3154–3158 CrossRef CAS.
- V. Katiyar and H. Nanavati, Polym. Chem., 2010, 1, 1491–1500 RSC.
- C. Wang, H. Li and X. Zhao, Biomaterials, 2004, 25, 5797–5801 CrossRef CAS PubMed.
- H. Li, C. Wang, J. Yue, X. Zhao and F. Bai, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3775–3781 CrossRef CAS.
- O. Coulembier, T. Josse, B. Guillerm, P. Gerbaux and P. Dubois, Chem. Commun., 2012, 48, 11695–11697 RSC.
- O. Coulembier, C. Delcourt and P. Dubois, Open Macromol. J., 2007, 1, 1–5 CrossRef CAS.
- Materials Studio Modelling v6.0.0, Accelrys Software Inc, San Diego, CA, 2011 Search PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- H. Sun, Macromolecules, 1995, 28, 701–712 CrossRef CAS.
- A. A. A. De Queiroz, É. J. França, G. A. Abraham and J. S. Román, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 714–722 CrossRef CAS.
- O. Coulembier, S. Moins, J.-M. Raquez, F. Meyer, L. Mespouille, E. Duquesne and P. Dubois, Polym. Degrad. Stab., 2011, 96, 739–744 CrossRef CAS.
- H. Qian, J. Bei and S. Wang, Polym. Degrad. Stab., 2000, 68, 423–429 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, J. Am. Chem. Soc., 2008, 130, 8714–8725 CrossRef CAS PubMed.
- J.-F. Gohy, in Block Copolymers II, ed. V. Abetz, Springer, Berlin Heidelberg, 2005, vol. 190, ch. 48, pp. 65–136 Search PubMed.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer characterisations, synthesis of statistical copolymer, modelization and physico-chemical properties. See DOI: 10.1039/c3ra47204c |
| This journal is © The Royal Society of Chemistry 2014 |