
Addition of carbon nucleophiles to hemiaminals promoted by a Lewis acidic polyoxotungstate†‡
Wen-Jing
Xuan
ab,
Candice
Botuha
ab,
Bernold
Hasenknopf
ab and
Serge
Thorimbert
*ab
aSorbonne Universités, UPMC Univ Paris 06, UMR 8232, Institut Parisien de Chimie Moléculaire, F-75005 Paris, France. E-mail: serge.thorimbert@upmc.fr
bCNRS, UMR 8232, IPCM, F-75005 Paris, France
First published on 10th September 2014
Abstract
A Lewis acidic hafnium(IV) ion incorporated in a polyoxotungstate (POM/Hf) was successfully employed as recoverable catalyst in the nucleophilic addition of carbon nucleophiles, such as silyl enol ethers, silyl ketene acetals, β-diketones and β-diketoesters, to unprotected hydroxy aminal at room temperature. The corresponding α-substituted heterocycles were obtained in good yields and the recovered POM catalyst could be reused up to three times without significant loss of activity.
In the synthesis of nitrogen-containing heterocycles, N-acyliminium ions have been widely used as efficient electrophiles.1 Most often these stabilized ionic intermediates are generated from precursors such as amino nitriles or N,O acetals in acidic medium in the presence of stoichiometric quantities of Brønsted acids or traditional Lewis acids, such as BF3·OEt2, TiCl4, SnCl4, InCl3, NbCl5 and Zn(OTf)2, which are environmentally harmful.2–4 More recently, catalytic versions have been reported that take advantage of the Lewis acidic properties of lanthanides triflates5 or N-trimethyl silyl-bis-(trifluoromethanesulfonyl)imide derivatives (HNTf2; Zn(NTf2)2)6 or Brønsted organocatalysts.7 Even if those conditions are efficient for synthetic purpose,8 the need of alkoxy- or acetoxy-aminals lengthen the overall process. Indeed, from the point of view of atom economy and environmental reasons, the direct use of unprotected hemiaminals, which are more easily available, would be better.9 Some results in this field, including those with activated benzylic alcohols have been reported9d but to our knowledge, none of them reports the recovery of the catalyst.
Polyoxometalates (POMs) as rapidly expanding green catalysts, are widely used in oxidation and acid catalyzed transformations.10 In a previous work, organic soluble Lanthanide complexes of POMs were synthesized by grafting Lewis acidic cations (Y3+, Yb3+, La3+, Hf4+, Sc3+etc.) onto monolacunary Dawson polyoxotungstates [P2W17O61]10− and used as Lewis acid catalysts for Mannich type reactions.11 We demonstrated that the coordination between the metal atoms and water molecules allows an indirect Brønsted acidity useful to catalyze Mukaiyama-aldol reactions.12 In order to expand the application of those catalysts, the hafnium(IV) complex TBA5K[α1-Hf(H2O)4P2W17O61] (POM/Hf) (Scheme 1) was selected as a representative species and used in the challenging nucleophilic addition reactions to in situ generated cyclic N-acyliminium ions from 5-N-Boc-2-hydroxypyrrolidine 1a and N-Boc-2-hydroxypiperidine 1b respectively.
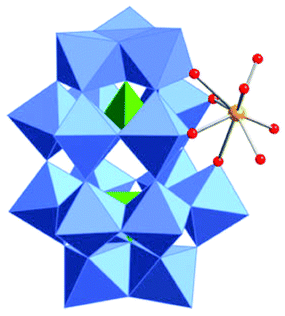 | ||
| Scheme 1 Representation of the anion of TBA5K[α1-Hf(H2O)4P2W17O61]. The phosphotungstic framework is shown as coordination polyhedra (blue W, green P). The Hf4+ ion (yellow) is coordinated to four terminal oxo ligands and four water molecules (red). | ||
The choice for the α1 isomer was dictated by steric and electronic considerations. Indeed, the Hf(IV) atom is a large cation that can be coordinated to the four oxido ligands in the lacunary site of the POM, and still has open coordination sites. It has been demonstrated that its α2 isomer was more prone to dimerization which may lead to less active catalysts.11a The chiral α1 framework is also of particular interest for its potential as chiral catalyst.
We report herein our results concerning the POM/Hf-mediated nucleophilic additions of silyl enol ethers, ketene-acetals and activated methylene C-nucleophiles to hemiaminals that provide an easy access to substituted pyrrolidines and piperidines derivatives via cyclic N-acyliminium ions at room temperature.
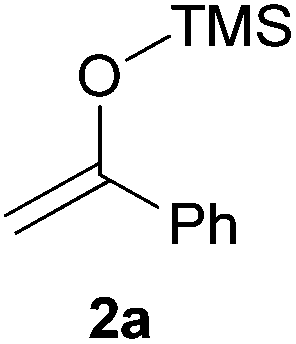
We initially chose to examine the addition of 1-phenyl-1-trimethylsiloxyethylene 2a to N-Boc-2-hydroxypyrrolidine 1a as a model reaction in the presence of 10 mol% catalyst at room temperature in CH3CN. The expected product 3a was isolated in 69% yield after 20 min (Table 1, entry 2).
|
|
|||
|---|---|---|---|
| Entry | POM/Hf (mol%) | Time | Yield (%) |
| 1 | — | 24 h | — |
| 2 | 10 | 20 min | 69 |
| 3 | 1 | 1 h | 72 |
| 4 | 0.25 | 17 h | 80 |
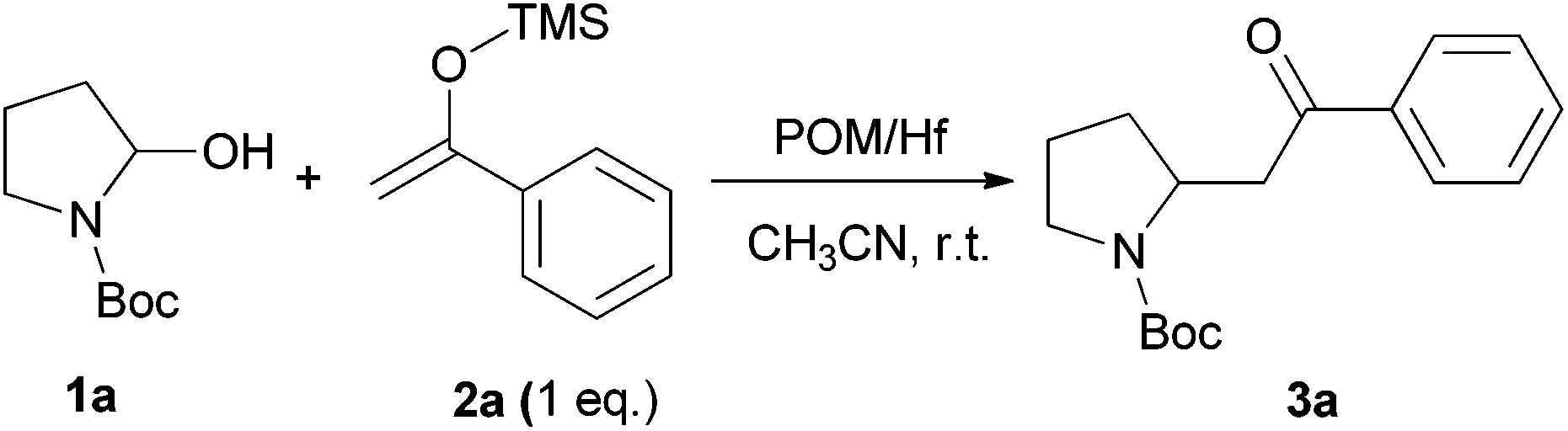
It is possible to decrease the loading of catalyst down to 0.25 mol% albeit the reaction time should be increased up to 17 h to achieve a similar yield (Table 1, entry 4). However, the amount of 1 mol% of POM/Hf was chosen to extend the scope of the nucleophilic addition as well as the use of N-Boc-2-hydroxypiperidine 1b as electrophile.
Unfortunately, the reaction of 1-phenyl-1-trimethylsiloxy-ethylene 2a with N-Boc-2-hydroxypiperidine 1b was not as efficient as for 1a. Indeed, in the previous conditions a low yield of the expected product 3b together with a total consumption of the starting material 1b was observed (Table 2, entry 2), a pattern that results from the elimination of water from the corresponding iminium. Actually, the in situ formed N-acyliminium intermediate resulting from the interaction of the N-Boc-2-hydroxypiperidine 1b and POM/Hf can lead to the relatively stable enecarbamate by-product 4b, or can undergo self-condensation to further form the “dimeric” enecarbamate 5b which proved to be inert to the nucleophile 2a.13,14 Both forms of enecarbamate cause the competitive loss of starting materials, which can explain the relatively low yield for all the 2-substituted N-piperidines (Scheme 2).
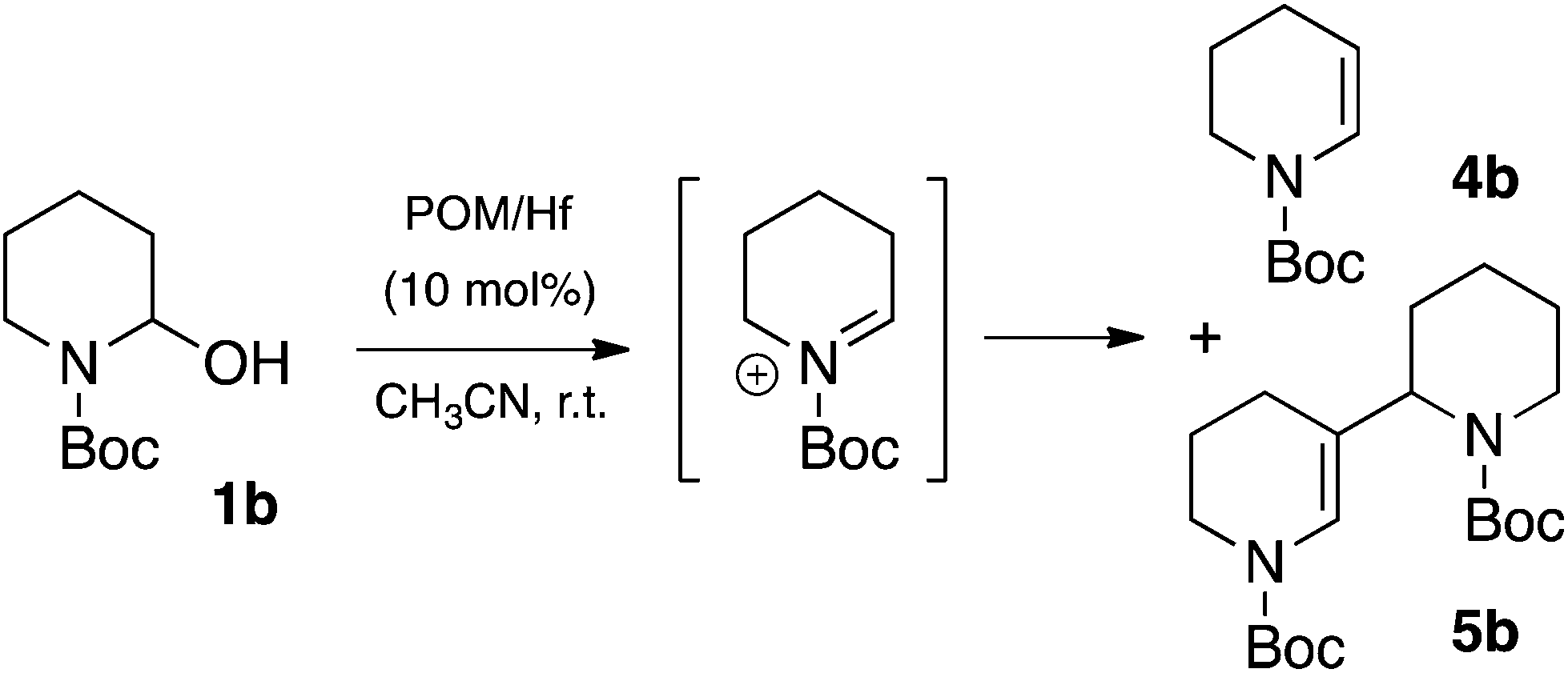 | ||
| Scheme 2 Competitive elimination and dimerization of N-Boc-2-hydroxypiperidine 1bvia N-acyliminium. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Nucleophile | Time (h) | Product | Yield (%) |
a 10 mol% catalyst was used.
b Diastereomeric ratio of 3e (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30).
c 98% yield after 18 min when 10 mol% catalyst was used.
d 24% yield after 1.5 h when 4 mol% catalyst was used. :30).
c 98% yield after 18 min when 10 mol% catalyst was used.
d 24% yield after 1.5 h when 4 mol% catalyst was used.
|
|||||
| 1 | 1a |
|
1 | 3a | 72 |
| 2 | 1b | 0.3 | 3b | 16 | |
| 3 | 1a |
|
18 | 3c | 58a |
| 4 | 1b | 16 | 3d | — | |
| 5 | 1a |
|
4 | 3e | 63c |
| 6 | 1b | 4 | 3f | — | |
| 7 | 1a |
|
24 | 3g | 69 |
| 8 | 1b | 15 | 3h | 11d | |
| 9 | 1a |
|
4 | 3i | 45a |
| 10 | 1b | 4 | 3j | 10a | |
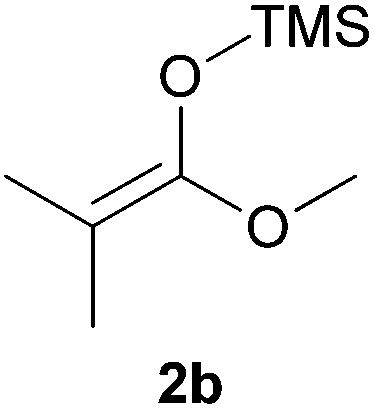
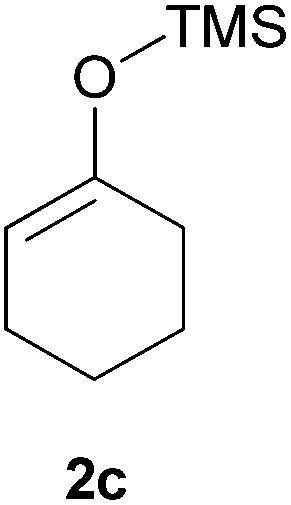
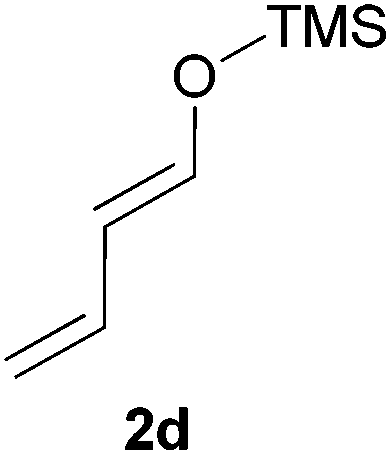
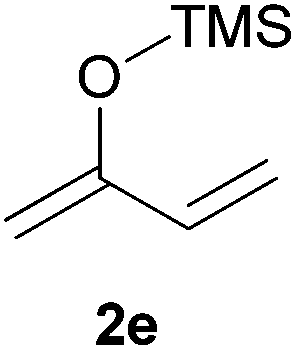
Thereafter, the scope of the reaction was investigated with different substrates N-Boc pyrrolidine 1a and N-Boc piperidine 1b and a series of nucleophiles such as silyl enol ethers or ketene-acetals. The 1-(trimethylsiloxy)-cyclohexene 2c acts as a good nucleophile with 1a and gives the corresponding 2-substituted N-Boc pyrrolidine 3e, in 63% yield after 4 h. Interestingly, an excellent yield of 98% as a 70:30 mixtures of diastereomers could be obtained by the use of 10 mol% of catalyst (Table 2, entry 5). Both 1- and 2-(trimethylsilyloxy) butadiene 2d and 2e reacted moderately with pyrrolidine 1a (Table 2, entries 7 and 9).
Attempts to employ methyl trimethylsilyl dimethylketene acetal 2b and 1-(trimethylsiloxy)-cyclohexene 2c as nucleophiles with N-Boc-2-hydroxypiperidine 1b failed, which might be attributed to the relative steric hindrance of the secondary or tertiary reactive C-atom whereas the less substituted nucleophiles 2d–e reacted in low yields. The competitive elimination of water on 1b (vide supra) simultaneously to the partial hydrolysis of the nucleophiles should be responsible of these unproductive results. In conclusion, our investigation on the addition of trimethylsilyl enol ethers to the 5-membered N-acyliminium precursor 1a under Lewis acidic POM/Hf revealed that moderate to high yields of the corresponding 2-substituted N-Boc pyrrolidines were obtained. Lower yields were observed for the reaction between the same nucleophiles and the 6-membered precursor 1b.
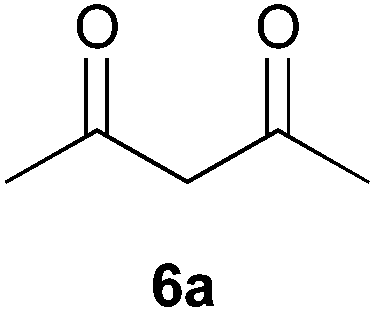
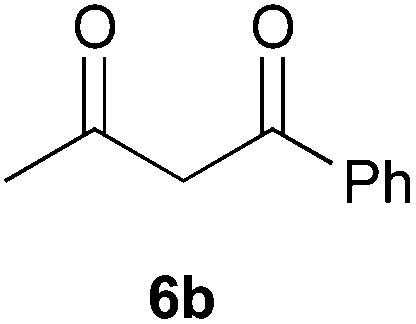
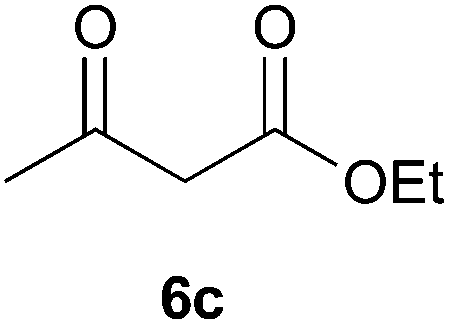
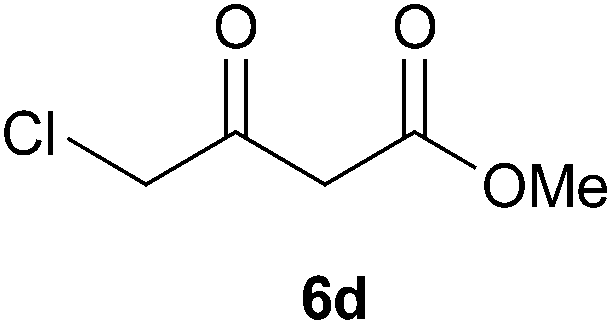
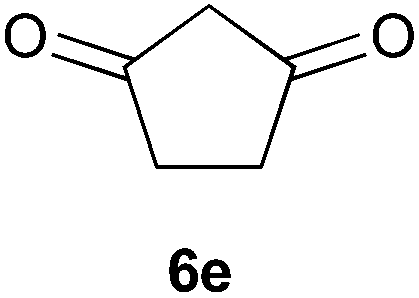
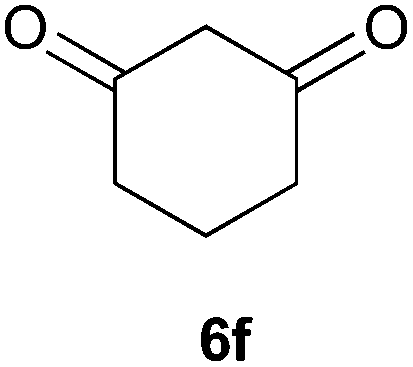
To further expand the scope of the reaction, the use of 1,3-dicarbonyl compounds 6 as nucleophiles was also investigated. Such enolizable C-nucleophiles are expected to react smoothly in such slightly acidic conditions. In the presence of POM/Hf, they provided the corresponding 2-substituted pyrrolidine adduct 7 in moderate to good yields as illustrated by the representative examples in Table 3. Acyclic diketones or keto esters reacted with electrophile 1a in yields ranging from 49 to 64% (Table 3, entries 1–6). Excellent yields were achieved in the reactions with 1,3-cyclopentanedione 6e or 1,3-cyclohexanedione 6f. The 2-substituted N-Boc-pyrrolidines 7i and 7k were isolated in 96 and 85% yields respectively with 1 mol% of POM/Hf as catalyst (Table 3, entries 9 and 11). The desired products 7i-k exist mainly in their corresponding enolic form as revealed by the corresponding 1H and 13C NMR spectra (see ESI‡). The products 7c, 7e and 7g were formed as difficultly separable mixture of diastereoisomers. While the purification of 7d allowed its isolation as a single, analytically pure diastereomer, 7h and 7l could not be obtained as pure products. Besides, the attempt to employ diethyl malonate as nucleophile failed so far. As previously observed with silylenol ethers as nucleophiles, lower yields were noticed with hydroxypiperidine 1b in comparison to hydroxypyrrolidine 1a (Table 3, compare entries 1,3,5,9 vs. 2,4,6,10). The lower reactivity of the 6-membered iminium in comparison to the 5-membered one, should be responsible for the presence of elimination products.13–16
The Hf4+ ion in the catalyst has four coordinated water molecules which could either act as proton donor or could exchange with the reactive organic molecules. Indeed, in previous work, the POM/Hf demonstrated Lewis acidity in Mannich type reactions (Nitrogen type electrophiles) and induced Brønsted acidity for Mukaiyama-aldol reactions (oxygen type electrophiles). We thus decided to explore the acidity involved in the present activation of hemiaminals 1a,b.
For that purpose, pyridine and 2,6-di-tert-butylpyridine were used as additives during the POM/Hf catalyzed nucleophilic addition.17 Both bases are strong enough to capture protons and should inhibit any Brønsted acid-catalyzed reaction. While pyridine can also bind to metal centers, thus inhibiting strongly Lewis acid-catalyzed reactions, 2,6-di-tert-butylpyridine is highly hindered and should less coordinate to the Lewis acidic Hafnium ion located in our bulky POM. Therefore its effect on Lewis acid catalysis is expected to be weak. In our experiments, when pyridine (20 mol%) was added, the reaction between 1-phenylvinyl trimethylsilyl ether 2a (5 equiv.) and N-Boc-2-hydroxypyrrolidine 1a was totally halted, while no influence was observed in the presence of 2,6-di-tert-butylpyridine (Table 4).
|
|
|||
|---|---|---|---|
| Entry | Additive | Time (h) | Yield (%) |
| 1 | — | 1 | 69 |
| 2 | 2,6-Di-tert-butylpyridine | 1 | 72 |
| 3 | Pyridine | 1 | — |
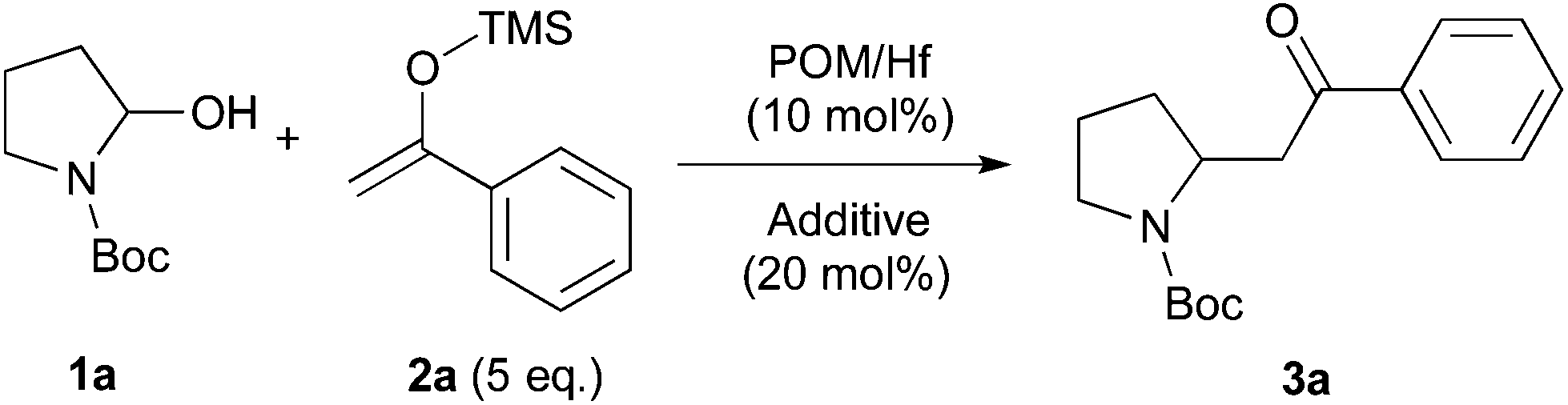
We conclude that the reaction is catalyzed by the POM/Hf in a Lewis acidic manner. First, one water molecule initially coordinated to the Hf atom is exchanged with the substrates. Then, the hydroxyaminal 1a,b that is coordinated to the POM/Hf via its hydroxy group is converted into its corresponding iminium by the departure of the anionic POM/Hf-OH that could itself activated the pronucleophiles 2 or 6.
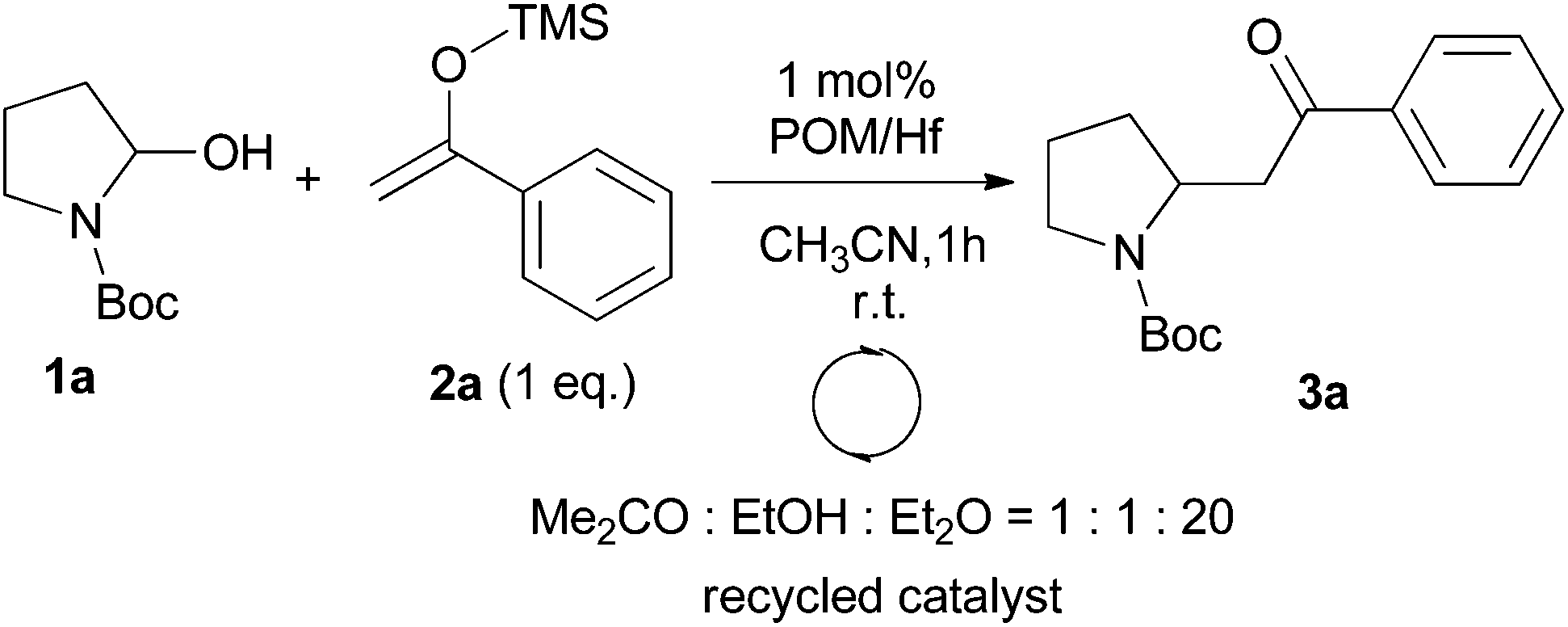
We also examined the possible recovery of our catalyst. Due to the specific solubility of POMs, the catalyst should be easily recovered. Indeed, at the end of the reaction the addition of a solvent mixture of acetone–ethanol–diethyl ether (1/1/20) resulted in the precipitation of the POM/Hf as a white powder (with a recovery of >95% yield). In this study, the recyclability of catalyst has been tested on the 1 mol% scale, and its purity was checked by 31P NMR after each round. The catalyst can be reused at least for three times without losing its reactivity. Slow decrease was then observed due to the few mg handled as well as probable slow decomposition and/or dimerization of the catalyst as time going (Table 5).18
|
|
||||
|---|---|---|---|---|
| Cycle | 1 | 2 | 3 | 4 |
| Yield (%) | 72 | 74 | 69 | 49 |
Finally, we also examined the addition of allyltrimethylsilane to the reagent 1a. By using 20 mol% of POM/Hf catalyst, a 17% yield of the expected 2-allyl carbamate product 8a was obtained after 120 h. Although the yield is relatively low, this is the first time such kind of polyoxometalate complex (POM/Hf) allows the nucleophilic addition of allyltrimethylsilane onto hemiaminal via the in situ formation of N-acyliminium ions (Scheme 3).
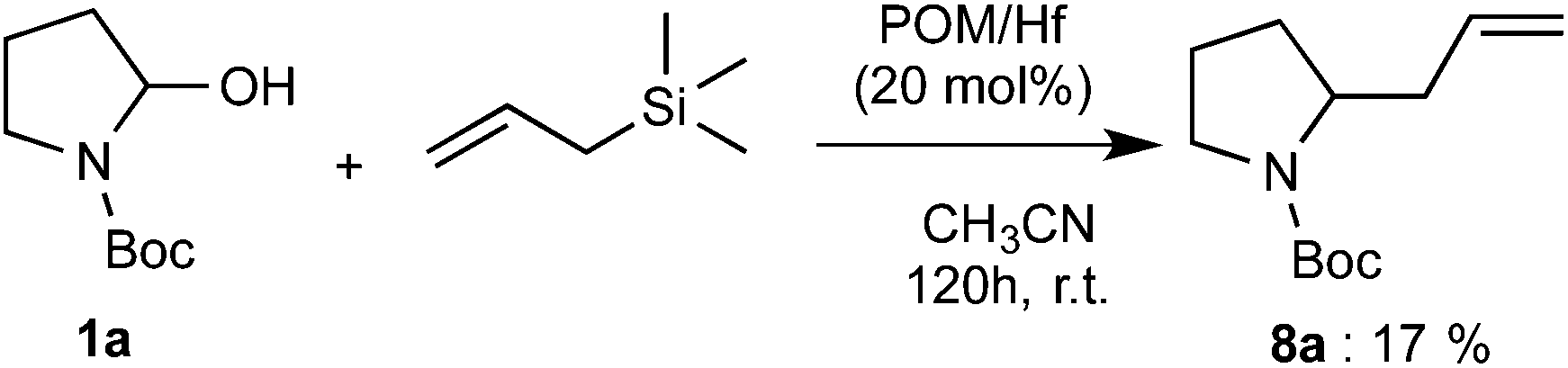 | ||
| Scheme 3 Reaction with allyl silane. | ||
In summary, we used a Hafnium containing Dawson polyoxometalate as soluble catalyst for the carbon nucleophilic addition of silyl ether and methylene activated pro-nucleophiles to cyclic hemiaminals. The in situ formed N-acyliminium ions reacted smoothly at room temperature giving the desired pyrrolidine and piperidine products in moderate to good yields. The POM/Hf has been proved to catalyze the reaction in a Lewis acidic pathway. It is playing a dual catalysis: activation of the nucleophile and the electrophile. It can be easily precipitated and removed taking advantage of its specific solubility. Recycling of the catalyst is possible for a limited number of cycles. Studies are still underway for its further application in other organic reactions and its asymmetric versions.
Acknowledgements
We thank the Université P. et M. Curie (UPMC) and CNRS for funding. The Fédération de Recherche (FR2769) provided technical access for analysis. M. Forêt Jacquard kindly tested the reproducibility of the reactions. W.J.X. acknowledges the China Scholarship Council (CSC) for a PhD fellowship.Notes and references
- Selected reviews: (a) B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS PubMed; (b) A. Yaziri and S. G. Pyne, Synthesis, 2009, 339 Search PubMed; (c) A. Yaziri and S. G. Pyne, Synthesis, 2009, 513 Search PubMed; (d) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trinidade, Chem. Rev., 2009, 109, 2703 CrossRef CAS PubMed; (e) U. Martinez-Estibalez, A. Gomez-SanJuan, O. Garceia-Calvo, E. Aranzamendi, E. Lete and N. Sotomayor, Eur. J. Org. Chem., 2011, 3610 CrossRef CAS; (f) P.-Q. Huang, Synlett, 2006, 1133 CrossRef CAS PubMed; P. M. Weintraub, J. S. Sabol, J. M. Kane and D. R. Borcherding, Tetrahedron, 2003, 59, 2953 Search PubMed; (g) D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC.
- (a) C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS.
- (a) D. Russowsky, R. Z. Petersen, M. N. Godoi and R. A. Pilli, Tetrahedron Lett., 2000, 41, 9939 CrossRef CAS; (b) C. K. Z. Andrade and R. A. F. Matos, Synlett, 2003, 1189 CrossRef PubMed.
- R. A. Pilli and L. G. Robello, Synlett, 2005, 2297 CrossRef CAS PubMed.
- (a) S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227 CrossRef CAS PubMed; (b) S. Kobayashi, Eur. J. Org. Chem., 1999, 15 CrossRef CAS; (c) For a recent application of Bi(OTf)3 see: A. E. Schneider, T. Beisel, A. Shemet and G. Manolikakes, Org. Biomol. Chem., 2014, 12, 2356 RSC.
- (a) H. Yamamoto, Proc. Jpn. Acad., Ser. B., 2008, 84, 134–146 CrossRef CAS; (b) B. Mathieu and L. Ghosez, Tetrahedron Lett., 1997, 38, 5497 CrossRef CAS; (c) R. Ben Othman, T. Bousquet, M. Othman and V. Dalla, Org. Lett., 2005, 7, 5335 CrossRef CAS PubMed.
- (a) Y. S. Lee, Md. M. Alam and R. S. Keri, Chem. – Asian J., 2013, 8, 2906 CrossRef CAS PubMed; (b) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (c) See a special issue of Chem. Rev., 2007, 107 , no. 12.
- Selected examples: (a) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796 CrossRef CAS PubMed; (b) A. C. Breman, J. Dijkink, J. H. van Maarseveen, S. S. Kinderman and H. Hiemstra, J. Org. Chem., 2009, 74, 6327 CrossRef CAS PubMed; (c) H. Sun, C. Martin, D. Kesselring, R. Keller and K. D. Moeller, J. Am. Chem. Soc., 2006, 128, 13761 CrossRef CAS PubMed; (d) U. Albrecht, H. Armbrust and P. Langer, Synlett, 2004, 143 CAS; (e) J.-C. Adelbrecht, D. Craig, B. W. Dymock and S. Thorimbert, Synlett, 2000, 467 CAS; (f) J.-C. Adelbrecht, D. Craig and S. Thorimbert, Tetrahedron Lett., 2001, 42, 8369 CrossRef CAS.
- (a) N. S. Camilo and R. A. Pilli, Tetrahedron Lett., 2004, 45, 2821 CAS; (b) R. Ben Othman, R. Affani, M.-J. Tranchant, S. Antoniotti, V. Dalla and E. Dunach, Angew. Chem., Int. Ed., 2010, 49, 776 CrossRef CAS PubMed; (c) For an original copper catalyzed version see: S.-L. Shi, X.-F. Wei, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2012, 134, 17019 CrossRef CAS PubMed; (d) M. Hamon, N. Dickinson, A. Devineau, D. Bolien, M.-J. Tranchant, C. Taillier, I. Jabin, D. C. Harrowven, R. J. Whitby, A. Ganesan and V. Dalla, J. Org. Chem., 2014, 79, 1900 CrossRef CAS PubMed; (e) M. Szostak, B. Sautier and D. J. Procter, Chem. Commun., 2014, 50, 2518 RSC.
- (a) M. Misono, Catal. Today, 2005, 100, 95 CrossRef CAS PubMed; (b) R. Neumann, Applications of Polyoxometalates in Homogeneous Catalysis, in Polyoxometalate Molecular Science, ed. J. J. Borras-Almenar, E. Coronado, A. Müller and M. T. Pope, Kluwer, Dordrecht, 2003, vol. 98, p. 327 Search PubMed; (c) I. V. Kozhevnikov, Heterogeneous Catalysis by Heteropoly Compounds, in Polyoxometalate Molecular Science, ed. J. J. Borras-Almenar, E. Coronado, A. Müller and M. T. Pope, Kluwer, Dordrecht, 2003, vol. 98, , p. 351 Search PubMed; (d) X. Fang and C. L. Hill, Angew. Chem., Int. Ed., 2007, 46, 3877 CrossRef CAS PubMed; (e) J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577 RSC. See also: (f) S. Luo, J. Li, H. Xu, L. Zhang and J. P. Cheng, Org. Lett., 2007, 9, 3675 CrossRef CAS PubMed; (g) J. Li, S. Hu, S. Luo and J. P. Cheng, Eur. J. Org. Chem., 2009, 132 CAS; (h) A. E. Kuznetsov, Y. V. Geletii, C. L. Hill, K. Morokuma and D. G. Musaev, Inorg. Chem., 2009, 48, 1871 CrossRef CAS PubMed; (i) A. Sartorel, M. Carraro, A. Bagno, G. Scorrano and M. Bonchio, Angew. Chem., Int. Ed., 2007, 119, 3319 CrossRef; (j) A. Sartorel, P. Miro, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. Di Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051 CrossRef CAS PubMed; (k) B. P. Burton-Pye and L. C. Francesconi, Dalton Trans., 2011, 40, 4421 RSC; (l) M. R. Antonio, J. Jing, B. P. Burton-Pye and L. C. Francesconi, Dalton Trans., 2010, 39, 7980 RSC; (m) S. Kosuke, M. Sugawa, Y. Kikukawa, K. Kamata, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2012, 51, 6953 CrossRef PubMed; (n) H. G. T. Ly, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2013, 42, 10929 RSC.
- (a) C. Boglio, G. Lenoble, C. Duhayon, B. Hasenknopf, R. Thouvenot, C. Zhang, R. C. Howell, B. P. Burton-Pye, L. C. Francesconi, E. Lacôte, S. Thorimbert, M. Malacria, C. Afonso and J.-C. Tabet, Inorg. Chem., 2006, 45, 1389 CrossRef CAS PubMed; (b) C. Boglio, G. Lemière, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Angew. Chem., Int. Ed., 2006, 45, 3324 CrossRef CAS PubMed; (c) C. Boglio, K. Micoine, P. Rémy, B. Hasenknopf, S. Thorimbert, E. Lacôte, M. Malacria, C. Alfonso and J.-C. Tabet, Chem. – Eur. J., 2007, 13, 5426 CrossRef CAS PubMed; (d) M. Bosco, S. Rat, N. Dupré, B. Hasenknopf, E. Lacôte, M. Malacria, P. Rémy, J. Kovensky, S. Thorimbert and A. Wadouachi, ChemSusChem, 2010, 3, 1249 CrossRef CAS PubMed. See also some other pioneer references: (e) Y. Kikukawa, S. Yamaguchi, K. Tsuchida, Y. Nakagawa, K. Uehara, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 5472 CrossRef CAS PubMed; (f) X. Zhang, J. Li, Y. Chen, J. Wang, L. Feng, X. Wang and F. Cao, Energy Fuels, 2009, 23, 4640 CrossRef CAS; (g) J. Li, X. Zhang, W. Zhu and F. Cao, ChemSusChem, 2009, 2, 177 CrossRef CAS PubMed; (h) K. Nomiya, K. Ohta, Y. Sakai, T. Hosoya, A. Ohtake, A. Takakura and S. Matsunaga, Bull. Chem. Soc. Jpn., 2013, 86, 80 CrossRef.
- (a) E. Derat, E. Lacôte, B. Hasenknopf, S. Thorimbert and M. Malacria, J. Phys. Chem. A, 2008, 112, 13002 CrossRef CAS PubMed; (b) N. Dupré, P. Rémy, K. Micoine, C. Boglio, S. Thorimbert, E. Lacôte, B. Hasenknopf and M. Malacria, Chem. – Eur. J., 2010, 16, 7256 CrossRef PubMed.
- D. F. Oliveira, P. C. M. L. Miranda and R. D. C. Carlos, J. Org. Chem., 1999, 64, 6646 CrossRef CAS PubMed.
- (a) A. R. Pinder, Nat. Prod. Rep., 1989, 515 RSC; (b) G. J. Koomen and M. J. Wanner, J. Org. Chem., 1996, 61, 5581 CrossRef.
- R. A. Pilli, L. G. Robello, N. S. Camilo, J. Dupont, A. A. Moreira Lapis and B. A. da Silveira Neto, Tetrahedron Lett., 2006, 47, 1669 CrossRef CAS PubMed.
- (a) R. A. Pilli, M. A. Böckelmann and C. F. Alves, J. Braz. Chem. Soc., 2001, 12, 634 CrossRef CAS PubMed; (b) M. G. M. D'Oca, L. A. B. Moraes, R. A. Pilli and M. N. J. Eberlin, J. Org. Chem., 2001, 66, 3854 CrossRef PubMed.
- (a) H. C. Brown and R. B. Johannesen, J. Am. Chem. Soc., 1953, 75, 16 CrossRef CAS; (b) A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS; (c) Y. Kikukawa, S. Yamaguchi, Y. Nakagawa, K. Uehara, S. Uchida, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15872 CrossRef CAS PubMed.
- (a) M. N. Sokolov, N. V. Izarova, E. V. Peresypkina, D. A. Mainichev and V. P. Fedin, Inorg. Chim. Acta, 2009, 362, 3756 CrossRef CAS PubMed; (b) Y. Saku, Y. Sakai and K. Nomiya, Inorg. Chim. Acta, 2010, 363, 967 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00193a |
| This journal is © the Partner Organisations 2014 |