Chopping unfunctionalized carbon–carbon bonds: a new paradigm for the synthesis of organonitriles†
Philippe
Bisseret
*,
Guillaume
Duret
and
Nicolas
Blanchard
*
Université de Strasbourg, Laboratoire de Chimie Moléculaire, CNRS UMR 7509, Ecole Européenne de Chimie, Polymères et Matériaux, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: philippe.bisseret@unistra.fr; n.blanchard@unistra.fr
First published on 20th June 2014
Abstract
In the last year, a new strategy for the synthesis of organonitriles, whether aliphatic or aromatic, has emerged based on the direct carbon–carbon double or triple bond cleavage of alkenyl or alkynyl precursors. This Highlight mainly focuses on the efforts made, often nearly simultaneously, by three research teams that shaped the implementation of this new paradigm, which should be of great value for the synthetic community facing the ever-growing demand for new organonitriles, especially in medicinal chemistry.
Introduction
Among the functionalities encountered in organic chemistry, the cyano group occupies a front rank position mainly because of its versatility, as highlighted by its potential for further transformation into amides, amines, aldehydes, acids, esters, and nitrogen-containing heterocycles.1 For these reasons, organonitriles have been widely implemented as prominent intermediates in pharmaceutical and natural product synthesis. Organonitriles themselves do occur naturally in a variety of plants, animals, and microorganisms, ranging from simple, long-chain alkanenitriles to architecturally complex polycyclic molecules which are often endowed with interesting antibiotic and/or antitumor properties.2 Arylnitriles, as well as to a lesser extent alkenyl- and alkylnitriles, are also a common motif for several dozen approved drugs as well as drugs in clinical development.3 Three examples of front rank pharmaceuticals, whose activity could be at least in part traced to their nitrile-containing pharmacophore, are displayed in Scheme 1: fadrozole monohydrochloride 14 and letrozole 25 are both prescribed for treatment of breast cancer, and rilpivirine 3 is touted as among the most potent anti-HIV agent ever discovered.6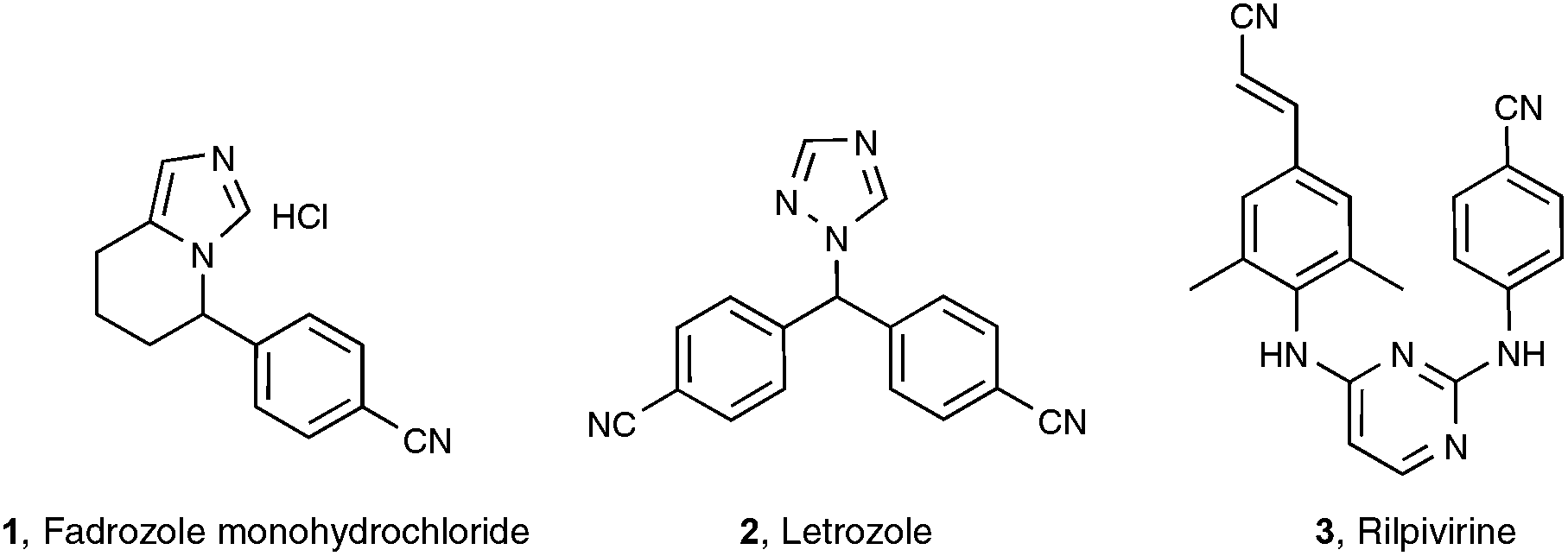 | ||
| Scheme 1 Examples of widely-prescribed nitrile-containing pharmaceuticals. | ||
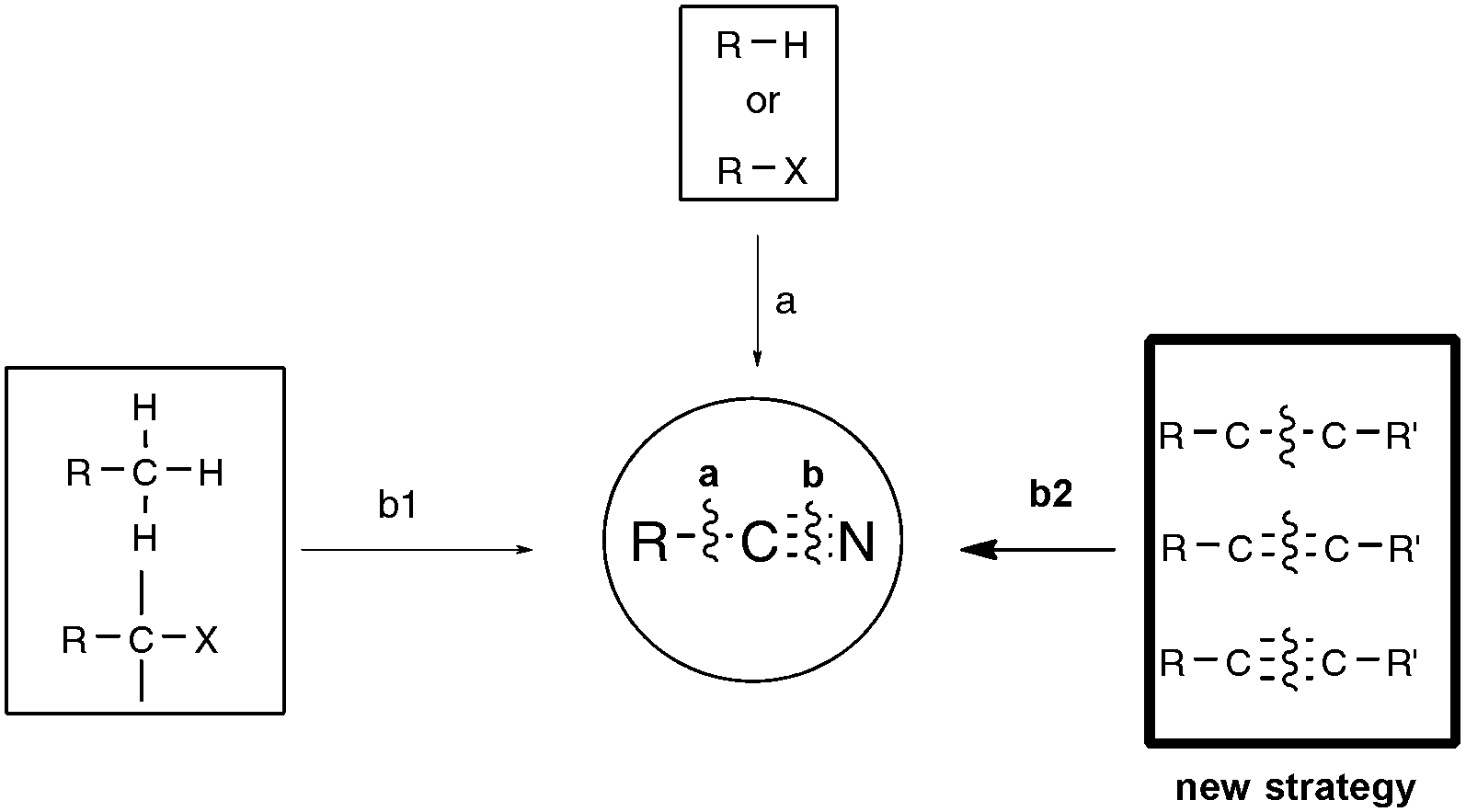
Concerning the synthesis of organonitriles, since the initial forays in the 19th century that included the now classical Sandmeyer and Rosenmund von Braun reactions,7 a plethora of methods have been made available which, until one year ago or so, could be combined into the two-pronged strategies a and b1 presented in Scheme 2.1 Until recently, the first strategy mainly encompassed metal-catalyzed cyanation procedures based on the utilization of metallic or metalloid cyanide donors “MCN” such as NaCN, KCN, CuCN, Zn(CN)2, or TMSCN.8 Although quite efficient in most cases, these methods are generally not favored due to the utilization of a toxic metal. In addition, they require careful handling in order to prevent any emission of hazardous HCN gas. Due to the rising concern by the chemist community for sustainable processes, much attention has been paid in the last decade to address the latter handicaps, highlighting the development of safe cyanide sources for the cyanation reaction. For that purpose, potassium ferrocyanide K4[Fe(CN)6], a user-friendly reagent introduced in 2004 by the Beller group,9 and non-metallic cyano-group sources have been employed, with a special focus on the cyanation of aryl halides, boronic acids, and arene C–H bonds. Not only organic sources that contained the “CN” unit within the molecule could be successfully applied to the cyanation reactions, but, more surprisingly, organic solvents such as DMF, which do not bear the latter unit, could also be tailored to the delivery of either both atoms of the cyano functional group or of its carbon atom only, the nitrogen atom being then generally supplied from ammonium salts.10 In spite of the innovative chemistry displayed in the latter non-“MCN” protocols, efforts are still to be made in order to improve their sustainability, as they are so far copper-mediated and pallado-catalyzed.
 | ||
| Scheme 2 Main strategies for the preparation of organonitriles. | ||
In contrast to the previous approach, the second general pathway leading to organonitriles does not require a carbon chain elongation step, as schematized in Scheme 2 (b1), but, in most of the cases, simply relies on straightforward chemical transformations of already functionalized substrates, such as dehydration or oxidative dehydration reactions.1,11 In some instances, unsophisticated aromatic nitriles of industrial relevance were also synthesized, simply by exposing unfunctionalized toluene precursors to an ammonia and oxygen atmosphere at a temperature higher than 600 K and in the presence of a vanadium-based catalyst.12 In 2009, by employing sodium azide as the nitrogen source and phenyliodonium diacetate (PIDA) as the oxidant, Jiao's group from the Peking University renewed interest in the latter so-called ammoxidation reaction, ensuring for the first time the direct transformation of a range of electron-rich methyl arenes into the corresponding aryl nitriles at room temperature, highlighting the implication of transient benzylic radicals.13,14 Capitalizing on related free-radical strategies, the group of Wang, at the same university, and the Togo group from Japan recently discovered mild ammoxidation conditions amenable to a wider range of functionalized methyl arenes.15,16 For their part, Jiao's group focused on the streamlining potential of preparing nitriles beyond the scope of methyl arenes as substrates, once again by combining NaN3 or TMSN3 with an oxidant, and eventually reported new syntheses through C–H bond cleavage, such as the direct oxidative transformation of allylarenes or alkenes to alkenyl nitriles,13,17 and the transformation of methyl imines to iminonitriles.13,18 Even more interestingly, they also reported about one year ago the first syntheses of organonitriles from terminal alkynes, at the reasonable temperature of 100 °C, through C–C triple bond cleavage.13,19 Shortly after Jiao's outstanding discovery, the group of Yanada from Hokkaido University reported the first syntheses of nitriles from internal alkynes,20 giving thus support to the exciting new possibility of synthesizing nitriles under mild conditions through C–C triple bond cleavage as shown in Scheme 2, b2. Extension of the strategy to alkenes in place of alkynes followed also closely with the research from both the Jiao group,13,21 once again, and Guo's group from Hunan University.22 All these efforts accomplished within the past ten months, and most often almost simultaneously, are presented here.
Synthesis of nitriles through carbon–carbon triple bond cleavage
Let us focus first on the syntheses of organonitriles through carbon–carbon triple bond cleavage, as they include the first investigations that shaped the new strategy presented in Scheme 2. These pioneering contributions also proved beneficial to the chemistry of alkynes.23 Indeed, the cleavage of carbon–carbon triple bonds has always ranked among the most challenging subjects in organic chemistry, being mainly limited to the metal-catalyzed alkyne metathesis24 or the oxidative conversion of alkynes into carboxylic acids.25 To the best of our knowledge, prior to the investigations highlighted here, only one example of preparation of a nitrile derivative through the cleavage of a C–C triple bond has been disclosed. As shown in Scheme 3, sodium nitrite was found to react with phenyl(phenylacetylenyl)iodonium toluene-4-sulfonate 4 to yield benzonitrile 5a upon phase transfer in H2O–CHCl3.26 In spite of its modest yield, this transformation has remained totally overlooked since its discovery and certainly deserves further attention, in order, not only to investigate more closely its mechanism, but even more to examine its scope. In spite of its potential, it relies on highly reactive alkynyliodonium derivatives and thus detracts in no way from the discovery of the Jiao group which simply implies terminal alkynes.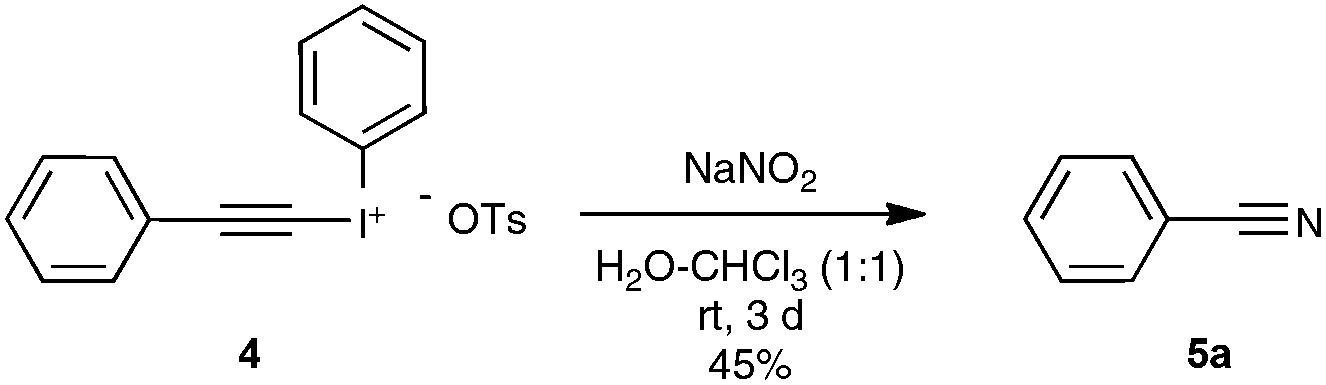 | ||
| Scheme 3 Preparation of benzonitrile from a phenylacetylenyliodonium salt. | ||
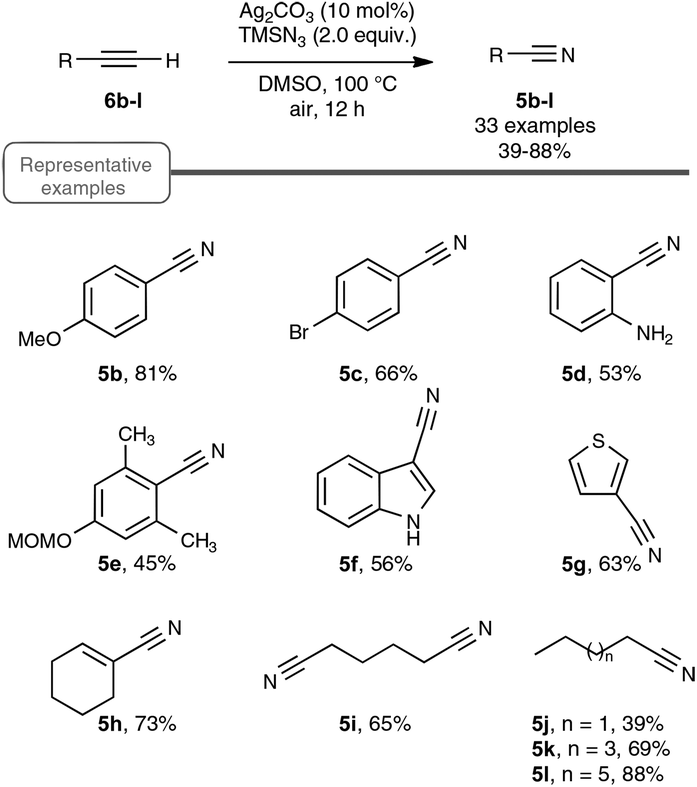
Inspired by their own experience in nitrogenation reactions, as well as by the very few reports in the literature of transformations of azido alkenes into nitriles by C–C cleavage,27 the Jiao group succeeded in converting, in a first set of experiments performed under air, para-methoxy phenylacetylene 6b into para-methoxy-benzonitrile 5b with a yield of 81%.13,19 As displayed in Scheme 4, the optimized conditions involve the utilization of azidotrimethylsilane (TMSN3) as the nitrogen source, DMSO as the solvent and Ag2CO3 as the catalyst. Surprisingly, the reaction did not proceed satisfactorily with any of the other π-acid catalysts examined, such as NiCl2, Cu(OAc)2, Pd(OAc)2, FeCl2 or even the alkynophilic AuCl3. In the same vein, other azide reagents such as diphenylphosphoryl azide, NaN3, and para-tosyl azide did not yield the desired product. As outlined in Scheme 4, under the previous conditions, both aryl- and alkyl terminal alkynes could be converted into the corresponding organonitriles in moderate to very good yields. The reaction also performed satisfactorily from 3-thiophenyl- and 3-indolyl alkynes 6g and 6f as well as from 1-cyclohexenylacetylene 6h, paving the way for exploring further the reactivity of other hetero-aryl- and alkenyl alkynes. In the case of aryl alkynes, it is worth noting that the protocol accommodates substrates with unprotected reactive groups, such as an amino function, and those bearing a halogen substituent.
 | ||
| Scheme 4 Direct conversion of terminal alkynes into nitriles. | ||
The proposed mechanism involves, as outlined in Scheme 5, a silver-catalyzed hydroazidation step of the starting alkyne, generating a vinylazide 8, with the participation of trace amounts of water in the DMSO solvent, which would undergo a cyclisation with HN3 to form an unstable highly nitrogenated intermediate 9, which would rearrange ultimately into the nitrile product, with the release of both HN3 and CH2N2. Although further work is still needed to decipher the latter unusual rearrangement, the authors firmly assessed, in a set of control experiments, the participation of the vinyl azide as a key intermediate, conversely ruling out notably the involvement of azirinyl- or tetrazolyl-based intermediates.
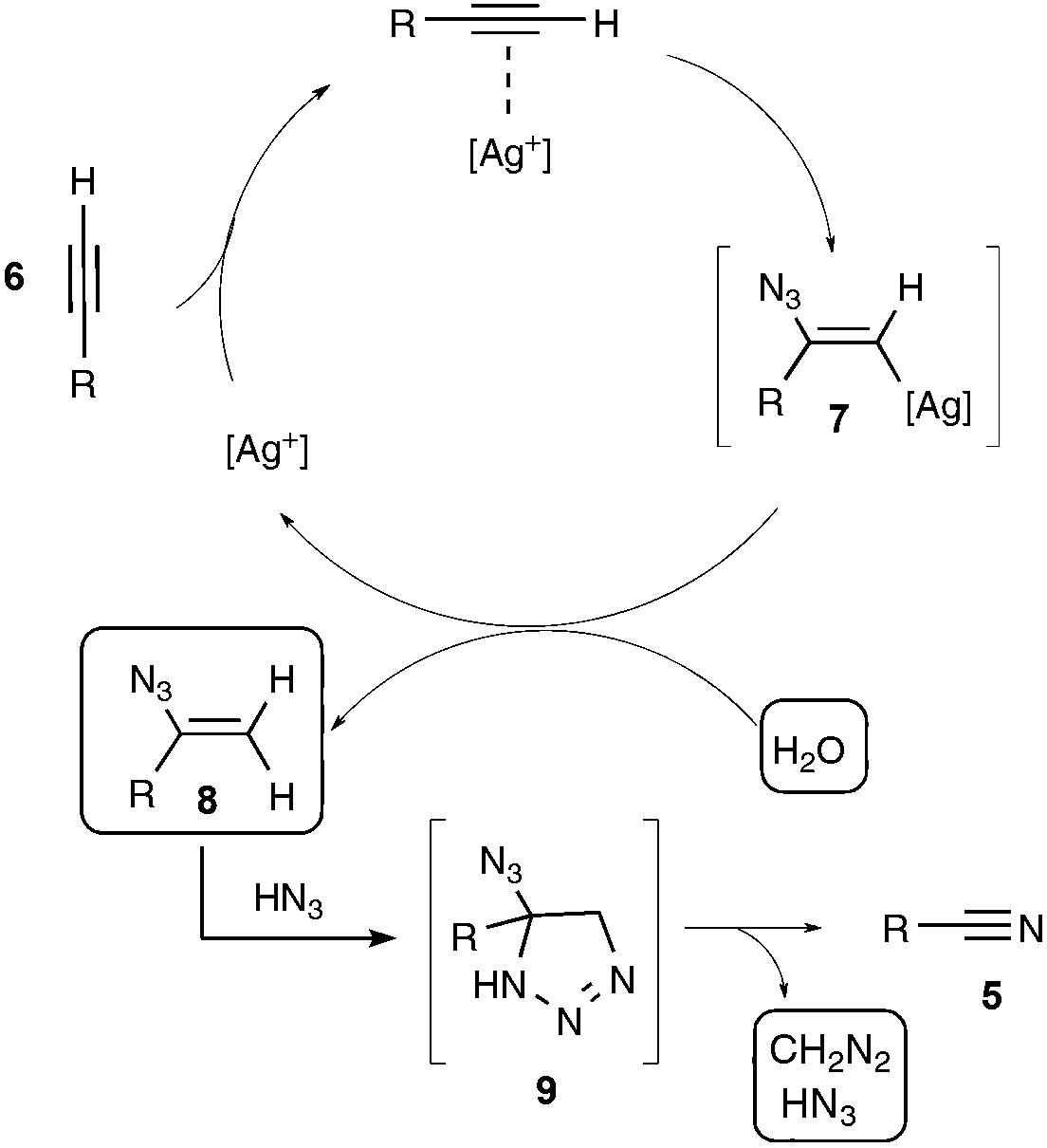 | ||
| Scheme 5 Proposed mechanism for the conversion of terminal alkynes into nitriles. | ||
With regard to the quite challenging transformations easily performed at a reasonable temperature, and notably without the need to exclude ambient air and moisture, Jiao's work is remarkable. The scope of the reaction at this stage remains somehow limited however, since the reactivity of phenylacetylenes bearing electron-withdrawing substituents was not investigated, probably reflecting their lack of reactivity. In addition, the authors did not probe in their study any internal alkynes, or did they clearly mention the possibility of exploring their reactivity in a future work.
A few weeks later, echoing the limitations of Jiao's method, Yanada and co-workers independently published an approach to nitrile synthesis from a palette of alkynes, encompassing mostly a wide range of internal alkynes, but also para-methoxy phenylacetylene, as an example of a terminal alkyne (Scheme 6).20 By contrast with the previous work, which highlighted a silver-catalyzed hydroazidation strategy, the Yanada group opted for a non-metal protocol, based on the haloazidation of the C–C alkyne triple bond. After extensive preliminary investigations performed on 1,2-bis(4-methoxyphenyl)ethyne 6bb, an optimized yield of para-methoxy-benzonitrile 5b could be obtained using 2.4 equivalents of NIS as the halogenating agent as well as 2.4 equivalents of TMSN3 as the nitrogen source. In particular, utilization of NBS or NCS, in place of NIS, resulted in a much lower yield of nitrile. The choice of the solvent also proved crucial and best results were obtained with DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN in equal proportion, starting at room temperature and rising to 70 °C. Having in hand the optimized reaction conditions, the authors explored the reactivity of an array of internal diaryl- and aryl-alkyl alkynes. As shown in Scheme 6, in the case of diaryl compounds, whether symmetrical or not, the reaction proceeded quite satisfactorily even in the case of aryl moieties equipped with an electron-withdrawing group. Internal alkynes, bearing aromatic–aliphatic substituents were also cleaved to furnish the desired pair of nitriles, in lower yield however. Besides internal alkynes, the authors also performed the reaction with para-methoxy phenylacetylene 6b, as an example of terminal alkyne. In this case, the yield of para-methoxy-benzonitrile 5b, which resulted from C–C cleavage, was however significantly lower than that obtained with Jiao's protocol.
:MeCN in equal proportion, starting at room temperature and rising to 70 °C. Having in hand the optimized reaction conditions, the authors explored the reactivity of an array of internal diaryl- and aryl-alkyl alkynes. As shown in Scheme 6, in the case of diaryl compounds, whether symmetrical or not, the reaction proceeded quite satisfactorily even in the case of aryl moieties equipped with an electron-withdrawing group. Internal alkynes, bearing aromatic–aliphatic substituents were also cleaved to furnish the desired pair of nitriles, in lower yield however. Besides internal alkynes, the authors also performed the reaction with para-methoxy phenylacetylene 6b, as an example of terminal alkyne. In this case, the yield of para-methoxy-benzonitrile 5b, which resulted from C–C cleavage, was however significantly lower than that obtained with Jiao's protocol.
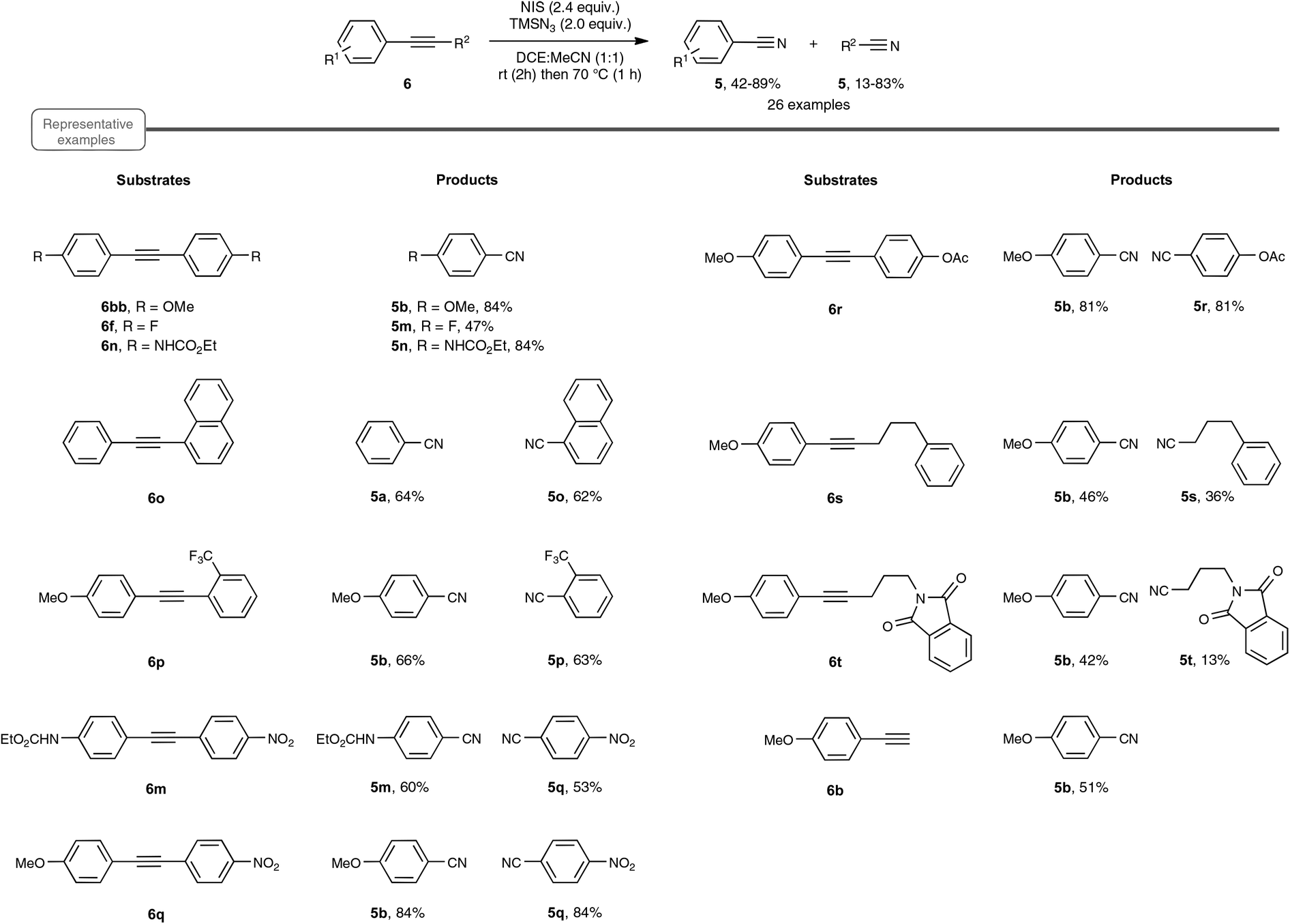 | ||
| Scheme 6 Triple bond cleavage reaction via iodoazidation. | ||
The postulated mechanistic pathway relies on the formation of an unstable iodo vinyl azide 10 as a key intermediate, which would generate, by releasing nitrogen gas, a strained iodo azirine 11 (Scheme 7). The iodo azirine would undergo halide displacement with the azide anion to form the corresponding azido azirine 12, which finally would experience a ring-opening reaction to yield the two nitrile products. In a control experiment performed with diphenylacetylene as the starting alkyne under the standard conditions, but with the omission of the heating period at 70 °C, the authors managed to isolate in 39% yield the corresponding iodo vinyl azide, which perfectly tallies with the proposed mechanism. Although based on precedents in the literature,28 the transformation into the nitrile of the key vinyl iodide derivative deserves further studies, in order to ascertain in particular the passage via an iodo azirine derivative.
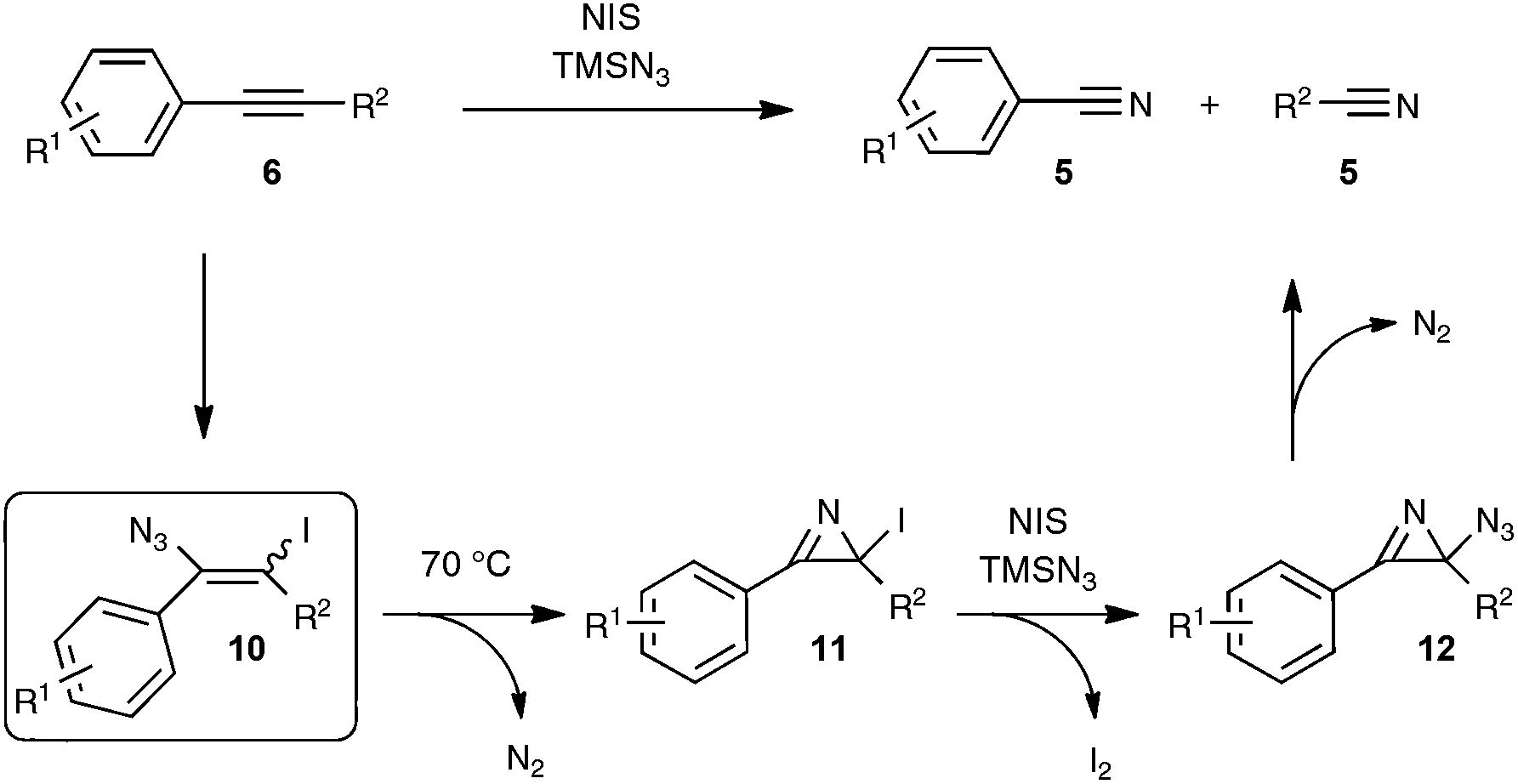 | ||
| Scheme 7 Proposed mechanism for the conversion of alkynes into nitriles via an azido-iodination strategy. | ||
Before closing this section, it is instructive to linger upon the wonderful complementarity of the two studies, which, taken together, possess quite a remarkable broad substrate scope.
Synthesis of nitriles through carbon–carbon double-bond cleavage
Let us turn now our attention to the recent findings aimed at synthesizing organonitriles directly through the oxidative double-bond cleavage of unfunctionalized alkenyl precursors. In the wake of their remarkable contribution to a novel synthesis of arylamines through the FeCl2-promoted direct cleavage of unactivated C–C single bond of alkylarenes, which makes use of alkylazides as the nitrogen source and DDQ as the oxidant, the Jiao group explored the reactivity of other types of substrates with a variety of nitrogenating and oxidizing agents, as well as non-metal catalysts. The most salient observation of a series of trials performed with catalytic amounts of TEMPO in place of the iron catalyst was the formation of acetophenone and HCN, when α-methylstyrene was submitted to TMSN3 under oxygen. In order to shed light on this quite intriguing transformation, which highlights a C–C double-bond cleavage, the authors selected 1H-indene 13a as the model substrate (Scheme 8). By making use of 1.5 equivalents of TMSN3 in MeCN as the solvent at the temperature of 80 °C, an optimized yield of the corresponding oxo-nitrile derivative 14a, resulting from C–C double-bond cleavage with oxygenation occurring at the benzylic site, was obtained.13,29 As outlined in Scheme 8, under these conditions, the reaction proceeded quite well with a range of benzylic cycloalkenes, excluding 1,2-dihydronaphthalene 13h which offered only a modest yield of the corresponding oxo-nitrile derivative 14h. At the slightly higher temperature of 95 °C, the reaction also performed notably well with 1-chloro-2-cycloheptenylbenzene 13b, giving a straightforward access to a key intermediate 14b in the synthesis of a widely used analgesic medicine named (S)-ketamine.30 In contrast, the reaction could not be accomplished under the previous reaction conditions with simple aliphatic cyclic olefins. In that case, the addition of a catalytic amount of phenyliododiacetate (PIDA) was necessary to achieve modest to good yields, although the precise role of PIDA remains to be elucidated. It is also worth noting that benzonitrile derivatives could not be prepared directly from styrenyl precursors by one of these two protocols, which yielded instead the corresponding benzaldehydes.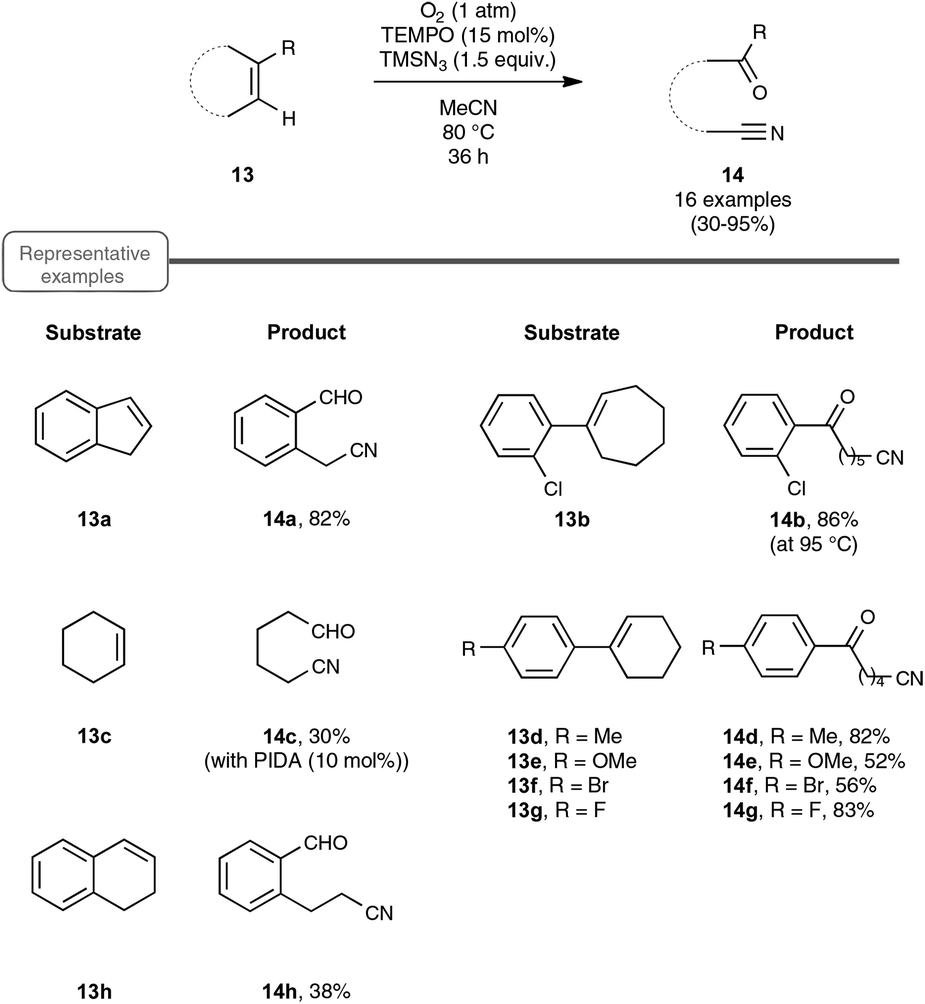 | ||
| Scheme 8 Direct transformation of cycloalkenes to oxo-nitriles. | ||
In order to probe the mechanism of this reaction, the authors performed a set of control experiments, which assigned in particular a pivotal dual role to oxygen, as an oxidant to reoxidize the catalyst and as an oxygen source for the oxygenation process (Scheme 9). By contrast, water was shown to play no role in the process, although no deleterious impact on the reaction outcome was observed in its presence in small amounts. As depicted in Scheme 9, the assumed mechanistic pathway relies on the generation of an azido radical (eqn (1)), which would subsequently attack the alkene and terminates with molecular oxygen to form a peroxyl radical 16 (eqn (2)). The latter radical would ultimately deliver the oxo-nitrile product 14 with the release of N2via an intricate rearrangement (eqn (3)), which certainly deserves further attention.
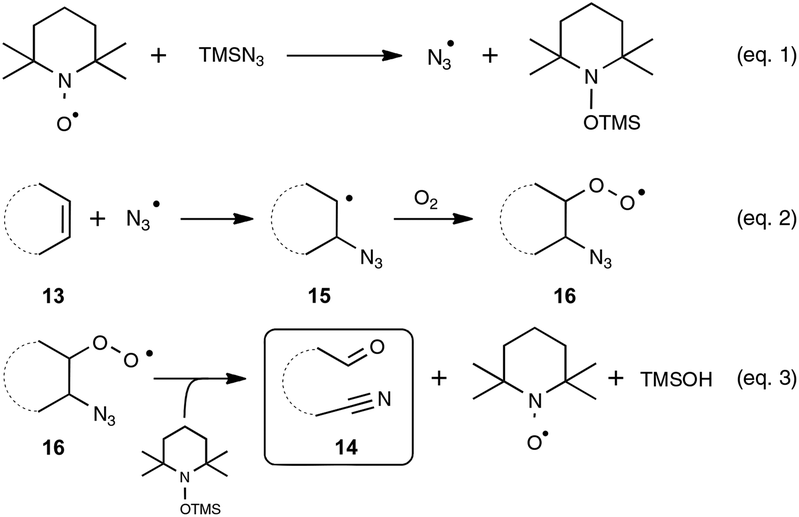 | ||
| Scheme 9 Proposed mechanism for the oxidative nitrogenation of alkenes. | ||
As a continuation of their work on the synthesis of nitriles through the direct C–C double-bond cleavage of unfunctionalized alkenes, the Jiao group focused on the possibility of preparing benzonitriles from styrenyl precursors (Scheme 10), as this transformation could not be realized by making use of the previously described TEMPO-catalyzed procedure. For that purpose, they implemented a strategy based on the formation of intermediate α-azido styrenes, amenable in principle to further transformation into the desired organonitriles, according to their own report on the cleavage of C–C triple bonds (see Scheme 5). In order to generate the azido olefins, they were inspired from studies dating back to the late sixties which involve a regioselective halo-azidation of the starting styrenes followed by a dehydrohalogenation step under basic conditions.31 After extensive preliminary experiments with para-methoxystyrene 17b as the test substrate, an optimized yield of para-methoxybenzonitrile 5b was obtained by running the reaction under air and by making use of 2.7 equiv. of TMSN3 as the azidation agent, 1.2 equiv. of NBS and Cu(OAc)2 as the catalyst and K3PO4·7H2O as the base.32 It ought to be noted that the transformation proceeded almost equally well when an oxygen atmosphere or even argon gas was used in place of ambient air and that the presence of the copper-salt catalyst was not essential, although a slightly lower yield was obtained when it was omitted. In contrast, the utilization of other base sources in place of potassium phosphate heptahydrate, such as pyridine, Et3N or K2CO3, was much less conclusive.
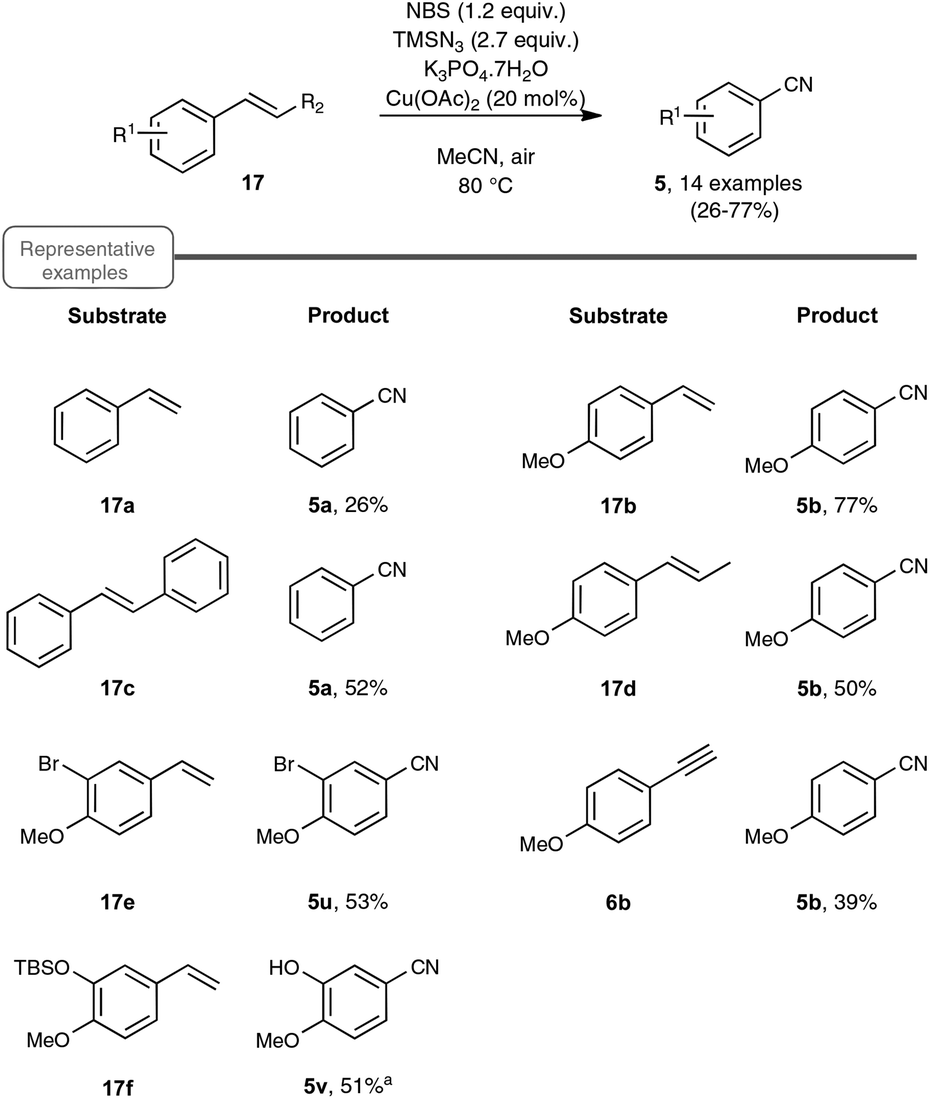 | ||
| Scheme 10 Preparation of benzonitriles from styrene derivatives. aLoss of TBS was observed in the corresponding benzonitrile. | ||
As indicated in Scheme 10, styrene derivatives equipped with an electro-donating substituent in the para position reacted satisfactorily under the optimized conditions, yielding the corresponding nitriles, generally not contaminated with any azido styrene derivatives. Apart from the latter substrates, the scope of the reaction appeared quite limited. For instance, styrene itself gave only a poor yield in benzonitrile and para-bromostyrene or para-chlorostyrene exclusively gave, in place of the desired nitriles, the corresponding diazide outlined in Scheme 11. The method was however applicable to para-methoxy phenyl acetylene 6b as the substrate, although only a modest yield of the corresponding nitrile was obtained in that case, which cannot compete with the much better yield resulting from the silver carbonate based protocol presented in Scheme 4.
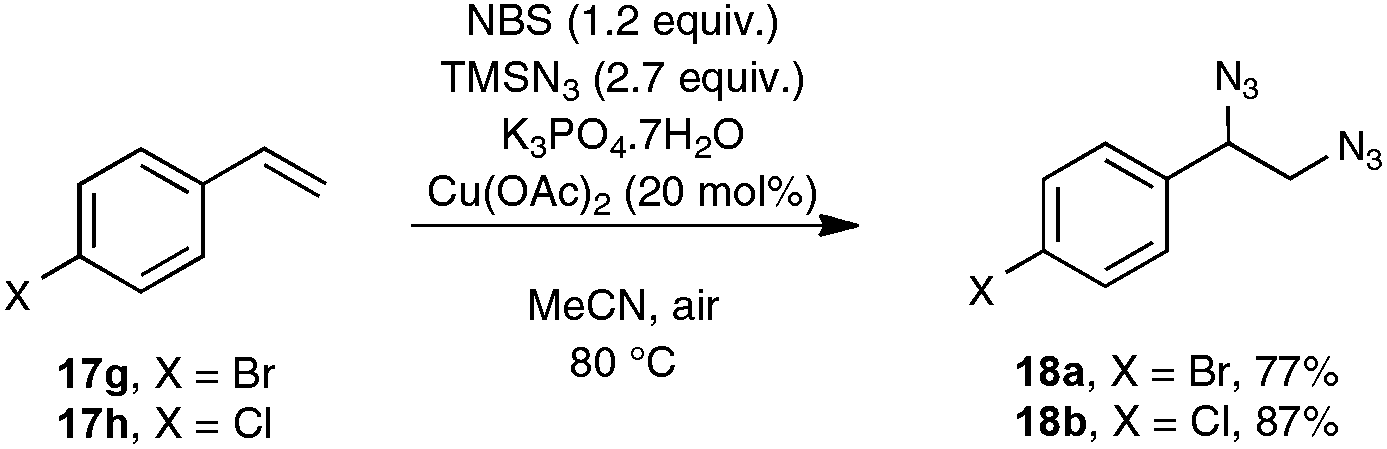 | ||
| Scheme 11 Nitrogenation reaction with para-bromo- and para-chlorostyrenes. | ||
As shown in Scheme 12, the mechanistic pathway highlights the formation of a transient azido alkene 20, whose conversion under copper catalysis into the nitrile product would involve a copper iminyl species 21. If the key participation of the azido alkene could be firmly established, the second part of the mechanism, which is based on the recently reported oxygen-mediated C–C bond cleavage of the iminyl copper species,33 is clearly a subject of debate, as the reaction was found to be also quite effective without metal catalysis and under an inert atmosphere, pointing to the type of azido-mediated rearrangement previously proposed by the authors in the case of the formation of nitriles through C–C triple bond cleavage (see Scheme 5).
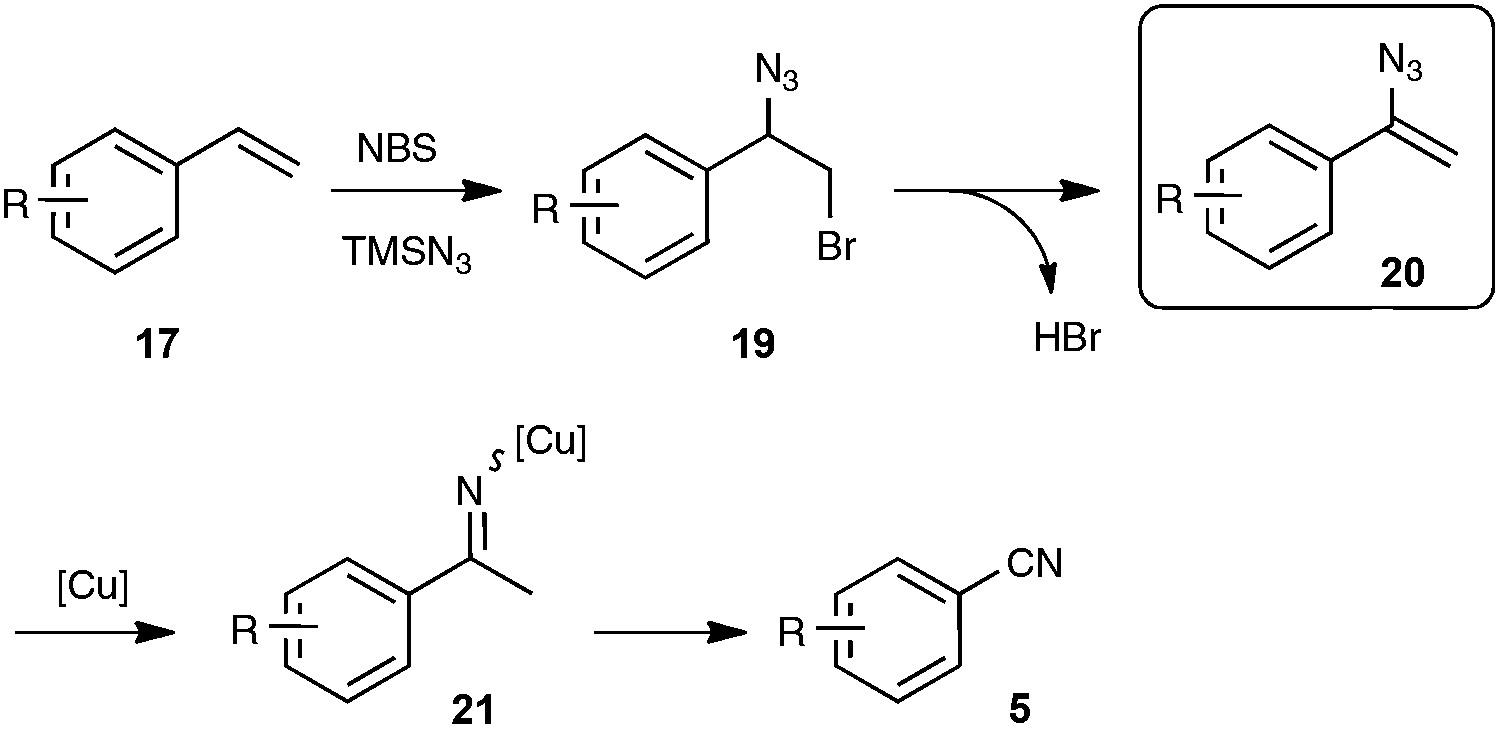 | ||
| Scheme 12 Postulated mechanism for the direct preparation of benzonitriles from styrenyl precursors. | ||
Simultaneous to the Jiao group's efforts aimed at improving the scope of their first work on the synthesis of nitriles through C–C double-bond cleavage, and which highlighted the combination of both NBS and TMSN3, the group of Guo independently developed, for similar purposes, a strategy based on the utilisation of ammonium hydrogenocarbonate as the nitrogen source and PIDA as the oxidizing agent (Scheme 13).22 The few reports in the literature on the utilization of PIDA as the oxidant, in conversion of alkenes to aldehydes, were clearly a source of inspiration for their work.34
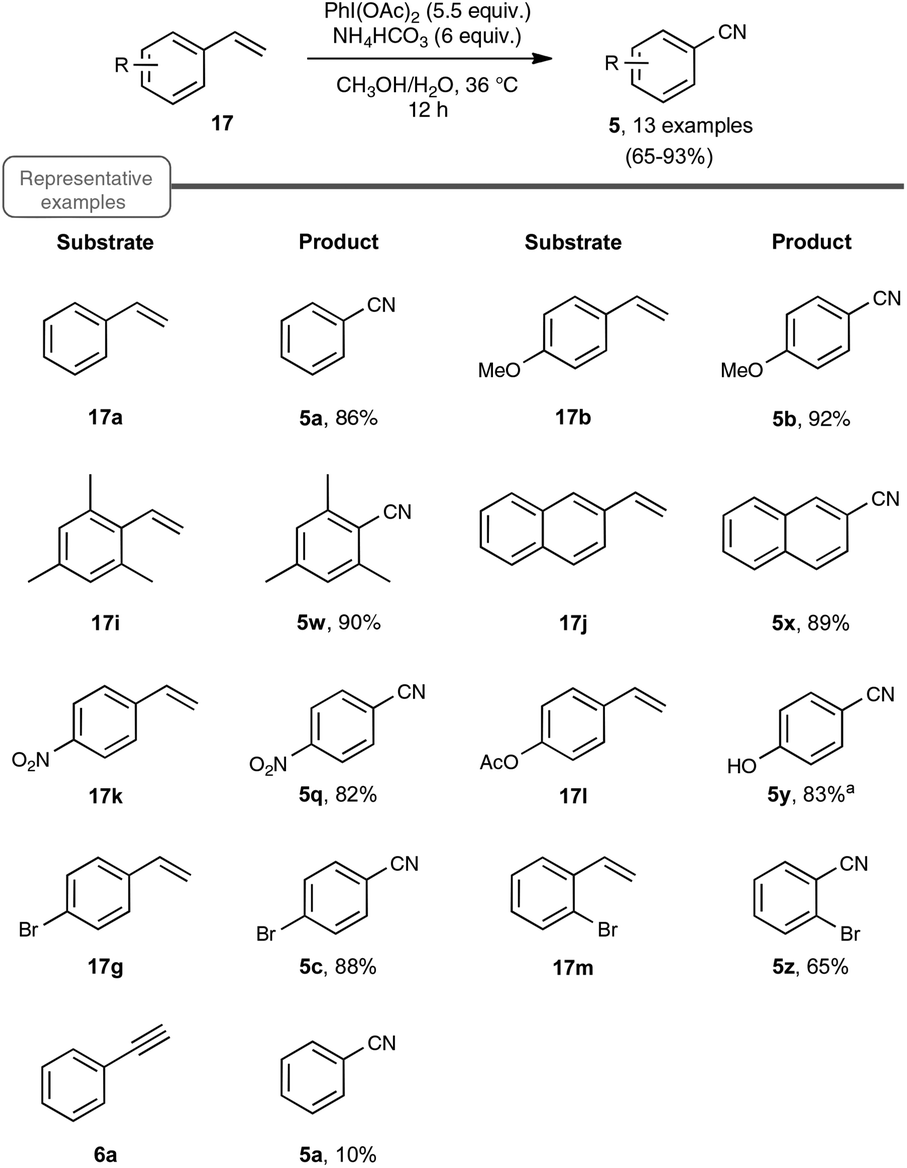 | ||
| Scheme 13 PIDA-mediated transformation of styrenes to nitriles. aLoss of acetate was observed in the corresponding benzonitrile. | ||
Choosing styrene as the test substrate, the optimized conditions involve a mixture of methanol and water as the solvent, 5.5 equiv. of PIDA and 6 equiv. of ammonium hydrogenocarbonate. The reaction performs best at 36 °C, a temperature just above the one at which the ammonium salt starts to decompose into NH3 and CO2. As shown in Scheme 13, the reaction worked very well for a wide variety of styrenes, even with those with electron-withdrawing substituents or with an halide atom on the phenyl ring, which enables a potential application in further functionalization. By curiosity, the authors also ran the reaction with phenylacetylene 6a as the starting material, but obtained in that case benzonitrile with a modest yield of only 10%. While the results of styrenes were impressive, the reaction did not perform satisfactorily with aliphatic alkenes, such as 1-octene.
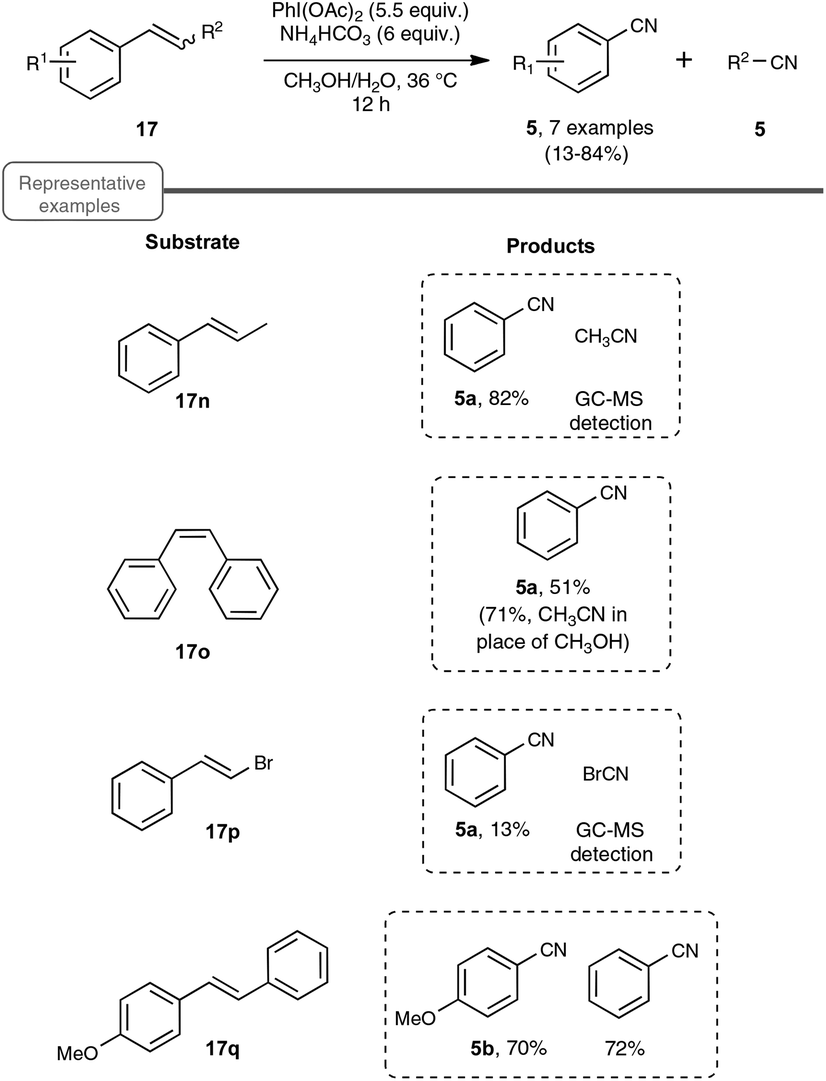
As shown in Scheme 14, the reaction could be extended to a palette of β-substituted styrenes, although the yields in nitriles were more contrasted, ranging from 13 to 84%. No explanation was provided to reflect the differences in yields.
 | ||
| Scheme 14 PIDA-mediated transformation of internal olefins to nitriles. | ||
In order to gain insight into the mechanistic pathway, reaction of styrene was carried out under the optimized conditions, but at the temperature of 0 °C, well under the temperature of decomposition of NH4HCO3 into NH3. Under these conditions, benzaldehyde was isolated in place of benzonitrile after the reaction, providing a solid basis to the mechanism outlined in Scheme 15.
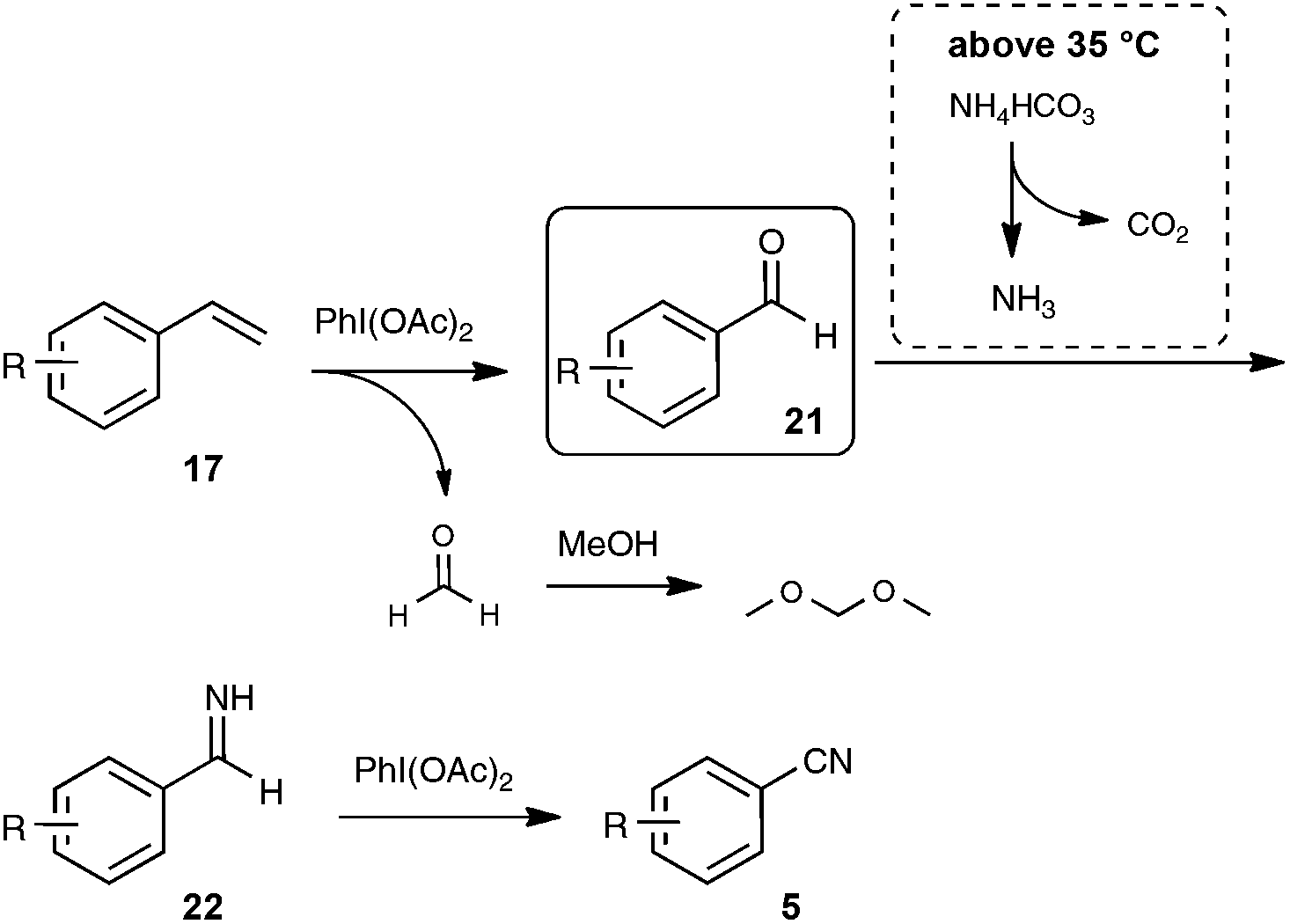 | ||
| Scheme 15 Proposed mechanism for the PIDA-mediated transformation of styrenes to nitriles. | ||
Synthesis of nitriles through carbon–carbon single bond cleavage
In contrast to the previously described transformations, the synthesis, under reasonably mild conditions, of organonitriles through the cleavage of unfunctionalized C–C single bonds is still unreported. Indeed, only C–C bonds equipped with at least one functionalized carbon atom could be cleaved to generate the nitrile function, as very recently exemplified by the preparation presented in Scheme 16 of a wide range of benzonitriles through copper-catalyzed decarboxylation under an O2 atmosphere of phenylacetic acids 23, at the rather forgiving temperature of 100–120 °C.35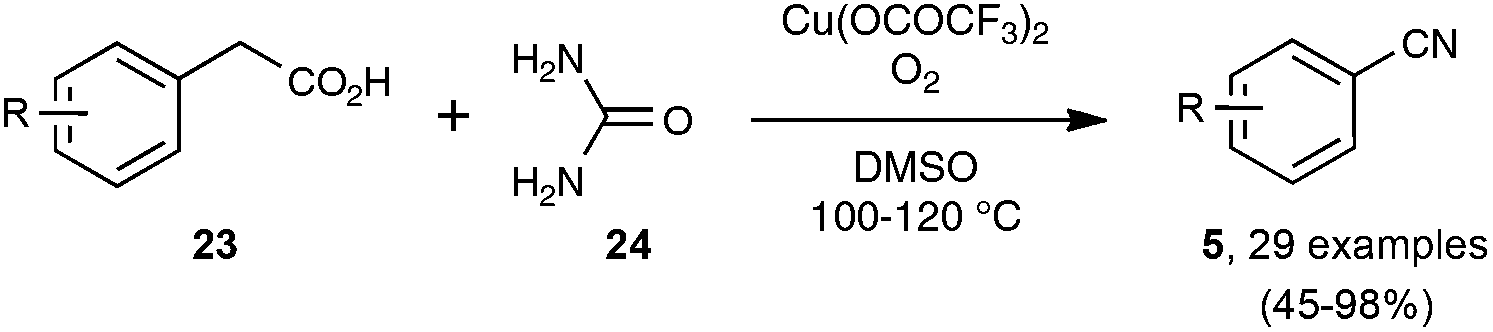 | ||
| Scheme 16 Synthesis of nitriles from phenylacetic acids. | ||
The splitting of non-functionalized C–C bonds surely represents a daunting task, taking into account their marked chemical inertness. It has been accomplished so far in a couple of instances only at high temperatures, such as in the conversion at 620 °C of ethylbenzene into benzonitrile by the ammoxidation procedure.12b In the course of research aimed at reducing the polluting emission of nitrogen trifluoride gas from the manufacture of silicon chips, Belter recently performed the same transformation at 400 °C, by exposing ethylbenzene to excess NF3.36 As outlined in Scheme 17, the author proposed a mechanism based on the thermal splitting of the reacting gas into two fluorinated radicals, which highlights the formation of an unstable fluorimine 27. The fluorimine is then supposed to undergo a Beckmann-type rearrangement, with subsequent fragmentation to deliver the nitrile function. In spite of its obvious limitations linked to the harsh conditions required, this transformation highlights an appealing mechanistic pathway which could be a source of inspiration for the discovery of related reactions under milder conditions.
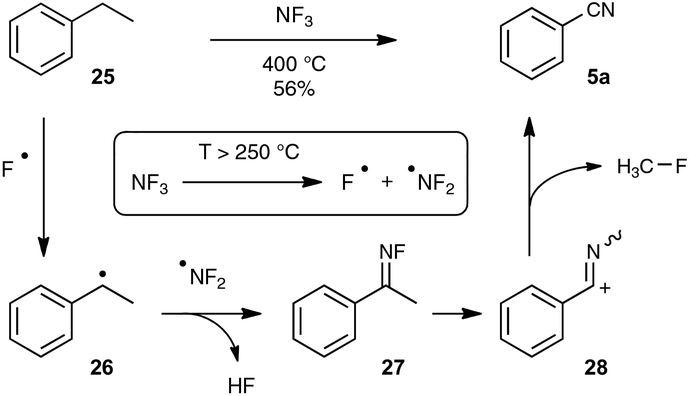 | ||
| Scheme 17 NF3-mediated synthesis of benzonitrile from ethylbenzene. | ||
Conclusion
In the last year or so, an important advancement has been gained in the field of the synthesis of organonitriles, whether aromatic or aliphatic, with the implementation of a new strategy based on the utilization of unfunctionalized olefins or alkynes as “progenitors” of the nitrile function directly through carbon–carbon C–C splitting of their unsaturated bond. This new approach stands in sharp contrast with the synthetic procedures reported so far, which could be essentially grouped either in cyanation procedures implying a carbon chain elongation step of the substrate, or in simple chemical transformations of already functionalized substrates. Particularly with alkynes, the approach to nitriles is quite innovative and may appear even counter-intuitive, if we keep in mind the nearly absence of alkyne transformations via C–C triple bond cleavage, beyond the alkyne metathesis and a few oxygenation procedures. Although the group of Jiao from Peking University was the first to usher into this new era for the synthesis of nitriles, with their seminal paper on the direct transformation of terminal alkynes to nitriles, they were closely followed by Yanada's group from Hokkaido University, who published their independent research activities targeting a similar transformation from internal alkynes as the substrates. The synthesis of nitriles from alkenes also benefited from complementary research activities run nearly at the same time by the group of Jiao, once again, and by Guo's group from Hunan University. In this context, the emergence of the new paradigm for the generation of the nitrile function through the direct cleavage of unfunctionalized carbon–carbon double- or triple bonds reflects, more than a competition, the productive joined efforts between the three teams. Although much work is still needed in order to discover new syntheses of organonitriles through the cleavage of unfunctionalized carbon–carbon C–C single bonds, the results highlighted here should find a wide acceptance from the synthetic community in the near future. They are indeed particularly welcome, taking into account for instance the ever-growing demand for new pharmaceuticals possessing either a nitrile function or those whose synthesis employs an organonitrile as a key intermediate.References
- (a) The chemistry of the cyano group, ed. Z. Rappoport, Wiley, London, 1970 Search PubMed; (b) Comprehensive organic transformations: A guide to functional group preparation, ed. R. C. Larock, VCH, New York, 1989 Search PubMed.
- (a) P. J. Scheuer, Acc. Chem. Res., 1992, 25, 433 CrossRef CAS; (b) F. F. Flemming, Nat. Prod. Rep., 1999, 16, 597 RSC.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed.
- L. J. Browne, C. Gude, H. Rodriguez, R. E. Steele and A. Bhatnager, J. Med. Chem., 1991, 34, 725 CrossRef CAS PubMed.
- A. S. Bhatnagar, Breast Cancer Res. Treat., 2007, 105, 7 CrossRef CAS PubMed.
- E. De Clercq, Expert Opin. Emerg. Drugs, 2005, 10, 241 CrossRef CAS PubMed.
- (a) T. Schareina and M. Beller, in Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Hoboken, 2013, ch. 9 Search PubMed; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (b) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC; (c) Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271 CrossRef CAS.
- T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 RSC.
- (a) For a recent review article see: J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed; (b) S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed.
- (a) C. Czekelius and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 612 CrossRef CAS PubMed; (b) V. N. Telvekar, A. Kulbhushan and K. A. Sasane, Synlett, 2010, 2778 CrossRef CAS; (c) H. Shargi and M. H. Sarvari, Synthesis, 2003, 243 CrossRef.
- (a) For a review article see: B. Lücke, K. V. Narayana, A. Martin and K. Jähnisch, Adv. Synth. Catal., 2004, 346, 1407 CrossRef; (b) P. B. Vorob'ev, T. P. Mikhailovskaya, L. F. Gabdulina and D. K. Sembaev, Russ. J. Gen. Chem., 2000, 70, 1331 Search PubMed.
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137 CrossRef CAS PubMed.
- W. Zhou, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7094 CrossRef CAS PubMed.
- Z. Shu, Y. Ye, Y. Deng, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 10573 CrossRef CAS PubMed.
- D. Tsuchiya, Y. Kawagoe, K. Moriyama and H. Togo, Org. Lett., 2013, 15, 4194 CrossRef CAS PubMed.
- C. Qin and N. Jiao, J. Am. Chem. Soc., 2010, 132, 15893 CrossRef CAS PubMed.
- F. Chen, X. Huang, Y. Cui and N. Jiao, Chem. – Eur. J., 2013, 19, 11199 CrossRef CAS PubMed.
- T. Shen, T. Wang, C. Qin and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 6677 CrossRef CAS PubMed.
- N. Okamoto, M. Ishikura and R. Yanada, Org. Lett., 2013, 15, 2571 CrossRef CAS PubMed.
- C. Qin, T. Shen, C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 6971 CrossRef CAS PubMed.
- J.-H. Xu, Q. Jiang and C.-C. Guo, J. Org. Chem., 2013, 78, 11881 CrossRef CAS PubMed.
- Further transformations of alkynes through the cleavage of their C–C triple bond were recently reported, see: (a) S. K. Rout, S. Guin, A. Gogoi, G. Majji and B. K. Patel, Org. Lett., 2014, 16, 1614 CrossRef CAS PubMed; (b) Q. Jiang, A. Zhao, B. Xu, J. Jia, X. Liu and C. Guo, J. Org. Chem., 2014, 79, 2709 CrossRef CAS PubMed; (c) J. Sun, F. Wang, H. Hu, X. Wang, H. Wu and Y. Liu, J. Org. Chem., 2014, 79, 3992 CrossRef CAS PubMed.
- For selected reviews see: (a) A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC; (b) H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55 RSC; (c) W. Zhang and J. S. Moore, Adv. Synth. Catal., 2007, 349, 93 CrossRef CAS.
- For selected examples, see: (a) T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 6646 CrossRef CAS PubMed; (b) S. Datta, C.-L. Chang, K.-L. Yeh and R.-S. Liu, J. Am. Chem. Soc., 2003, 125, 6646 CrossRef PubMed; (c) A. Wang and H. Jiang, J. Am. Chem. Soc., 2008, 130, 5030 CrossRef CAS PubMed.
- C. J. Woltermann and H. Shechter, Helv. Chim. Acta, 2005, 88, 354 CrossRef CAS.
- K. Banert, J. R. Fotsing, M. Hagedorn, H. P. Reisenauer and G. Maier, Tetrahedron, 2008, 64, 5645 CrossRef CAS.
- (a) C. R. Alonso-Cruz, A. R. Kennedy, M. S. Rodríguez and E. Suárez, Tetrahedron Lett., 2007, 48, 7207 CrossRef CAS; (b) C. R. Alonso-Cruz, A. R. Kennedy, M. S. Rodríguez and E. Suárez, J. Org. Chem., 2008, 73, 4116 CrossRef CAS PubMed; (c) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 12169 CrossRef CAS PubMed.
- T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692 CrossRef CAS PubMed.
- R. Yokoyama, S. Matsumoto, S. Nomura, T. Higaki, T. Yokoyama and S.-I. Kiyooka, Tetrahedron, 2009, 65, 6181 CrossRef.
- (a) A. Hassner and F. W. Fowler, J. Org. Chem., 1968, 33, 2686 CrossRef CAS; (b) A. Hassner and F. Boerwinkle, J. Am. Chem. Soc., 1968, 90, 216 CrossRef CAS.
- X. Zong, Q.-Z. Zheng and N. Jiao, Org. Biomol. Chem., 2014, 12, 1198 CAS.
- S. Chiba, L. Zhang, G. Y. Ang and B. W.-Q. Hui, Org. Lett., 2010, 12, 2052 CrossRef CAS PubMed.
- See for example: K. C. Nicolaou, V. A. Adsool and C. R. H. Hale, Org. Lett., 2010, 12, 1552 CrossRef CAS PubMed.
- Q. Feng and Q. Song, Adv. Synth. Catal., 2014, 356, 1697 CrossRef CAS.
- R. K. Belter, J. Fluorine Chem., 2011, 132, 318 CrossRef CAS.
Footnote |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| This journal is © the Partner Organisations 2014 |