Sonogashira coupling in natural product synthesis
Dan
Wang
and
Shuanhu
Gao
*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China. E-mail: shgao@chem.ecnu.edu.cn; Fax: (+)86-21-62604784; Tel: (+)86-21-62604784
First published on 20th March 2014
Abstract
This review will focus on selected applications of Sonogashira coupling and subsequent transformations as key steps in the total synthesis of natural products. A brief introduction to the history and development of Sonogashira coupling will be presented. The organization of the synthetic applications is based on the structure of target molecules and the transformations followed by the Sonogashira coupling, which includes (1) preparation of natural products containing conjugated enynes or enediynes; (2) Sonogashira coupling followed by stereoselective reduction and (3) Sonogashira coupling followed by regioselective annulations.
 Dan Wang | Dan Wang was born in Hubei Province, China, in 1989. She received her bachelor's degree in 2012 from Lanzhou University. In 2012, she joined Prof. Shuanhu Gao's group to pursue her doctoral studies at the Department of Chemistry, East China Normal University. Her research projects focus on total synthesis of bioactive natural products. |
 Shuanhu Gao | Shuanhu Gao was born in Ningxia Province, China, in 1979. He received his B.S. degree from Lanzhou University in 2001. In 2006 he earned his Ph.D. from Lanzhou University under the direction of Professor Yongqiang Tu. From 2007 to 2010, he was a postdoctoral fellow in Professor Chuo Chen's group at the UT Southwestern Medical Center at Dallas. He started his independent career as a professor in the Department of Chemistry at East China Normal University in October 2010. His current research interests are primarily focused on the synthesis of complex natural products and medicinal chemistry. |
1. Introduction
Significant advances have been made over the past four decades in palladium (Pd)-catalyzed cross-coupling reactions to form carbon–carbon (C–C) bonds.1 As a result, Pd-catalyzed inter- and intramolecular reactions and cyclizations are widely used to synthesize natural products, therapeutic agents and organic materials.2 These reactions dramatically improve the efficiency of organic syntheses, such as those involving the formation of sp–sp2 bonds. These bonds occur in arylalkynes, conjugated enynes and enediynes, synthons that occur in such natural products as phorbaside A (1),3 pyrrhoxanthin (2),4 dynemicin A (3),5 kedarcidin chromophore (4)6 and calicheamicin γ1 (5)7 (Fig. 1). The value of such natural products as medicinal chemistry targets and as inspiration for methodological advances makes sp–sp2 bond formation particularly important in organic synthesis.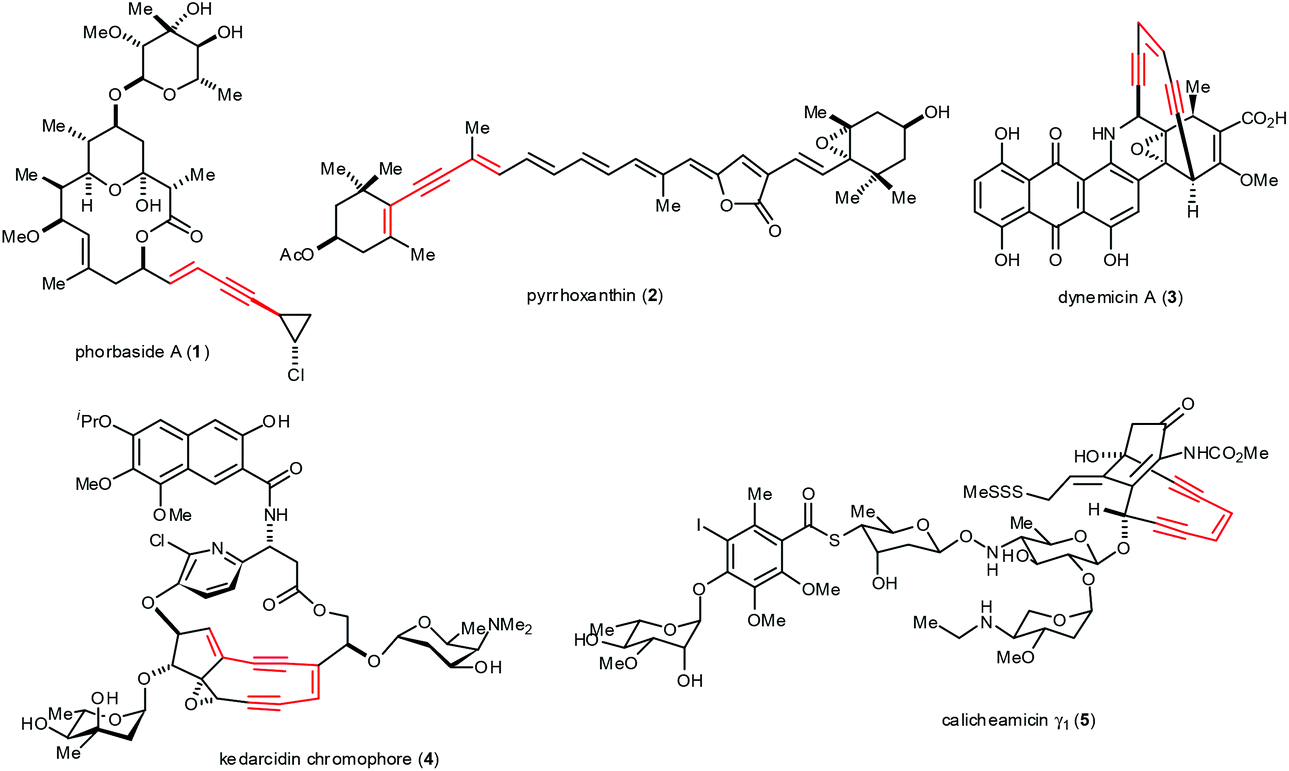 | ||
| Fig. 1 Selective natural products containing conjugated enynes. | ||
In 1975, Heck8 and Cassar9 independently reported the Pd-catalyzed arylation and alkenylation of alkynes in the presence of organic or inorganic bases at temperatures around 100 °C. Subsequently, Sonogashira and Hagihara demonstrated that adding a catalytic amount of copper(I) iodide dramatically improved the rate of this alkynylation, even allowing the cross-coupling to be performed at room temperature.10 The core of the Sonogashira–Hagihara protocol, known as Sonogashira coupling, has become a generally accepted method; in this reaction, Pd and Cu co-catalyze the construction of sp–sp2 bonds involving terminal alkynes and aryl or alkenyl halides or triflates (Scheme 1). Since its discovery, Sonogashira coupling has been extensively studied in an effort to understand its mechanism as well as explore different catalysts, ligands, and additives.2
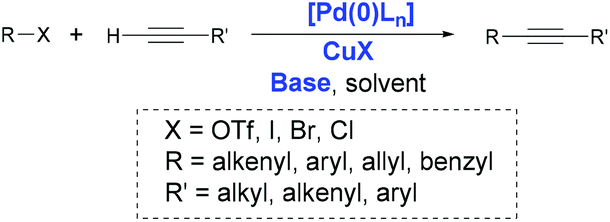 | ||
| Scheme 1 Sonogashira coupling. | ||
Despite these efforts, the mechanism of Pd/Cu-cocatalyzed cross-coupling remains unclear. It is assumed to proceed through the combination of Pd-catalyzed and Cu-catalyzed cycles (Scheme 2). In the Pd-catalyzed cycle, in situ reduction of Pd(II) complexes may give rise to a catalytically active species Pd(0)L2, which facilitates the fast oxidative addition of R–X. The rate of oxidative addition of R–X depends on the electronic properties of the R–X bond as well as on the reactivity of X (I ≥ OTf ≥ Br > Cl). Normally, electron-withdrawing groups activate the R–X bond by decreasing its electron density. Next, in the Cu-catalyzed cycle, the copper acetylide undergoes transmetalation. This step, which is the rate-determining reaction in the overall coupling, generates a RPd(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR′)L2 species, which then undergoes reductive elimination to yield the final coupled alkyne and regenerate the Pd(0)L2 catalyst.
CR′)L2 species, which then undergoes reductive elimination to yield the final coupled alkyne and regenerate the Pd(0)L2 catalyst.
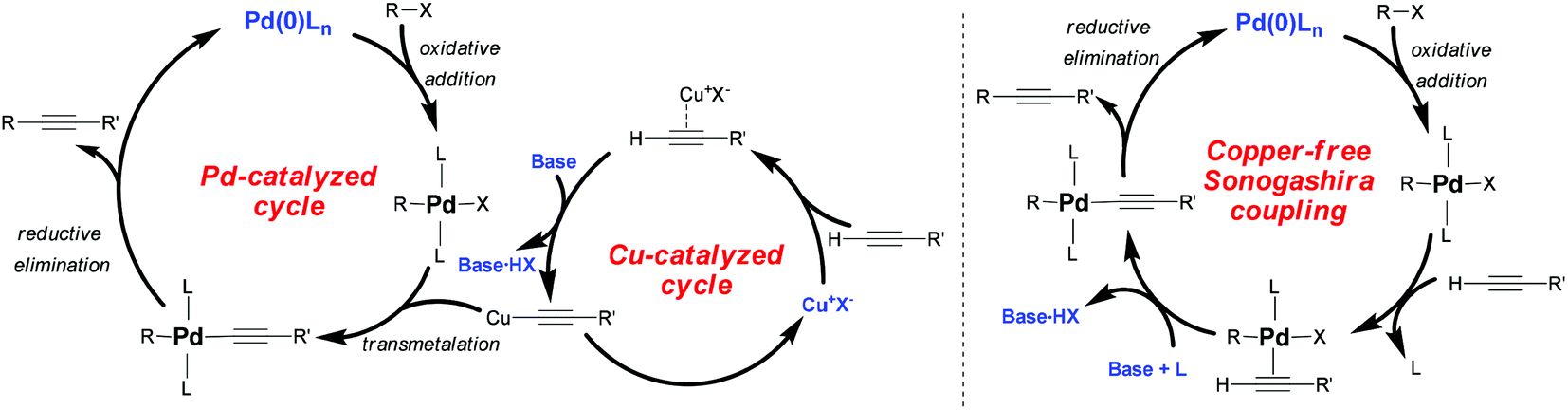 | ||
| Scheme 2 The proposed mechanism of Sonogashira coupling. | ||
One problem with Pd/Cu-cocatalyzed Sonogashira coupling is that the alkyne can undergo homocoupling in the presence of oxygen via the Hay/Glaser reaction.11 To avoid this side reaction, researchers have explored Cu-free Sonogashira coupling.2 While the mechanism of this reaction is poorly understood, it is thought that reversible π-coordination and deprotonation, analogous to those in the Cu-catalyzed cycle of Pd/Cu-cocatalyzed coupling, generate an alkyne–Pd(II) complex [RPd(–CCR′)L2].
Sonogashira coupling efficiently produces arylalkynes and conjugated enynes that can participate in a broad variety of transformations. For example, reduction or regioselective annulation of Sonogashira products can generate diverse building blocks and heterocycles, such as alkyl, alkene, benzofuran, indole, oxyindole, isoquinolinone, isochromenone systems and related structures (Scheme 3). Comprehensive reviews of the Sonogashira reaction which covered the catalyst system, mechanism studies and applications have been published in recent years,2 which indicated a trend of its rapid expansion and applications. In this review, we focus on selected applications of Sonogashira coupling and subsequent transformations as key steps in the total synthesis of natural products.
 | ||
| Scheme 3 Transformations of arylalkynes and conjugated enynes generated by Sonogashira coupling. | ||
2. Synthetic applications
Efficiency is essential in organic synthesis, particularly natural product synthesis. Designing efficient reactions means satisfying several constraints, including the need for chemoselectivity,12 atom economy,13 step economy14 and redox economy.15 Achieving all these things is made more challenging by the fact that total synthesis of complex natural products nearly always requires several steps.16,17 In many cases, these steps can be carried out by a straightforward linear procedure, but this sometimes leads to low yields because of relatively poor material balance and supply, which suppress its practicability.17 In these and other cases, a convergent approach can prove much more efficient than a linear one because it couples the synthesis of individual fragments. Its advantages include: (1) convergent synthesis is usually simpler to execute than linear synthesis; (2) it involves better material balance and supply and (3) it gives higher overall yields.17 On the other hand, convergent synthesis requires designing effective coupling steps involving efficient reactions that are tolerant of diverse functional groups. Sonogashira couplings enable convergent synthesis strategies, which are of value due to their advantages in efficiency over more linear synthesis strategies. Here we describe examples where the Sonogashira reaction serves as a key coupling step in the convergent synthesis of natural products.2.1. Natural products containing conjugated enynes
Natural products containing conjugated enynes or enediynes have attracted considerable attention from organic chemists and biologists because of their sensitive structural features and their potential as bioactive compounds and as starting compounds in chemical biology studies.3–7 The total synthesis of these natural products is challenging mostly because of the difficulty in efficiently constructing the sensitive motif containing the conjugated enynes and enediynes. Inter- or intramolecular Sonogashira reactions to construct the sp–sp2 bond have proven useful for achieving this step. In pioneering work in 1992, Nicolaou and co-workers demonstrated the power of Sonogashira coupling in the total synthesis of calicheamicin γ1 (5).7 The Pd/Cu-cocatalyzed coupling of alkyne 6 with vinyl chloride 7 gave the enediyne product 8 in 91% yield (Scheme 4). This reaction proceeded smoothly under mild conditions at low temperature, preserving the sensitive protecting groups as well as the Z conformation of the alkene.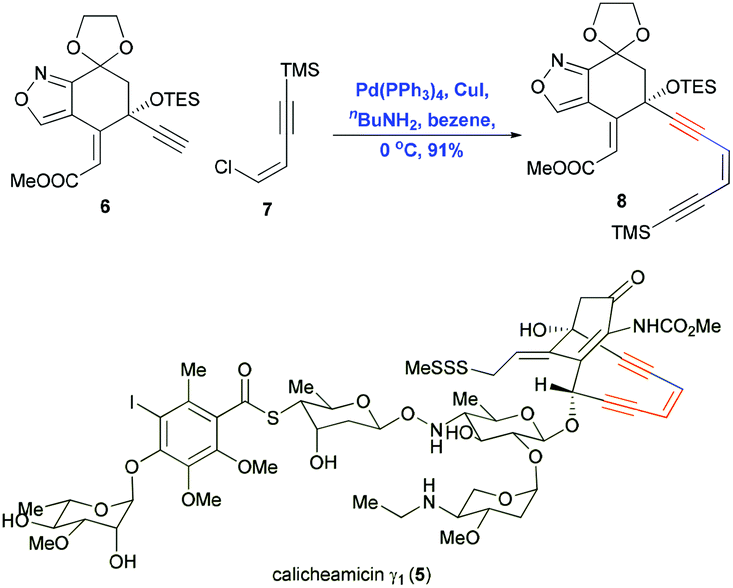 | ||
| Scheme 4 Construction of the enediyne motif using a Sonogashira coupling in the total synthesis of calicheamicin γ1 (5). | ||
Schreiber and co-workers performed Sonogashira coupling followed by a transannular Diels–Alder reaction to construct the core skeleton of dynemicin A (3),5 a potent enediyne-containing antibiotic. Coupling enediyne 9 with vinyl bromide 10 in the presence of Pd(PPh3)4 and CuI gave the polyunsaturated ester 11, which was transformed through alkaline hydrolysis into the corresponding carboxylic acid 12 (Scheme 5). Subsequent Yamaguchi macrocyclization gave the pentacyclic product 14 in 50% yield. The proposed mechanism involves formation of the macrocyclic ring 13, followed by a spontaneous transannular Diels–Alder reaction. Dramatically, Schreiber and co-workers showed that intramolecular Sonogashira coupling of 15 in the presence of Pd(PPh3)4 and CuI yielded 14 directly in 25% yield. This impressive one-pot cascade reaction efficiently constructed three rings with a highly strained enediyne motif and four contiguous stereocenters.
 | ||
| Scheme 5 Sonogashira coupling followed by the transannular Diels–Alder reaction to construct the core skeleton of dynemicin A. | ||
Sonogashira coupling also proved useful for synthesizing the highly complex maduropeptin chromophore (21), which contains a bicyclo[7.3.0]enediyne and a 15-membered ansa-macrolactam with atropisomerism. This compound exhibits potent antitumor and antibacterial activity through a mechanism that differs from that of other enediyne natural products. In 2007, Inoue and co-workers used a convergent approach including Sonogashira coupling to synthesize the aglycon of 21 (Scheme 6).18 Coupling vinyl triflate 16 with the densely functionalized terminal alkyne 17 under mild Pd/Cu-cocatalytic conditions yielded the corresponding product 18 in quantitative yield, and this was converted to aldehyde 19 through deprotection and oxidation. Remarkably, scaling up this Sonogashira coupling (7.35 mmol) gave excellent yield in only 20 min, helping ensure sufficient substrate for the next step of the synthesis. Ring closure of the strained nine-membered diyne ring was achieved by acetylide–aldehyde condensation promoted by LiN(SiMe2Ph)2 and CeCl3.
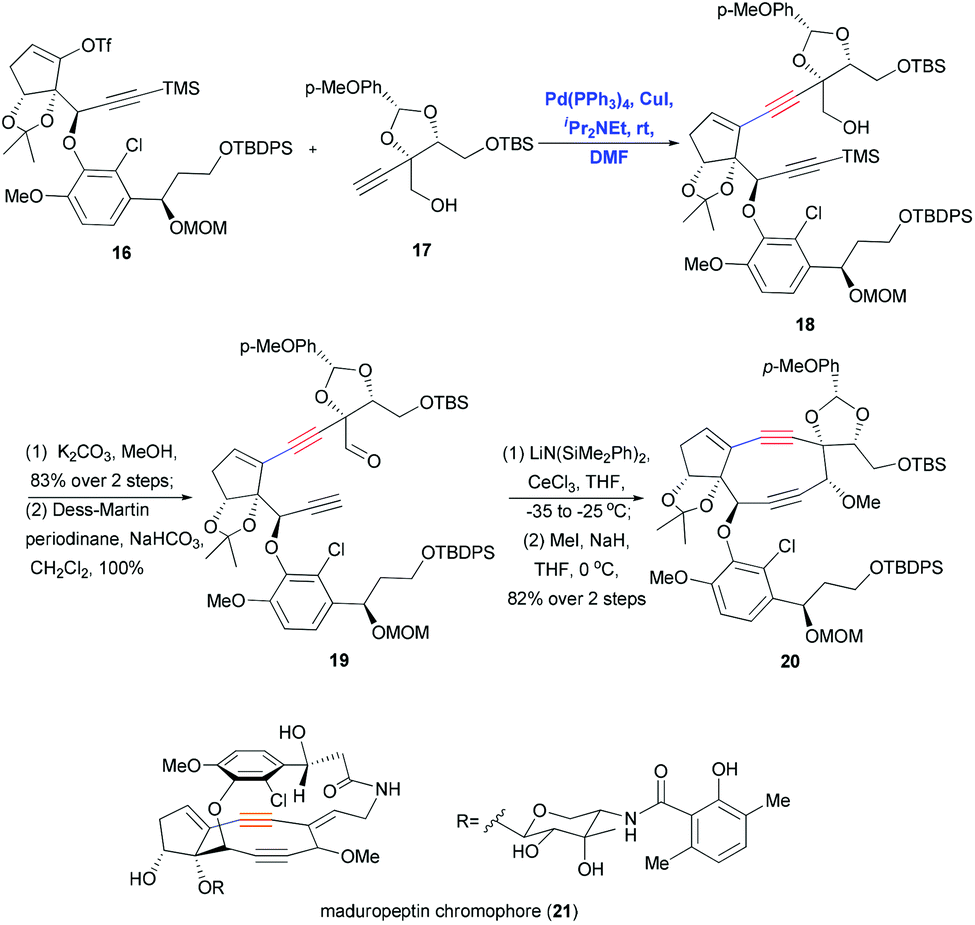 | ||
| Scheme 6 Sonogashira coupling of fragments to synthesize the aglycon of maduropeptin chromophore (21). | ||
The Sonogashira reaction is also used as a key coupling step in the convergent syntheses of pyrrhoxanthin (2)4 and callipeltoside A (28),3 both of which contain conjugated enynes (Scheme 7). Coupling a trisubstituted dienyl iodide 22 with terminal alkyne 23 in the presence of Pd(PPh3)4 and CuI in nBuNH2 in benzene gave the desired product 24 in 56% yield. In contrast, reacting dienyl iodide 25 with terminal alkyne 26 under the same conditions failed to provide the desired product. Andrus et al. reported the same problem during the synthesis of stipiamide and related polyenes.19 Those authors screened reaction conditions of the Sonogashira coupling and found Pd(II) chloride [(PPh3)2PdCl2] to be a much better catalyst than Pd(PPh3)4, especially for couplings involving the electron-deficient alkynyl amides. Using this improved catalyst together with CuI and iPr2NH in ethyl acetate, they obtained the protected aglycon 27 in 93% yield with retention of double-bond geometry.
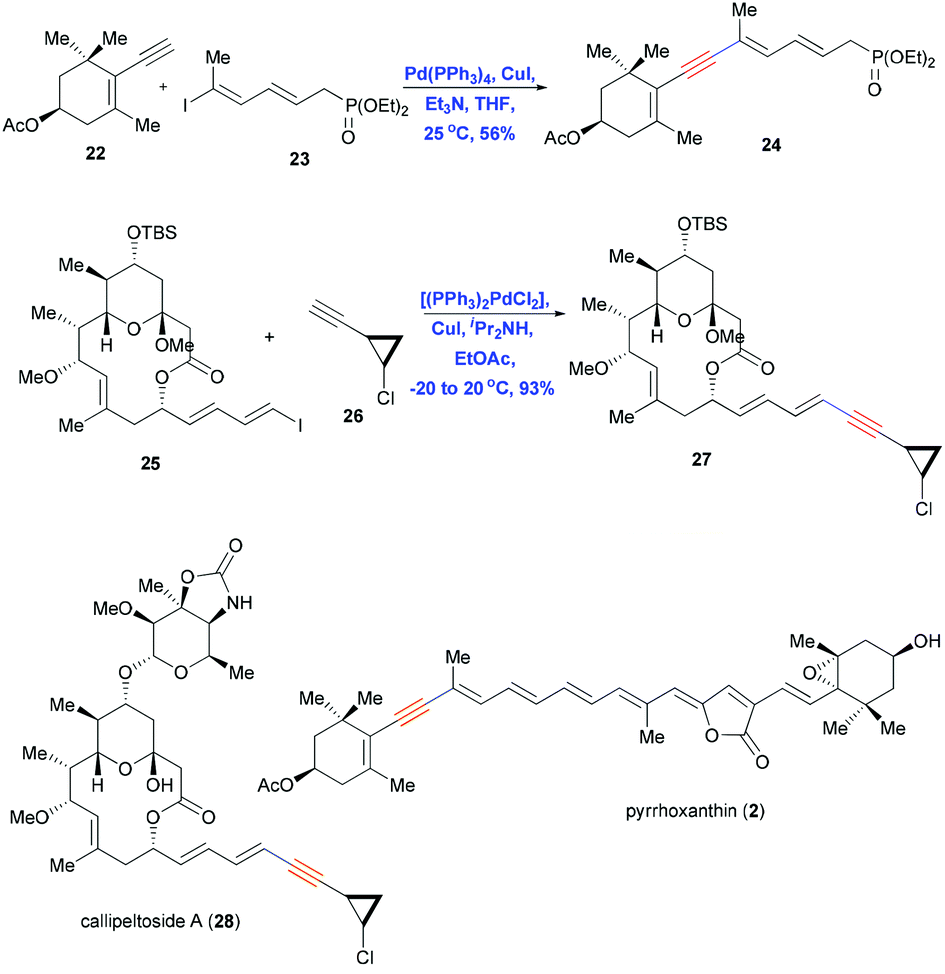 | ||
| Scheme 7 Sonogashira coupling in the total syntheses of pyrrhoxanthin (2) and callipeltoside A (28). | ||
2.2. Sonogashira coupling followed by reduction
Since Sonogashira coupling provides a reliable and effective method of constructing inter- and intramolecular sp–sp2 C–C bonds, it can be combined with reduction reactions to convert the triple bonds of alkyne–alkene systems into sp2–sp2, sp2–sp3 and sp3–sp3 bonds in a regio- and stereoselective fashion. In addition, reaction conditions can be adjusted to control the E or Z geometry of the alkene. This combination of Sonogashira coupling followed by stereoselective reduction has been used in many convergent syntheses of natural products, including polyketides, alkaloids and polycyclic xanthones.In 2004, Wipf and co-workers used a convergent, stereoselective approach to achieve the total synthesis of disorazole C1 (35),20 which has significant cytotoxic and antitubulin activities at nanomolar concentrations. To generate 35, which has a dimeric structure composed of a macrocyclic–heterocyclic ring with labile polyene systems (E, Z, Z), the authors assembled four fragments using double Sonogashira coupling, esterification and Yamaguchi macrocyclization (Scheme 8). Coupling terminal alkyne 29 with vinyl iodide 30 in the presence of (PPh3)2PdCl2 and CuI gave the coupling product in 94% yield, which was then esterified with fragment 32 to achieve vinyl iodide 33. A second Sonogashira coupling under the same conditions was performed to introduce segment 29. A subsequent series of simple transformations, including saponification, Yamaguchi lactonization, deprotection and stereoselective reduction, completed the total synthesis of 35. Notably, this approach allowed the labile triene to remain protected until late in the synthesis, when the Lindlar catalyst was used to construct Z olefins stereoselectively.
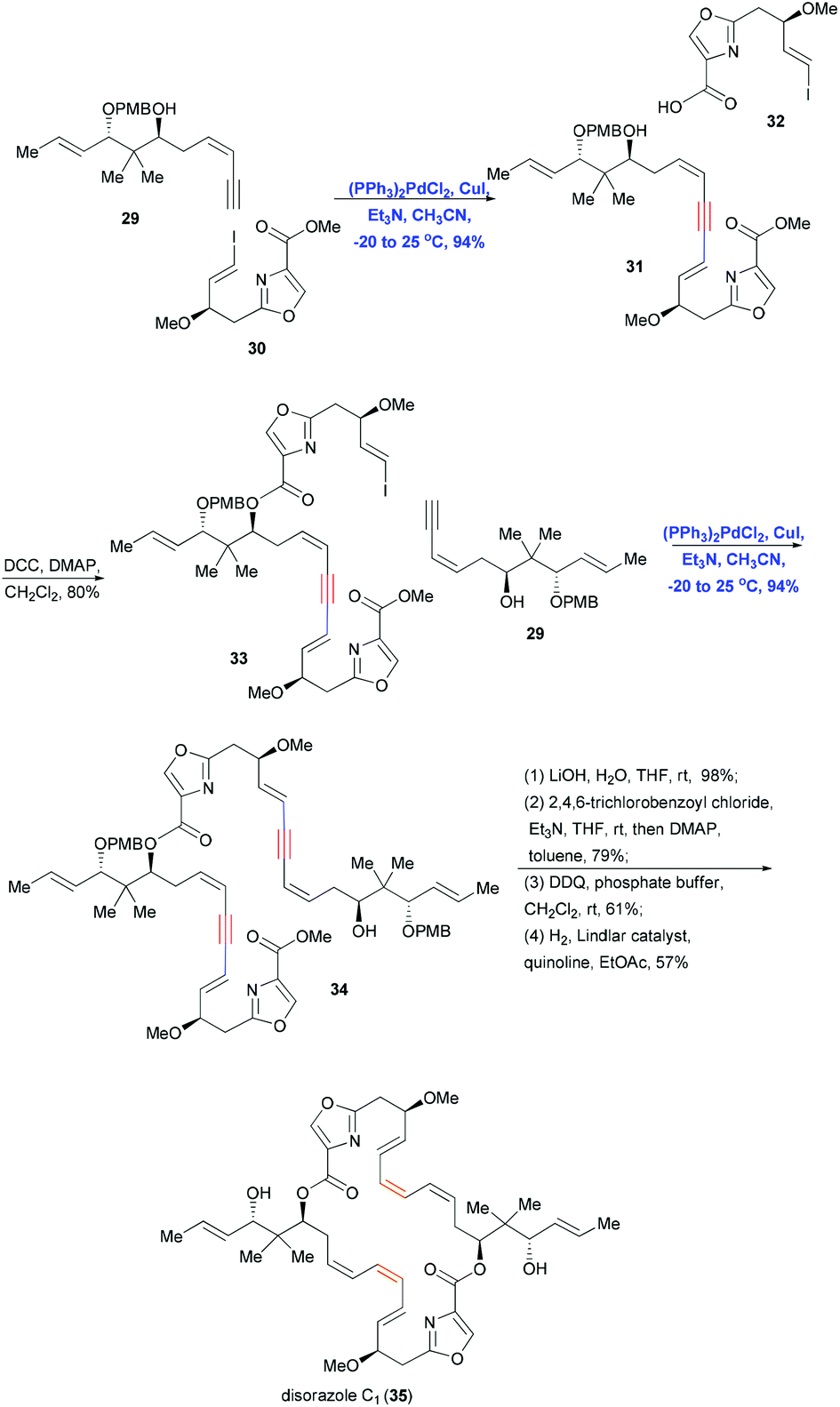 | ||
| Scheme 8 Convergent total synthesis of disorazole C1 (35). | ||
Intramolecular Sonogashira coupling has been reported less often in total syntheses, partly because it is effective only for constructing macrocycles with more than approximately 15 members as the conjugated enyne produces ring strain. One of the few examples of intramolecular Sonogashira coupling in total synthesis is the strategy reported by Mohapatra and co-workers in 2008 for making penarolide sulfate A1 (41) (Scheme 9).21 Fragments 36 and 37 were connected in high yield using EDC/HOBt-mediated coupling, and then an intramolecular Sonogashira reaction catalyzed by Pd(PPh3)4 and CuI in Et2NH solvent furnished the 30-membered macrocyclic ring. Finally the conjugated enyne was hydrogenated using RANEY® Ni in ethanol to generate the saturated ring system.
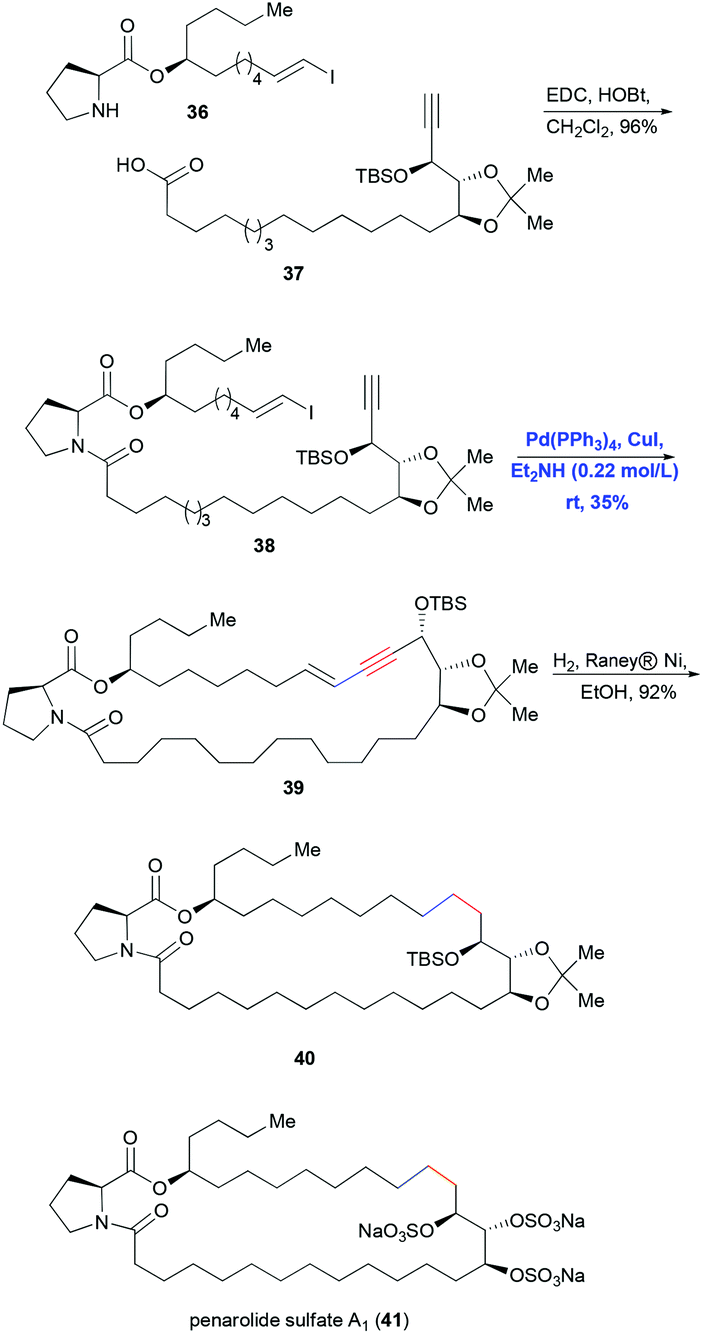 | ||
| Scheme 9 Total synthesis of penarolide sulfate A1 (41) using intramolecular Sonogashira coupling. | ||
Selectivity of the Sonogashira coupling of terminal alkynes to different electrophilic groups is normally controlled by adjusting the reactivity of the C–X bond (I ≥ OTf ≥ Br > Cl). If the steric effects of specific substrates are adjusted as well, the coupling can be carried out in a stepwise, regioselective fashion. One example is the total synthesis of desmosine (50) reported by Usuki and co-workers in 2012.22 This compound contains four amino acid side chains of different lengths. In their approach, those authors performed the regioselective Sonogashira coupling of 42 or 43 with terminal alkyne 44 at the 3- and 5-positions in the presence of Pd(PPh3)4 and CuI in DMF and iPr2NEt (Scheme 10). Oxidative addition of I or Br at the C-4 position was avoided because of steric hindrance from the neighboring 3,5-diiodo groups. For the same reason, achieving the third Sonogashira coupling at the sterically hindered C-4 required a more reactive catalyst system. The authors created this catalyst by reacting Pd2(dba)3 with the electron-rich phosphine ligand P(2-furyl)3 in the presence of CuI in DMF and iPr2NEt. This catalyst facilitated the conversion of either 45 or 46 into the desired 3,4,5-trialkynyl pyridine 48 in good yield. Finally, Pd/C-catalyzed hydrogenation saturated the trialkynyl motif, generating the corresponding alkyl side chain.
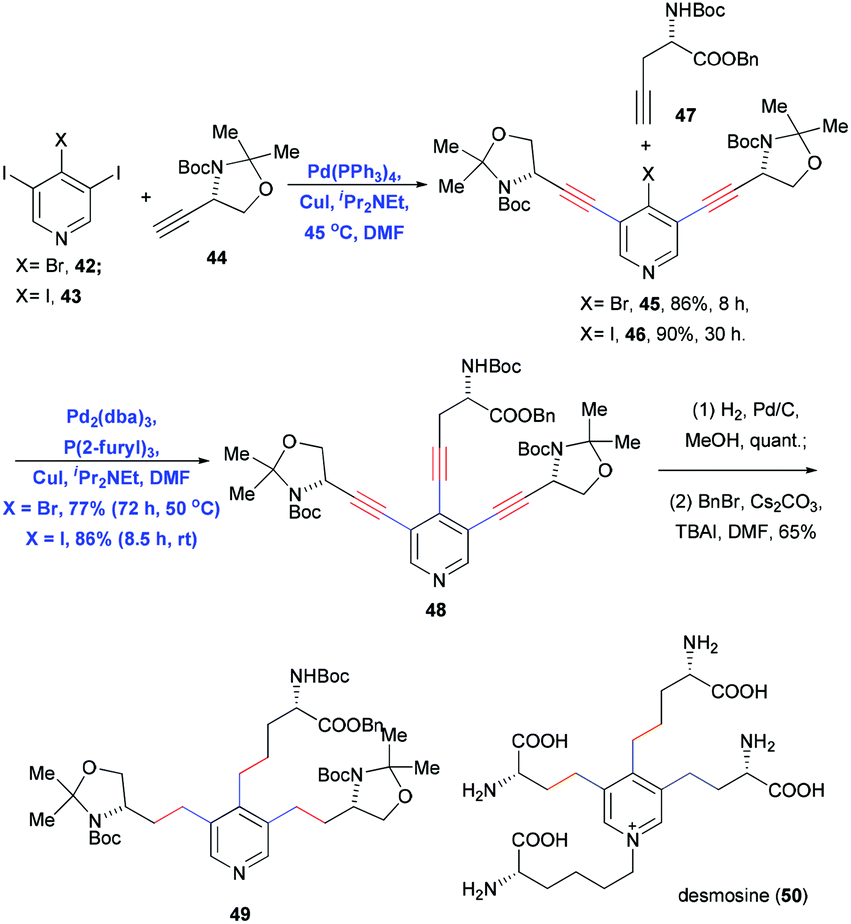 | ||
| Scheme 10 Total synthesis of desmosine (50). | ||
Regioselective Sonogashira coupling was also used by Baran and Shenvi in 2006 to combine the doubly halogenated indole 51 and imidazole-containing alkyne 52 in the total synthesis of chartelline C (55).23 The active C–I bond facilitated the Pd/Cu-cocatalyzed cross-coupling and gave the desired indole–imidazole product 53 in 85% yield, leaving the C–Br bond intact (Scheme 11). These results indicate that Sonogashira coupling can tolerate imidazoles, which should be useful for synthesizing alkaloids. The alkyne motif was then selectively reduced with RANEY® nickel to olefin 54 with Z geometry and subsequently transformed by macrocyclization.
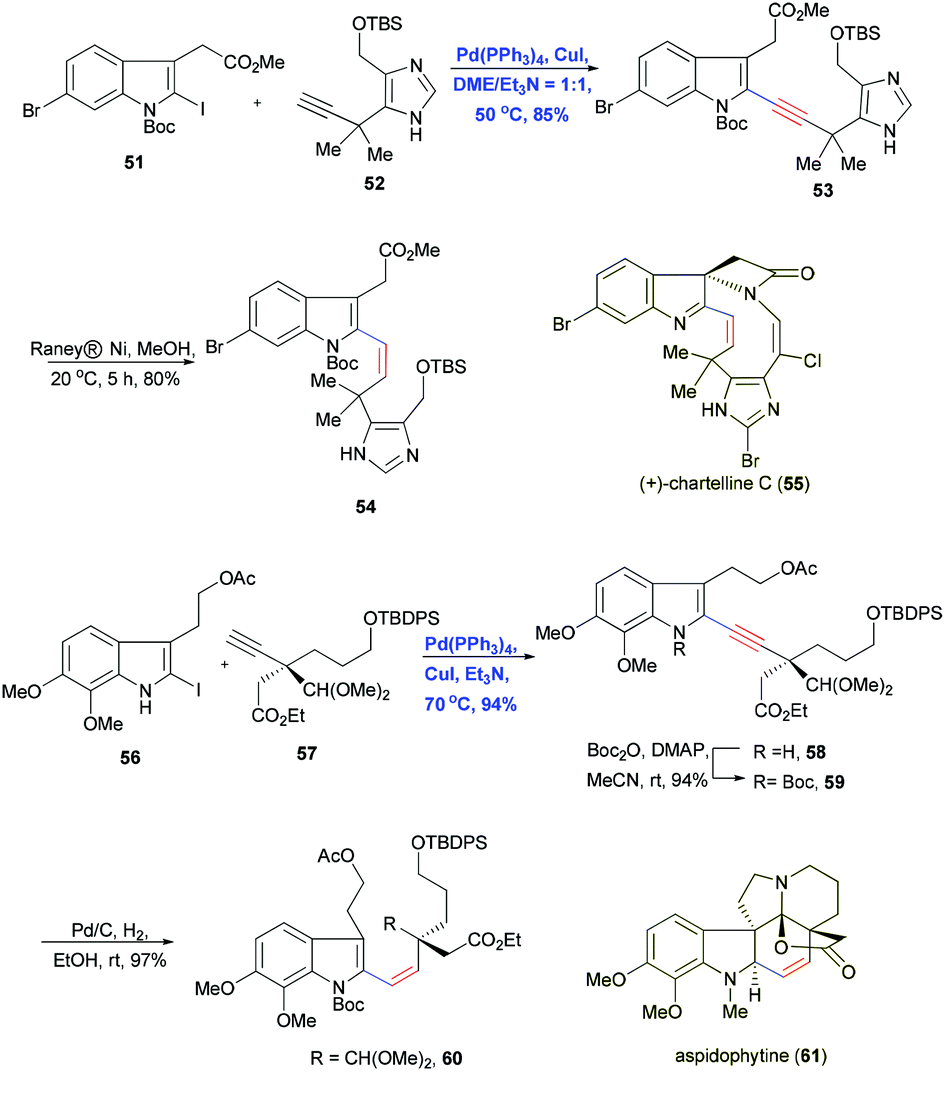 | ||
| Scheme 11 Total synthesis of chartelline C (55) and aspidophytine (61). | ||
Another example of Sonogashira coupling followed by stereoselective reduction was reported by Tokuyama and Fukuyama and co-workers in 2003 with the convergent total synthesis of indole-type alkaloid aspidophytine (61).24 Pd(PPh3)4 and CuI in Et3N solvent reliably cross-coupled 56 and terminal alkyne 57, which contains an all-carbon quaternary center, furnished the corresponding product in 94% yield (Scheme 11). The conjugated enyne allowed subsequent construction of the Z olefin in aspidophytine (61).
Silyl acetylene carrying either the TMS or TIPS group is a two-carbon alkyne building block that can act as a mono- or double cross-coupling component to elongate a carbon chain or couple fragments. This building block has been used by Ready and co-workers in the total synthesis of kibdelone C (71) (Scheme 12).25 Cross-coupling of 62 and (triisopropyl)acetylene 63 followed by desilylation generated another terminal alkyne 65 in 85% yield. A second Sonogashira coupling of 65 with 66 gave the desired coupling product in nearly quantitative yield; this reaction was carried out in the absence of Cu and with slow addition of the alkyne in order to avoid oxidative dimerization. Subsequent hydrogenation, iodination and protection generated 69, which underwent ring closure via intramolecular C–H bond arylation.
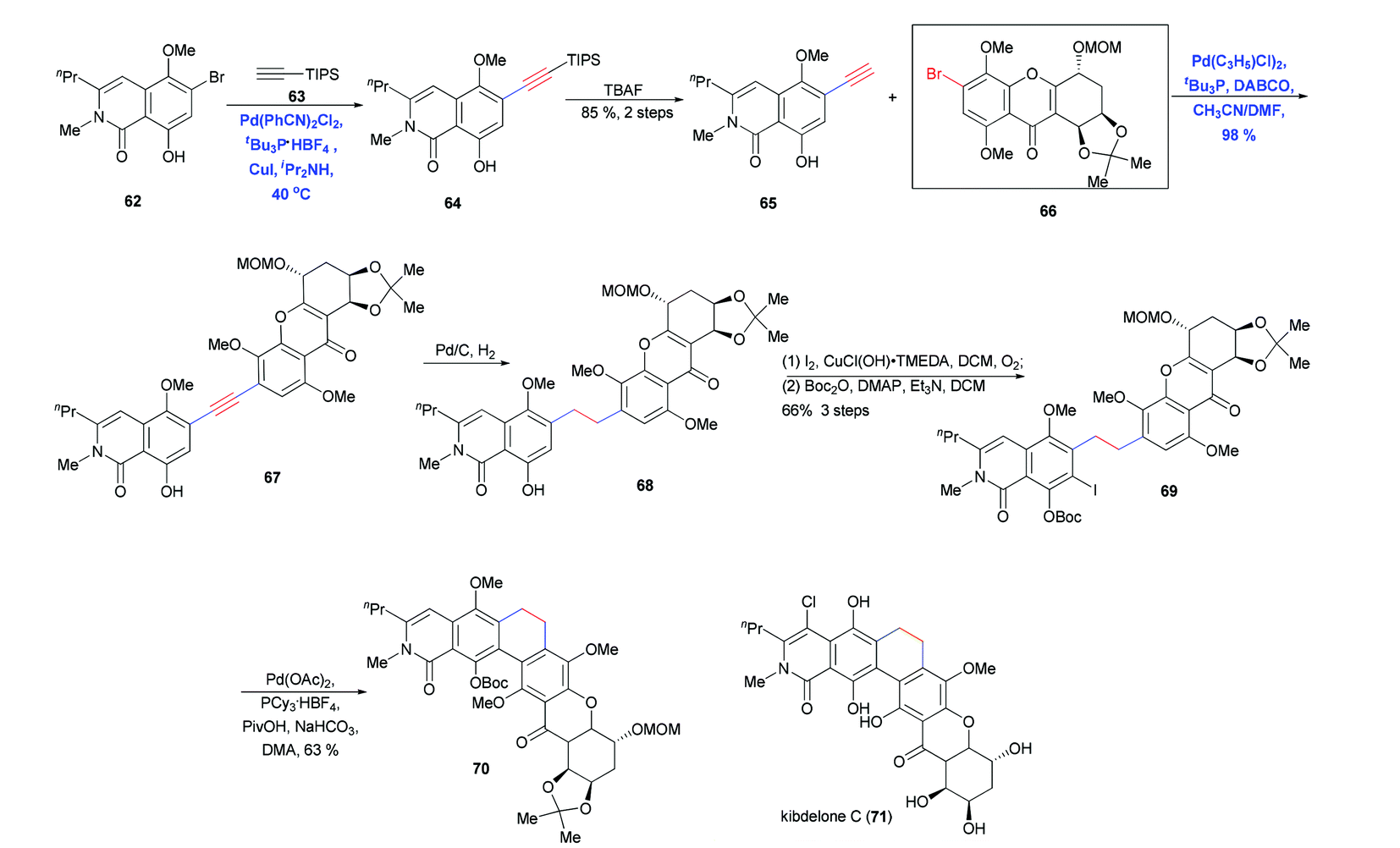 | ||
| Scheme 12 Total synthesis of kibdelone C (71). | ||
2.3. Sonogashira coupling followed by regioselective annulation
Heterocycles are ubiquitous in natural products, pharmaceuticals, dyes and organic materials. “Of the more than 20 million chemical compounds currently registered, about one half contain heterocyclic systems.”26 As a result, the efficient construction of heterocycles is of great significance in organic synthesis. Sonogashira coupling to generate substituted alkynes followed by their regioselective hetero-annulation can be used to construct furan, benzofuran, indole, oxyindole, isoquinolinone, isochromenone rings and related structures. An even greater diversity of hetero-annulation products can be achieved by adding a base to increase the nucleophilicity of hetero-nucleophiles or by using transition metal catalysts to activate the alkynes. Here we describe some examples of Sonogashira coupling in the total synthesis of natural products in which the alkyne is transformed into a heterocyclic ring.In 2006, Pan, She and co-workers reported using Sonogashira coupling followed by regioselective hetero-annulation to construct a benzofuran ring as part of the asymmetric synthesis of (+)-machaeriol D (76).27 Cross-coupling 72 with ortho-iodophenyl acetate 73 in the presence of Pd–Cu cocatalyst gave intermediate 74 in 89% yield (Scheme 13). Subsequent saponification and cyclization of phenolic acetate 74 afforded the desired benzofuran 75 in high yield. A similar example is the total synthesis of frondosin B reported by Danishefsky and co-workers,28 who used a one-pot, Pd/Cu-cocatalyzed Sonogashira coupling and hetero-annulation sequence to combine a terminal alkyne and ortho-iodophenol. Coster and co-workers used this approach in the formal total synthesis of lithospermic acid (82) (Scheme 13).29 In this synthesis, Sonogashira coupling of fragments 77 and 78 generated 79 in good yield, during which the aldehyde group was protected as acetal. Deacetylation was carefully performed at low temperature to avoid direct annulation. Carbonylative annulation of ortho-hydroxydiarylalkyne 80 was catalyzed with PdCl2 and CuCl2 in the presence of CO, which not only installed the benzofuran but also introduced a carbomethoxy functionality.
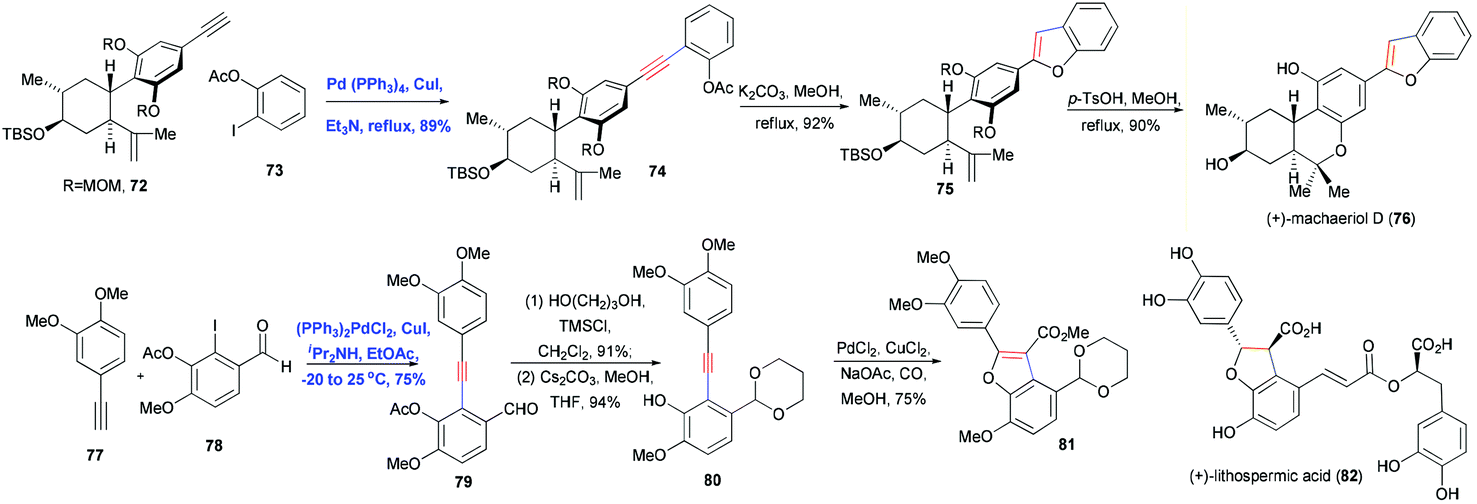 | ||
| Scheme 13 Total synthesis of (+)-machaeriol D (76) and (+)-lithospermic acid (82). | ||
A particularly impressive example of Sonogashira coupling followed by hetero-annulation was reported by Shair and co-workers in the total synthesis of cephalostatin 1 (88).30 In this approach, Pd-catalyzed Sonogashira coupling and Au-catalyzed hetero-annulation were used to construct the dihydrofuran ring (Scheme 14). Cross-coupling of vinyl triflate 83 and terminal alkyne 84 in the presence of Pd(PPh3)4, CuI and iPr2NEt in DMF produced the enyne 85 in 94% yield. After a two-step sequence in which Sharpless dihydroxylation and oxidation generated an α-hydroxy cyclopentenone, which was then reduced in a stereocontrolled manner, the trans-diol 86 was obtained in 36% yield. Activation of the alkyne motif with Au(I) and subsequent nucleophilic attack by a hydroxyl group led to the formation of dihydrofuran 87 in 88% yield through 5-endo-dig cyclization. This dihydrofuran was then used to install the spiroketal moiety in the right fragment of the natural cephalostatin 1 (88).
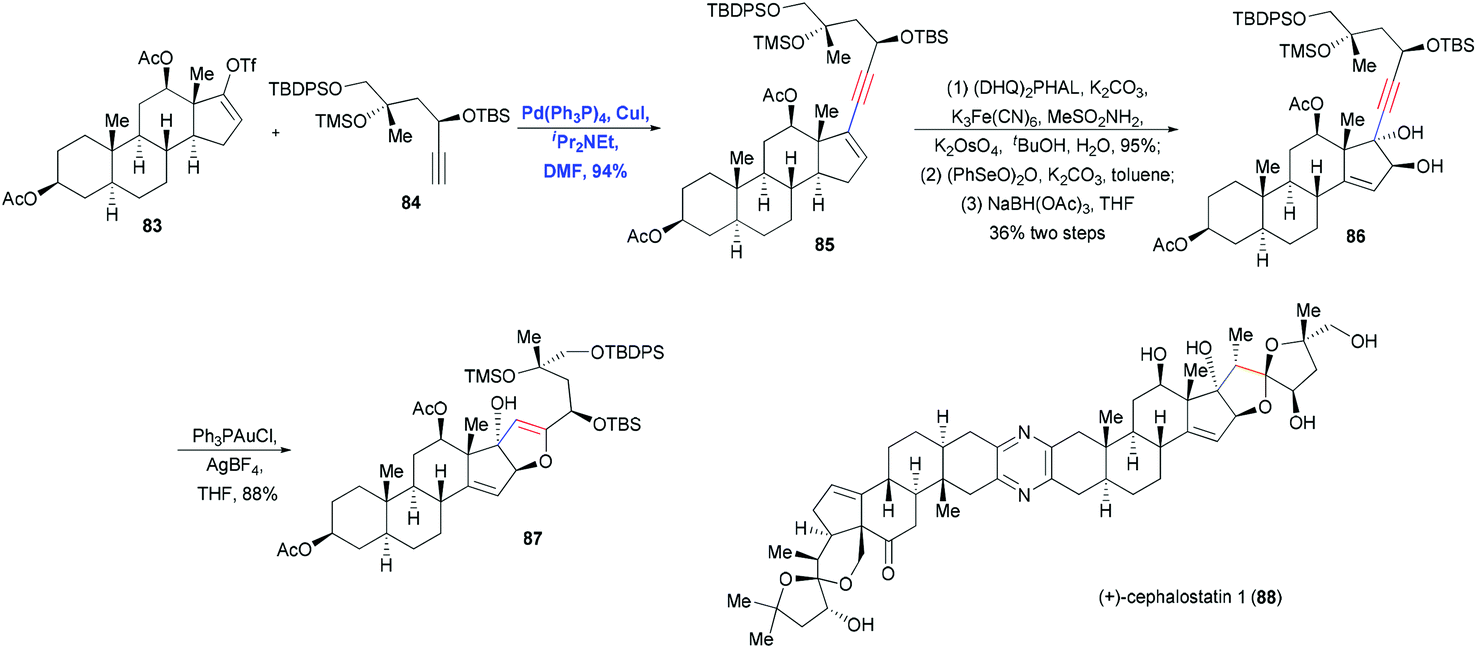 | ||
| Scheme 14 Total synthesis of cephalostatin (88). | ||
Tetrahydroisoquinoline-type alkaloids are a large family of isoquinoline natural products that have attracted considerable attention from chemists and biologists for their fascinating structures and remarkable biological activities.31 The isoquinoline core of these compounds has traditionally been constructed using Pictet–Spengler condensation32 and asymmetric imine hydrogenation.33 Fujii, Ohno and co-workers reported a convergent synthesis of quinocarcin (96) using Sonogashira coupling and Au(I)-catalyzed hydroamination (Scheme 15).34 Cross coupling of 89 and 90 at 80 °C in the presence of Pd(PPh3)4, CuSO4, and sodium ascorbate in DMF and Et3N gave the coupling product 91 in 92% yield. Using equimolar amounts of CuSO4 and sodium ascorbate effectively prevented the homocoupling side reaction. After extensively surveying reaction conditions and catalysts, the authors found 93 to be the most effective Au catalyst for hydroaminating the free amine-containing 92. The desired 6-endo-dig annulation proceeded smoothly, giving 94 in high yield; this compound was then reduced with NaBH3CN in a stereocontrolled fashion to form the tetrahydroisoquinoline core 95.
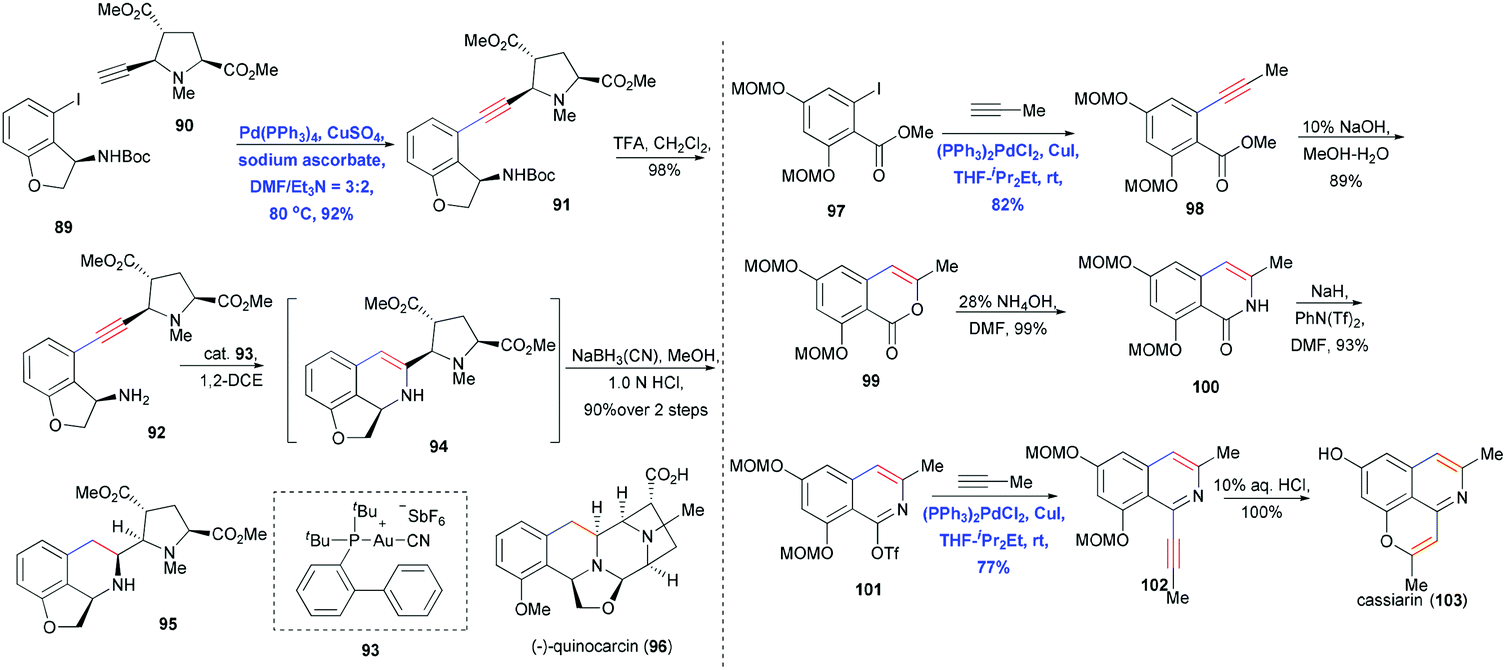 | ||
| Scheme 15 Total synthesis of quinocarcin (96) and cassiarin A (103). | ||
An alternative way to generate the isoquinoline core via Sonogashira coupling was reported by Honda and co-workers, who prepared isoquinoline from isocoumarine to achieve the total synthesis of alkaloid cassiarin A (103) (Scheme 15).35 The first Sonogashira coupling of iodide 97 and in situ-generated propyne was performed in the presence of Pd/Cu cocatalyst, yielding 98 in 82% yield. Hydrolysis followed by acidification generated the isocoumarine 99, which was transformed into isoquinolone 100 after treatment with ammonium hydroxide in DMF and cyclization of a ketoamide intermediate. Impressively, this isoquinolone was converted into its active triflate form 101, which underwent a second Sonogashira coupling with propyne to furnish isoquinoline 102. Finally deprotection using HCl promoted the 6-endo-dig annulation and provided cassiarin A (103) in quantitative yield.
In 2012, Ramana and co-workers reported the total synthesis of (−)-isatisine A (111)36 using four consecutive metal-mediated transformations: Sonogashira coupling, Pd-catalyzed nitroalkyne cycloisomerization, InCl3-promoted indole addition and Rh-catalyzed oxidative N-heterocyclization (Scheme 16). The standard Sonogashira coupling furnished the nitroalkyne 106 in 90% yield, which underwent nitroalkyne cycloisomerization catalyzed by 5 mol% of Pd(CH3CN)2Cl2, yielding isatogen 108 in 72% yield. Then the basic skeleton of 110 was efficiently constructed using another two metal-mediated transformations.
 | ||
| Scheme 16 Total synthesis of (−)-isatisine A (111). | ||
A selection of recent reports of total syntheses of natural products with intriguing structural features and involving Sonogashira coupling are shown in Fig. 2. They include moracin P (112, Kyeong Lee and co-workers, 2009);37 indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (113, Thomas G. Minehan and co-workers, 2012);38 smyrindiol (114, Dieter Enders and co-workers, 2012);39 7′,8′-dihydroaigialospirol (115, Margaret A. Brimble and co-workers, 2012);40 linoxepin (116, Lutz F. Tietze and co-workers, 2013),41 borrelidin (117, James P. Morken and co-workers, 2003),42 murisolin (118, Tetsuaki Tanaka and co-workers, 2004);43 cylindramide (119, Sabine Laschat and co-workers, 2005),44 leucascandrolide A (120, James S. Panek and co-workers, 2007);45 thuggacin B (121, Andreas Kirschning and co-workers, 2008);46 neopeltolide (122, Martin E. Maier, 2008);47 cortistatin J (123, K. C. Nicolaou, David Y.K. Chen, and co-workers, 2009);48 bryostatin 7 (124, Michael J. Krische and co-workers, 2011);49 8-deshydroxyajudazol B (125, Mark A. Rizzacasa and co-workers, 2011)50 and psymberin = irciniastatin A (126, Jef K. De Brabander and co-workers, 2012).51
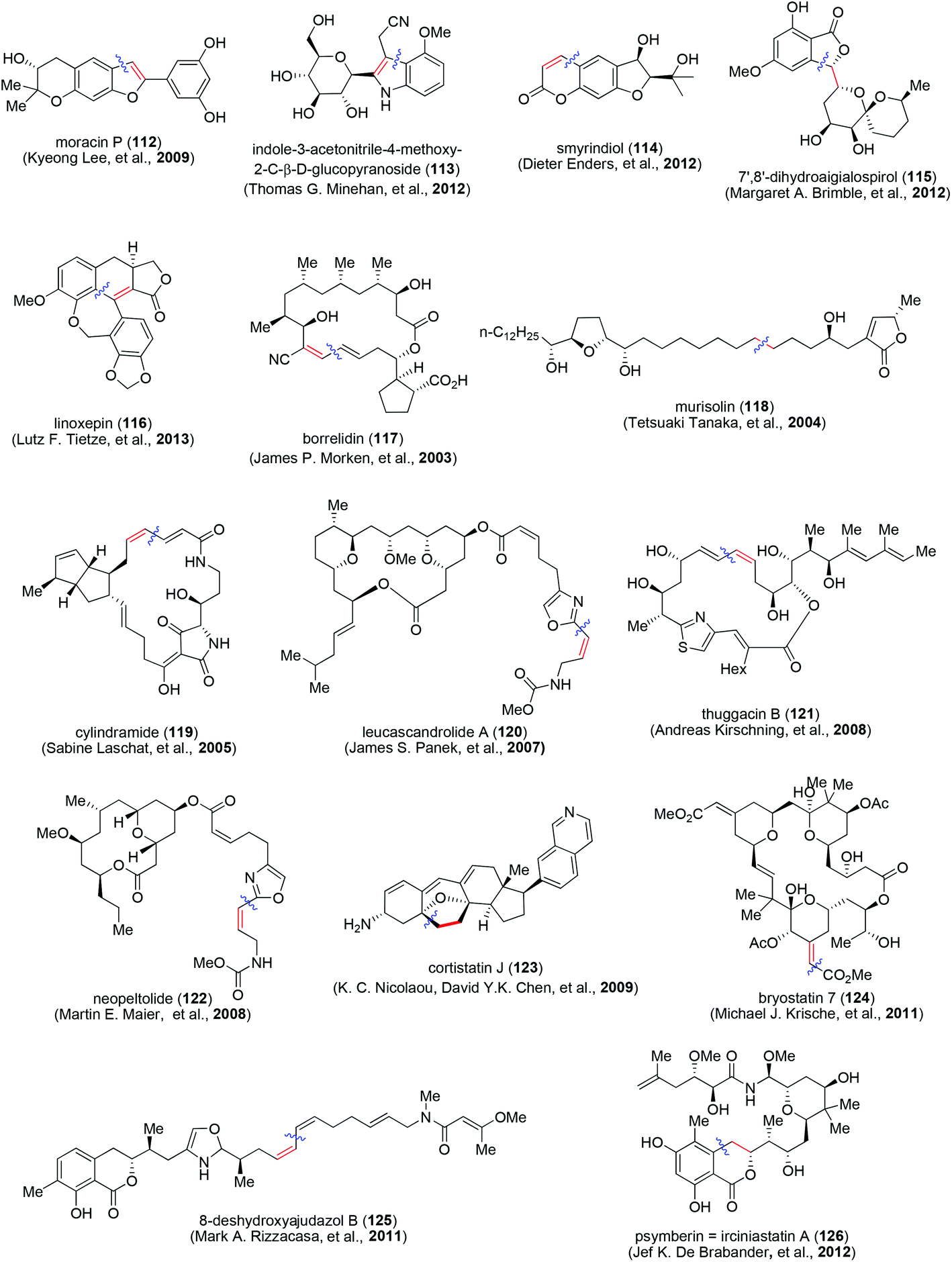 | ||
| Fig. 2 Total synthesis of natural products involving Sonogashira coupling. | ||
3. Conclusion
Palladium-catalyzed inter/intramolecular cross-coupling reactions have dramatically improved the efficiency of organic synthesis. As we demonstrate herein, Sonogashira coupling provides an effective methodology for the construction of C–C bonds (sp–sp2) which has been wildly used in the synthesis of natural products. Rational design using Sonogashira coupling and transformations based on the Sonogashira products facilitates the fragment coupling and construction of heterocycles in convergent approaches. At present, the traditional reaction procedure using a Pd–Cu co-catalyzed system in the presence of an amine predominates in most cases. There is no doubt that more synthetic applications will be reported with the improvement and development of catalysts, ligands and reaction conditions.Acknowledgements
Financial support was provided by the NSFC (21102045, 21272076), the program for professor of special appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) and the Program for New Century Excellent Talents in University (NCET).References
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley Interscience, New York, 2002 Search PubMed; (c) Cross-Coupling Reactions: A Practical Guide, ed. N. Miyaura, Springer, Berlin, 2002 Search PubMed; (d) Metal-Catalyzed Cross Coupling Reactions, ed. P. J. Stang and F. Diederick, Wiley-VCH, Weinheim, 1998 Search PubMed; For reviews of the Heck reaction, see: (e) A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2379–2411 Search PubMed; (f) M. Shibasaki and E. M. Vogl, J. Organomet. Chem., 1999, 576, 1–15 CrossRef CAS; (g) I. P. Belestskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef PubMed; (h) M. Shibasaki, C. D. J. Boden and A. Kojima, Tetrahedron, 1997, 53, 7371–7393 CrossRef CAS; For reviews of the Stille reaction, see: (i) V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS; (j) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef PubMed; (k) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS; For reviews of the Suzuki reaction, see: (l) A. Suzuki, Acc. Chem. Res., 1982, 15, 178–184 CrossRef CAS; (m) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; For reviews of Negishi reactions, see: (n) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2018 CrossRef CAS PubMed; For reviews of the Tsuji-Trost reaction, see: (o) B. M. Trost, Acc. Chem. Res., 1980, 13, 385–393 CrossRef CAS; (p) J. Tsuji, Tetrahedron, 1986, 42, 4361–4401 CrossRef CAS; For reviews of the palladium-catalyzed asymmetric alkylation reaction, see: (q) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed; (r) B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS PubMed; For reviews of palladium-catalyzed cross-coupling reactions in total synthesis, see: (s) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 42, 4442–4489 CrossRef PubMed.
- (a) R. R. Tykwinski, Angew. Chem., Int. Ed., 2003, 42, 1566–1568 CrossRef CAS PubMed; (b) R. Rossi, A. Carpita and F. Bellina, Org. Prep. Proced. Int., 1995, 27, 127–160 CrossRef CAS; (c) K. Sonogashira, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, p. 521 Search PubMed; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (e) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (f) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- (a) I. Paterson, R. D. M. Davies and R. Marquez, Angew. Chem., Int. Ed., 2001, 40, 603–607 CrossRef CAS; (b) I. Paterson and T. Paquet, Org. Lett., 2010, 12, 2158–2161 CrossRef CAS PubMed.
- B. Vaz, L. Otero, R. Álvarez and Á. R. de Lera, Chem. – Eur. J., 2013, 19, 13065–13074 CrossRef CAS PubMed.
- (a) J. Taunton, J. L. Wood and S. L. Schreiber, J. Am. Chem. Soc., 1993, 115, 10378–10379 CrossRef CAS; (b) J. L. Wood, J. A. Porco Jr., J. Taunton, A. Y. Lee, J. Clardy and S. L. Schreiber, J. Am. Chem. Soc., 1992, 114, 5898–5900 CAS; (c) H. Chikashita, J. A. Porco Jr., T. J. Stout, J. Clardy and S. L. Schreiber, J. Org. Chem., 1991, 56, 1692–1694 CrossRef CAS; (d) J. A. Porco Jr., F. J. Schoenen, T. J. Stout, J. Clardy and S. L. Schreiber, J. Am. Chem. Soc., 1990, 112, 7410–7411 CrossRef.
- (a) A. G. Myers, P. C. Hogan, A. R. Hurd and S. D. Goldberg, Angew. Chem., Int. Ed., 2002, 41, 1062–1067 CrossRef CAS; (b) Y. Koyama, M. J. Lear, F. Yoshimura, I. Ohashi, T. Mashimo and M. Hirama, Org. Lett., 2005, 7, 267–270 CrossRef CAS PubMed, and references therein.
- K. C. Nicolaou, C. W. Hummel, E. N. Pitsinos, M. Nakada, A. L. Smith, K. Shibayama and H. Saimoto, J. Am. Chem. Soc., 1992, 114, 10082–10084 CrossRef CAS.
- H. Dieck and F. Heck, J. Organomet. Chem., 1975, 93, 259–263 CrossRef CAS.
- L. Cassar, J. Organomet. Chem., 1975, 93, 253–257 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- (a) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (b) P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS.
- (a) B. M. Trost and T. N. Salzmann, J. Am. Chem. Soc., 1973, 95, 6840–6842 CrossRef CAS; (b) B. M. Trost, Science, 1983, 219, 245–250 CAS; (c) R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530–541 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS PubMed.
- (a) P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef CAS PubMed; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed; (c) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
- (a) J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS PubMed; (b) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- (a) Classics in Total Synthesis, ed. K. C. Nicolaou and E. J. Sorensen, Wiley-VCH, Weinheim, 1996 Search PubMed; (b) Classics in Total Synthesis II, ed. K. C. Nicolaou and S. A. Snyder, Wiley-VCH, Weinheim, 2003 Search PubMed; (c) Classics in Total Synthesis III, ed. K. C. Nicolaou and J. S. Chen, Wiley-VCH, Weinheim, 2010 Search PubMed.
- The Logic of Chemical Synthesis, ed. E. J. Corey and X.-M. Cheng, Wiley-VCH, Weinheim, 1995 Search PubMed.
- K. Komano, S. Shimamura, M. Inoue and M. Hirama, J. Am. Chem. Soc., 2007, 129, 14184–14186 CrossRef CAS PubMed.
- M. B. Andrus, S. D. Lepore and T. M. Turner, J. Am. Chem. Soc., 1997, 119, 12159–12169 CrossRef CAS.
- P. Wipf and T. H. Graham, J. Am. Chem. Soc., 2004, 126, 15346–15347 CrossRef CAS PubMed.
- D. K. Mohapatra, D. Bhattasali, M. K. Gurjar, M. I. Khan and K. S. Shashidhara, Eur. J. Org. Chem., 2008, 6213–6224 CrossRef CAS PubMed.
- T. Usuki, H. Yamada, T. Hayashi, H. Yanuma, Y. Koseki, N. Suzuki, Y. Masuyama and Y. Y. Linb, Chem. Commun., 2012, 48, 3233–3235 RSC.
- P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028–14029 CrossRef CAS PubMed.
- T. Miyazaki, S. Yokoshima, S. Simizu, H. Osada, H. Tokuyama and T. Fukuyama, Org. Lett., 2007, 9, 4737–4740 CrossRef CAS PubMed.
- J. R. Butler, C. Wang, J. Bian and J. M. Ready, J. Am. Chem. Soc., 2011, 133, 9956–9959 CrossRef CAS PubMed.
- The Chemistry of Heterocycles, ed. T. Eicher and S. Hauptmann, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
- Q. Wang, Q. Huang, B. Chen, J. Lu, H. Wang, X. She and X. Pan, Angew. Chem., Int. Ed., 2006, 45, 3651–3653 CrossRef CAS PubMed.
- M. Inoue, M. W. Carson, A. J. Frontier and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 1878–1889 CrossRef CAS PubMed.
- J. Fischer, G. P. Savage and M. J. Coster, Org. Lett., 2011, 13, 3376–3379 CrossRef CAS PubMed.
- K. C. Fortner, D. Kato, Y. Tanaka and M. D. Shair, J. Am. Chem. Soc., 2010, 132, 275–280 CrossRef CAS PubMed.
- (a) D. Jack and R. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC; (c) Q. Y. Zhang, G. Z. Tu, Y. Y. Zhao and T. M. Cheng, Tetrahedron, 2002, 58, 6795–6798 CrossRef CAS; (d) A. J. Aladesanmi, C. J. Kelly and J. D. Leary, J. Nat. Prod., 1983, 46, 127–131 CrossRef CAS; (e) A. Zhang, J. L. Neumeyer and R. J. Baldessarini, Chem. Rev., 2007, 107, 274–302 CrossRef CAS PubMed; (f) K. Ye, Y. Ke, N. Keshava, J. Shanks, J. A. Kapp, R. R. Tekmal, J. Petros and H. C. Joshi, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1601–1606 CrossRef CAS.
- (a) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341–3370 CrossRef CAS PubMed; (b) J. Stçckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef PubMed.
- (a) Y. G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed; (b) D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- (a) H. Chiba, S. Oishi, N. Fujii and H. Ohno, Angew. Chem., Int. Ed., 2012, 51, 9169–9172 CrossRef CAS PubMed; (b) H. Chiba, Y. Sakai, A. Ohara, S. Oishi, N. Fujii and H. Ohno, Chem. – Eur. J., 2013, 19, 8875–8883 CrossRef CAS PubMed.
- M. Rudyanto, Y. Tomizawa, H. Morita and T. Honda, Org. Lett., 2008, 10, 1921–1922 CrossRef CAS PubMed.
- P. Patel and C. V. Ramana, J. Org. Chem., 2012, 77, 10509–10515 CrossRef CAS PubMed.
- N. Kaur, Y. Xia, Y. L. Jin, N. T. Dat, K. Gajulapati, Y. Choi, Y.-S. Hong, J. J. Leea and K. Lee, Chem. Commun., 2009, 1879–1881 RSC.
- A. Yepremyan and T. G. Minehan, Org. Biomol. Chem., 2012, 10, 5194–5196 CAS.
- D. Enders, J. Fronert, T. Bisschops and F. Boeck, Beilstein J. Org. Chem., 2012, 8, 1112–1117 CrossRef CAS PubMed.
- T.-Y. Yuen and M. A. Brimble, Org. Lett., 2012, 14, 5154–5157 CrossRef CAS PubMed.
- L. F. Tietze, S.-C. Duefert, J. Clerc, M. Bischoff, C. Maaβ and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 3191–3194 CrossRef CAS PubMed.
- M. O. Duffey, A. LeTiran and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 1458–1459 CrossRef CAS PubMed.
- N. Maezaki, H. Tominaga, N. Kojima, M. Yanai, D. Urabe and T. Tanaka, Chem. Commun., 2004, 406–407 RSC.
- (a) N. Cramer, S. Laschat, A. Baro, H. Schwalbe and C. Richter, Angew. Chem., Int. Ed., 2005, 44, 820–822 CrossRef CAS PubMed; (b) N. Cramer, M. Buchweitz, S. Laschat, W. Frey, A. Baro, D. Mathieu, C. Richter and H. Schwalbe, Chem. – Eur. J., 2006, 12, 2488–2503 CrossRef CAS PubMed.
- Q. B. Su, L. A. Dakin and J. S. Panek, J. Org. Chem., 2007, 72, 2–24 CrossRef CAS PubMed.
- M. Bock, R. Dehn and A. Kirschning, Angew. Chem., Int. Ed., 2008, 47, 9134–9137 CrossRef CAS PubMed.
- V. V. Vintonyak, B. Kunze, F. Sasse and M. E. Maier, Chem. – Eur. J., 2008, 14, 11132–11140 CrossRef CAS PubMed.
- K. C. Nicolaou, X. S. Peng, Y. P. Sun, D. Polet, B. Zou, C. S. Lim and D. Y. K. Chen, J. Am. Chem. Soc., 2009, 131, 10587–10597 CrossRef CAS PubMed.
- Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876–13879 CrossRef CAS PubMed.
- S. Birkett, D. Ganame, B. C. Hawkins, S. Meiries, T. Quach and M. A. Rizzacasa, Org. Lett., 2011, 13, 1964–1967 CrossRef CAS PubMed.
- Y. Feng, X. Jiang and J. K. De Brabander, J. Am. Chem. Soc., 2012, 134, 17083–17093 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |