Catalytic asymmetric allylation of carbonyl compounds and imines with allylic boronates
Hua-Xing
Huo
a,
Jeremy R.
Duvall
b,
Meng-Yuan
Huang
a and
Ran
Hong
*a
aCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: rhong@sioc.ac.cn; Web: http://honggroup.sioc.ac.cn Fax: (+86)21-6416-6128
bTherapeutics Platform, The Broad Institute, 7 Cambridge Center, Cambridge, MA 02142, USA Web: http://www.broadinstitute.org
First published on 28th February 2014
Abstract
Enantioselective allylation is a highly used organic reaction to prepare chiral homoallylic alcohols and amines, which serve as important building blocks in the synthesis of a variety of natural products and pharmaceuticals. In particular, catalytic asymmetric allylation of carbonyl compounds and imines with organoboronates has seen rapid development in the past decade and is the focus of this review.
1. Background
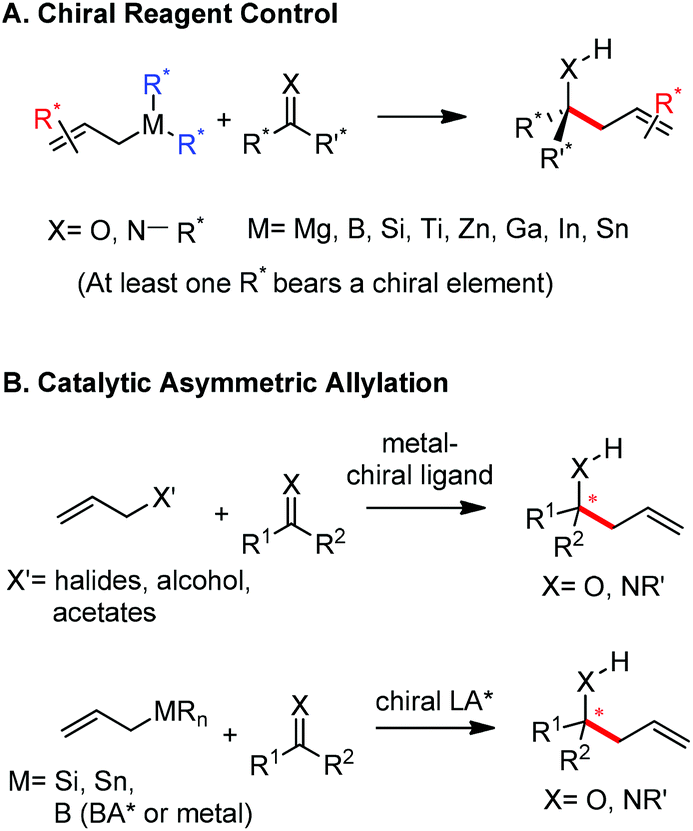
The allylation reaction is widely applied to prepare homoallylic alcohols and amines, which serve as common building blocks for the synthesis of a variety of natural products and pharmaceutically relevant compounds (Scheme 1).1 In this reaction, besides the alcohol or amine being introduced, the carbon–carbon double bond serves as a versatile motif and is readily transformed into other functional groups or used in a a carbon chain elongation.2 When a ketone or its imine derivative is chosen as the electrophile, asymmetric allylation results in a chiral tetrasubstituted carbon, a long standing challenge in synthetic organic chemistry.3 For these reasons, this field has attracted wide interest in the last decade. A lot of effort and some impressive progress have been made in the development of stereoselective allylation of carbonyl compounds or imines.1,2,4 The use of stoichiometric chiral inducing reagents (including substrate control and reagent control) is a common approach to access key intermediates in the synthesis of natural products (Scheme 2).4d The major diastereoisomer in most of these examples can be rationalized through the existing working models.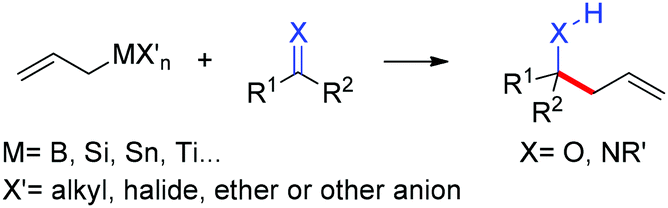 | ||
| Scheme 1 Allylation reaction. | ||
 | ||
| Scheme 2 Enantioselective allylation reaction. | ||
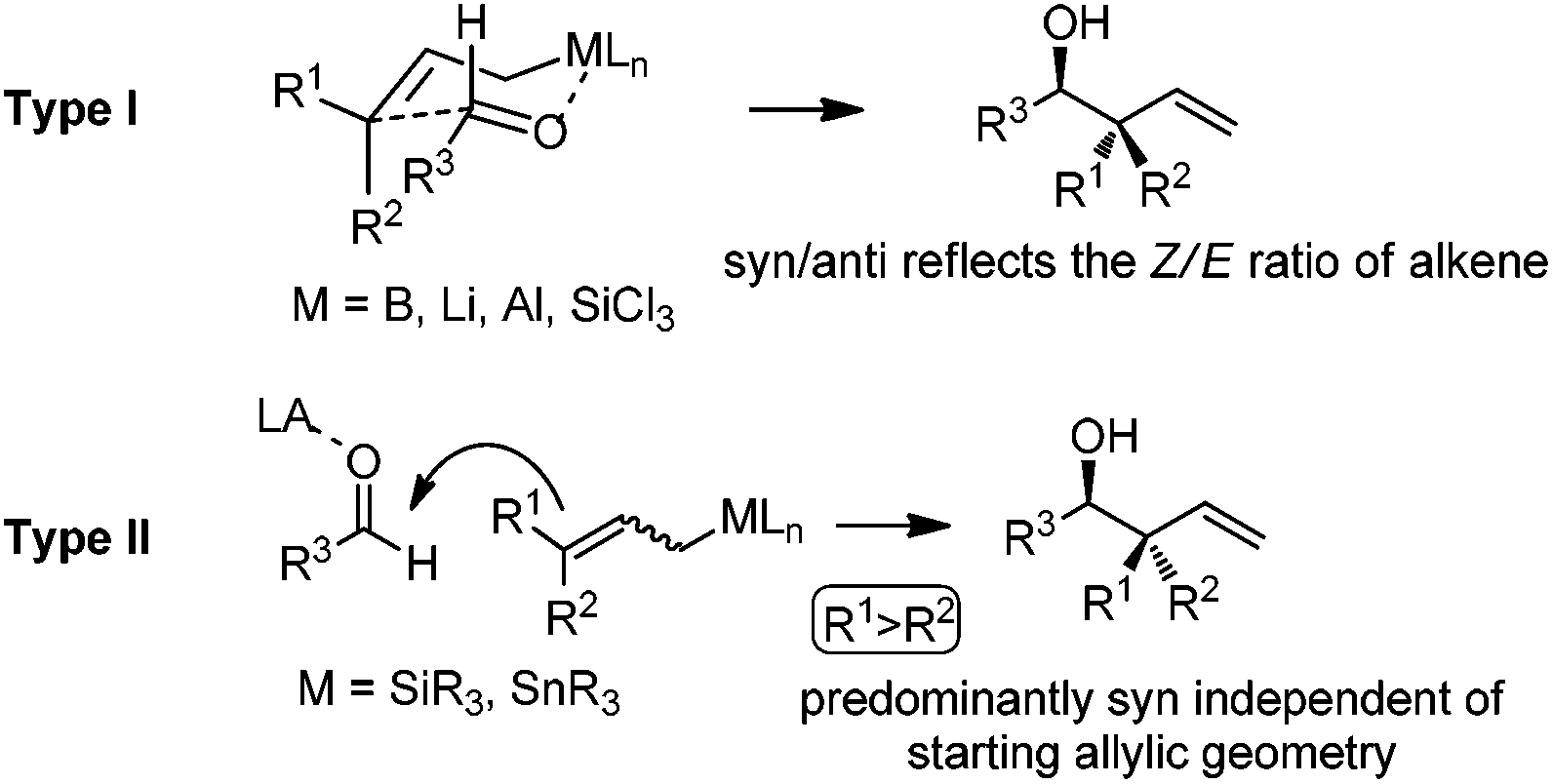
Allylboration was originally documented in a 1964 paper contributed by Mikhailov and Bubnov.5,6 Triallylborane reacted with aldehydes or ketones to give homoallylic alcohols. In 1966, Gaudemar and co-workers utilized allylic boronate for the allylation of aldehydes.7 In the late 1970s, the regio- and diastereospecific addition of crotylboronate with aldehydes was rationalized by Hoffmann's model.8 In 1983, Denmark and co-workers classified two modes of addition for different allylation reagents (Scheme 3).9 In the Type I class, allylic boron reagents can activate the carbonyl to form a closed six-membered chair-like transition state which yields a γ-allylation (Scheme 3).10 On the other hand, as shown in the Type II class, allyl trialkylsilanes and allyl trialkylstannanes generally react with aldehydes under the activation of an external Lewis acid through an open transition state (Scheme 3). The regioselectivity and diastereoselectivity are generally higher via the Type I mechanism than via Type II.
 | ||
| Scheme 3 Mechanistic models for allyl reagents. | ||
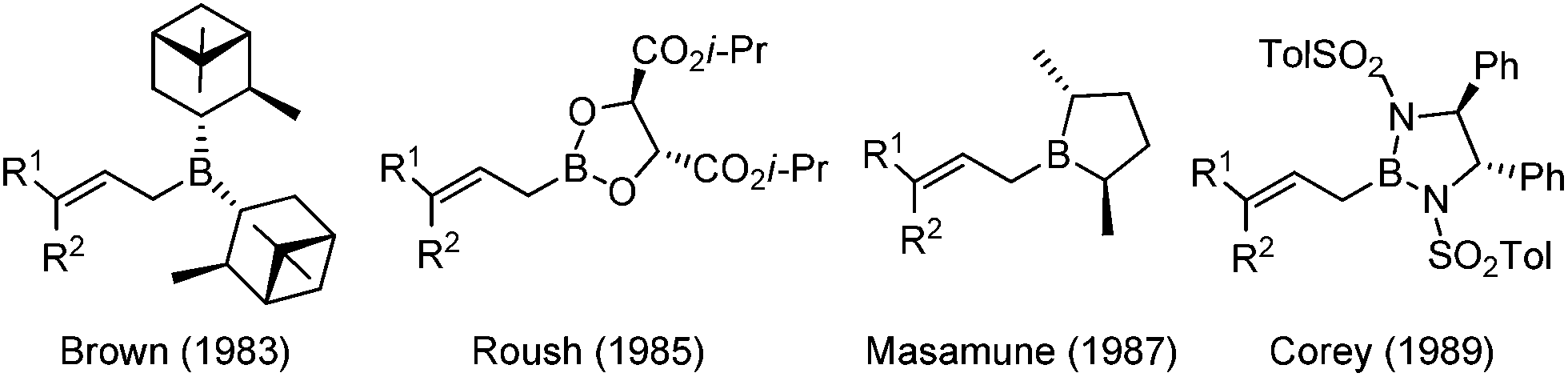
In the 1980s, highly efficient allylboration reactions were reported with excellent enantioselectivity by introducing chiral boron reagents. In the first twenty years of development, several C2 symmetric chiral boron reagents were disclosed and received wide appreciation in this field.11 Representative examples of the advancements include pinane-derived borane (Brown),12 tartrate boronates (Roush),13 borolane derivatives (Masamune),14 and bis(sulfonamide) derivatives (Corey)15 (Scheme 4).
 | ||
| Scheme 4 Representative chiral borane reagents for enantioselective allylboration. | ||
Unfortunately, the development of enantioselective allylboration has long been restricted to chiral auxiliary approaches, requiring stoichiometric amounts of chiral reagent which are difficult to recycle. Lewis acids which potentially induce a changeover from a Type I mechanism toward the open transition structures (Type II) was considered unfeasible for the catalytic approach. One of the early examples to address this challenge was disclosed by Miyaura and co-workers in 2002. They reported the catalytic enantioselective allylation of an aldehyde with allylic boronates by using a catalytic amount of Et2AlCl/BINOL complex.16 The corresponding homoallylic alcohols were obtained in excellent diastereoselectivity albeit in moderate yield and enantioselectivity (Scheme 5). Since the continuing research was not followed, it is still not certain whether optimized conditions for higher enantioselectivity could be achieved.
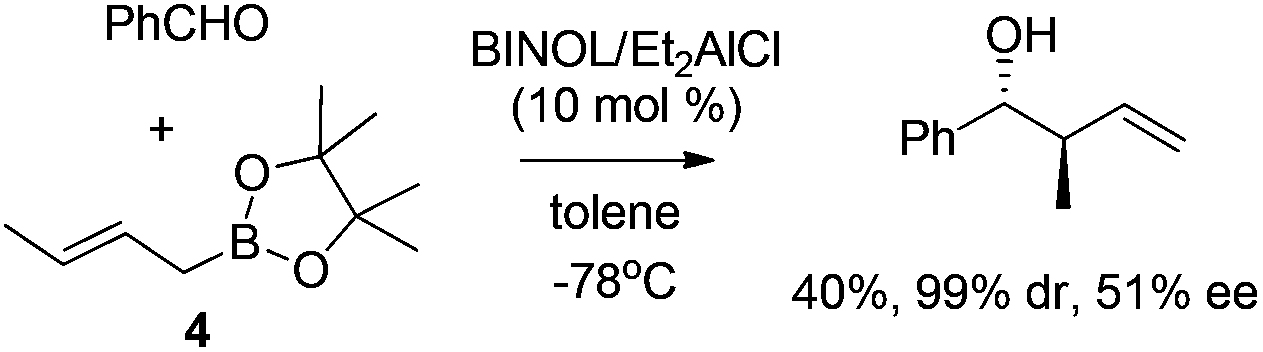 | ||
| Scheme 5 Early example of catalytic enantioselective allylation. | ||
Hall and co-workers reported a comprehensive study on Lewis acid-catalyzed addition of 2-alkoxycarbonyl allylboronates 1 to aldehydes with high diastereocontrol (Scheme 6).17a Based on extensive experimental and kinetic studies,17b Hall and co-workers subsequently suggested that the Lewis acid most likely coordinated to one of the boronate oxygens, probably the most accessible pseudo-axial one, instead of to the carbonyl oxygen of the aldehyde as generally proposed. The transition state still follows Denmark's classification of a closed chair-like transition state. This model was further refined by Sakata's computational study (B3LYP level),18 whereas AlCl3 chelates to the boronate oxygen atom, strengthening the electrophilicity of the boron center to accelerate the allylboration of the aldehyde (Scheme 7).
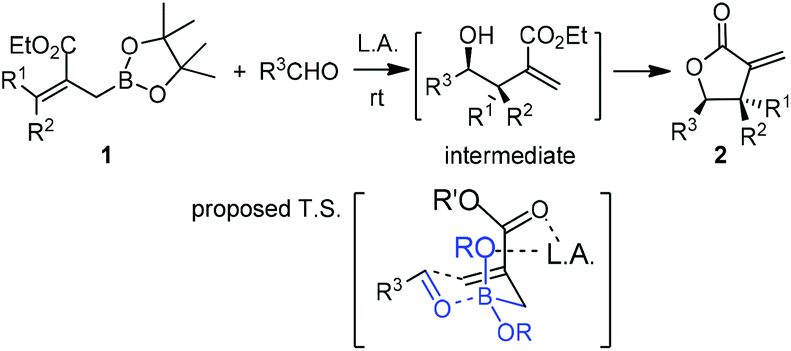 | ||
| Scheme 6 Lewis acid-catalyzed allylborations. | ||
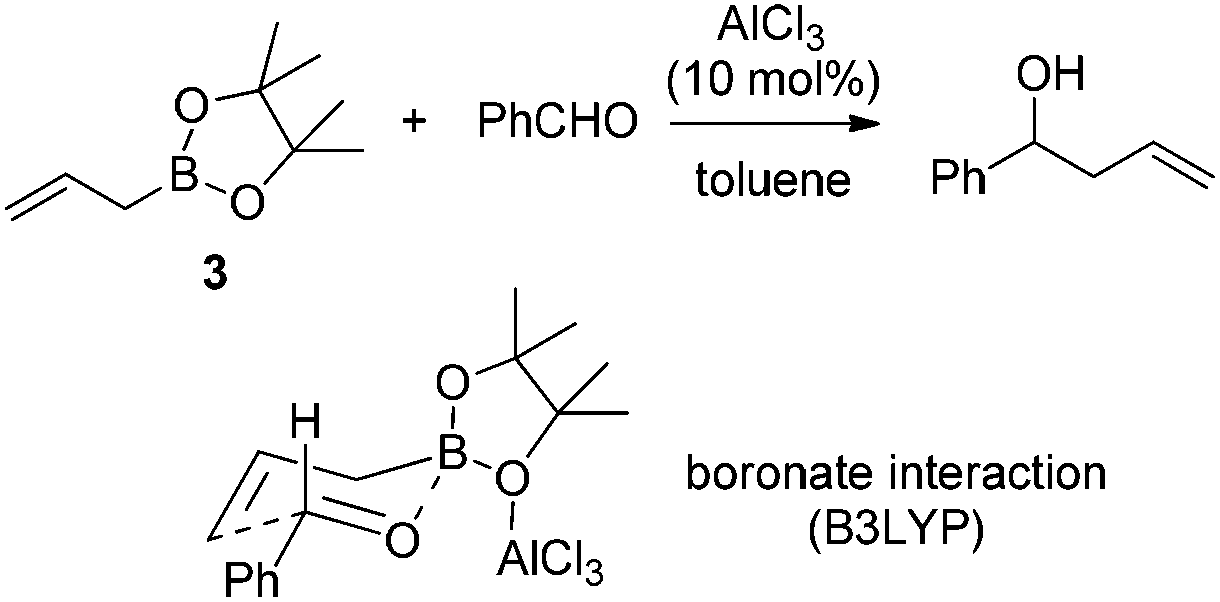 | ||
| Scheme 7 Theoretical study of Lewis acid-catalyzed allylboration. | ||
Due to their ease of preparation, functional group tolerance, stability, low toxicity, and overall operational simplicity of the addition reaction, allylboronate has been intensively studied in recent years. In this review, we are not intending to cover all allylations since several excellent reviews have appeared in recent years.1,4 The achievement related to the nature of the boronate–Lewis acid complex which leads to the rapid development of a catalytic enantioselective allylation is discussed. There is no doubt that the catalytic asymmetric version is the focus of current research due to its sustainability and application.19
In the following sections, according to the catalyst applied, we will introduce enantioselective catalysis of allylation in three categories including metal complex-catalyzed asymmetric allylation, acid-catalyzed asymmetric allylation by activating boronates and catalyzed asymmetric allylation by ligand exchange of boronate.
2. Metal complex-catalyzed allylation
In this category, the stereoselective allylation with allylboronates is carried out in the presence of metal salts and chiral ligands. The reaction mode involves the critical ligand exchange (allyl group) from boron to the metal and the stereoselectivity is controlled by chiral ligands associated with the metal (Scheme 8). Because of the rapid transmetallation to form the active species and the fast regeneration of catalyst, asymmetric allylation proceeds efficiently with a broad substrate scope including aldehydes, ketones, and imines. However, the aldehyde is prone to react with allylboronates even without a catalyst, which therefore decreases the enantioselectivity of allylation. As a result, the reaction with aldehydes is always performed with a relatively higher catalyst loading within a shorter time.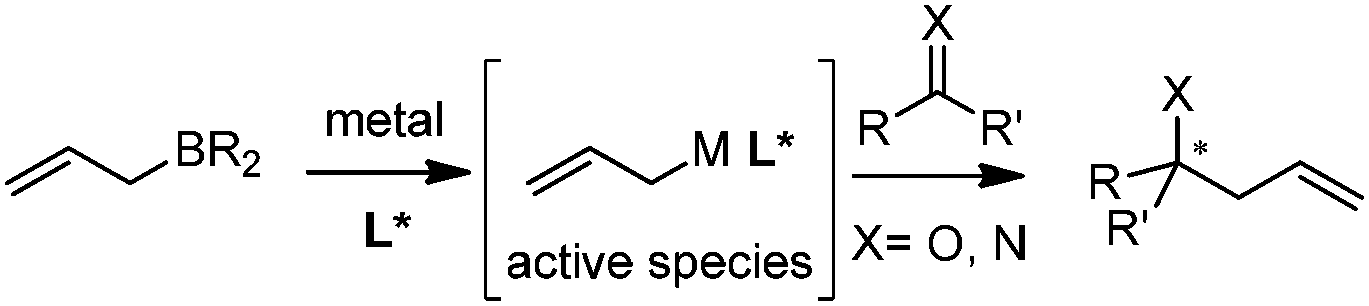 | ||
| Scheme 8 General reaction model for metal-mediated allylboration. | ||
2.1 Cu-mediated system
In 2004, Kanai, Shibasaki, and co-workers found that combination of 3 mol% of CuF2·2H2O, 6 mol% of (R,R)-i-Pr-DuPHOS (5) as a chiral ligand and 4.5 mol% of La(Oi-Pr)3 as a co-catalyst was able to catalyze enantioselective allylboration of ketones.20,21 Substrates bearing aromatic, heteroaromatic, cyclic, and aliphatic ketones were investigated, and they all proceeded in a short reaction time (1 h) resulting in excellent yields and high enantioselectivities (condition A in Table 1). The preliminary mechanistic study indicated that La(Oi-Pr)3 facilitates the transmetallation rather than acting as Lewis acid to activate allylboronates.|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Substrate | Condition A | Condition B | ||
| Yield (%) | ee (%) | Yield (%) | ee (%) | ||
| 1 |
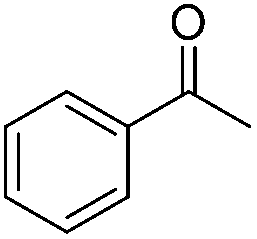
|
94 | 82 | 99 | 89 |
| 2 |
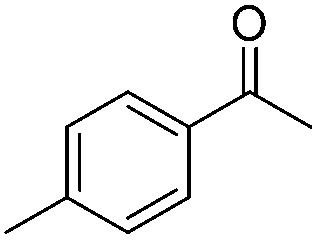
|
89 | 84 | 98 | 92 |
| 3 |
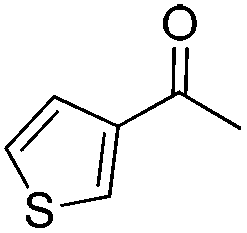
|
84 | 85 | 94 | 93 |
| 4 |
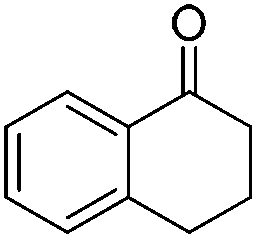
|
88 | 84 | 99 | 98 |
| 5 |
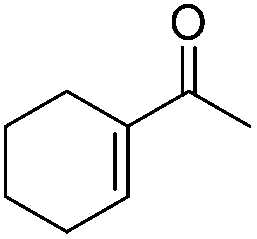
|
87 | 90 | 91 | 90 |
| 6 |
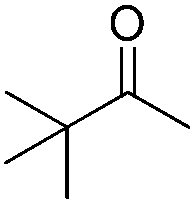
|
99 | 91 | 88 | 83 |

In 2010, they further synthesized a new chiral phosphine 6 for CuOAc-catalyzed allylation of ketones.22 Under the optimized conditions, enantioselective allylation proceeded in better yield and enantioselectivity (condition B versus A in Table 1). To gain preliminary insight into the origin of the high catalytic activity and enantioselectivity, a single crystal of the CuOAc–6 complex was collected, and the corresponding X-ray structure revealed a rigid folded conformation of the core macrocycle. This chiral space provided by the linker and wing modules may be responsible for the high stereoselectivities.22 Experiments also showed that chiral ligand 6 offered better diastereo- and enantioselectivities in spite of E- or Z-allylboronate being utilized as the allyl transfer reagent (Scheme 9).20,22
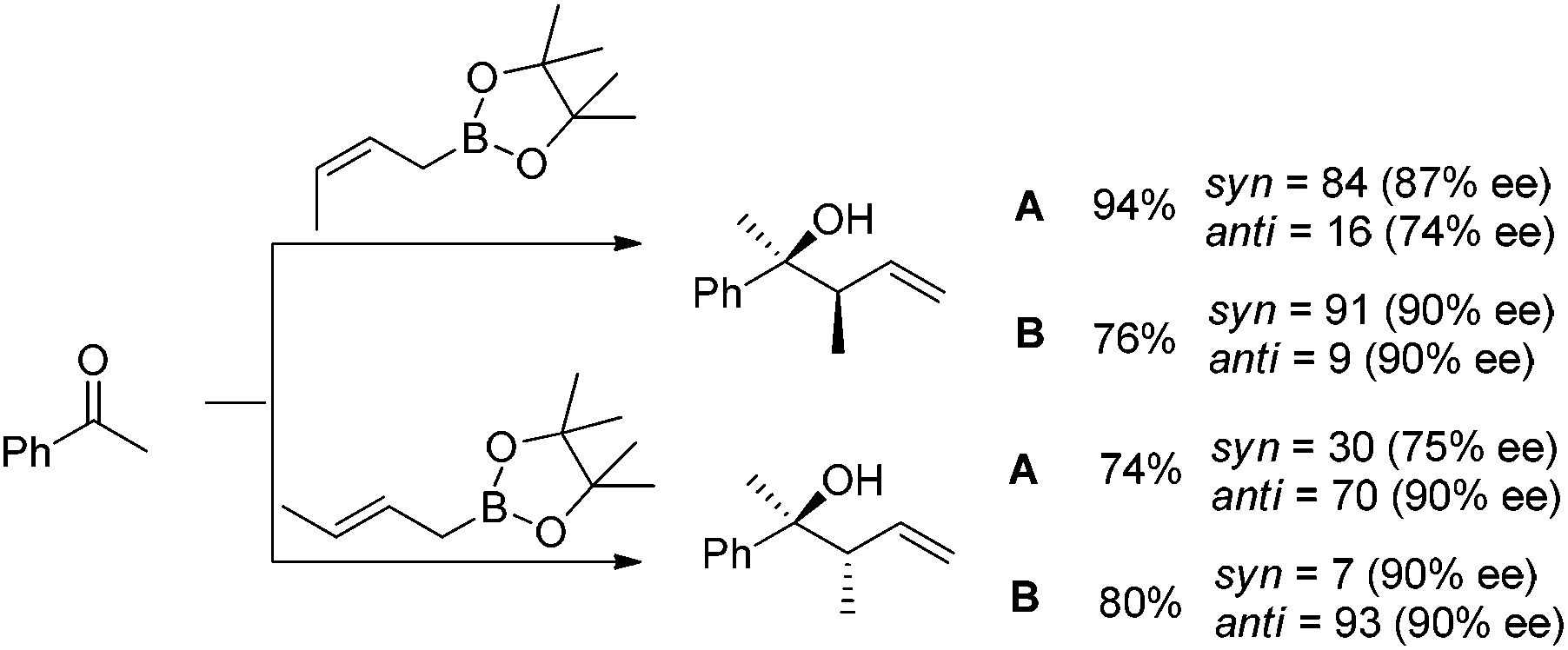 | ||
| Scheme 9 Catalytic enantioselective crotylation of ketone. | ||
In addition to carbonyl compounds as substrates, Shibasaki and co-workers also disclosed the first catalytic enantioselective allylation of ketimines,23 where N-benzylketimines reacted with allylboronate in the presence of CuF, LiOi-Pr, and the ligand, (R,R)-cyclopentyl-DuPHOS (7). LiOi-Pr was found to accelerate the reaction rate better than La(Oi-Pr)3. Good yields and enantioselectivities were generally obtained for a series of aromatic ketimines, but aliphatic ketimines were not optimal substrates under the reaction conditions (entry 8, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R′ | t (h) | Yield (%) | ee (%) |
| 1 |
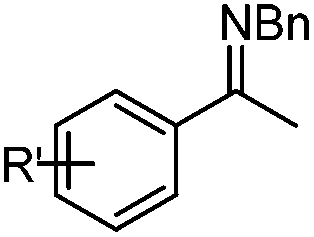
|
Ph | 0.5 | 92 | 89 |
| 2 | 3-MeC6H4 | 1 | 96 | 91 | |
| 3 | 3-MeOC6H4 | 1 | 97 | 93 | |
| 4 | 3-FC6H4 | 1 | 89 | 87 | |
| 5 | 4-MeOC6H4 | 24 | 76 | 85 | |
| 6 | 4-CIC6H4 | 24 | 82 | 81 | |
| 7 |

|
12 | 88 | 92 | |
| 8 |
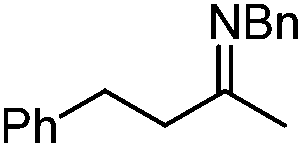
|
2 | 98 | 23 | |
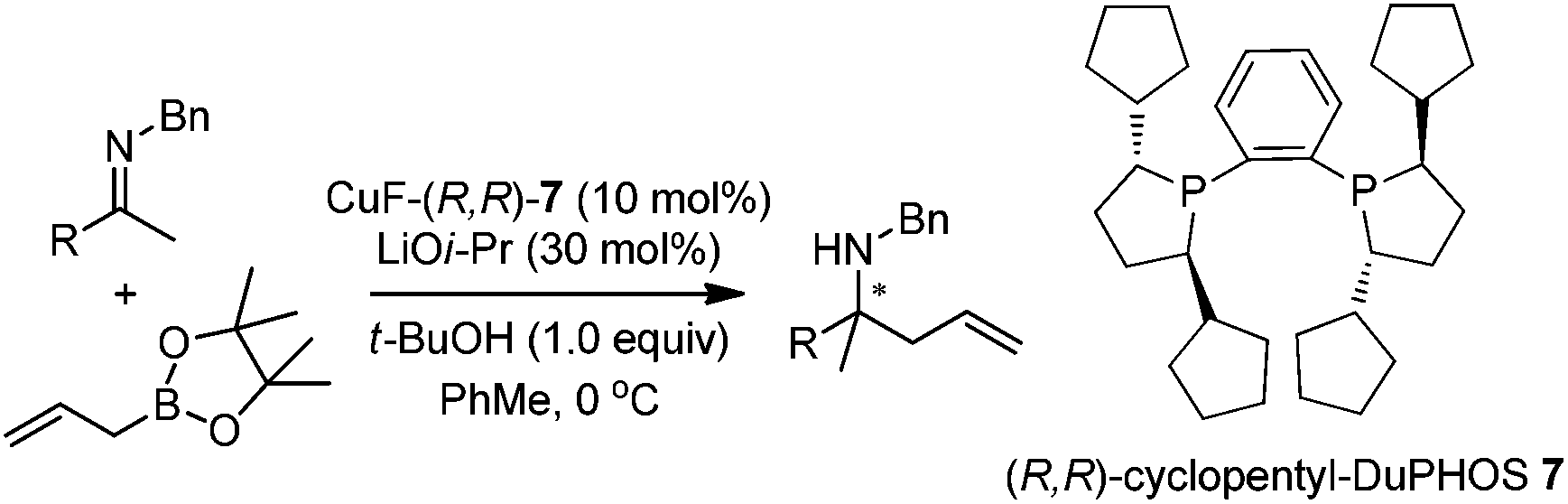
Based on kinetic and NMR studies, the following reaction mechanism was proposed (Scheme 10). First, LiOi-Pr facilitates the transmetalation to generate a reactive allylcopper species, which further reacts with ketamine to deliver a copper amide intermediate. After the ligand exchange, the homoallyl amine was released and the tBuO anion then associates with allylboronate to form an active boronate species which rapidly transmetallates to regenerate the allylcopper nucleophile in the catalytic cycle.23
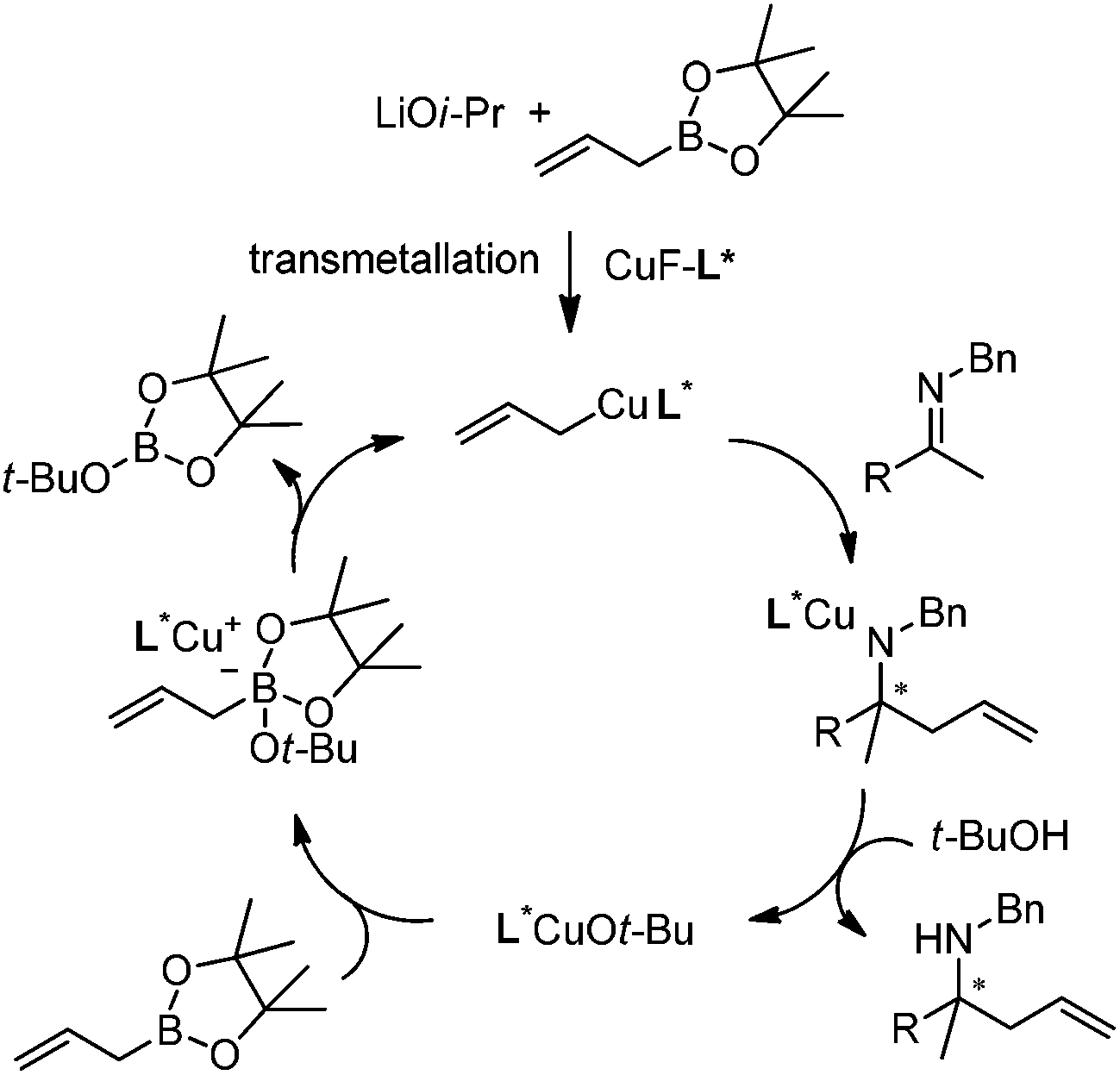 | ||
| Scheme 10 Proposed catalytic cycle. | ||
2.2 Zn-mediated system
Kobayashi and co-workers developed a ZnF2/chiral diamine (9)-catalyzed allylboration of hydrazono ester 8 in 2008.24 Using water and acetone as co-solvents, the products were obtained in high yields and good enantioselectivities (Scheme 11). Water was found to be crucial to maintain high conversion and enantioselectivity. Using either allylboronic acid pinacol ester or cyclic boronates, similar yield and enantioselectivity were achieved.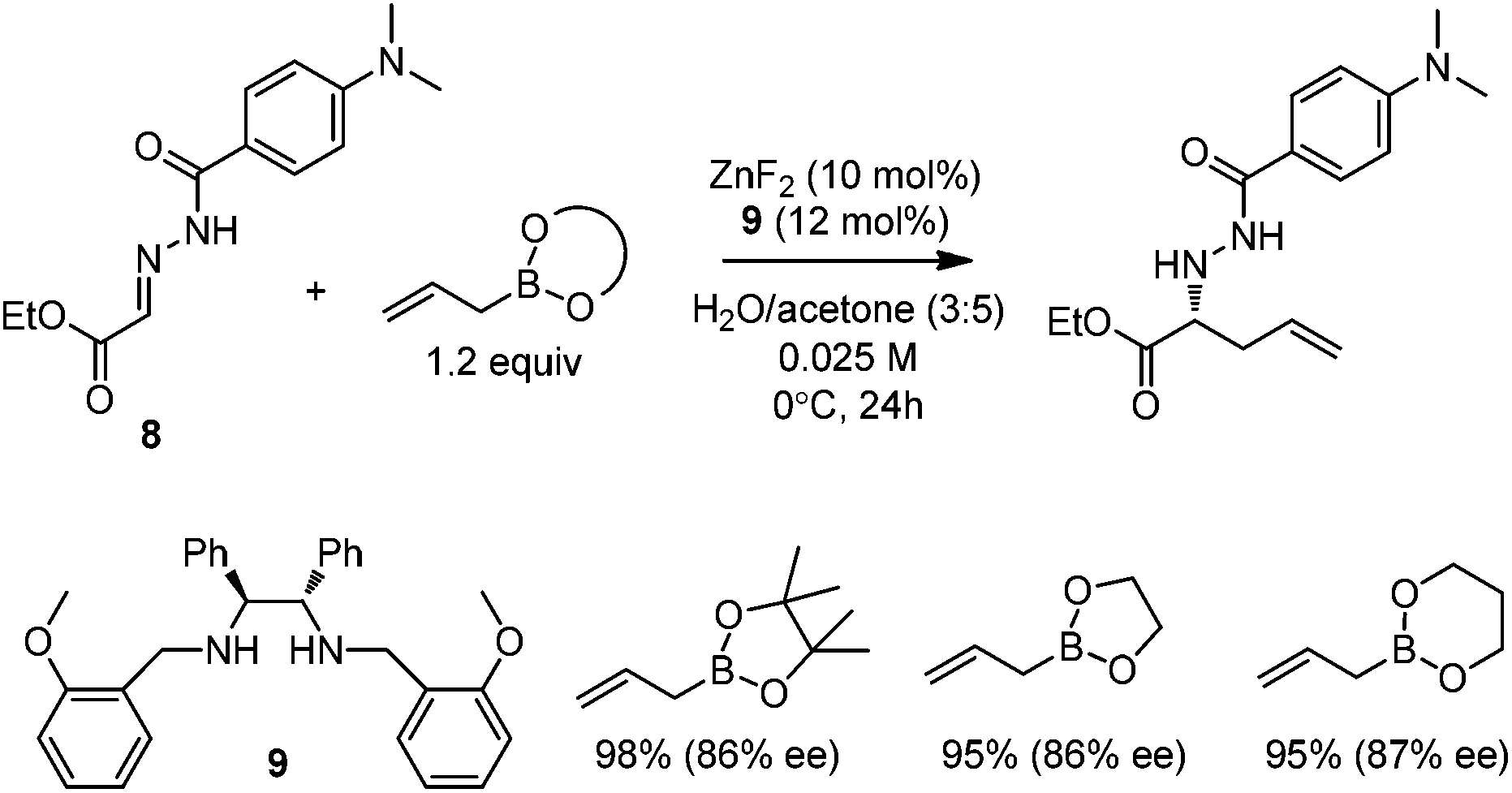 | ||
| Scheme 11 Allylboration of hydrazono esters 8. | ||
However, under the optimal conditions, crotylboronation of hydrazono ester with (Z)-crotylboronate only afforded the adduct in 25% yield and 14% ee. Interestingly, when α-branched allylboronates were employed, the allylboration proceeded in high yields. In all cases, only α-addition products were found with high anti-selectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and high enantioselectivities (Table 3).24 Kobayashi and co-workers proposed a double γ-allylation to afford the α-addition product (Scheme 12). The γ-substituted (Z)-allylzincate (confirmed by NMR when R = H) was formed by reacting allylboronate with [L*ZnF2] through a six-membered chair-like transition state. The allylzincate species then underwent the allylation of the hydrazono ester with anti-selectivity to yield the adduct via another chair-like transition state. Finally, the corresponding α-addition product was formed after hydrolysis.24
:1) and high enantioselectivities (Table 3).24 Kobayashi and co-workers proposed a double γ-allylation to afford the α-addition product (Scheme 12). The γ-substituted (Z)-allylzincate (confirmed by NMR when R = H) was formed by reacting allylboronate with [L*ZnF2] through a six-membered chair-like transition state. The allylzincate species then underwent the allylation of the hydrazono ester with anti-selectivity to yield the adduct via another chair-like transition state. Finally, the corresponding α-addition product was formed after hydrolysis.24
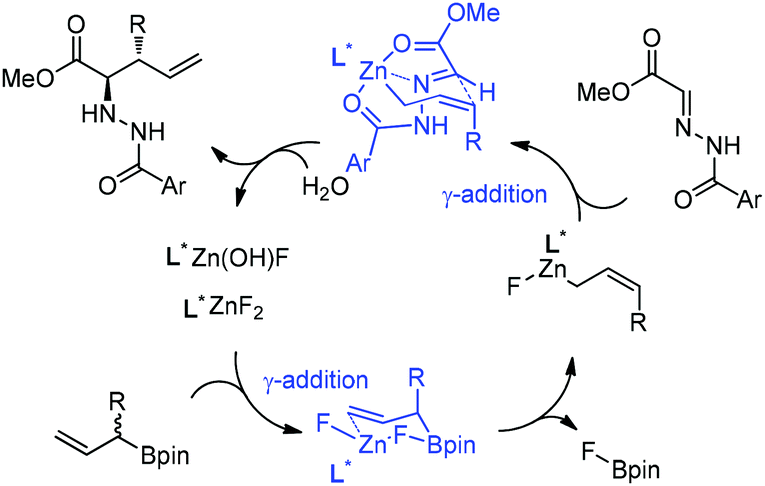 | ||
| Scheme 12 Proposed catalytic cycle. | ||
|
|
|||
|---|---|---|---|
| Entry | R | Yield (%) | ee (%) |
| 1 | Me | >99 | 88 |
| 2 | Et | 98 | 87 |
| 3 | n-Bu | 88 | 87 |
| 4 | (CH3)2CHCH2 | 76 | 87 |
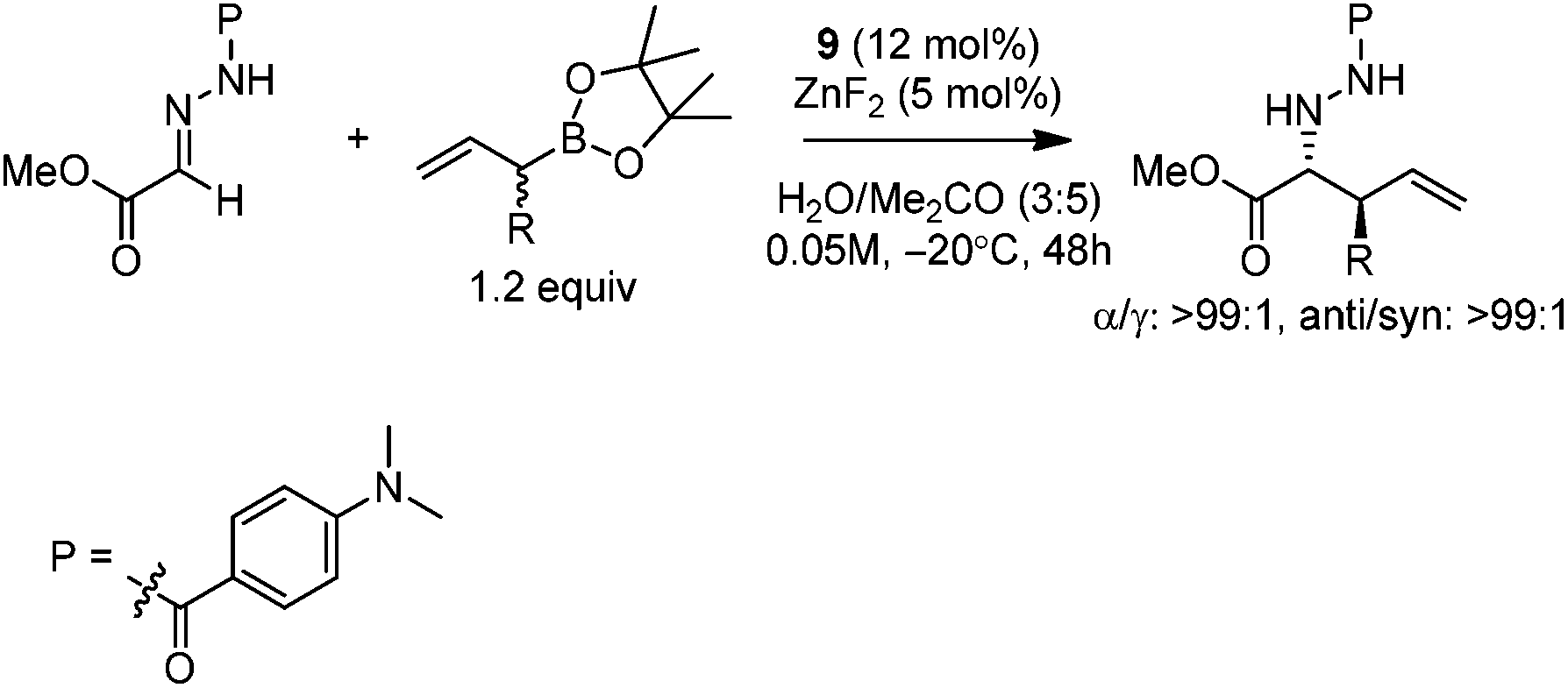
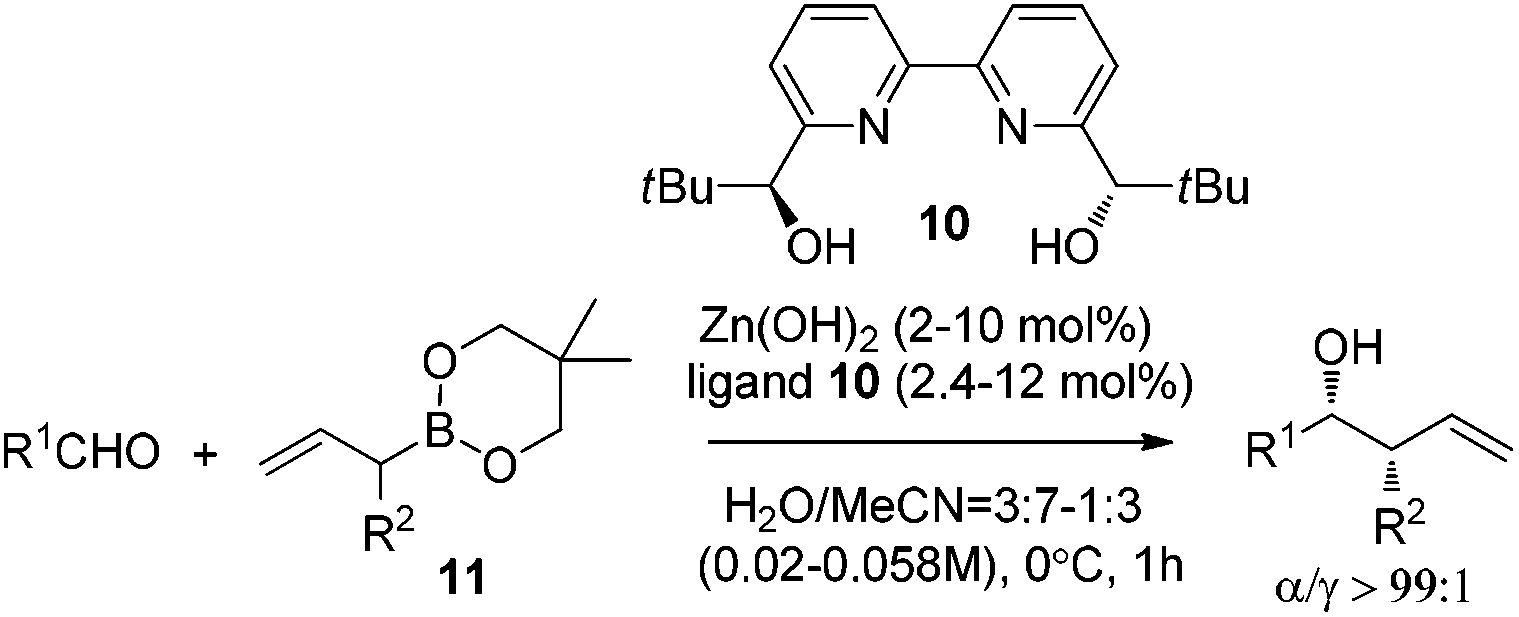
Kobayashi and co-workers further investigated the catalytic asymmetric allylation of aldehydes. They found that the catalytic system of Zn(OH)2 and chiral bipyridine ligand 10 promoted the addition of allylboronic acid 2,2-dimethyl-1,3-propanediol ester (11) with aldehydes to give excellent results under the optimized reaction conditions.25 Similar to allylation of hydrazono esters, the α-addition products were afforded and the favorable syn-adduct was generated with good enantioselectivity (Table 4). This catalytic system was also applied to α-methylallylation and other α-alkylallylations, giving moderate to excellent syn-selectivities and high to excellent enantioselectivities for both aromatic and aliphatic aldehydes.25
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Loading (mol%) | Yield (%) | ee (%) (syn) | syn/anti |
| 1 | Ph | Me | 10 | 92 | 81 | 10/1 |
| 2 | PhCH2CH2 | Me | 10 | 94 | 88 | 6/1 |
| 3 | CH3(CH2)8 | Me | 3 | 94 | 82 | 7/1 |
| 4 | PhCH2CH2 | Et | 5 | 96 | 91 | 3/1 |
| 5 | PhCH2CH2 | nBu | 5 | 97 | 90 | 3/1 |
| 6 | Ph | OBn | 10 | 82 | 88 | 24/1 |
| 7 | Ph | Cl | 5 | 92 | 88 | 24/1 |
| 8 | CH3(CH2)10 | Cl | 2 | 92 | 91 | 13/1 |
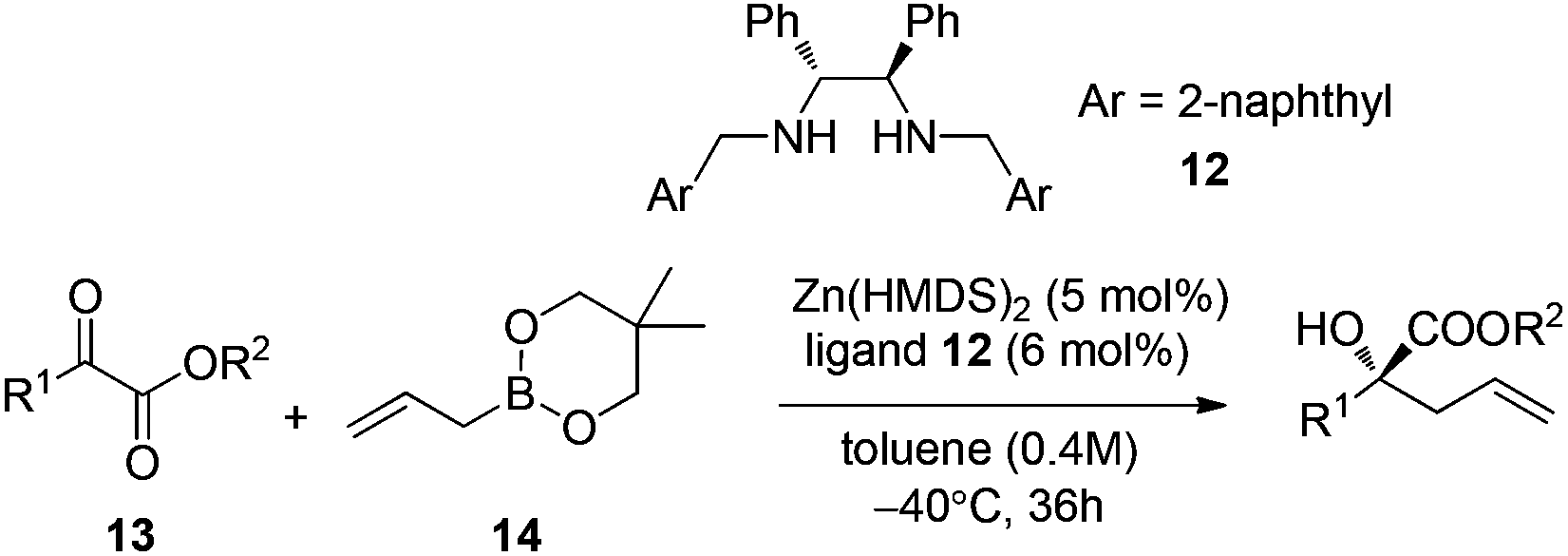
Further progress of enantioselective allylation of ketones required intense screening of zinc salts and chiral ligands.26 A 1,2-diphenylethylene-diamine derived chiral ligand 12 and Zn(HMDS)2 were identified for realizing the allylation of a few α-keto esters in high enantioselectivities under the optimal reaction conditions (Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yield (%) | ee (%) |
| 1 | Ph | Me | 86 | 97 |
| 2 | Ph | Bn | 78 | 83 |
| 3 | Me | Bn | 72 | 89 |
2.3 In-mediated system
In 2008, Kobayashi and co-workers reported a catalytic asymmetric allylation of acetophenone with allylboronate in the presence of 5 mol% In(0)-chiral bis(oxazoline) ligand 15 in water.27 Although it was the first example of In-mediated asymmetric allylboronation in water, the 68% yield and 52% ee are far from satisfactory (Scheme 13).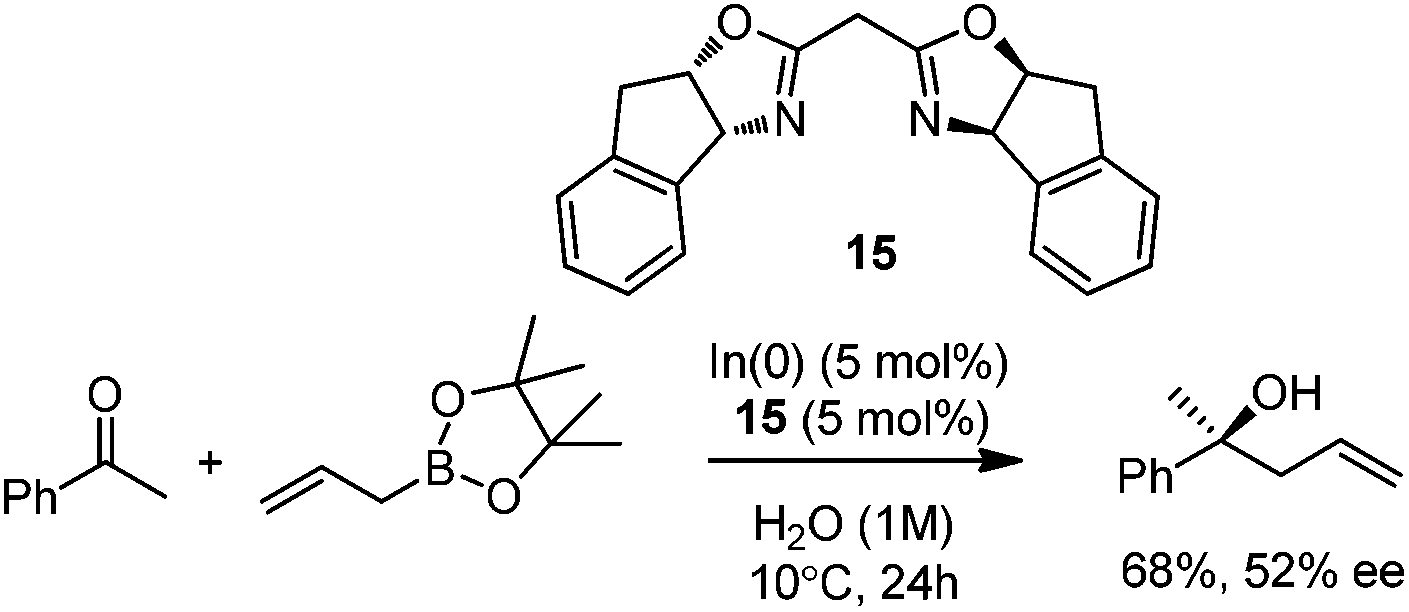 | ||
| Scheme 13 Asymmetric In(0)-catalysis in water. | ||
Kobayashi and co-workers later disclosed another example of applying indium catalysis in enantioselective allylation, crotylation, and α-chloroallylation of hydrazones with boronates.28 An in situ generated chiral indium(I)–semicorrin catalyst 16 could give high yields and excellent enantioselectivities when different aryl hydrazone substrates were used. The reaction tolerates functionalities at the arene, such as hydroxy, methoxy, tertiary amino, and nitro groups (Table 6). Crotylation of racemic α-methyl or α-chloroallyl boronic acid pinacol ester (17) produced exclusively an α-adduct with excellent anti/syn ratios and good enantioselectivities. However, reaction conditions were not suitable for cyclohexane or other aliphatic carbaldehyde imine derivatives (entry 4). Mechanistically, it was assumed that hydrazone acts as a Lewis base to activate the allylic boronate for transmetallation, and the resulting active species, a chiral allylindium reagent, undergoes the nucleophilic addition to the imine derivatives via a cyclic chair-like transition state. Because of the gauche interaction between R and R′ in the transition state when the (E)-isomer was employed, the anti-product afforded by the allylation of (Z)-allyl indium species was predominant. The authors also concluded that an equilibrium between (E) and (Z)-isomers through π-allyl indium species existed (Scheme 14).
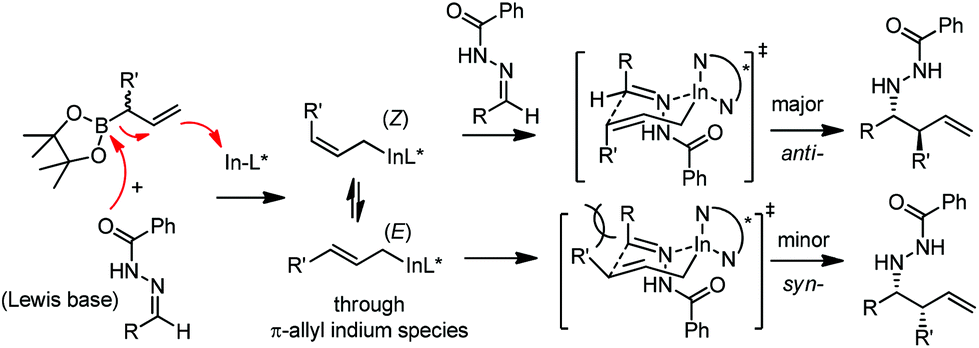 | ||
| Scheme 14 Proposed mechanism. | ||
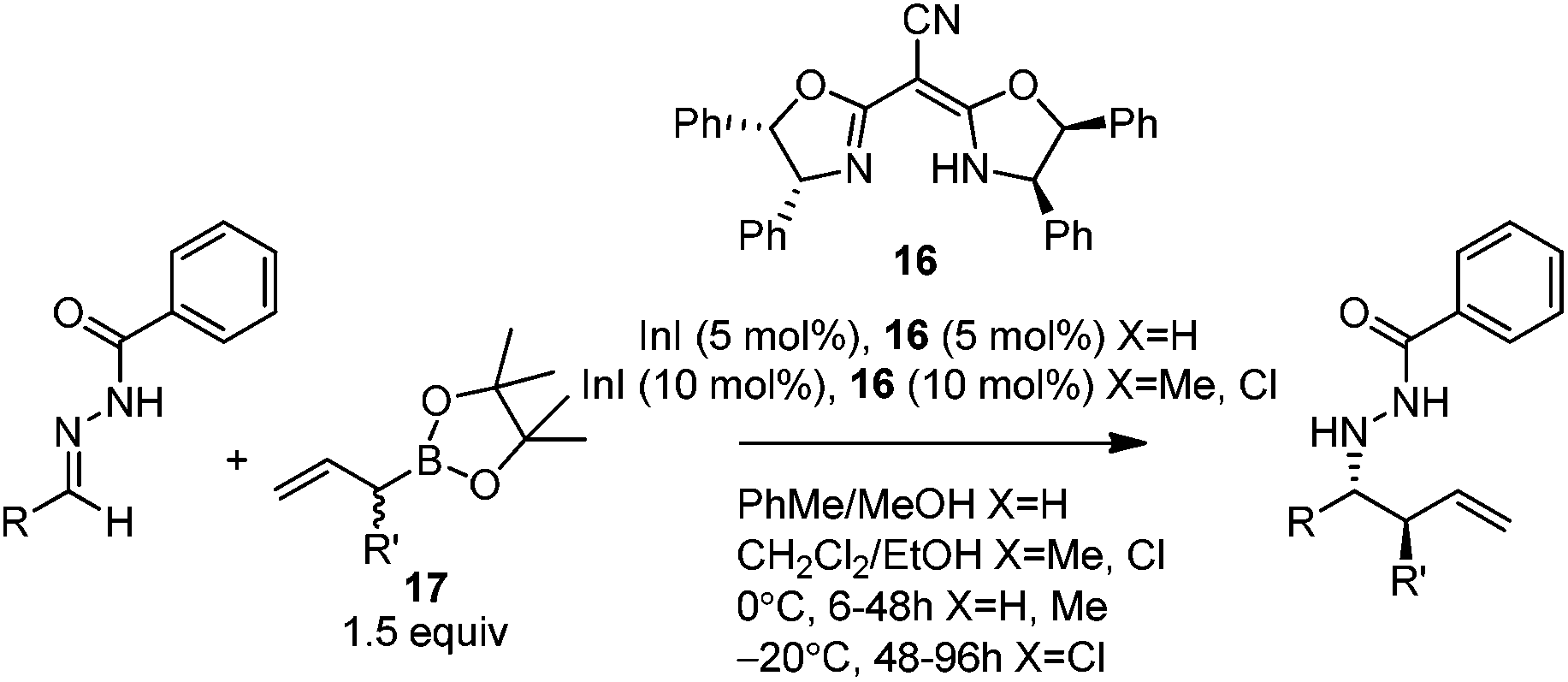
2.4 Ni-mediated system
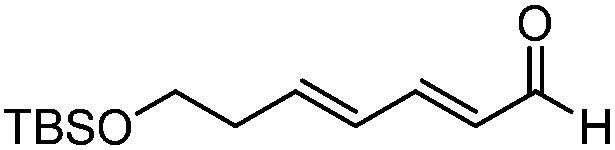
In 2009, Morken and co-workers reported a unique catalytic enantioselective allylation of dienals, which was proposed to occur by 3,3′-reductive elimination in the presence of Ni(cod)2 and chiral phosphonite (R,R)-18.29 Both δ-aromatic and aliphatic substituted dienals gave the predominant E,Z-adducts in high enantioselectivities (Table 7). Interestingly, the minor E,E-adduct was determined to be racemic and assumed to arise from a non-catalyzed reaction that occurs at room-temperature. Morken et al. further suggested that the following transmetallation consists of the boron Lewis acid-promoted electron transfer from Ni(0) to the dienal, which subsequently forms an allyl nickel species. At last a 3,3′-reductive elimination leads to the E,Z-allylation product (Scheme 15).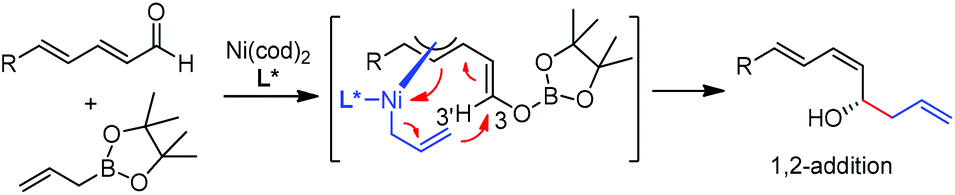 | ||
| Scheme 15 Proposed mechanism. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | (E,Z)/(E,E) | Yield (%) | ee (%) |
| 1 |
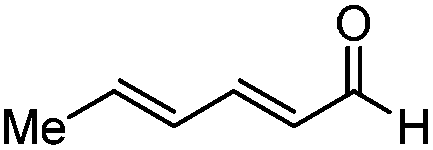
|
>20:1 |
84 | 88 |
| 2 |
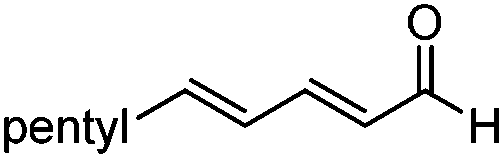
|
>20:1 |
84 | 87 |
| 3 |
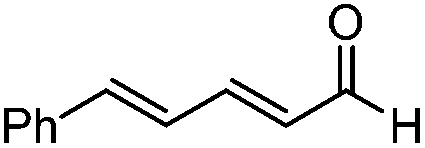
|
>20:1 |
68 | 91 |
| 4 |
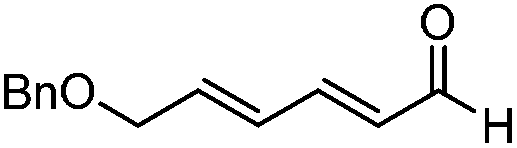
|
15:1 |
86 | 73 |
| 5 |
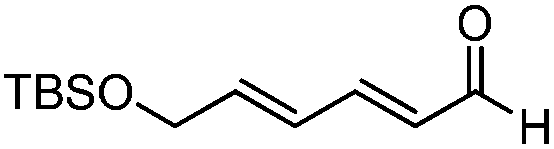
|
7:1 |
81 | 85 |
| 6 |
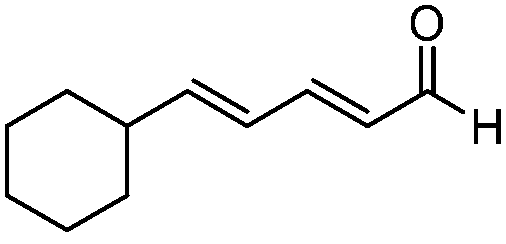
|
>20:1 |
92 | 93 |
| 7 |
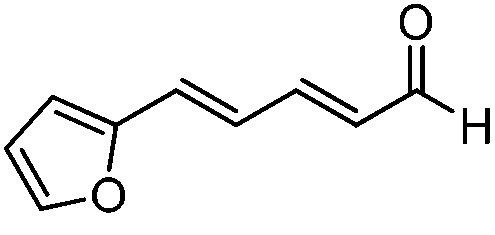
|
15:1 |
73 | 94 |
| 8 |
|
16:1 |
83 | 90 |
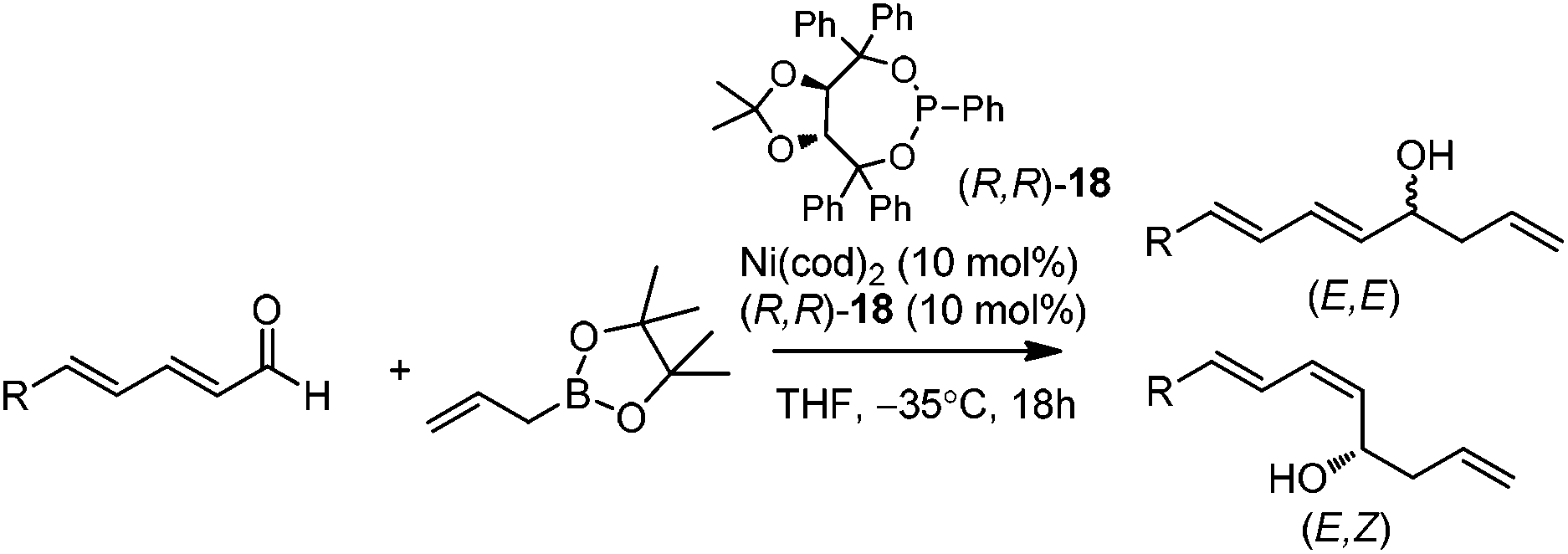
2.5 Rh-mediated system
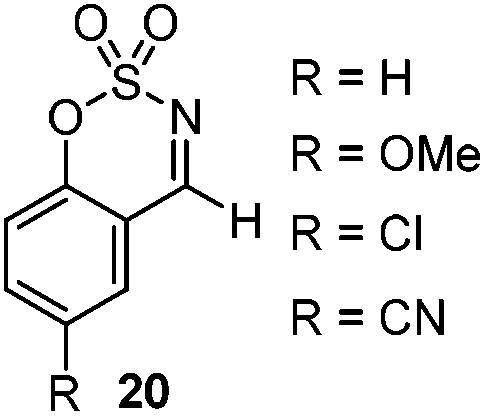
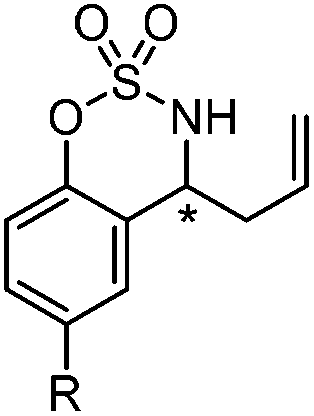
Very recently, Lam and co-workers reported an enantioselective allylation of cyclic aldimines and ketimines with potassium allyltrifluoroborates by using a chiral diene-ligated rhodium complex.30 In this study, only cyclic imines are effective substrates for allylation. Under the optimized reaction conditions, using diene 19 as the chiral ligand, high enantioselectivities (90–99% ee) and generally good yields were obtained for a wide range of cyclic imines (Table 8). Both benzoxathiazine-2,2-dioxides 20 bearing methyl, methoxy, halogen, cyano, and dioxole groups and other cyclic aldimines and ketimines delivered excellent results.|
|
||||
|---|---|---|---|---|
| Entry | Starting material | Product | Yield (%) | ee (%) |
| 1 |
|
|
87 | 96 |
| 2 | 92 | 91 | ||
| 3 | 96 | 96 | ||
| 4 | 97 | 98 | ||
| 5 |

|
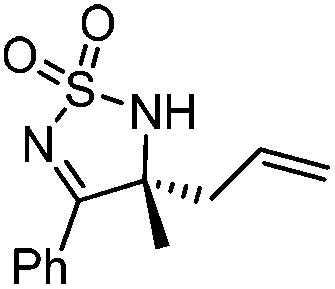
|
88 | 97 |
| 6 |
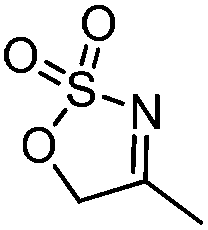
|
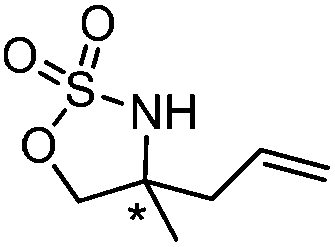
|
83 | 93 |
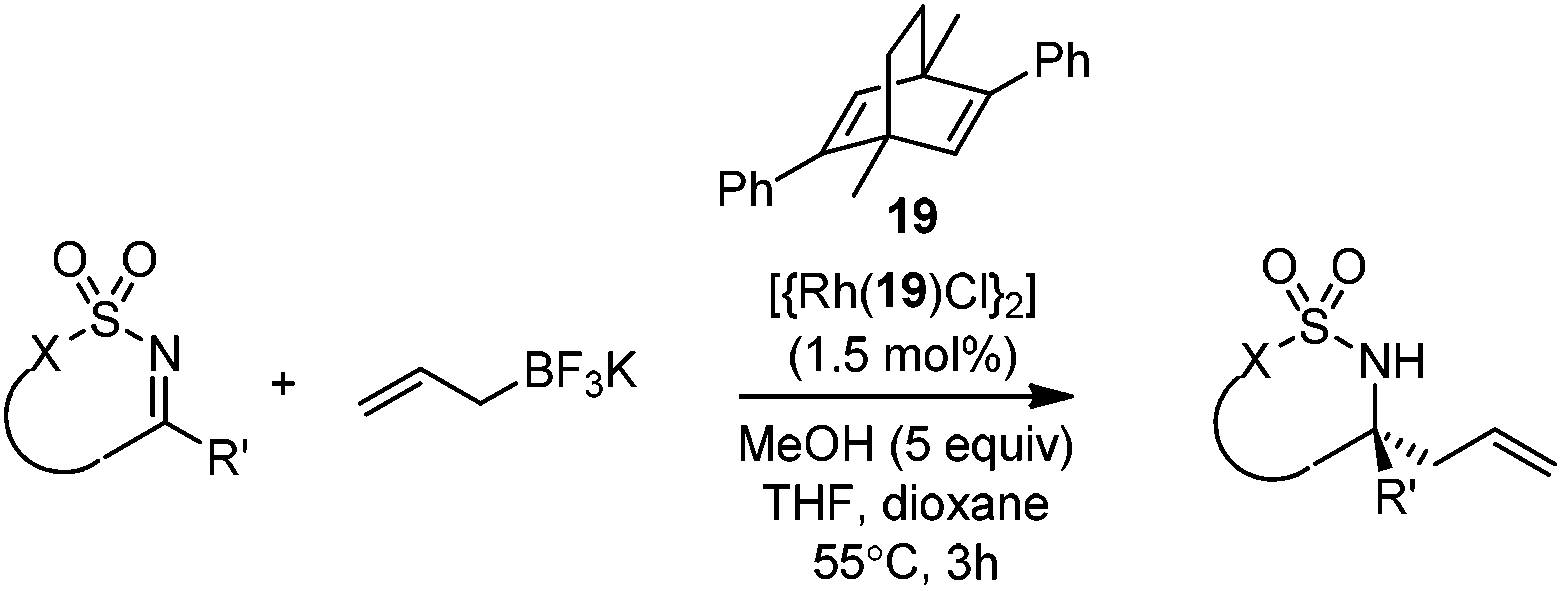
The crotylation of cyclic imine 21 was realized with high diastereo- and enantioselectivity (Table 9). E-Crotyltrifluoroborate 22 and Z-crotyltrifluoroborate 23 afforded anti-product and syn-product, respectively.30 To gain insights into the mechanism, ketimine 21 was subjected to the allylation with bisdeuterated potassium allyltrifluoroborate 24 (Scheme 16). A mixture of products 25 and 26 (ratio 1/1) suggested that a rapid interconversion between two σ-allyl haptomers might be feasible when allylrhodium(I) species 27a/27b were generated after transmetallation (Schemes 16 and 17). The subsequent allylation proceeded with excellent stereocontrol via a cyclic chair-like transition state. After the protonation with HX (X = Cl, F, or OMe) of the rhodium amide, the corresponding product is released and the active species 28 was re-generated to complete the catalytic cycle (Scheme 17).
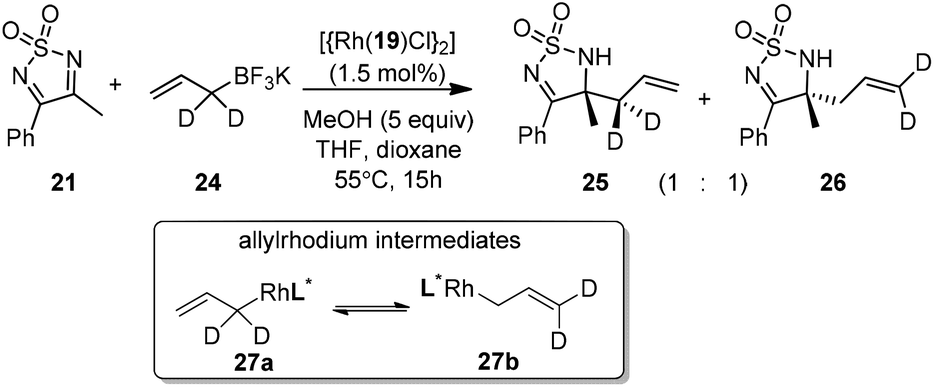 | ||
| Scheme 16 Deuterium-labeling experiment. | ||
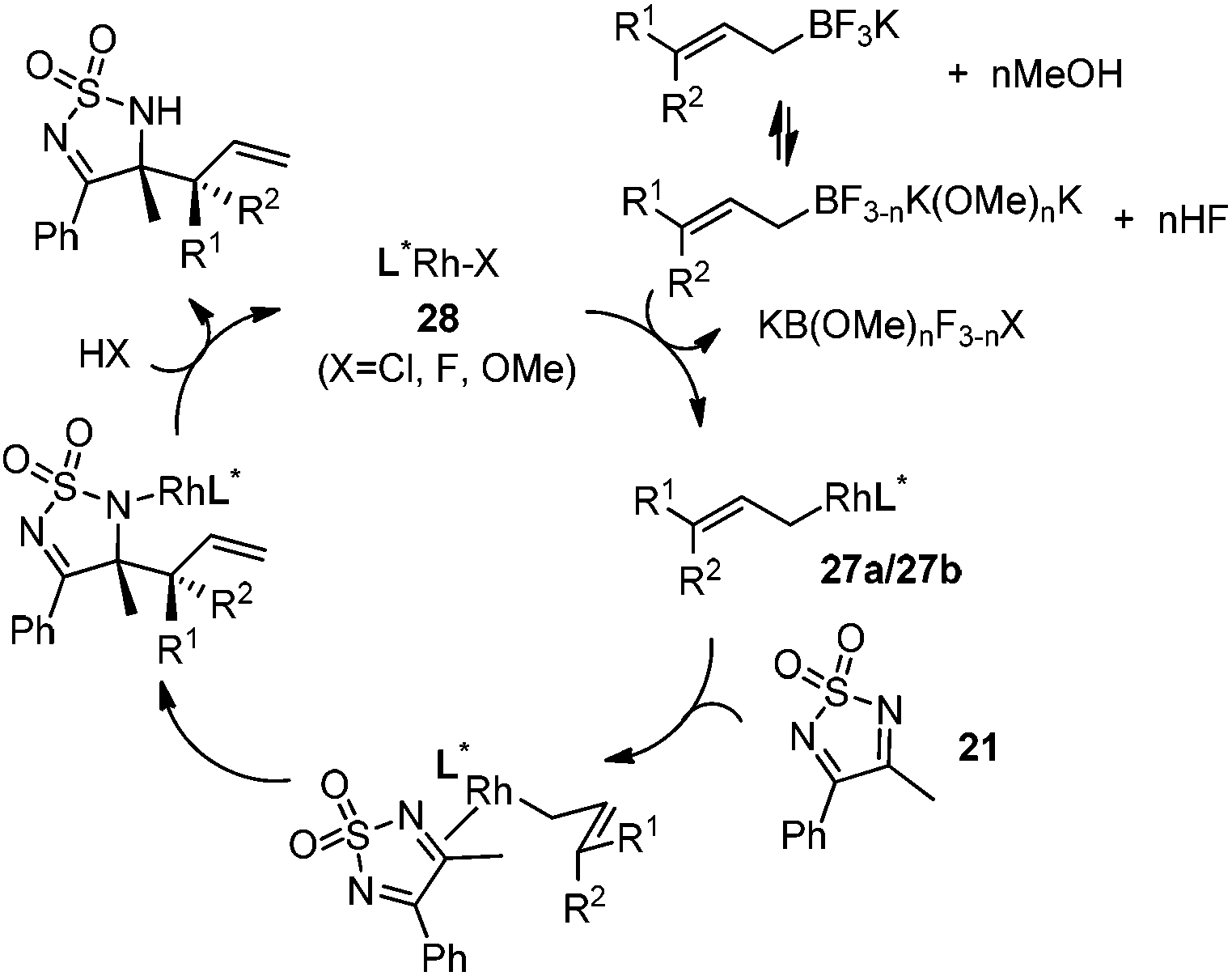 | ||
| Scheme 17 Possible catalytic cycle. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Allyltrifluoroborate | syn/anti | Yield (%) | ee (%) |
| 1 |
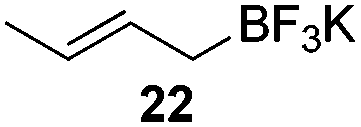
|
<1:19 |
68 | 97 |
| 2 |
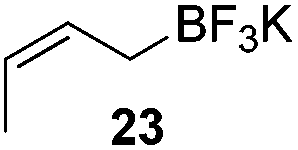
|
>19:1 |
89 | 99 |
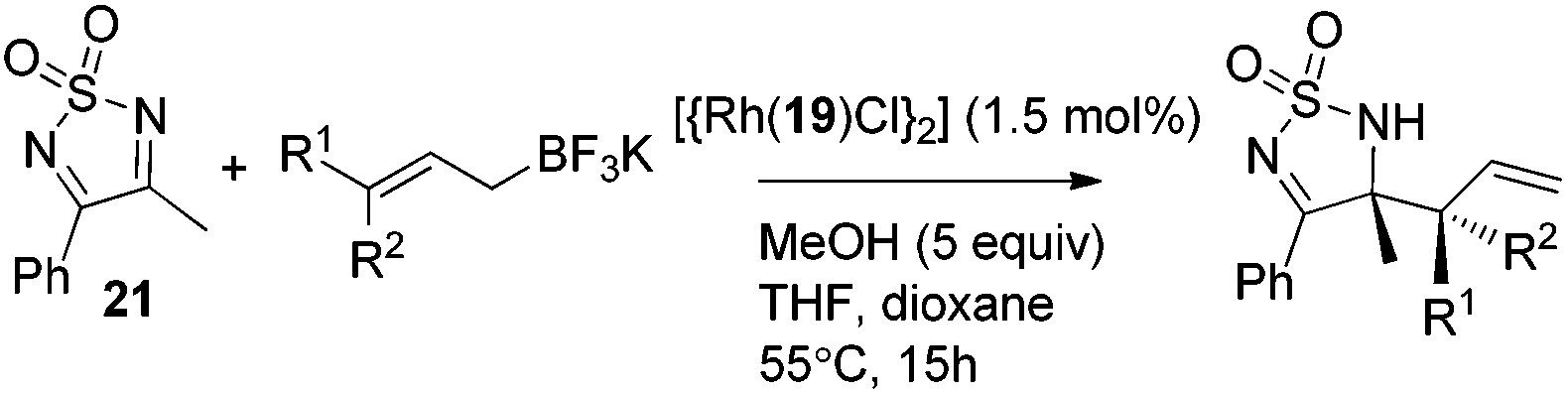
3. Brønsted acid-catalyzed asymmetric allylation
Recently, organocatalysis has served impressively in many asymmetric transformations. The reaction modes mainly lie in hydrogen bonding and covalent bond formation which differ from the major interaction in metal-catalyzed reactions. The unique interactions between substrates and organocatalysts offer new reactivities and novel transformations. In the presence of Brønsted acids, allylboronate reacts with aldehydes in a highly diastereoselective and enantioselective manner. Although Lewis acid-assisted Brønsted acid catalysis (LBA) is a metal catalysis, its mode of action is reminiscent of a hydrogen-bond donor like organocatalyst. The chiral acid activates one of the boronate oxygens and accelerates the subsequent enantioselective allylation via a pre-organized transition state (Scheme 18). With the proper acid or LBA in hand, the asymmetric allylation proceeds in high yield and enantioselectivity. Concerning phosphoric acid catalyzed allylation, the Lewis base center of the phosphoryl oxygen in the catalyst interacts with the formyl hydrogen of aldehyde (the Lewis acid center) which may be critical for the well-organized transition state.31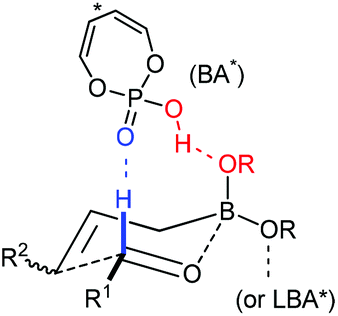 | ||
| Scheme 18 Interaction mode of acid-catalyzed asymmetric allylboration. | ||
3.1 Lewis acid-assisted Brønsted acid catalysis (LBA) system
Lewis acid-assisted Brønsted acid catalysis (LBA) was generally conceptualized by Yamamoto and co-workers. In the original catalyst system, tin chloride coordinates with the oxygen atoms of BINOL to increase the acidity of the hydroxylic proton, which is oriented in a particular direction.32 The catalyst system has been shown to be a particularly useful tool for asymmetric transformations (Scheme 19).33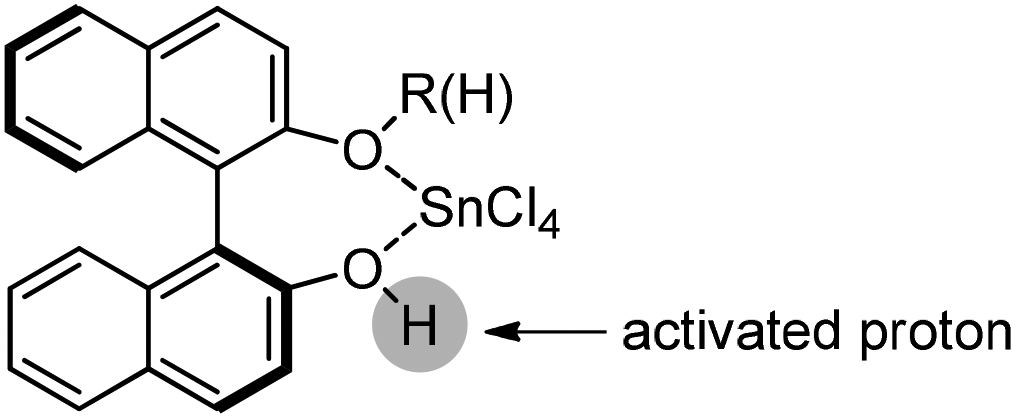 | ||
| Scheme 19 Yamamoto's LBA catalytic system. | ||
Following previous work on Lewis acid-catalyzed allylboration,15–17 Hall and co-workers screened several chiral ligand systems including scandium, copper, and many other metals as Lewis acids in the asymmetric allylation. Unfortunately, all of these attempts only provided either low ee values or no rate acceleration over the background reaction. It was assumed that these chiral metal complexes were simply too sterically bulky to coordinate effectively to the hindered boronic ester.34a Interestingly, when different C2-symmetric chiral diols combined with SnCl4 were screened, good to excellent asymmetric induction was obtained in the allylboration of aldehydes by the allylboronic acid pinacol.32 After extensive experiments, catalysts derived from diols (R,R)-29 and (R,R)-3034c were identified as the most efficient (Table 10).
|
|
|---|
|
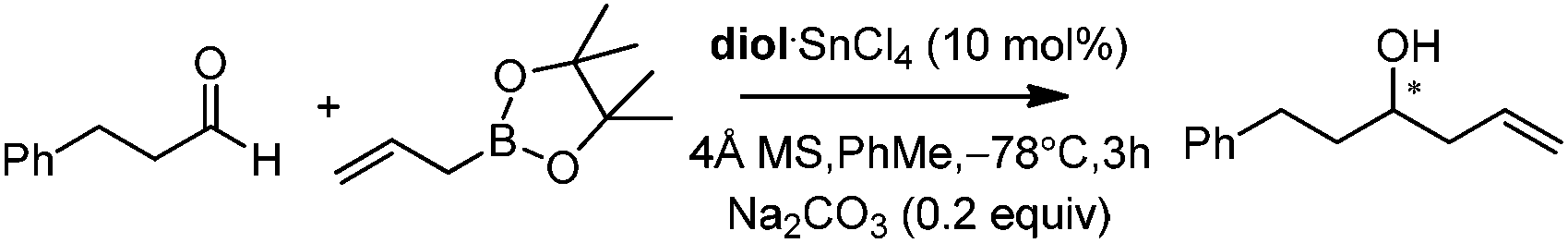
With catalytic allylboration, aliphatic aldehydes usually proceed in high enantioselectivities while aromatic aldehydes and enals show moderate enantioselectivity (Table 11).34a–e Under the optimized conditions, the crotylation of aldehydes with E-crotyl boronate was superior to the reaction with the (Z)-isomer (entry 7 vs. 8). This catalyst system was also efficient for the catalytic allylation of propargylic aldehydes (entries 9–12).34f Hall et al. also investigated the double diastereoselection of allylboration to chiral aldehyde34a (Scheme 20). In the presence of diol (R,R)-31, the allylboration resulted in a ratio of 84:16 favoring the anti-isomer (matched) while the usage of diol (S,S)-31 led to a lower selectivity (anti:syn = 35:64, mismatched). The moderate discrimination effect for chiral substances requires further optimization to explore the LBA approach.
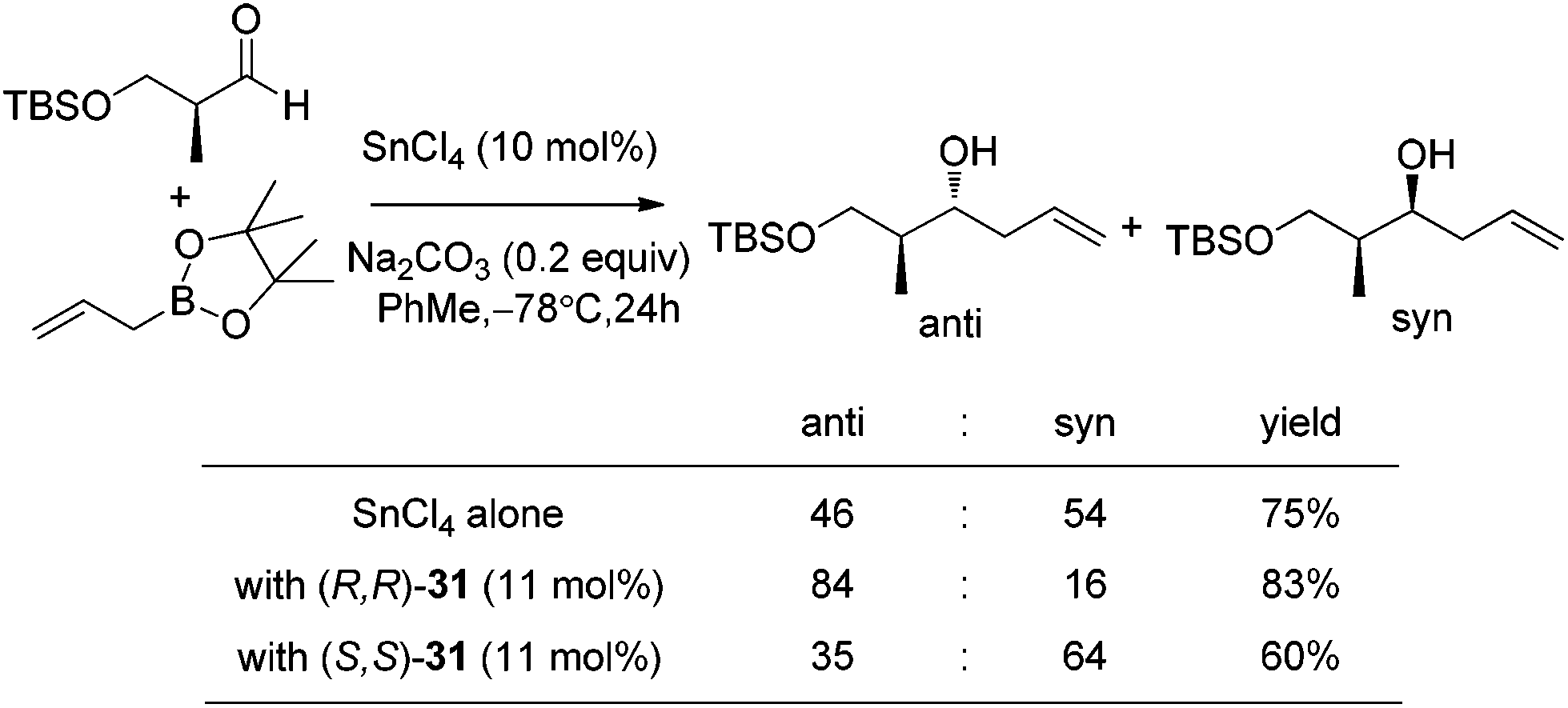 | ||
| Scheme 20 Double diastereoselection of allylboration to chiral substrate. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Diol | Yield (%) | ee (%) |
a Boronate =  .
b dr > 98:2. .
b dr > 98:2.
|
||||||
| 1 | Ph | H | H | 29 | 99 | 71 |
| 2 | PhCH2CH2 | H | H | 29 | 99 | 95 |
| 3 | TBSO(CH2)2 | H | H | 29 | 98 | 95 |
| 4 | PhCH2CH2 | H | H | 30 | 99 | 97 |
| 5 | TBSO(CH2)2 | H | H | 30 | 99 | 96 |
| 6 | 3,5-(CF3)2C6H3 | H | H | 29 | 99 | 94 |
| 7b | PhCH2CH2 | Me | H | 29 | 93 | 96 |
| 8b | PhCH2CH2 | H | Me | 29 | 78 | 84 |
| 9a | C4H9C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C C |
H | H | 30 | 84 | 71 |
| 10a | PhCC |
H | H | 30 | 80 | 69 |
| 11a,b | Ph(CH2)2CC |
Me | H | 30 | 88 | 91 |
| 12a,b | TMSCC |
Me | H | 30 | 83 | 82 |
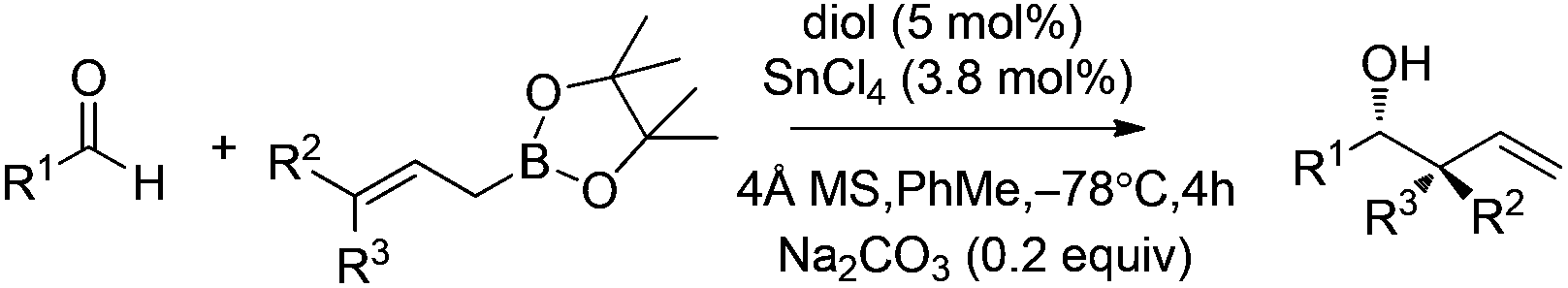
Concerning the mechanistic proposal of LBA-catalyzed allylation, the function of the diol·SnCl4 system is much more complex than a Brønsted acid or bisalkoxy-dichlorotin species alone.34c Based on the previous concerns of mechanism on Lewis acid-catalyzed allylborations and several controlled experiments,15–17 it was proposed that allylboration of aldehydes occurred by the coordination of one of the boronate oxygens with the “super” acidic proton which was formed from the combination of diols with SnCl4. The chiral diols determined the facial selectivity for the addition to aldehyde through a closed six-membered chair-like transition state with high levels of asymmetric induction (Scheme 21).
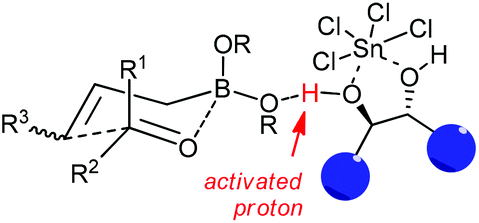 | ||
| Scheme 21 Proposed asymmetric allylation intermediate. | ||
3.2 Brønsted acid catalysis system
Organocatalysis always devises elegant solutions for the most challenging problems in modern asymmetric synthesis.35 In 2010, Antilla and co-workers reported the first high-yielding and highly enantioselective chiral phosphoric acid-catalyzed allylboration of aldehydes.36a The stability and availability of allylic boronates as well as chiral catalysts was astonishing for the organocatalytic allylation reaction. The reaction was proved to be highly general with a broad substrate scope covering aryl, heteroaryl, α,β-unsaturated, and aliphatic aldehydes. With the catalysts screened, the authors found that phosphoric acid (R)-TRIP 32 was the best catalyst for allylation of aldehydes (condition A in Table 12).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Condition A | Condition B | ||
| Yield (%) | ee (%) | Yield (%) | ee (%) | ||||
| a Reaction was run at 0 °C. | |||||||
| 1 | Ph | H | H | 99 | 98 | 97 | 99 |
| 2 | 4-MeOC6H4 | H | H | 95 | 98 | 95 | 99 |
| 3 | 4-CO2MeC6H4 | H | H | 96 | 96 | 96 | 99 |
| 4 | Cy | H | H | 98 | 73 | 90 | 91 |
| 5 | PhCH2 | H | H | 96 | 90 | 86 | 98 |
| 6 | 9-Anthracene | H | H | 94 | 91 | 92 | 97 |
| 7a | Ph | Me | H | 96 (anti:syn = 98:2) |
99 | 98 (anti:syn = 99:1) |
99 |
| 8 | Ph | H | Me | 95 (anti:syn = 2:98) |
94 | 99 (anti:syn = 1:99) |
99 |
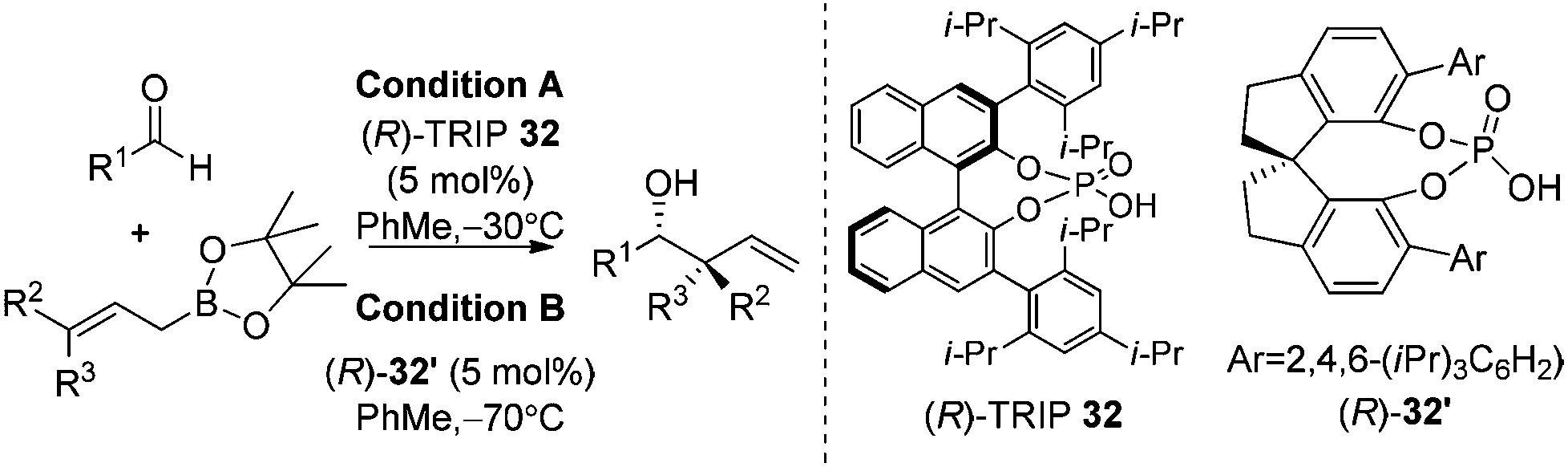
Further work by Hu and co-workers found that the SPINOL-based phosphoric acid (R)-32′ is a superior catalyst to achieve higher enantioselectivities on substrates used in Antilla's original report (condition B in Table 12). The new catalyst clearly maintains a high reactivity at lower temperature.36b
The reaction conditions were also shown to be effective for the enantioselective crotylation of aldehydes with high diastereoselectivities (entries 7 and 8). It indicates that the allylboration proceeds via a Type I mechanism involving a six-membered chair-like transition state. Similar to previous work on allylboration,15–17,34 Antilla et al. believed that activation via the protonation of the pseudo-equatorial oxygen in boronate by the chiral phosphoric acid catalyst is the most plausible transition state (Model A, Scheme 22).36a
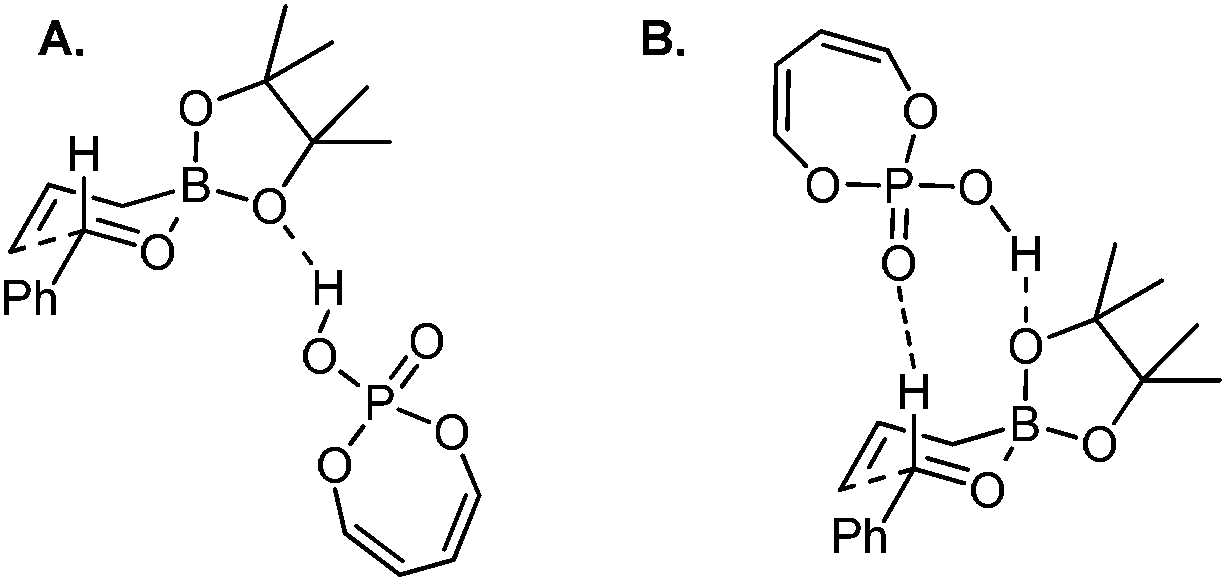 | ||
| Scheme 22 Optimized transition states with buta-1,3-diene-1,4-diol-phosphoric acid (the simpler model for TRIP). | ||
However, Goodman's group utilized DFT and QM/MM hybrid calculations to comment that the reaction actually adopts a transition state involving two crucial hydrogen-bonding interactions (Model B, Scheme 22). One is from the hydroxyl group in PA to the pseudo-axial oxygen of the cyclic boronate; another is a stabilizing interaction between the phosphoryl oxygen of the catalyst (Lewis base site) to the formyl hydrogen of the aldehyde (Lewis acid site).31 This alternative transition state has the lowest energy and extra rigidity accounting for the excellent asymmetric control.
Subsequently, Antilla and Houk reinvestigated the original TRIP-catalyzed allylboration with density functional theory (B3LYP and B3LYP-D3) besides several levels of DFT calculations in 2013.37 The lowest energy transition state of chiral BINOL-phosphoric acid catalyzed allylboration is indeed following Goodman's axial model. Moreover, the equatorial model originally proposed by Antilla accounted for the generation of the minor enantiomer (Scheme 23).
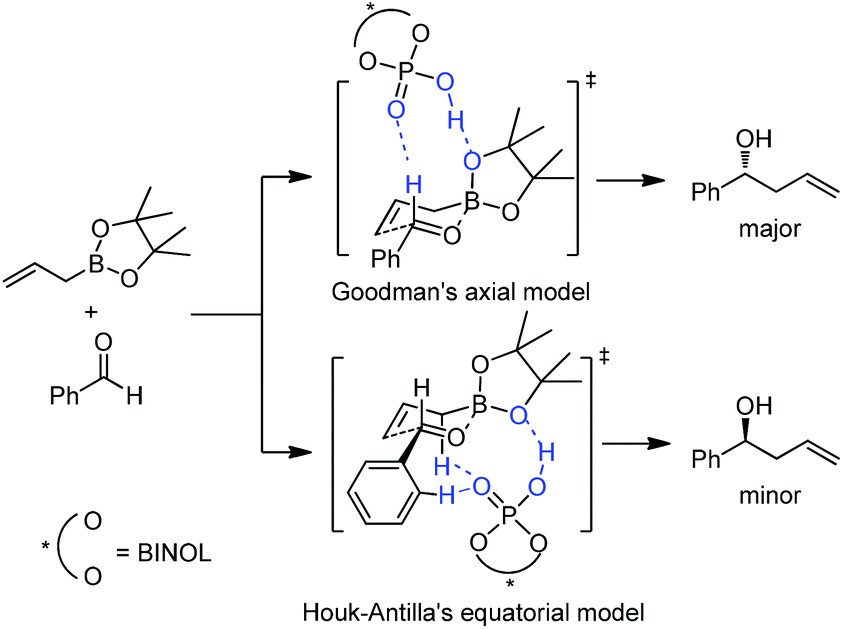 | ||
| Scheme 23 Transition state of chiral BINOL-phosphoric acid catalyzed allylboration. | ||
In 2013, Malkov et al. developed a chiral phosphoric acid-catalyzed kinetic resolution of racemic allylboronates 33 in a face- and Z-selective allylation of aldehydes (Scheme 24).38 The R-enantiomer of 33 was found to readily react with aldehydes to deliver adducts in good enantioselectivity, while the S-enantiomer remained behind. The enantioselective process was again interpreted by Goodman's axial model.
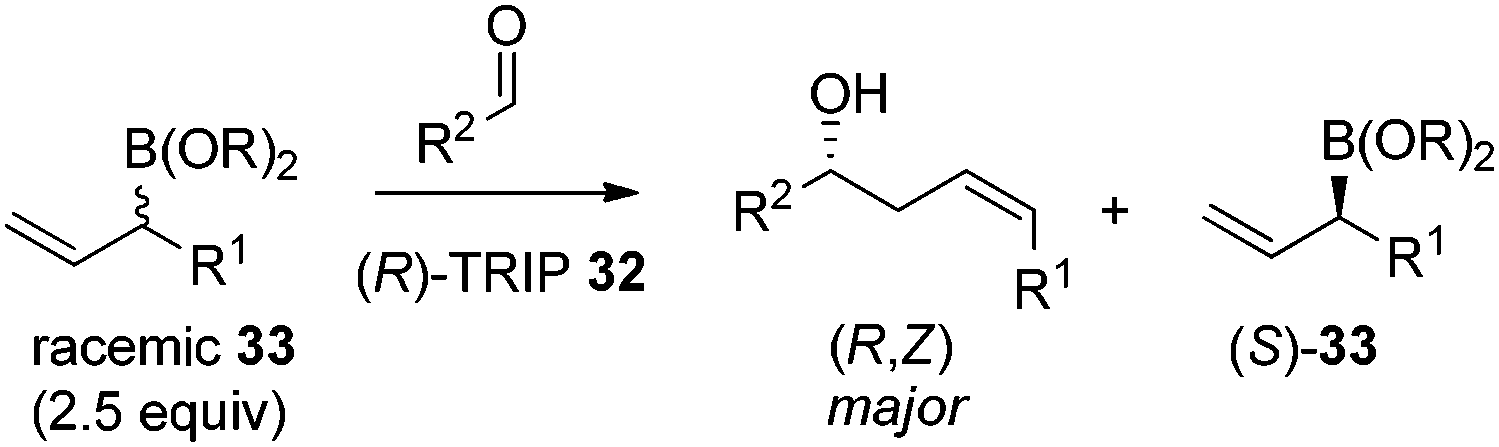 | ||
| Scheme 24 Kinetic allylboration of aldehydes. | ||
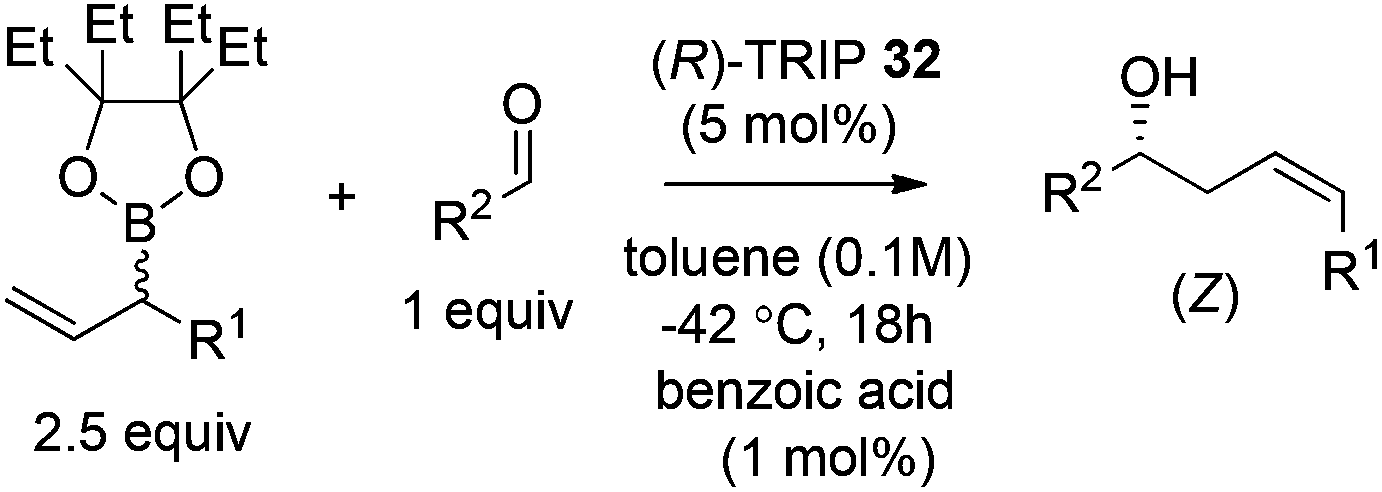
Hoffmann,39 Pietruszka and Schone40 illustrated that the E/Z ratio of the homoallylic alcohol products in the allylation with secondary alkyl allyl boronates was determined by the steric hindrance of the boronate fragment. The Z-isomer was more likely generated with larger groups, such as pinacolate or benzopinacolate in the boronate were used. Malkov et al. also performed DFT level calculations to understand the influence of the steric size of the cyclic boronate moiety on the E/Z ratio in the corresponding products.38 The computation revealed that the transition states involved a two-point activation mode in accord with Goodman's work. Importantly, the calculation also predicted that the tetraethylethylene glycol derivative (Epin) should give a better Z/E ratio, which indeed guided the authors to locate the optimal boronates. Under the optimal conditions, the allylation of both aromatic aldehydes and aliphatic aldehydes proceeded in high yields and enantioselectivities with an impressive Z selectivity of >25:1 (Table 13).38
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yield (%) | eea (%) |
|
a The Z/E ratio was >25:1.
b The product was assigned as S-configuration as a result of the change in the preference of the substituents in the Cahn–Ingold–Prelog system.
c The Z/E ratio was 13:1.
d Reaction time was 60 h.
|
||||
| 1 | Me | Ph | 96 | 97 |
| 2 | Me | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
84 | 97 |
| 3 | Me | 4-FC6H4 | 80 | 85 |
| 4 | Me | c-C6H11 | 72 | 88 |
| 5 | Me | PhCH2CH2 | 81 | 91b |
| 6 | nPr | Ph | 90 | 94 |
| 7 | nPr | PhCHCH |
97d | 93 |
| 8 | nPr | PhCH2CH2 | 80d | 87b,c |
In 2013, Murakami and co-workers reported a highly diastereo- and enantioselective synthesis of anti-homoallylic alcohols from terminal alkynes and aldehydes with a cationic iridium(I) complex/chiral phosphoric acid relay system (Scheme 25).41 The cationic iridium(I) complex-catalyzed olefin transposition of (E)-1-alkenylboronates, generated from hydroboration of the corresponding terminal alkynes, afforded (E)-2-alkenylboronates which further participated in the enantioselective allylboration of aldehydes catalyzed by (R)-TRIP 32 (Scheme 25). The iridium(I) catalyst system does not interfere with (R)-TRIP 32, which is used for the asymmetric allylation. While screening the scope of aldehydes, it was discovered that an electronically and sterically diverse array of aromatic aldehydes and aliphatic aldehydes generally proceeded in 85–99% yield with excellent diastereoselectivities and high enantioselectivities (except entry 5, Table 14).41
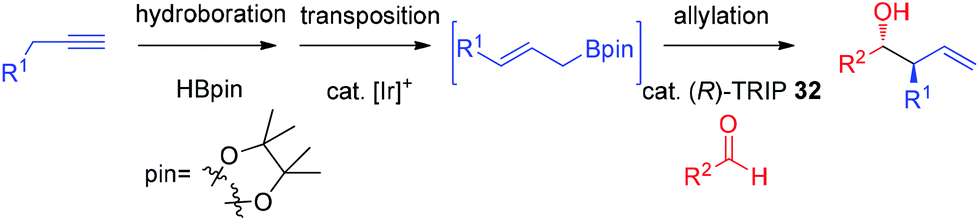 | ||
| Scheme 25 Allylboration with Ir(I)-complex/TRIP relay system. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Y | T (°C) | Yield (%) (anti:syn) |
ee (%) |
|
a Conditions: aldehydes (0.40 mmol), (E)-alkenylboronates (0.80 mmol), [Ir(cod)2]BF4–PCy3 (Ir:P = 2:5), MS 4 Å (50 mg) in 1,2-DCE (1 mL).
|
|||||||
| 1 | Ph | Et | 5.0 | 10 | 28 | 90 (>98:2) |
93 |
| 2 | Ph | Ph | 10 | 20 | −15 | 83 (>98:2) |
88 |
| 3 | Ph | (CH2)3OTBS | 7.5 | 20 | 28 | 85 (>98:2) |
90 |
| 4 | Ph | (CH2)3CO2Me | 7.5 | 20 | 28 | 86 (98:2) |
93 |
| 5 | Ph | OTBS | 7.5 | 10 | 28 | 97 (92:8) |
17 |
| 6 | 4-MeOC6H4 | Et | 7.5 | 15 | 28 | 99 (>98:2) |
92 |
| 7 | 4-NO2C6H4 | Et | 5.0 | 10 | 28 | 85 (>98:2) |
95 |
| 8 | 2-Furyl | Et | 5.0 | 10 | 28 | 91 (>98:2) |
92 |
| 9 | PhCH2CH2 | Et | 10 | 20 | 5 | 88 (>98:2) |
91 |
| 10 | Cy | Et | 10 | 20 | 5 | 82 (97:3) |
88 |
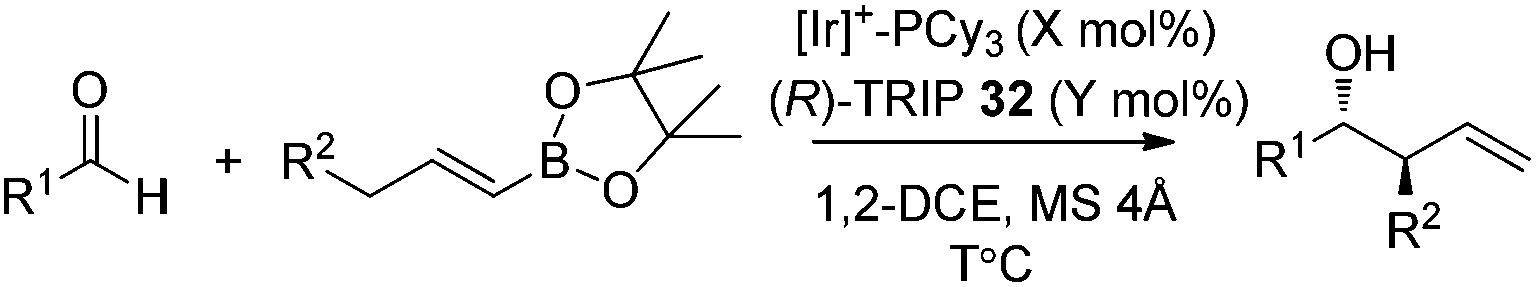
4. Catalytic asymmetric allylation by ligand exchange
Catalytic asymmetric allylation of ketones and imines can be realized through a crucial ligand-exchange step involving the allylboronate. After the ligand exchange on the borane, the corresponding chiral boronate proceeds through the allylation and regenerates the chiral ligand which participates in the subsequent ligand-exchange process (Scheme 26). Restricted to the speed of ligand exchange, this approach may not be feasible for aldehydes since the background reaction is highly prone to occur before the requisite ligand-exchange process.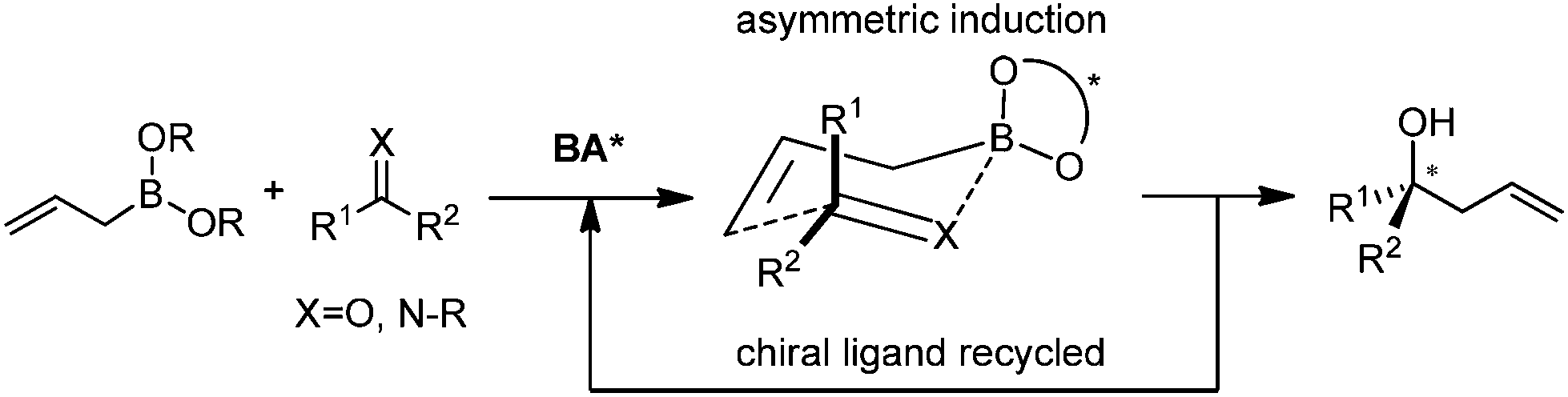 | ||
| Scheme 26 General reaction mode of ligand-exchange of boronates. | ||
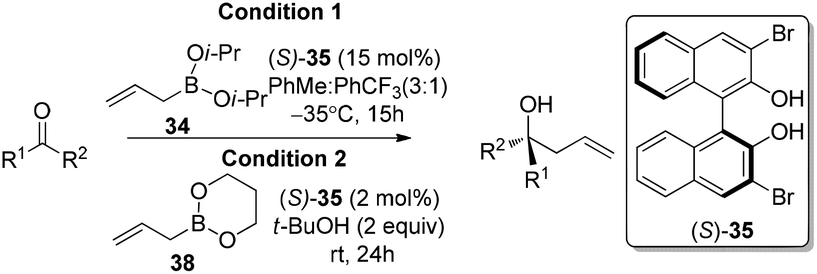
4.1 Exchange of chiral diols system
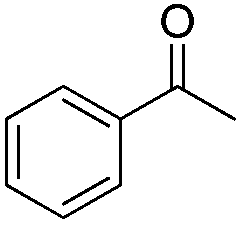
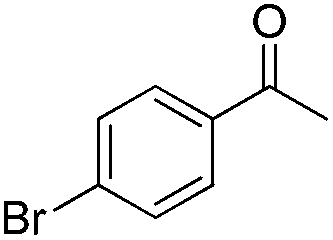
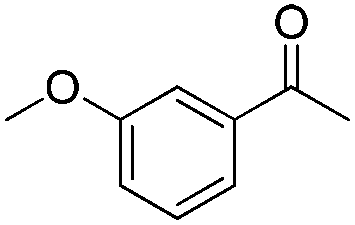
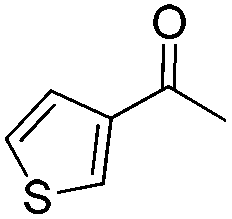
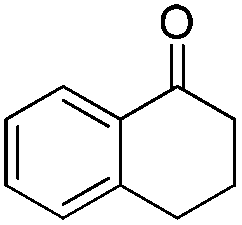
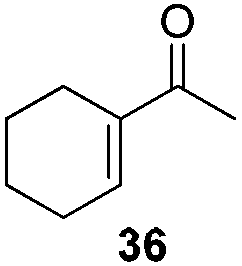
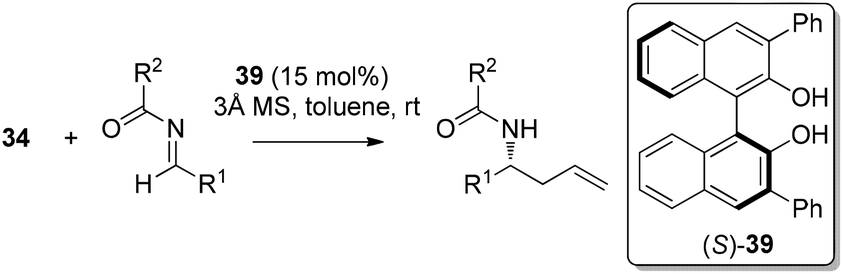
In 2006, Schaus and co-workers reported a class of chiral diols which catalyzes an enantioselective and diastereoselective allylboration of ketones with allyldiisopropoxyborane 34.42 It was found that 3,3′-Br2-BINOL 35 was the most effective catalyst to promote the asymmetric reaction of a variety of ketones with high enantioselectivities in PhCF3–toluene (1:3 ratio) at −35 °C (condition 1, Table 15). All electron-rich and electron-deficient aromatic ketones and heteroaromatic ketones proceeded with good results. Enones 36 and 37 also underwent 1,2-addition as the regioselective products. Allylboronate 38 was comparable under the standard allylation conditions.
Preliminary mechanistic experiments revealed that the ligand exchange process between one isopropoxy ligand of boronate and chiral diol was observed by 1H NMR and ESI-MS analysis during the reaction of 34 with 35. The catalyst-associated boronate complex reacts with ketones via a six-membered chair-like transition state which is responsible for the high enantioselectivity. Finally, another ligand-exchange process takes place to liberate the chiral diol and allylic alcohol (Scheme 27).42
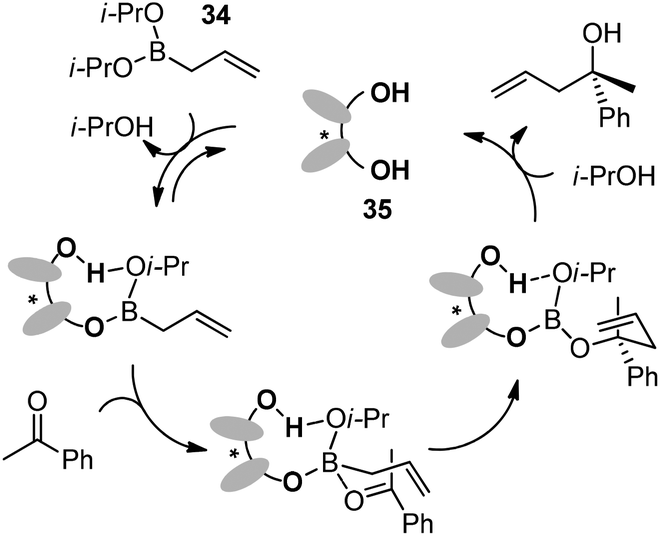 | ||
| Scheme 27 Possible catalytic cycle. | ||
Cyclic boronates such as dioxaborolane and dioxaborinane were further identified as better boronate resources since they are easier to prepare, stable during purification and can be stored for longer than acyclic boronates. In addition to the enhancement of stability, the tethered diol used to generate the cyclic boronate also facilitates ligand exchange at the end of a reaction cycle. As a result, Schaus et al. alternatively employed allyldioxaborinane 38 for the allylation of ketones with chiral 3,3′-Br2-BINOL 35.43 Under the optimized reaction conditions, allylation of ketones proceeded in excellent yields and enantioselectivities at room temperature (condition 2, Table 15). Moreover, the crotylboration of acetophenone with both diisopropoxy acyclic boronate and cyclic allyldioxaborinane was performed to give high yields, diastereoselectivities and enantioselectivities (Scheme 28).
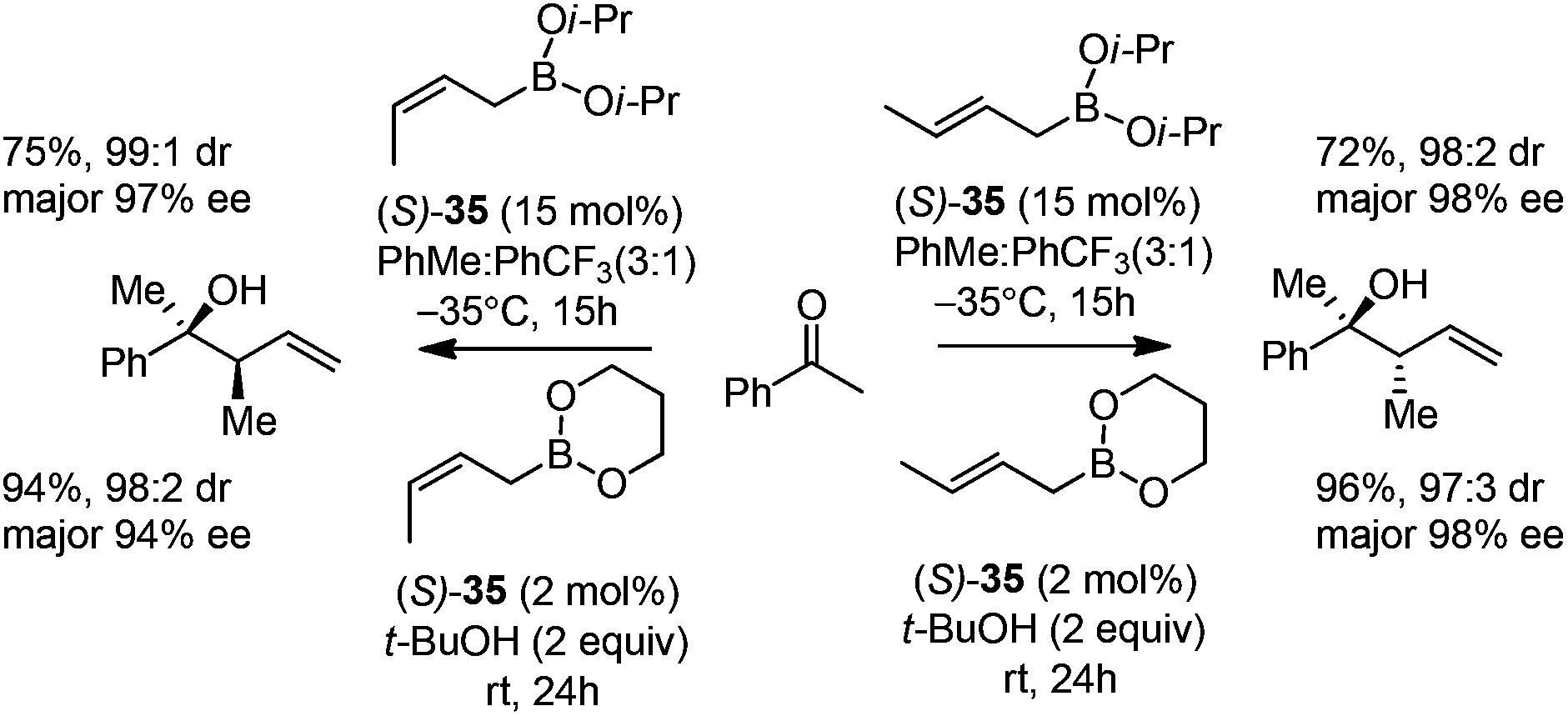 | ||
| Scheme 28 Crotylboration of acetophenone with diisopropoxy acyclic boronate and cyclic allyldioxaborinane. | ||
In 2007, Schaus and co-workers further explored the concept of ligand-exchange to allylboration of acyl imines.44 Gratifyingly, allylation of imines was achieved in good yields (75–94%) and high enantioselectivities (90–99% ee) with 15 mol% of 39 and allyldiisopropoxyborane 34. The reaction can tolerate both aromatic and aliphatic imines with examples such as aryl, cinnamoyl, and cyclohexyl carboxamide imine proceeding in good yield and enantioselectivity (Table 16). However, methyl and methoxyl carbamoyl imine (entries 8 and 10, Table 16) were exceptions to this broad substrate scope. Interestingly, either (E)-crotylboronate or (Z)-crotylboronate in the reaction resulted in the anti-addition product 41 (Scheme 29). The high degree of anti-selectivity afforded by (E)-crotylboronate can be rationalized by a chair-like transition state. For (Z)-crotylboronate, a boat-like transition state may be adopted to deliver the same product. A preferred conformer is organized by the pseudo-trans-diaxial interaction of the methyl group of the (Z)-boronate and the acyl substituent of the imine arising from the chair transition state.44
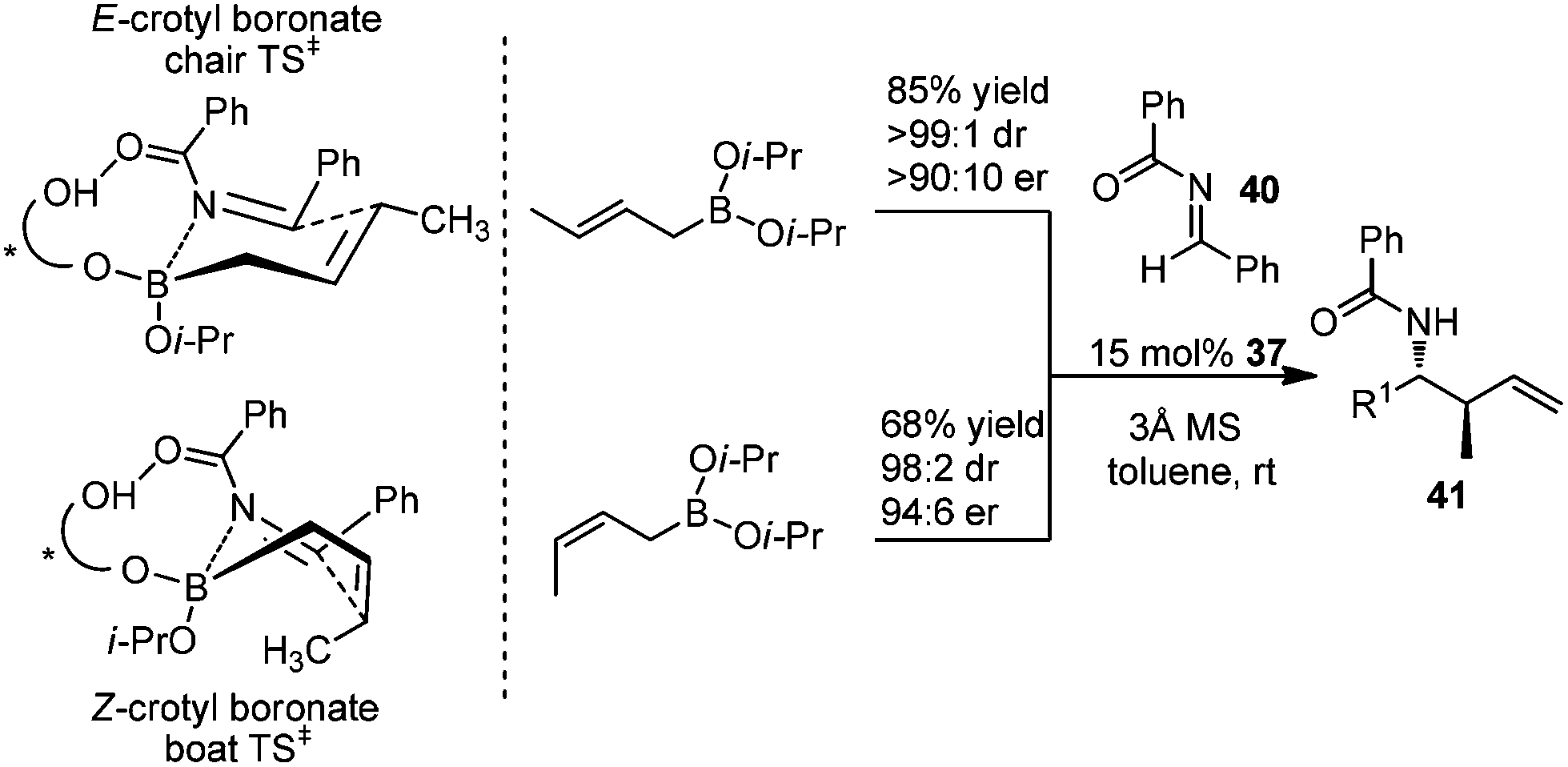 | ||
| Scheme 29 Crotylboration of imine 40 and proposed transition states. | ||
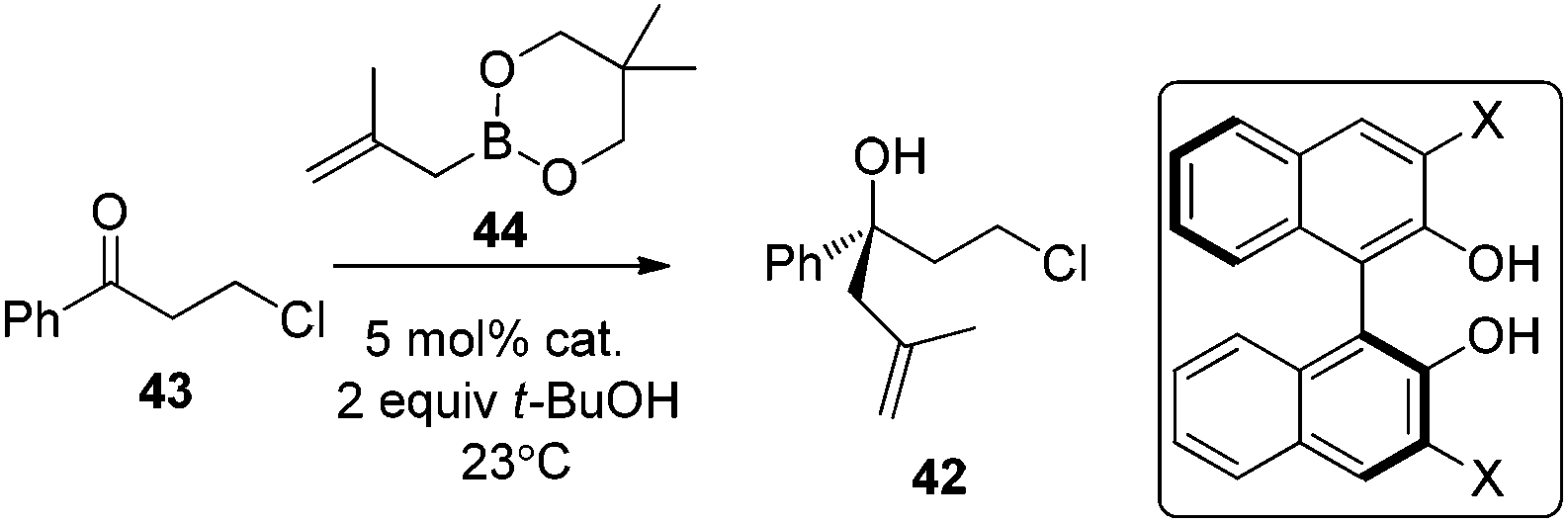
Following Schaus's seminal work, Zhang et al. reported asymmetric allylboration of ketones to prepare chiral tertiary alcohol 42, a precursor for the synthesis of a pharmaceutical agent.45 By using 3,3′-Br2-BINOL 35, moderate enantioselectivity and conversion were obtained for the allylation of ketone 43 with cyclic boronate 44 (entry 1, Table 17). When catalyst 3,3′-F2-BINOL 45 was used, the reaction could reach 98% conversion with 74% ee in 10 h (entry 6). To further explore the scope of reaction using 45, a variety of ketones were examined and good results were achieved under the optimized reaction conditions (Table 18). Most notably, the sterically hindered boronates (R3 = Et, Bu) were also tolerated to afford the corresponding adducts in high yields and enantioselectivities (entries 6 and 7 in Table 18).
|
|
||||
|---|---|---|---|---|
| Entry | X | t (h) | Conv. (%) | ee (%) |
| 1 | Br (35) | 10 | 61 | 78 |
| 2 | CO2Me | 16 | 84 | 2 |
| 3 | SO2CF3 | 20 | 88 | 0 |
| 4 | CF3 | 16 | 82 | 4 |
| 5 | Cl | 10 | 70 | 82 |
| 6 | F (45) | 10 | 98 | 74 |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | T (°C) | Yield (%) | ee (%) |
| 1 | Ph | CH2Ph | Me | 40 | 98 | 86 |
| 2 | 4-ClC6H4 | CH2Ph | Me | 40 | 97 | 92 |
| 3 | 2-Thienyl | CH2Ph | Me | 23 | 97 | 94 |
| 4 | Ph | (CH2)3Ph | Me | 40 | 96 | 80 |
| 5 | Ph | Me | Me | 23 | 96 | 56 |
| 6 | Ph | CH2Ph | Et | 23 | 93 | 90 |
| 7 | Ph | CH2Ph | Bu | 23 | 95 | 86 |
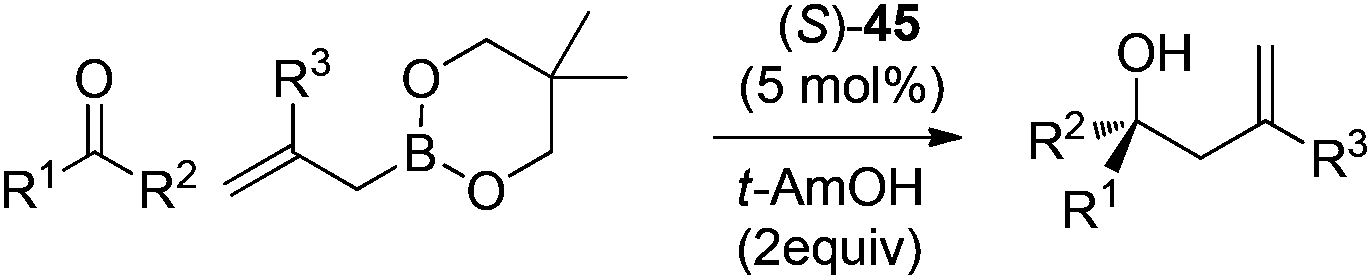
4.2 Exchange of boron–valine derivative system
In 2013, the Hoveyda group made truly ground-breaking progress in this field. They reported a class of small organic molecules that could catalyze asymmetric allylation of imines and carbonyls.46 The reactions were conducted with as little as 0.25–0.3 mol% of catalyst to generate products in more than 85% yield and ≥97:3 enantiomeric ratio. Furthermore, the catalysts, which were derived from abundant valine, were stable to air and moisture and could be easily prepared in large quantities in four steps.
With catalysts screened, aminophenol 46 was identified as the best candidate to promote the allylation reaction of N-phosphinoylimine with allylboronic acid pinacol ester. They used a phosphorus-based protecting group due to its facile preparation and products that were likely to be crystalline (chromatography avoided). Another reason was the inexpensive and mild conditions for removal of the protecting group. Under the optimized conditions, a vast array of aromatic imines was examined to provide excellent yields and enantioselectivities (Table 19). Even alkenyl-, alkynyl- and alkyl-substituted aldimines were tolerated. Moreover, 2-substituted allylboronate proceeded smoothly with equally good yields and enantioselectivities.46a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Y | Yield (%) | ee (%) |
| 1 | Ph | H | 3.0 | 2.5 | 95 | 93 |
| 2 | 2-FC6H4 | H | 3.0 | 2.5 | 91 | 96 |
| 3 | 4-MeOC6H4 | H | 3.0 | 2.5 | 98 | 93 |
| 4 |
|
H | 3.0 | 2.5 | 84 | >98 |
| 5 |
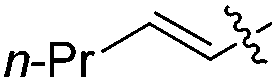
|
H | 2.5 | 2.5 | 96 | 96 |
| 6 |
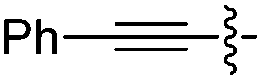
|
H | 3.0 | 2.5 | 95 | 76 |
| 7 |
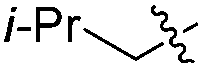
|
H | 6.0 | 5.0 | 50 | >98 |
| 8 |
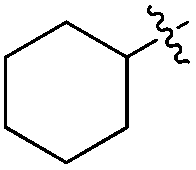
|
H | 6.0 | 8.5 | 51 | >98 |
| 9 | Ph | Me | 2.5 | 2.5 | 96 | 95 |
| 10 | Ph | Ph | 2.5 | 2.5 | 98 | 95 |

Interestingly, when 1-substituted allylboron reagents were examined, α-selectivity of allylation was found for all the reactions. Allylboronate 47 bearing an α-stereogenic quaternary carbon (95:5 er) gave product 48 with the chiral center reversed in 70% yield (for pure diastereomer), 89:11 dr and 95:5 er (for major isomer) (Scheme 30).46a No γ-addition was observed and the reversal in the stereochemistry implicated that the reactions involved double γ-allylation to afford the final α-addition products. To gain further insight into the mechanism, they carried out kinetic studies which concluded the rate determining step was the C–C bond forming step. They also found MeOH and NaOt-Bu or other bases were necessary to complete the transformation and phenol deprotonation of 46 respectively. Based on this evidence, the mechanism (allylboronate 47 as an example) was proposed with the formation of 49 which was derived from product 48, where the Lewis basic amide group stabilized the boron centre (e.g., 54). After ligand exchange with MeOH to release active boronate 50, the following step used substrate 47 to form chiral allylboron species 52 through a synclinal (cyclic) transition state 51. The γ-allylated species 52 participates in a stereoselective γ-allylation of the imine through the six-membered chair-like transition 53. The key proton embedded within its structure was crucial to form the rigid intermediate ensuring a high selectivity. The critical hydrogen-bonding interaction was also verified by computational studies.
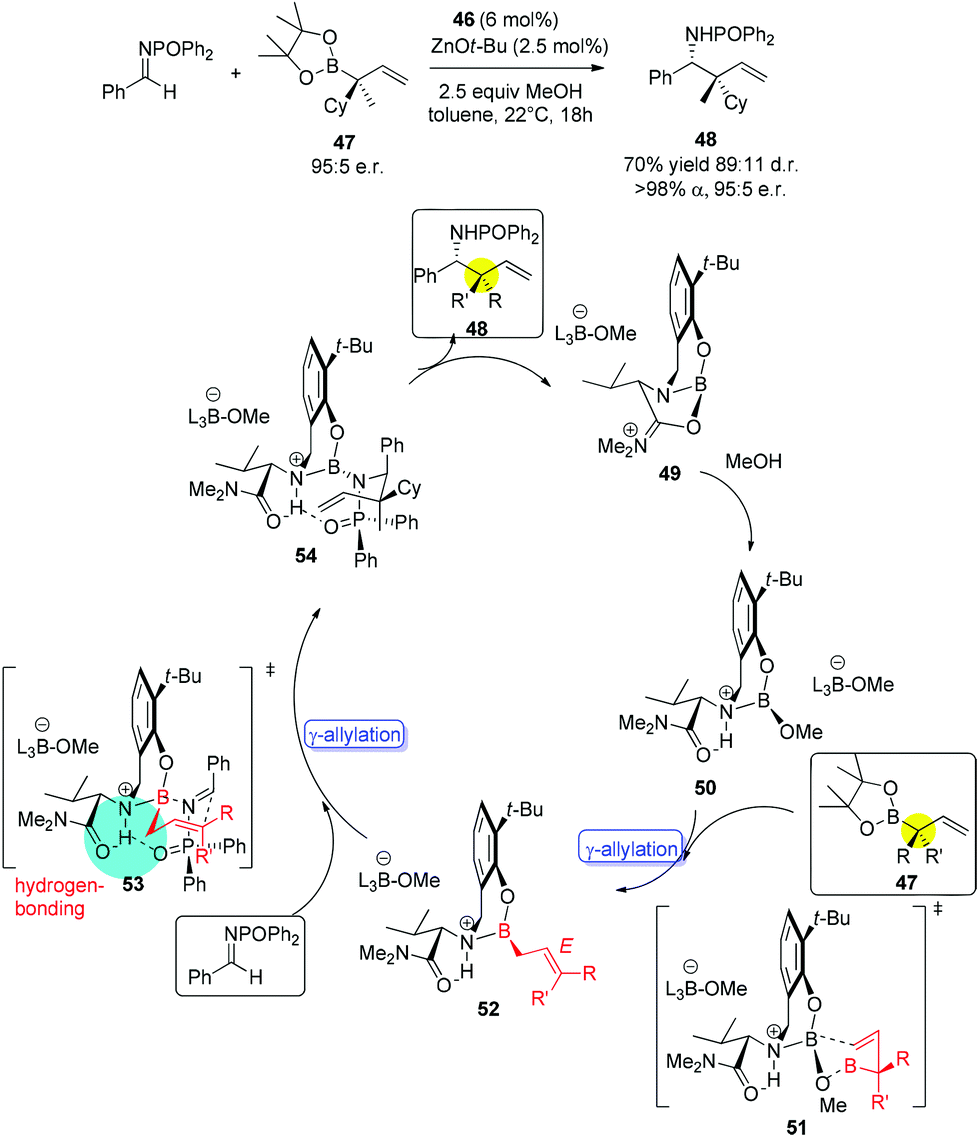 | ||
| Scheme 30 The proposed mechanism. | ||
The catalytic strategy was also effective for carbonyl-containing substrates.46a Asymmetric allylation of N-protected isatins proceeded smoothly at 22 °C within 2 hours in the presence of 0.5–2.0 mol% 46 and 1.5 equiv. of allylboronic acid pinacol ester. Homoallylic alcohols47 were obtained in 84–98% yield and 91.5:8.5–98.5:1.5 er (Scheme 31).
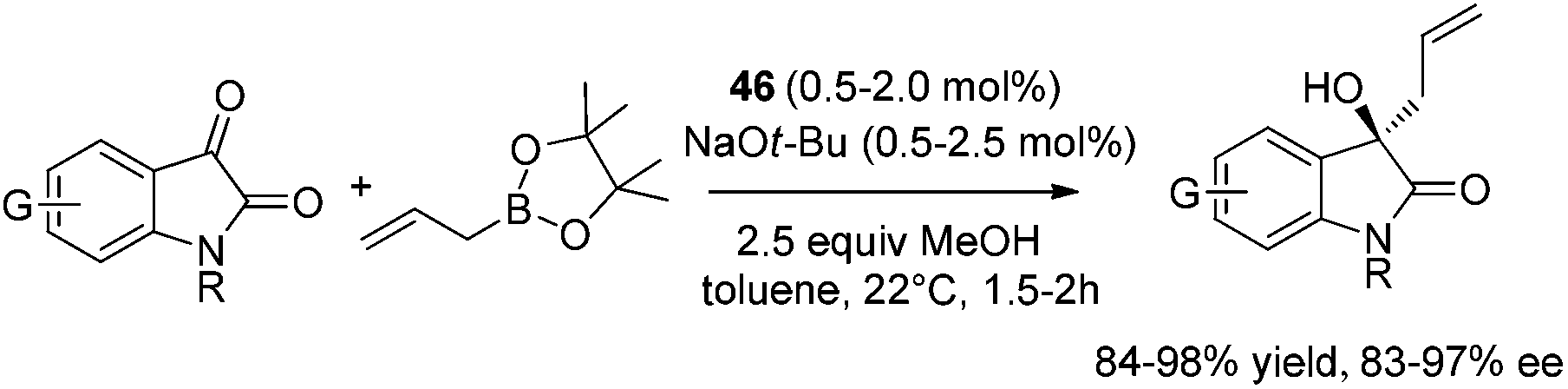 | ||
| Scheme 31 Allylation of N-protected isatins. | ||
5. Applications in total synthesis
Catalytic asymmetric allylation of carbonyl compounds and imines with organoboronates has been used in the synthesis of a number of pharmaceutical drugs and natural products. Selected examples presented here are focused on the synthesis of complex molecules employing catalytic enantioselective allylation as a key step. In principle, those allylboration steps enabled by the chiral auxiliary can be developed catalytically as illustrated in this review.In 2005, Maraviroc, a new CCR5 entry inhibitor, had been fast-tracked through clinical trials as a new compound class in HIV therapy.48 Schaus et al. applied the asymmetric allylation of difluorocyclohexane carboximide imine 55 as the key step to accomplish the synthesis of Maraviroc (Scheme 32).44 The enantioselective allylation proceeds efficiently under standard reaction conditions. This route featured fewer steps than Price's approach in which β-phenylalanine acid was introduced as the source of chirality for the synthesis,49 and advantageously diminished the manipulation of the amine protecting group.
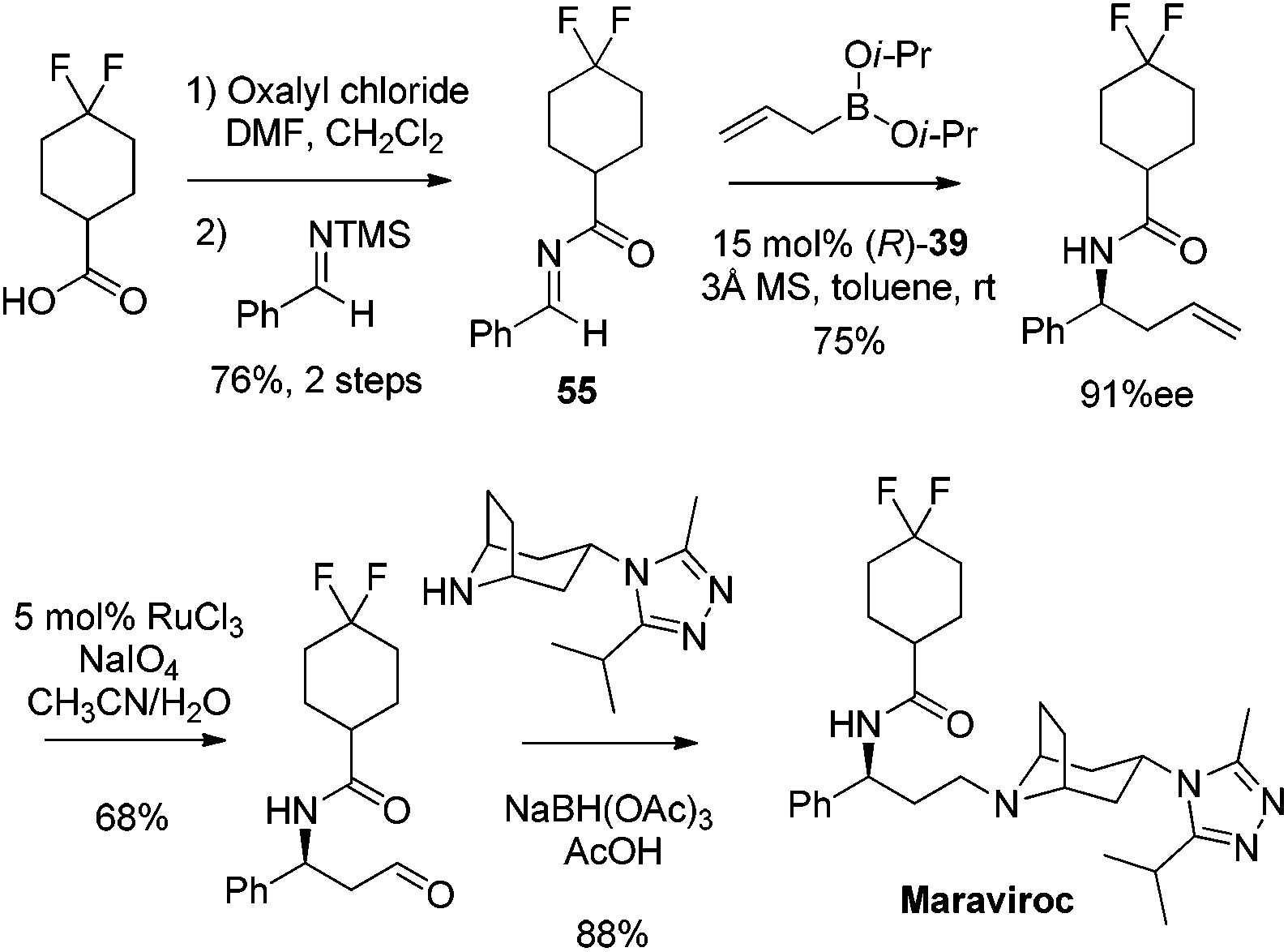 | ||
| Scheme 32 Synthesis of Maraviroc (Schaus, 2005). | ||
Hall et al. selected (+)-dodoneine as a target to demonstrate the efficiency of a catalytic asymmetric allylation in the presence of a chiral diol catalyst developed by his group.53 Dodoneine was isolated from a parasitic plant in Burkina Faso and displays a vasorelaxant effect on preconstricted rat aortic rings, thus suggesting a potential treatment toward cardiovascular disorders.50 (+)-Dodoneine has been synthesized by Marco51 and Cossy52 using an established allylation of an aldehyde. Hall devised similar routes to allow a direct comparison with the p-F-vivol (30)·SnCl4-catalyzed allylboration.53 Two subsequent aldehyde allylations were designed to afford homoallyl alcohols 56 and 57 respectively in almost quantitative yields and high enantioselectivities (Scheme 33). In comparison, Marco carried out a Brown allylation, albeit in lower ee (90%).51 The Keck allylation of the same aldehyde in Cossy's work also proceeded in a lower yield (77%).52 The following steps involving O-acylation with acryloyl chloride, ring-closing metathesis, and desilylation were carried out to complete the synthesis of (+)-dodoneine.
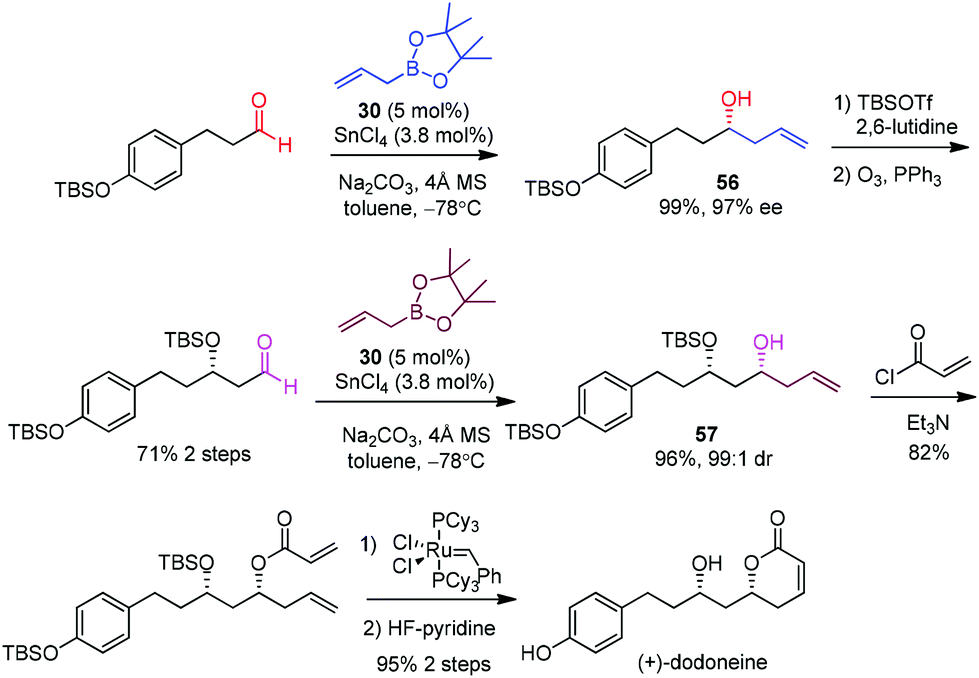 | ||
| Scheme 33 Synthesis of (+)-dodoneine (Hall, 2009). | ||
In 2013, Zhang and co-workers applied Schaus's method in the allylation of ketone 43.45 The corresponding chiral tertiary alcohol 42 could be further converted to the key chiral building block 58, an intermediate for a pharmaceutical agent. They utilized 3,3′-F2-BINOL as a highly active organocatalyst for the first time. The process of asymmetric allylation was successfully carried out on a kilogram scale in 95% yield with 74% ee after simple workup. Cyclic carbamate product 58 was obtained by reacting with isocyanate in 62% yield after crystallization. After another crystallization process, the enantiomeric purity of 58 was readily enriched to 99.4:0.6 (Scheme 34).
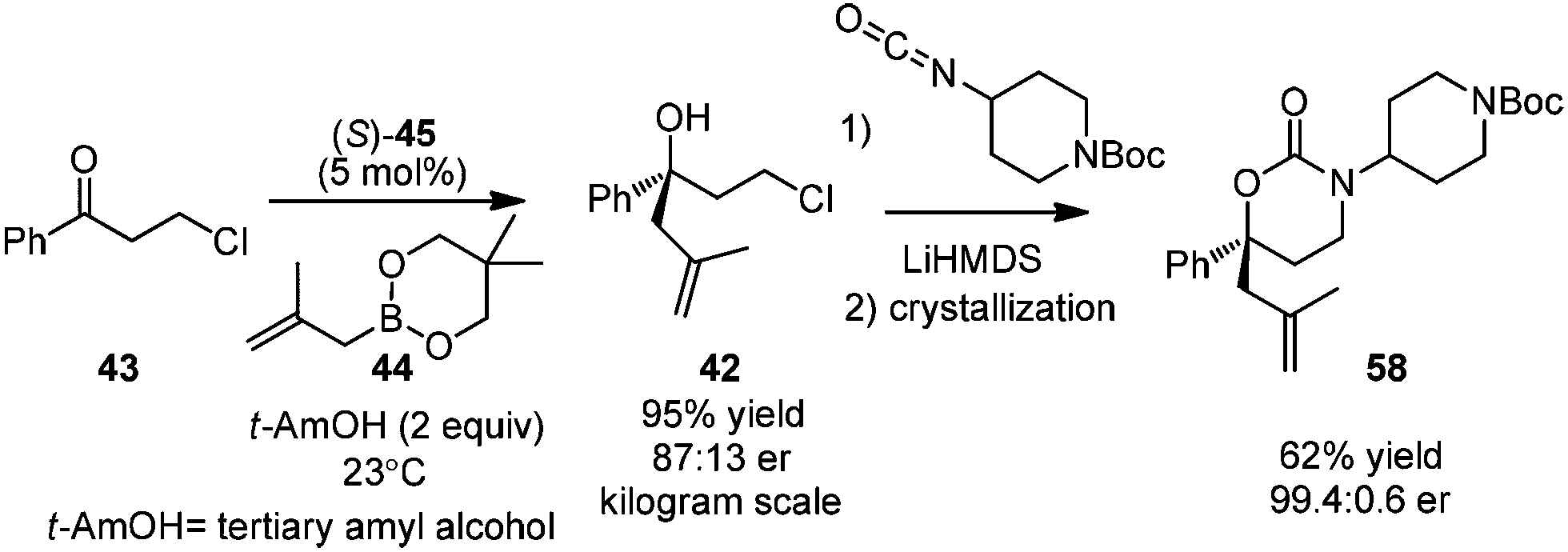 | ||
| Scheme 34 Synthesis of pharmaceutical intermediate 58 (Zhang, 2013). | ||
Isolated from the fermentation broth of Streptomyces nitrosporeus K93-0711 by Omura et al., madindoline A was found as a selective inhibitor of interleukin-6.54 In 2013, Hoveyda et al. employed the organocatalytic enantioselective allylation to construct homoallyl carbinol 59, the key intermediate for madindoline A.46a The allylation proceeded efficiently under the optimized conditions with as little as 0.5 mol% catalyst in 1.5 hours at room temperature. Homoallyl carbinol 59 was achieved in 94% yield and 96% ee46a and was readily converted to madindoline A in several steps through a previously reported sequence (Scheme 35).55 The same protocol was applied to prepare compound 60,46a an important intermediate to convolutamydines.56 The catalytic asymmetric reaction had a good enantioselectivity, while avoiding the use of chiral auxiliary in Palmisano's route (Scheme 35).56
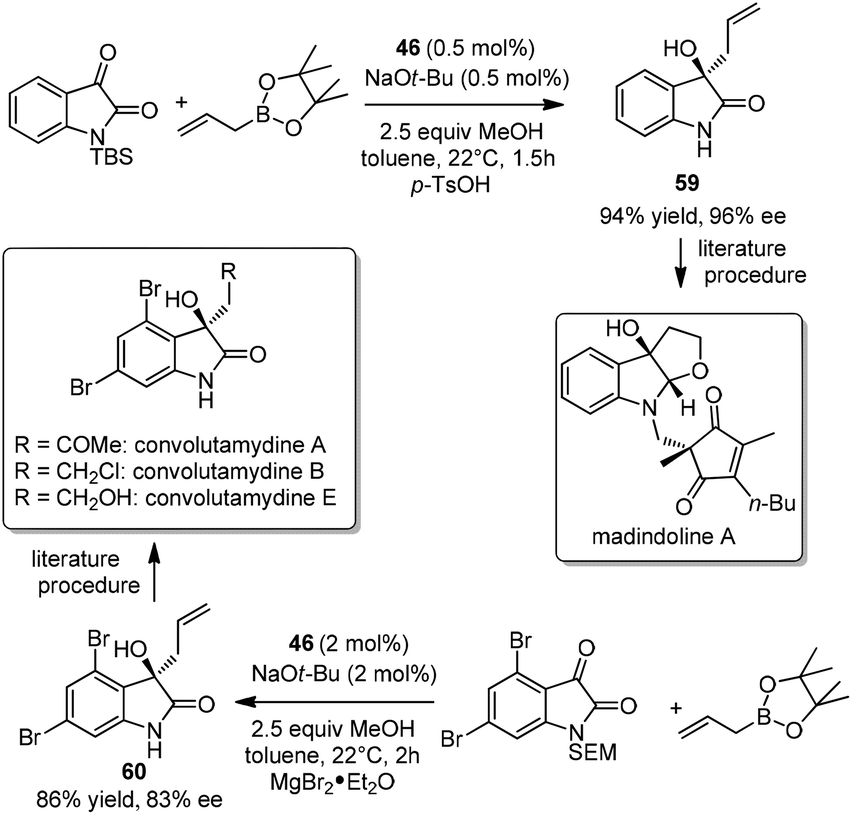 | ||
| Scheme 35 Formal syntheses of madindoline A and convolutamydines (Hoveyda, 2013). | ||
6. Conclusions and outlook
Catalytic asymmetric allylation of carbonyl compounds and imines with allylic boronates has witnessed a rapid development in the past decade. The achievements in this field have made it a powerful tool for constructing homoallylic alcohols or amines, which serve as common building blocks and important precursors for the synthesis of a variety of different pharmaceutically relevant compounds and natural products.According to the activation mode, enantioselective catalysis of allylation is divided into three categories including metal-mediated asymmetric allylation, acid catalyzed asymmetric allylation by activating boronates and catalytic asymmetric allylation by ligand exchange of boronate. Among them, metal mediated allylation is most exhaustively explored. With the facile transmetallation, swift ligand exchange and catalyst regeneration, this asymmetric allylation proceeds efficiently and covers a broad range of substrates such as ketones, imines, and aldehydes. Of course, under the growing concerns of environmental impact and atom-economy, decreasing the catalyst loading or a metal-free approach is more likely to expand in future developments, and the most recent asymmetric organocatalysis arouses a wide appreciation. As an example, BINOL-derived compounds and chiral phosphorus acid were first developed as simple and highly efficient catalysts which already have had an impact in process chemistry, although they both are restricted in substrate scope (only for aldehydes or ketones, imines respectively). Hoveyda and co-workers devised a novel class of easily prepared and low cost catalysts to promote the allylation smoothly and efficiently under very mild conditions. This protocol will find a wide application in organic synthesis.
Moreover, catalytic asymmetric approaches discussed in this review can be extended to propargylation when allenyl boronates are used (Scheme 36). The activation mode of propargylation is similar to the allylboration with a substrate derived from allylboronate to allenylboronate. The recent progress was beautifully exemplified57–60 and surveyed in a recent review.61 Although so far sporadic examples and only selected aldehydes and ketones have been investigated, this field is expected to have a promising future ahead.
 | ||
| Scheme 36 Propargylation with allenylboronate. | ||
In addition, catalytic asymmetric allylation of carbonyl compounds and imines with 3,3′-disubstituted allylic boronates affording two continuous quaternary/tertiary chiral centers remains a formidable challenge for catalyst development (Scheme 37). So far, there are only a few works on allylation with 3,3′-disubstituted allylboronates or trichlorosilane to construct the chiral quaternary carbon in homoallylic alcohols. Hoffmann and Hara both used chiral allylboronates to accomplish the stereocontrolled allylation of aldehydes. Hoffmann found that the homoallylic alcohol was achieved with good enantioselectivity with a chiral α-branched boronate while α-unsubstituted ones gave racemic products.62 Hara applied Roush's boronate to obtain a moderate enantioselectivity.63 Denmark disclosed the only enantioselective addition of trisubstituted allyltrichlorosilane to benzaldehyde with catalytic chiral phosphoramide.64 This field is primed for further exploration in terms of the challenge of constructing vicinal quaternary/tertiary chiral centers.
 | ||
| Scheme 37 Allylation with 3,3′-disubstituted allylic boronates or trichlorosilane. | ||
In short, significant advancements in catalytic asymmetric allylation of carbonyl compounds and imines with allylic boronates have occurred over the past decade. The continuing focus on method development will strengthen the field of research and build the confidence of chemists looking to construct chiral homoallyl alcohols and imines in a highly enantioselective manner.
Addendum (March 2014)
After this manuscript was accepted, Hoveyda and co-workers further explored the asymmetric allylation of imines with allenyl boronate.65 The use of Boc-imines resulted in α-selectivity for the allylation reaction, which afforded homoallenylamide as the major product. With 0.1–3.0 mol% of chiral catalyst 46, enantioselective allyl additions proceeded in 66–91% yield and 68–99% ee at ambient temperature. The substrate scope covers aryl-, heteroaryl-, and alkylsubstituted imines. In addition, they utilized this method to use enantiopure homoallenylamides (up to 94% ee) as key chiral intermediates to accomplish the synthesis of anisomycin and epi-cytoxazone efficiently on a significant laboratory scale. Overall they offered an efficient, practical, and enantioselective method for the synthesis of homoallenylamides.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21290184 and 21172236), the National Basic Research Program of China (2010CB833200), and Chinese Academy of Sciences is highly appreciated. We also thank Mr Xianwei Mi for helpful discussions during the manuscript preparation.Notes and references
- For reviews, see: (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (b) P. V. Ramachandran, Aldrichimica Acta, 2002, 35, 23 CAS; (c) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef CAS PubMed; (d) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (e) P. Merino, T. Tejero, J. I. Delso and V. Mannucci, Curr. Org. Synth., 2005, 2, 479 CrossRef CAS; (f) H. Ding and G. K. Friestad, Synthesis, 2005, 2815 CAS; (g) R. B. Kargbo and G. R. Cook, Curr. Org. Chem., 2007, 11, 1287 CrossRef CAS; (h) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (i) H. Yamamoto and M. Wadamoto, Chem.–Asian J., 2007, 2, 692 CrossRef CAS PubMed; (j) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541 CrossRef CAS PubMed; (k) M. Kanai, R. Wada, T. Shibuguci and M. Shibasaki, Pure Appl. Chem., 2008, 80, 1055 CrossRef CAS; (l) L. F. Tietze, T. Kinkel and C. C. Brazel, Acc. Chem. Res., 2009, 42, 367 CrossRef CAS PubMed; (m) J. Li and D. Menche, Synthesis, 2009, 2293 CAS; (n) J. L. Leighton, Aldrichimica Acta, 2010, 43, 3 CAS; (o) T. G. Elford and D. G. Hall, Synthesis, 2010, 893 CAS.
- (a) S. E. Denmark and N. G. Almstead, Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, p. 299 Search PubMed; (b) S. R. Chemler and W. R. Roush, Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, 403 Search PubMed; (c) M. B. Smith and J. March, March's Advanced Organic Chemistry Reactions, Mechanisms and Structure, Wiley-Interscience, Hoboken, NJ, 6th edn, 2007, p. 1251 Search PubMed.
- (a) E. J. Corey and A. Guzmán-Pérez, Angew. Chem., Int. Ed., 1988, 37, 388 CrossRef; (b) K. Fuji, Chem. Rev., 1993, 93, 2037 CrossRef CAS; (c) D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2004, 43, 284 CrossRef PubMed; (d) Quaternary Stereocenters, ed. J. Chistoffers and A. Baro, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- For reviews, see: (a) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed; (b) T. R. Ramadhar and R. A. Batey, Synthesis, 2011, 1321 CAS; (c) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed; (d) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed; (e) M. Sugiura and M. Nakajima, Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, 2012, 214 Search PubMed.
- B. M. Mikhailov and Y. N. Bubnov, Izv. Akad. Nauk. SSSR, Ser. Khim., 1964, 1874 CAS.
- B. M. Mikhailov and Y. N. Bubnov, Organoboron Compounds in Organic Synthesis, OPA, Amsterdam B. V., 1984, 571 Search PubMed.
- E. Favre and M. C. C. Gaudemar, C. R. Seances Acad. Sci., Ser. C, 1966, 263, 1543 CAS.
- (a) R. W. Hoffmann and H.-J. Zeiss, Angew. Chem., Int. Ed., 1979, 18, 306 CrossRef; (b) R. W. Hoffmann and H.-J. Zeiss, J. Org. Chem., 1981, 46, 1309 CrossRef CAS.
- S. E. Denmark and E. J. Weber, Helv. Chim. Acta, 1983, 66, 1655 CrossRef CAS.
- (a) Y. Li and K. N. Houk, J. Am. Chem. Soc., 1989, 111, 1236 CrossRef CAS; (b) A. Vulpetti, M. Gardner, C. Gennari, A. Bernardi, J. M. Goodman and I. Paterson, J. Org. Chem., 1993, 58, 1711 CrossRef CAS; (c) C. Gennari, E. Fioravanzo, A. Bernardi and A. Vulpetti, Tetrahedron, 1994, 50, 8815 CrossRef CAS; (d) K. Omoto and H. Fujimoto, J. Org. Chem., 1998, 63, 8331 CrossRef CAS; (e) J. J. Gajewski, W. Bocian, N. L. Brichford and J. L. Henderson, J. Org. Chem., 2002, 67, 4236 CrossRef CAS PubMed.
- A seminal contribution by the Soderquist group in the development of new chiral borane reagent, see: (a) J. A. Soderquist, K. Matos, C. H. Burgos, C. Lai, J. Vaquer, J. R. Medina and S. D. Huang, ACS Symposium Series 783, ed. P. V. Ramachandran and H. C. Brown, American Chemical Society, Washington, DC, 2000, 13, p. 176 Search PubMed; (b) J. A. Soderquist, K. Matos, C. H. Burgos, C. Lai, J. Vaquer and J. R. Medina, Contemporary Boron Chemistry, ed. M. G. Davidson, K. Wade, T. B. Marder and A. K. Hughes, RSC Publications, 2000, 472 Search PubMed; (c) C. Lai and J. A. Soderquist, Org. Lett., 2005, 7, 799 CrossRef CAS PubMed; (d) C. H. Burgos, E. Canales, K. Matos and J. A. Soderquist, J. Am. Chem. Soc., 2005, 127, 8044 CrossRef CAS PubMed; (e) E. Canales, K. G. Prasad and J. A. Soderquist, J. Am. Chem. Soc., 2005, 127, 11572 CrossRef CAS PubMed; (f) E. Canales, E. Hernandez and J. A. Soderquist, J. Am. Chem. Soc., 2006, 128, 8712 CrossRef CAS PubMed; (g) E. Hernández, E. Canales, E. González and J. A. Soderquist, Pure Appl. Chem., 2006, 7, 1389 Search PubMed; (h) J. G. Román and J. A. Soderquist, J. Org. Chem., 2007, 72, 9772 CrossRef PubMed; (i) E. Canales, A. Z. Gonzalez and J. A. Soderquist, Angew. Chem., Int. Ed., 2007, 46, 397 CrossRef CAS PubMed; (j) A. Z. González, J. G. Román, E. Alicea, E. Canales and J. A. Soderquist, J. Am. Chem. Soc., 2009, 131, 1269 CrossRef PubMed; (k) L. Muñoz-Hernández and J. A. Soderquist, Org. Lett., 2009, 11, 2571 CrossRef PubMed; (l) J. R. González, A. Z. González and J. A. Soderquist, J. Am. Chem. Soc., 2009, 131, 9924 CrossRef PubMed.
- (a) H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092 CrossRef CAS; (b) H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 293 CrossRef CAS; (c) P. V. Ramachandran, Aldrichimica Acta, 2002, 35, 23 CAS.
- (a) W. R. Roush, A. E. Walts and L. K. Hoong, J. Am. Chem. Soc., 1985, 107, 8186 CrossRef CAS; (b) W. R. Roush, K. Ando, D. B. Powers, A. D. Palkowitz and R. L. Halterman, J. Am. Chem. Soc., 1990, 112, 6339 CrossRef CAS.
- (a) J. Garcia, B. Kim and S. Masamune, J. Org. Chem., 1987, 52, 4831 CrossRef CAS; (b) R. P. Short and S. Masamune, J. Am. Chem. Soc., 1989, 111, 1892 CrossRef CAS.
- (a) E. J. Corey, C.-M. Yu and S. S. Kim, J. Am. Chem. Soc., 1989, 111, 5495 CrossRef CAS; (b) E. J. Corey, C.-M. Yu and D.-H. Lee, J. Am. Chem. Soc., 1990, 112, 878 CrossRef CAS.
- T. Ishiyama, T.-A. Ahiko and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 12414 CrossRef CAS PubMed.
- (a) J. W. J. Kennedy and D. G. Hall, J. Am. Chem. Soc., 2002, 124, 11586 CrossRef CAS PubMed; (b) V. Rauniyar and D. G. Hall, J. Am. Chem. Soc., 2004, 126, 4518 CrossRef CAS PubMed.
- K. Sakata and H. Fujimoto, J. Am. Chem. Soc., 2008, 130, 12519 CrossRef CAS PubMed.
- (a) Stereodirected Synthesis with Organoboranes, ed. B. M. Trost, Springer, Berlin, 1995 Search PubMed; (b) Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005 Search PubMed.
- R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8910 CrossRef CAS PubMed.
- M. Kanai, R. Wada, T. Shibuguchi and M. Shibasaki, Pure Appl. Chem., 2008, 80, 1055 CrossRef CAS.
- S.-L. Shi, L.-W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638 CrossRef CAS PubMed.
- R. Wada, T. Shibuguchi, S. Makino, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7687 CrossRef CAS PubMed.
- M. Fujita, T. Nagano, U. Schneider, T. Hamada, C. Ogawa and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 2914 CrossRef CAS PubMed.
- S. Kobayashi, T. Endo and M. Ueno, Angew. Chem., Int. Ed., 2011, 50, 12262 CrossRef CAS PubMed.
- (a) Y. Cui, Y. Yamashita and S. Kobayashi, Chem. Commun., 2012, 48, 10319 RSC; (b) Y. Cui, L. Wei, T. Sato, Y. Yamashita and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 1193 CrossRef CAS.
- U. Schneider, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13824 CrossRef CAS PubMed.
- A. Chakrabarti, H. Konishi, M. Yamaguchi, U. Schneider and S. Kobayashi, Angew. Chem., Int. Ed., 2010, 49, 1838 CrossRef CAS PubMed.
- P. Zhang and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 12550 CrossRef CAS PubMed.
- Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309 CrossRef CAS PubMed.
- M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2012, 134, 2716 CrossRef CAS PubMed.
- S. Nakamura, M. Kaneeda, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 8120 CrossRef CAS.
- H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed.
- (a) V. Rauniyar and D. G. Hall, Angew. Chem., Int. Ed., 2006, 45, 2426 CrossRef CAS PubMed; (b) D. G. Hall, Synlett, 2007, 1644 CrossRef CAS PubMed; (c) V. Rauniyar and D. G. Hall, Synthesis, 2007, 3421 CAS; (d) V. Rauniyar, H. Zhai and D. G. Hall, J. Am. Chem. Soc., 2008, 130, 8481 CrossRef CAS PubMed; (e) V. Rauniyar and D. G. Hall, J. Org. Chem., 2009, 74, 4236 CrossRef CAS PubMed; (f) U. Bhakta, E. Sullivan and D. G. Hall, Tetrahedron, 2014, 70, 678 CrossRef CAS PubMed.
- Monographs and reviews, see: (a) A. Berkessel and H. Groger, Asymmetric Organocatalysis From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) R. M. de Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 2575 CrossRef CAS; (d) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed; (e) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (f) Organocatalysis, ed. M. T. Reetz, B. List, S. Jaroch and H. Weinmann, Springer Verlag, Berlin, 2008 Search PubMed; (g) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS PubMed; (h) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed; (i) A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922 CrossRef CAS PubMed; (j) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (k) E. Marqués-López, R. P. Herrerabc and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC; (l) F. Giacalone, M. Gruttadauria, P. Agrigentoa and R. Noto, Chem. Soc. Rev., 2012, 41, 2406 RSC; (m) R. C. Wendea and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC.
- (a) P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884 CrossRef CAS PubMed; (b) C.-H. Xing, Y.-X. Liao, Y. Zhang, D. Sabarova, M. Bassous and Q.-S. Hu, Eur. J. Org. Chem., 2012, 1115 CrossRef CAS.
- P. Jain, H. Wang, K. N. Houk and J. C. Antilla, J. Org. Chem., 2013, 78, 1208 CrossRef PubMed.
- C. A. Incerti-Pradillos, M. A. Kabeshov and A. V. Malkov, Angew. Chem., Int. Ed., 2013, 52, 5338 CrossRef CAS PubMed.
- R. W. Hoffmann and U. Weidmann, J. Organomet. Chem., 1980, 195, 137 CrossRef CAS.
- J. Pietruszka and N. Schone, Eur. J. Org. Chem., 2004, 5011 CrossRef CAS.
- T. Miura, Y. Nishida, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2013, 135, 11497 CrossRef CAS PubMed.
- S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2006, 128, 12660 CrossRef CAS PubMed.
- D. S. Barnett, P. N. Moquist and S. E. Schaus, Angew. Chem., Int. Ed., 2009, 48, 8679 CrossRef CAS PubMed.
- S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2007, 129, 15398 CrossRef CAS PubMed.
- Y. Zhang, N. Li, B. Qu, S. Ma, H. Lee, N. C. Gonnella, J. Gao, W. Li, Z. Tan, J. T. Reeves, J. Wang, J. C. Lorenz, G. Li, D. C. Reeves, A. Pesmasiri, N. Grinberg, N. Haddad, B. Z. Lu, J. J. Song and C. H. Senanayake, Org. Lett., 2013, 15, 1710 CrossRef CAS PubMed.
- (a) D. L. Silverio, S. Torker, T. Pilyugina, E. M. Vieira, M. L. Snapper, F. Haeffner and A. H. Hoveyda, Nature, 2013, 494, 216 CrossRef CAS PubMed; (b) Y. Cui and S. Kobayashi, ChemCatChem, 2013, 5, 2805 CrossRef CAS.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
- (a) A. Wood and D. Armour, Prog. Med. Chem., 2005, 43, 239 CrossRef CAS; (b) P. Dorrr, M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Armour, D. Price, B. Stammen, A. Wood and M. Perros, Antimicrob. Agents Chemother., 2005, 49, 4721 CrossRef PubMed.
- D. Price, S. Gayton, M. D. Selby, J. Ahman, S. Haycock-Lewandowski, B. L. Stammen and A. Warren, Tetrahedron Lett., 2005, 46, 5005 CrossRef CAS PubMed.
- M. Ouedraogo, H. Carreyre, C. Vandebrouck, J. Bescond, G. Raymond, I.-P. Guissou, C. Cognard, F. Becq, D. Potreau, A. Cousson, J. Marrot and J.-M. Coustard, J. Nat. Prod., 2007, 70, 2006 CrossRef CAS PubMed.
- P. Alvarez-Bercedo, E. Falomir, J. Murga, M. Carda and J. A. Marco, Eur. J. Org. Chem., 2008, 4015 CrossRef CAS.
- A. Dittoo, V. Bellosta and J. Cossy, Synlett, 2008, 2459 CAS.
- V. Rauniyar and D. G. Hall, J. Org. Chem., 2009, 74, 4236 CrossRef CAS PubMed.
- M. Hayashi, M.-C. Rho, A. Enomoto, A. Fukami, Y.-P. Kim, Y. Kikuchi, T. Sunazuka, T. Hirose, K. Komiyama and S. Omura, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14728 CrossRef CAS PubMed.
- T. Itoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2009, 11, 3854 CrossRef CAS PubMed.
- G. Cravotto, G. B. Giovenzana, G. Palmisano, A. Penoni, T. Pilati, M. Sistic and F. Stazi, Tetrahedron: Asymmetry, 2006, 17, 3070 CrossRef CAS PubMed.
- S.-L. Shi, L.-W. Xu, K. Oisaki and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638 CrossRef CAS PubMed.
- P. Jain, H. Wang, K. N. Houk and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 1391 CrossRef CAS PubMed.
- A. S. Tsai, M. Chen and W. R. Roush, Org. Lett., 2013, 15, 7 Search PubMed.
- E. R. Jarvo, B. L. Kohn and N. Ichiishi, Angew. Chem., Int. Ed., 2013, 52, 4413 Search PubMed.
- X.-L. Hou and C.-H. Ding, Chem. Rev., 2011, 111, 1914 CrossRef PubMed.
- R. W. Hoffmann and A. Schlapbach, Liebigs Ann. Chem., 1991, 1203 CrossRef CAS.
- Y. Yamamoto, S. Hara and A. Suzuki, Synlett, 1996, 883 CrossRef CAS PubMed.
- S. E. Denmark, J. Fu and M. J. Lawler, J. Org. Chem., 2006, 71, 1523 CrossRef CAS PubMed.
- H. Wu, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014 DOI:10.1021/ja500374p.
| This journal is © the Partner Organisations 2014 |