Influence of B-ring modifications on proton affinity, transmembrane anion transport and anti-cancer properties of synthetic prodigiosenes†
Received
4th July 2014
, Accepted 8th August 2014
First published on 8th August 2014
Abstract
Prodigiosin is the parent compound of the tripyrrolic natural products known as the prodigiosenes. Some of these natural products and their synthetic analogs show anti-cancer, immunosuppressive and antimicrobial actions, amongst other biological activities. One mechanism put forth to explain their biological activity is that since prodigiosenes are typically protonated at physiological pH they can alter intracellular pH via HCl co-transport (or Cl−/OH− exchange) across cell membranes. In this study we synthesized a series of prodigiosene analogs with different –O-aryl substituents attached to the B-ring of the tripyrrolic skeleton. NMR studies showed that these analogs can exist as a mixture of two stable α and β conformers in acidic solution, and that both conformers can bind anions in solution. We found that the electronic nature of the O-aryl substituent on the B-ring influences the rate at which these prodigiosenes catalyze transmembrane anion transport, i.e. the prodigiosenes with the higher pKa had greater Cl−/NO3− exchange rates. Four of the synthetic prodigiosenes were tested for their in vitro anti-cancer activities in the NCI60 human tumour panel. Despite their promising in vitro anti-cancer activity (GI50 values ranging from 18 to 74 nM), there was no evidence that this activity is influenced by the extent of protonation of these synthetic prodigiosenes.
Introduction
In the last decade various compounds that catalyze transmembrane transport of anions have been identified.1 Much of this activity has been driven by supramolecular chemists interested in systems that work at the water-lipid interface.2 Challenges in transmembrane anion transport are to extract an anion from water and then move it across a hydrophobic membrane. Such transport is facilitated by (1) amphiphilic “carriers” that bind anions and diffuse through the membrane or (2) compounds that self-assemble into channels that span the membrane and provide a low-energy pathway.1,2 Efforts in this area have led to a better understanding of the factors that control anion transport. Thus, non-covalent interactions like hydrogen bonding, electrostatic effects, anion-π and halogen bonds have been used to effect anion transport.3 Recent work has also clarified how ligand conformation,4 flexibility,5 and lipophilicity6 can be used to catalyze transmembrane anion transport. Another important factor promoting this research is the possibility that anion transporters might be developed into therapeutics for diseases, like cystic fibrosis, that are caused by defective transport of Cl−.7
Some synthetic transporters have been developed after considering how nature catalyzes the process. For example, peptides that contain domains of the Cl− channel can transport anions across lipid membranes,8 while modification of natural amphiphiles like cholic acid has provided potent anion carriers.9 The tripyrrolic prodigiosenes are known to catalyze transmembrane anion transport.10 The parent compound, prodigiosin (1), is a natural product isolated from Serratia marcescens (Fig. 1) that has much biological potential11 as it is able to permeabilize cell membranes, alter intracellular pH and trigger apoptosis.12 Early studies indicated that prodigiosin is too cytotoxic for healthy cells and, for this reason, it was not initially pursued as a drug candidate.13 There has been a renewed interest in prodigiosenes since some synthetic analogs have promising anti-cancer,14 immunosuppressive,11b,15 anti-malarial,16 and antimicrobial17 activities at concentrations below where they are cytotoxic to healthy cells.
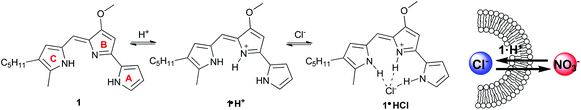 |
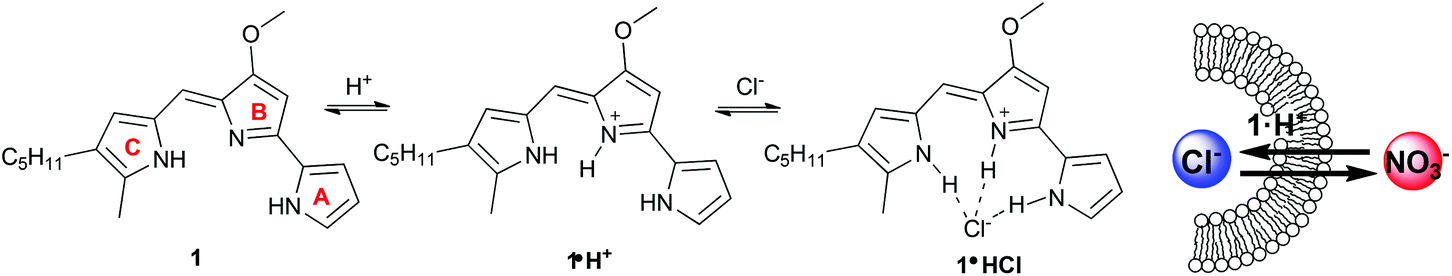
| | Fig. 1 Protonation of prodigiosin (1) and subsequent binding of chloride anion by 1·H+. The schematic at the right depicts the protonated prodigiosin 1·H+ catalyzing the exchange of anions across a lipid bilayer. | |
As depicted in Fig. 1 protonated prodigiosin 1·H+ has a binding pocket for anions, using hydrogen bonds and electrostatic interactions for anion coordination. The resulting amphiphilic neutral salt 1·H+Cl− can then readily diffuse through the cell membrane. We have previously shown that prodigiosin 1·H+Cl− catalyzes the transmembrane exchange of anions such as Cl−, NO3− and HCO3−.10d–f Prodigiosenes also alter intracellular pH, presumably through catalyzing co-transport of H+Cl− or the exchange of Cl−/OH− across cell membranes. This ability to change intracellular pH is one potential mechanism by which prodigiosenes trigger apoptosis.11a,12a,18 Other possible modes of actions include stranded-DNA cleavage in the presence of Cu2+,19 as well as protein kinase20 and Bcl-2 inhibition.21 The effect of the electronic properties of the A-ring upon the proton affinity and the anti-cancer potency of some prodigiosenes has previously been reported, with no identified correlation between pKa and the inhibition of cell proliferation.22
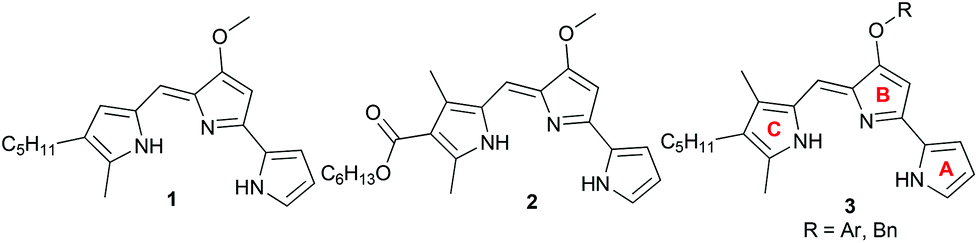
We recently found that ester 2 (Fig. 2), with an electron-withdrawing carbonyl group appended to the C-ring, was less basic by two orders of magnitude than the parent prodigiosin (1) (prodigiosin 1·H+, pKa = 8.2; 2·H+, pKa = 6.5) and also a much less effective anion transporter than 1.10f We suggested that the ease of protonation of the carrier and, thus it's the consequent anion transport activity could be modulated by changing the electronic properties of the C-ring substituents. Herein, we describe a systematic study on some B-ring analogs that confirms transmembrane anion transport rates can be modulated by tuning the pKa of the prodigiosene skeleton. We describe the synthesis, anion transport and in vitro anti-cancer properties of prodigiosenes 3 with different –OAr substituents attached to the tripyrrolic B-ring. The synthetic prodigiosenes (3, Fig. 2) used in this study all have an A-ring pyrrole, a B-ring pyrrole substituted with various aryl groups to modulate pKa, and a C-ring pyrrole that differs from the natural compound 1 by an extra methyl group on the heterocyclic unit.
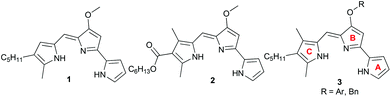 |
| | Fig. 2 Natural prodigiosin 1 and synthetic prodigiosenes 2 and 3. | |
Synthesis of B-ring modified prodigiosenes
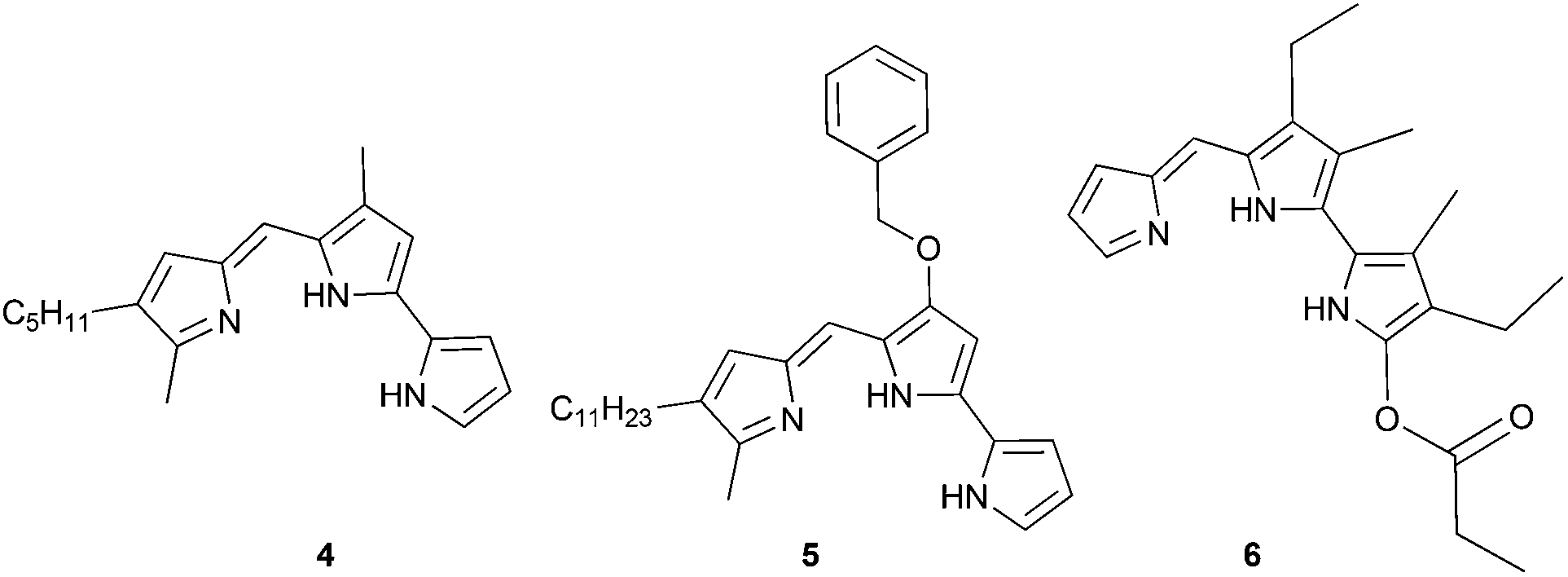
There are few reports regarding synthetic modification of the prodigiosene B-ring.10a,15,23 Moreover, the impact of B-ring modifications on anti-cancer activity seems mixed. Indeed, the desmethoxy analog 4 (Fig. 3) was relatively inactive as an anti-cancer agent, as compared to the parent prodigiosin (1), suggesting that the –OMe group is essential for anti-cancer activity.23a However, some analogs without an –OMe group demonstrated reasonable anti-cancer activity. For example, PNU-156804 5, with an –OBn group, exhibited nM IC50 values against leukemia and melanoma cells,15 and the synthetic analog 6 was active against lung cancer cell lines.10a In this study we prepared novel derivatives substituted with different –OAr groups on the B-ring, alongside common features at the A and C-ring, so as to probe the influence of electronic effects upon pKa values, transmembrane anion exchange rates and anti-cancer activity. This is, to our knowledge, the first report of prodigiosenes substituted with –OAr groups on the B-ring.
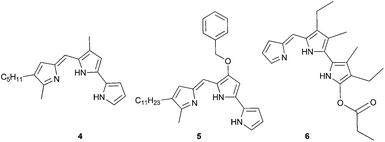 |
| | Fig. 3 Examples of B-ring modified prodigiosin analogs. | |
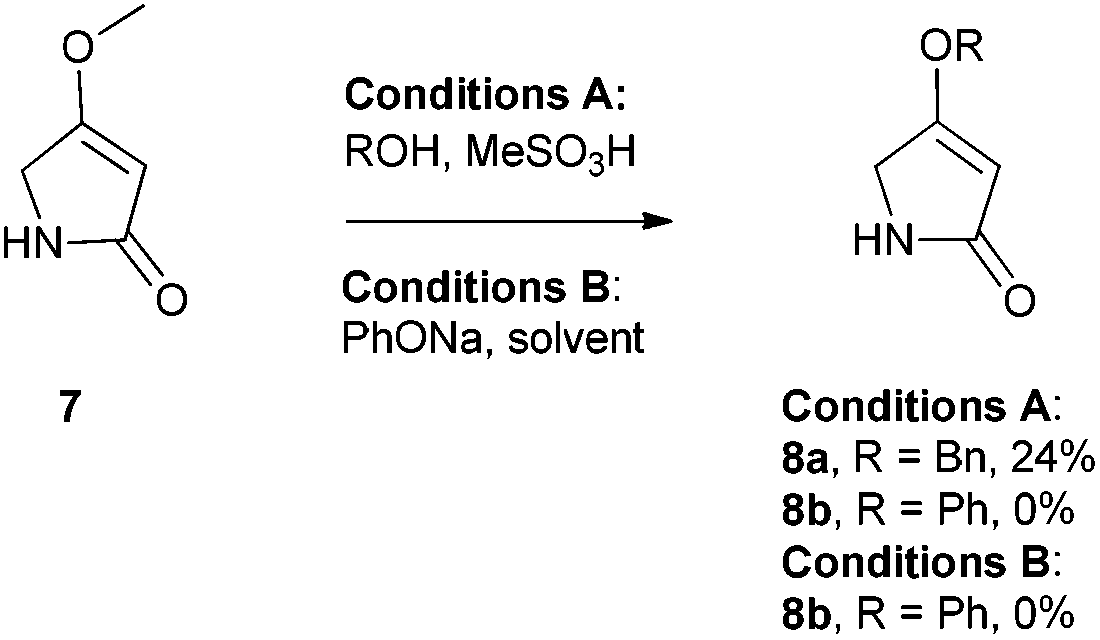
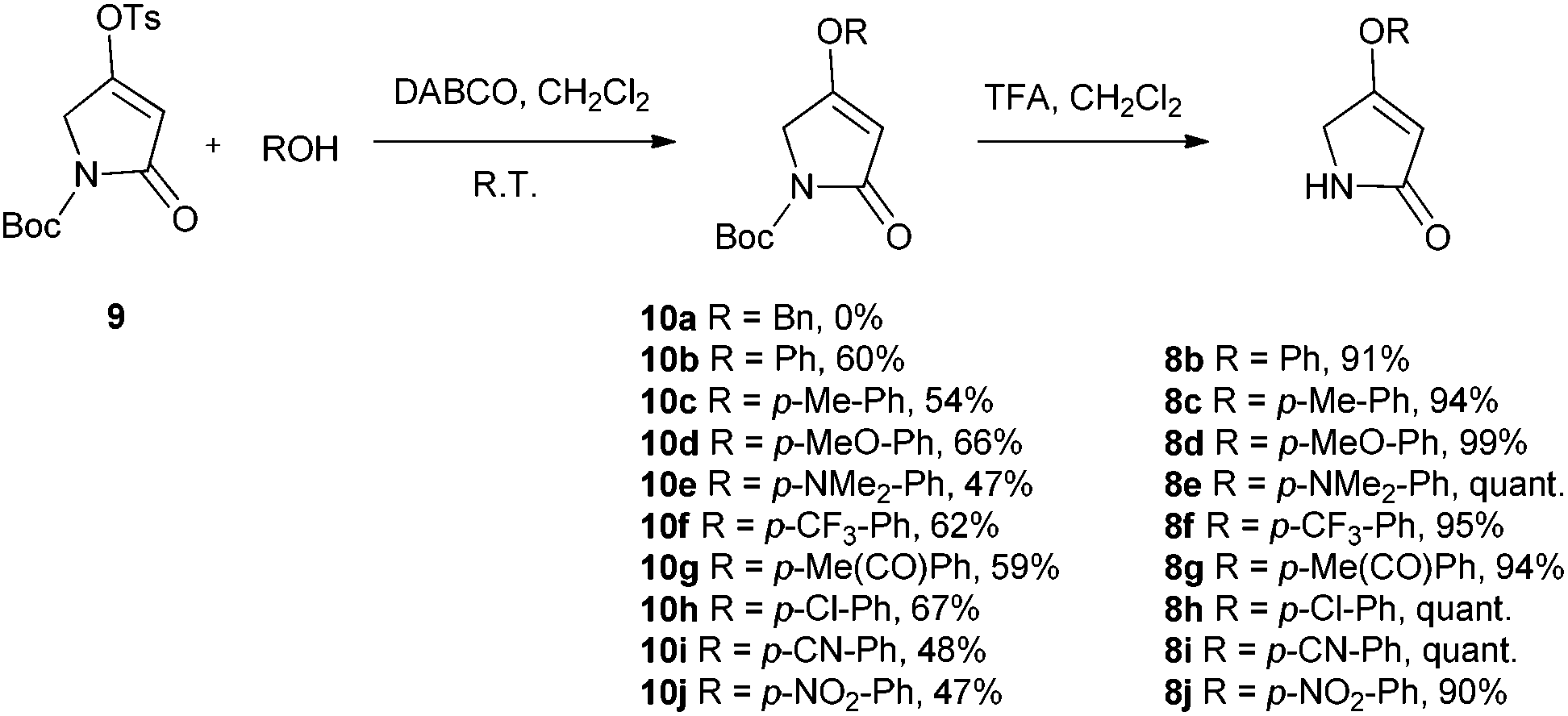
We first required a reliable method to obtain the O-substituted pyrrolinones 8 (Scheme 1), as this moiety would become the B-ring of the ultimate prodigiosenes. Following a patented procedure24 the –OBn pyrrolinone 8a was successfully obtained from the –OMe pyrrolinone 7 (Scheme 1). However, using phenol as a nucleophile instead of benzyl alcohol failed to give pyrrolinone 8b.25 Attempts using sodium phenoxide were also unsuccessful (Conditions B).
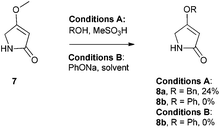 |
| | Scheme 1 Direct substitution of pyrrolinone 7. | |
To accomplish the key substitution with phenolates, we decided to use the –OTs pyrrolinone 9,26 bearing a competent leaving group (Scheme 2). By varying the nature and amount of the base and solvent, as well as the concentration of the pyrrolinone, we found a suitable method for the formation of 10b using 2.2 equivalents of DABCO in CH2Cl2 at room temperature with the tosylate 9 at a concentration of 0.02 M (see ESI Table S1†). Using these optimized conditions, we prepared nine pyrrolinones, 10b–j, in moderate-good yields (47–62%), regardless of whether the phenol contained an electron-withdrawing or an electron-donating substituent. Acid-catalyzed deprotection of the N-Boc group then gave the pyrrolinones 8b–8j in excellent yields.
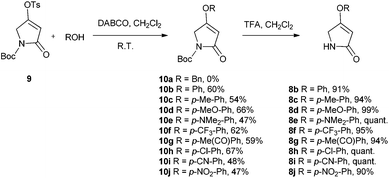 |
| | Scheme 2 Preparation of –OAr pyrrolinones 8. | |
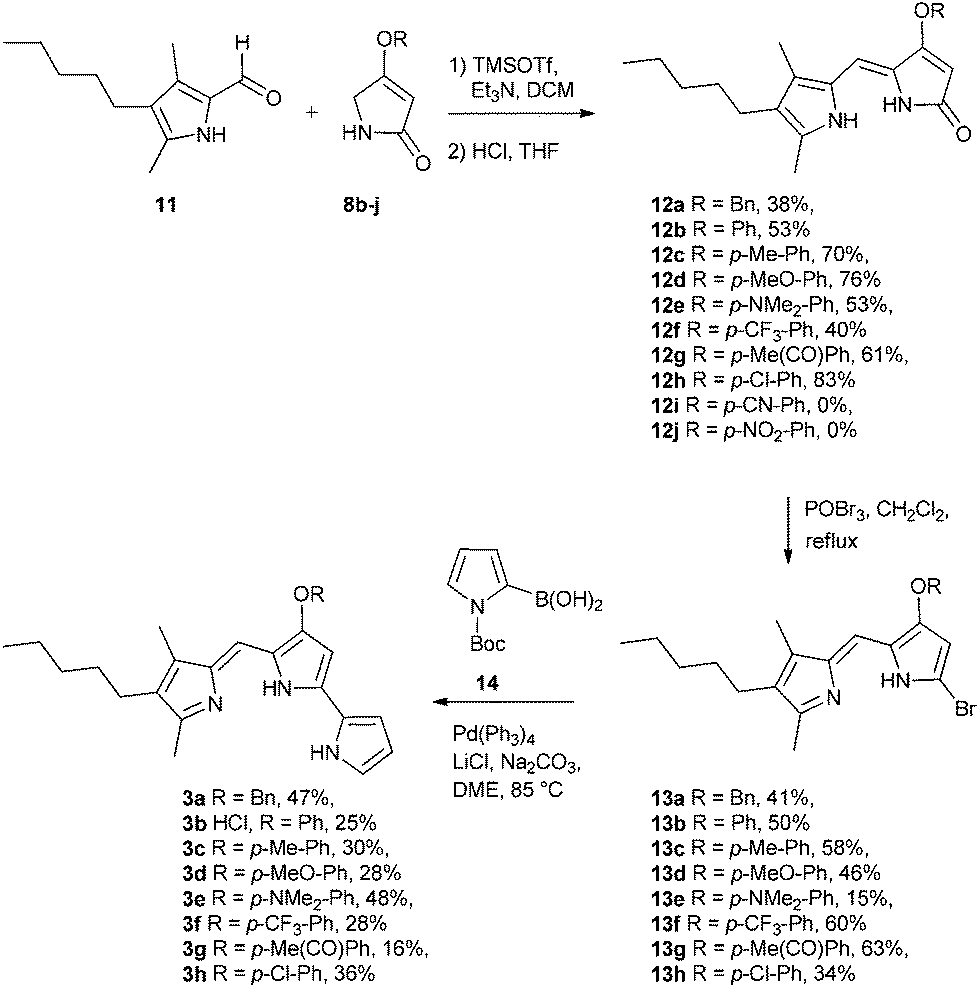
With the B-ring precursors 8 in hand we then prepared dipyrrinones 12a–h (intermediates that contain the B- and C-rings of the ultimate prodigiosene targets, Scheme 3). Aldol condensation of aldehyde 1110f with pyrrolinones 8b–j, promoted by TMSOTf and Et3N, proceeded in good yield, except when the O-aryl group was substituted with strong electron-withdrawing groups (8i R = Ph-pCN and 8j R = Ph-pNO2). Apparently, the presence of a strong electron-withdrawing group in 8i and 8j makes the ether prone to hydrolysis or substitution, as we always detected the corresponding phenol in the crude mixture (TLC) after the reaction. Bromination of dipyrrinones 12a–h, followed by Suzuki–Miyaura coupling using (1-Boc-1H-pyrrol-2-yl)boronic acid 14,27 gave the prodigiosenes 3a–h in moderate yields. As described below, we next conducted mechanistic studies to investigate prodigiosene conformation in solution, proton affinity, transmembrane anion transport activity and anti-cancer properties for this class of B-ring adducts.
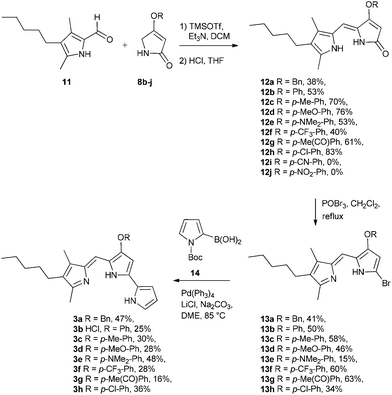 |
| | Scheme 3 Synthesis of prodigiosenes 3. | |
NMR experiments reveal that both α and β conformations of 3b·H+ bind and exchange anions
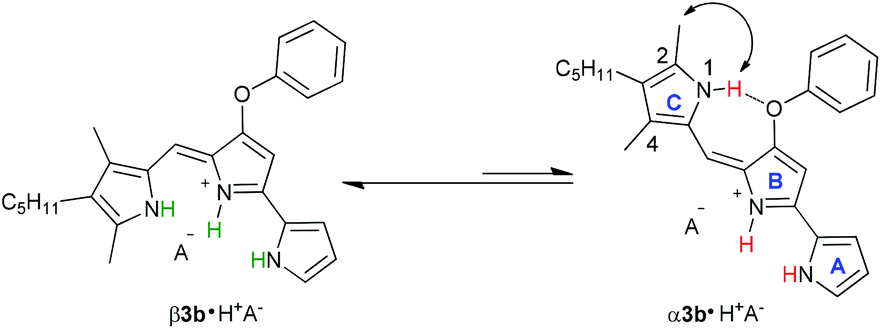
Transmembrane exchange of Cl− and NO3− anions by prodigiosin (1) involves anion binding by the protonated transporter (1·H+). This is followed by diffusion of this neutral complex 1·H+Cl− across the lipid membrane, and Cl−/NO3− anion exchange at the lipid–water interface.10d The anion binding and transport processes are complicated by the fact that protonated prodigiosenes often exist as two isomers (Scheme 4). This α/β conformational equilibrium can vary as a function of ligand structure and environmental conditions.23b,28 As depicted in Scheme 4, the prodigiosene α and β conformations should both bind anions.
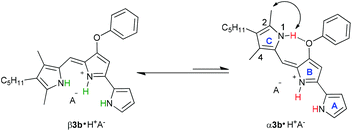 |
| | Scheme 4 β and α conformational isomers of prodigiosene 3b. Both the major β and minor α isomers should be able to bind anions. | |
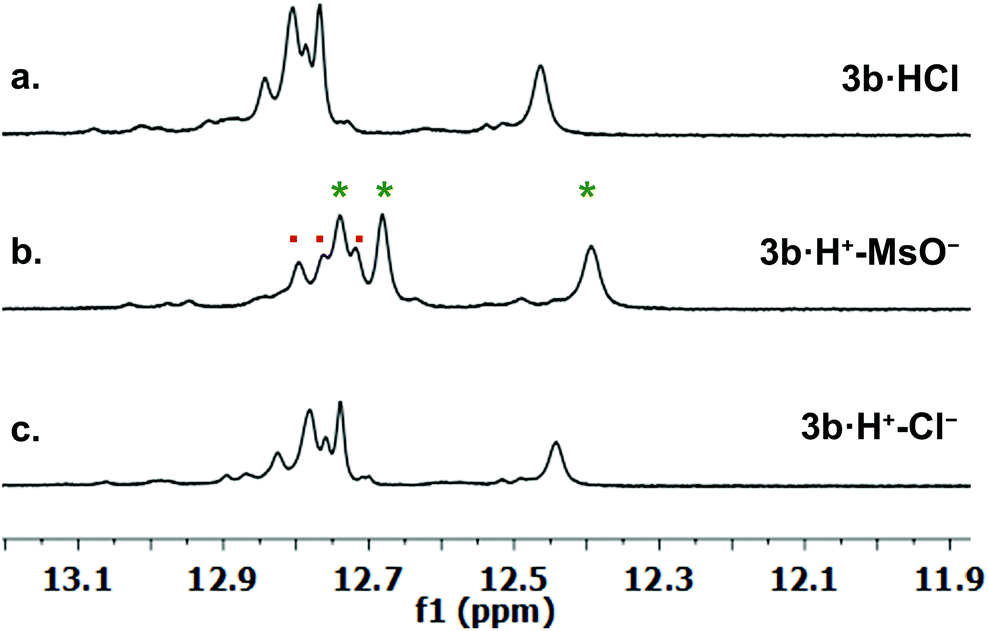
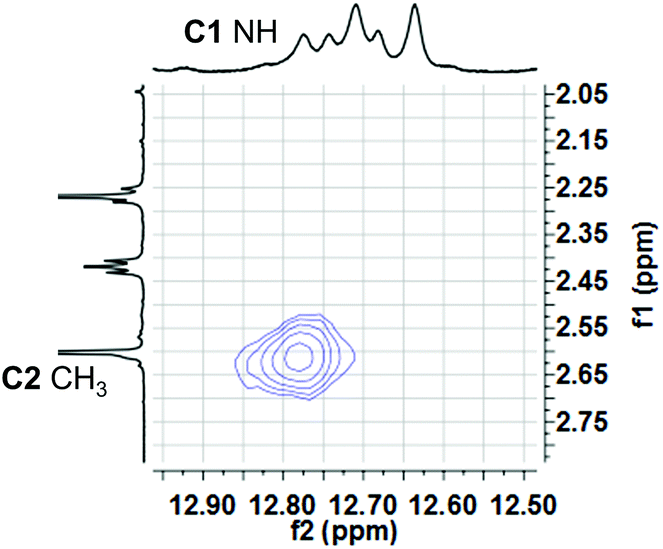
We investigated qualitative aspects of the solution-state conformation of the –OPh derivative 3b·H+. Our goals were (1) to determine whether 3b·H+ exists in both α and β conformations in solution and (2) to obtain evidence that 3b·H+ undergoes anion exchange in solution. The complex 3b·HCl (1 mM) showed one set of signals, consistent with the β conformation, when initially dissolved in CDCl3 (see spectra on p. 56 of ESI†). However, after being in CDCl3 solution over time the same sample showed two sets of 1H NMR signals, in an approximate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Based on analogy with previous NMR studies of other prodigiosenes in CDCl3, by Rizzo et al. and by Quesada et al.,23b,28 we assigned the major set of NMR signals to the β isomer of 3b·HCl and the minor set of signals to the α isomer. We next investigated whether both conformers of 3b·H+ undergo anion binding and exchange in solution. Thus, we titrated methanesulfonic acid (MsOH) into a solution of 3b·HCl in CDCl3 and followed changes in 1H NMR signals for the pyrrolic NH protons (δ 12–13 ppm). We reasoned that changes in this region of the spectrum would reflect anion binding and exchange between chloride and methanesulfonate anions, since it is the pyrrole NH atoms that hydrogen bond with the anion. Fig. 4 shows representative spectra from the titration. The NH region of the 1H NMR spectrum for the 3b·HCl salt (Fig. 4a) indicates a mixture of isomers, but the NMR signals were poorly resolved. However, this overlap for the pyrrolic NH protons was removed upon the addition of MsOH to a solution of 3b·HCl (Fig. 4b). As shown in Fig. 4b, we observed two well-resolved sets of three pyrrolic NH signals for the 3b·HOMs complex. There are two important findings here. First, the NH chemical shifts change upon addition of the MsOH, suggesting that both the α and β isomers bind anions. Thus, as shown in Fig. 4b chemical shifts for the six NH protons moved upfield when MsOH was added to a solution of 3b·HCl. These changes in NH chemical shifts indicate anion exchange, as Cl− is replaced by the softer methanesulfonate anion in the binding site of the prodigiosene. Second, the α/β isomer ratio for 3b·HCl and 3b·HOMs does not change significantly with a change in bound anion (Fig. 4c). A 2D NOESY NMR experiment of the 3b·HOMs complex allowed us to assign signals for the three NH protons in the major β isomer and for the three NH protons in the α isomer. The NOE correlation that best supports our assignments can be observed between one of the minor pyrrole NH signals and the signal corresponding to the C2 methyl group of the α isomer (Fig. 5). For this α isomer, the C-ring NH proton forms an intramolecular hydrogen bond with the –OPh group on the B-ring (Scheme 4). Presumably, this hydrogen-bonded NH proton exchanges much more slowly with residual water in the CDCl3 solvent and, therefore, shows a NOE correlation with its neighbouring C2-methyl group.
:1 ratio. Based on analogy with previous NMR studies of other prodigiosenes in CDCl3, by Rizzo et al. and by Quesada et al.,23b,28 we assigned the major set of NMR signals to the β isomer of 3b·HCl and the minor set of signals to the α isomer. We next investigated whether both conformers of 3b·H+ undergo anion binding and exchange in solution. Thus, we titrated methanesulfonic acid (MsOH) into a solution of 3b·HCl in CDCl3 and followed changes in 1H NMR signals for the pyrrolic NH protons (δ 12–13 ppm). We reasoned that changes in this region of the spectrum would reflect anion binding and exchange between chloride and methanesulfonate anions, since it is the pyrrole NH atoms that hydrogen bond with the anion. Fig. 4 shows representative spectra from the titration. The NH region of the 1H NMR spectrum for the 3b·HCl salt (Fig. 4a) indicates a mixture of isomers, but the NMR signals were poorly resolved. However, this overlap for the pyrrolic NH protons was removed upon the addition of MsOH to a solution of 3b·HCl (Fig. 4b). As shown in Fig. 4b, we observed two well-resolved sets of three pyrrolic NH signals for the 3b·HOMs complex. There are two important findings here. First, the NH chemical shifts change upon addition of the MsOH, suggesting that both the α and β isomers bind anions. Thus, as shown in Fig. 4b chemical shifts for the six NH protons moved upfield when MsOH was added to a solution of 3b·HCl. These changes in NH chemical shifts indicate anion exchange, as Cl− is replaced by the softer methanesulfonate anion in the binding site of the prodigiosene. Second, the α/β isomer ratio for 3b·HCl and 3b·HOMs does not change significantly with a change in bound anion (Fig. 4c). A 2D NOESY NMR experiment of the 3b·HOMs complex allowed us to assign signals for the three NH protons in the major β isomer and for the three NH protons in the α isomer. The NOE correlation that best supports our assignments can be observed between one of the minor pyrrole NH signals and the signal corresponding to the C2 methyl group of the α isomer (Fig. 5). For this α isomer, the C-ring NH proton forms an intramolecular hydrogen bond with the –OPh group on the B-ring (Scheme 4). Presumably, this hydrogen-bonded NH proton exchanges much more slowly with residual water in the CDCl3 solvent and, therefore, shows a NOE correlation with its neighbouring C2-methyl group.
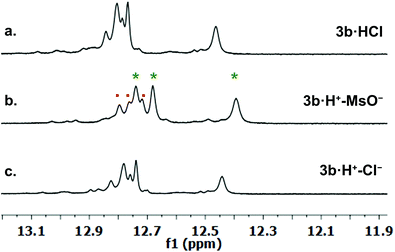 |
| | Fig. 4
1H NMR spectra in CDCl3 for the pyrrolic NH region of: (a) 3b·HCl; (b) after addition of 6 eq. of MsOH to sample a, two sets of NH signals were observed for each α (·) and β (*) conformer; (c) after addition of 1 eq. of tetrabutylammonium chloride (TBACl) to sample b. | |
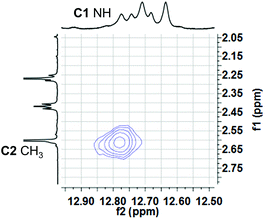 |
| | Fig. 5 A region of the 1H–1H NOESY spectrum of 3b·OMs in CDCl3 shows a NOE correlation between one of the “minor” pyrrole NH protons and C2 methyl hydrogens. This correlation is consistent with the pyrrolic NH of the C-ring being involved in an intramolecular H-bond with the –OPh group of the B-ring (see Scheme 4). | |
These NMR titrations allowed us to conclude that (1) both the major β and minor α isomer of 3b·H+ bind and exchange anions and (2) Cl− interacts more strongly than OMs− with 3b·H+. We next determined the apparent pKa values for some O-aryl analogs with the intent to correlate their ease of protonation with their rates of transmembrane anion transport.
Determination of apparent pKa values for synthetic prodigiosenes
We hypothesized that prodigiosene basicity would correlate with its efficiency as an anion transporter, since the protonated prodigiosene 3b·H+ is the likely transporter. The concept that transporter efficiency can be tuned by modulating the acidity of hydrogen bond donors has been demonstrated with other systems.29 To learn how B-ring substitution influences the acid-base properties of these synthetic prodigiosenes we used a spectrophotometric method, described by Manderville,22 to measure the apparent pKa values for five B-ring analogs: the –OPh derivative (3b·H+), two analogs with electron-donating groups on the –OAr ring (3d·H+ R = Ph-pOMe, 3e·H+ R = Ph-pNMe2), and two analogs with electron-withdrawing groups on the B-ring (3f·H+ R = Ph-pCF3, 3h·H+ R = Ph-pCl).
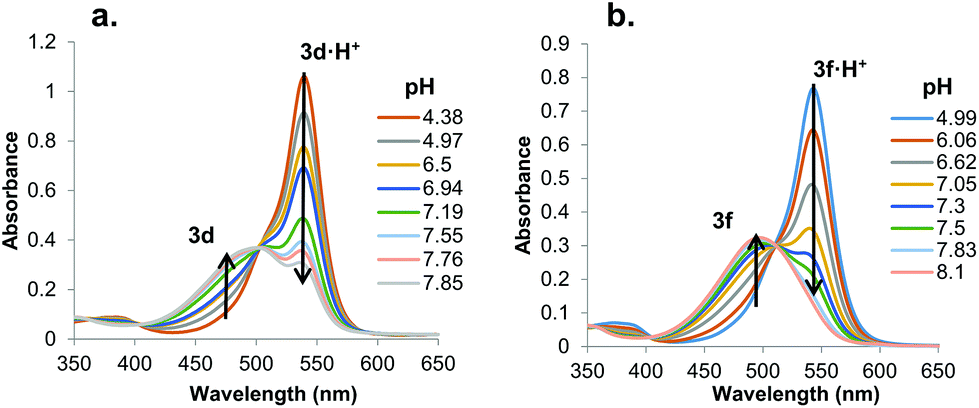
In solution, protonated prodigiosenes are pink with absorbance maxima above 500 nm. The free-base absorbs below 500 nm. Fig. 6 shows representative pH-dependent spectra for 3d and 3f in 1:1 acetonitrile–water at 25 °C. Absorption maxima for the free-base of the Ph-pOMe analog 3d (λmax = 484 nm) and its protonated form 3d·H+ (λmax = 541 nm) are well separated (Fig. 6a). The Ph-pCF3 derivative showed similar properties, with λmax = 450 nm for the free-base 3f and λmax = 545 nm for 3f·H+ (Fig. 6b). Furthermore, the ionization states of 3d/3d·H+ and 3f/3f·H+ change smoothly as a function of pH, both revealing isosbestic points. The apparent pKa values, which correspond to values for the equilibrium mixture of α and β isomers, for these B-ring prodigiosenes were determined from plots of the log(ionization ratio) vs. pH.22 As seen in Table 1, analogs with electron-donating groups on the B-ring, namely 3d·H+ (R = Ph-pOMe, pKa = 7.3) and 3e·H+ (R = Ph-pNMe2, pKa = 7.4) are weaker acids than are analogs with electron-withdrawing groups, 3f·H+ (R = Ph-pCF3, pKa = 6.7) and 3h·H+ (R = Ph-pCl, pKa = 6.8). This means that the free-bases of 3d (R = Ph-pOMe) and 3e (R = Ph-pNMe2) are more basic than 3f (R = Ph-pCF3) and 3h (R = Ph-pCl). The increased basicity of 3d and 3e means that a greater percentage of these analogs would be protonated at physiological pH, which should enhance their ability to extract anions from water and transport these bound anions as neutral complexes 3·HA across lipid membranes.
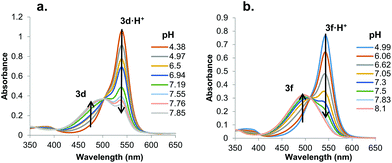 |
| | Fig. 6 UV-Vis absorbance spectra for a. 3d and b. 3f as a function of pH in 1:1 CH3CN–H2O (v/v) at 25 °C (0.1 M NaCl). | |
Table 1 pKa values and transmembrane anion exchange rates for some synthetic prodigiosenes with different –OAr groups on the B-ring. Transport experiments were repeated in triplicate and all traces reported in Fig. 7 are the average of those 3 trials
|
3·H+
|
pKa |
Initial rate for Cl−/NO3− exchange (s−1) |
t
1/2 for Cl− efflux (s) |
|
3d (Ph-pOMe) |
7.3 ± 0.1 |
15.1 ± 0.2 |
65 ± 2 |
|
3e (Ph-pNMe2) |
7.4 ± 0.1 |
10.8 ± 0.3 |
87 ± 3 |
|
3b (Ph) |
7.2 ± 0.1 |
10.8 ± 0.3 |
90 ± 5 |
|
3h (Ph-pCl) |
6.8 ± 0.1 |
3.4 ± 0.1 |
168 ± 3 |
|
3f (Ph-pCF3) |
6.7 ± 0.1 |
1.0 ± 0.1 |
287 ± 2 |
Correlation between anion transport rates and transporter basicity
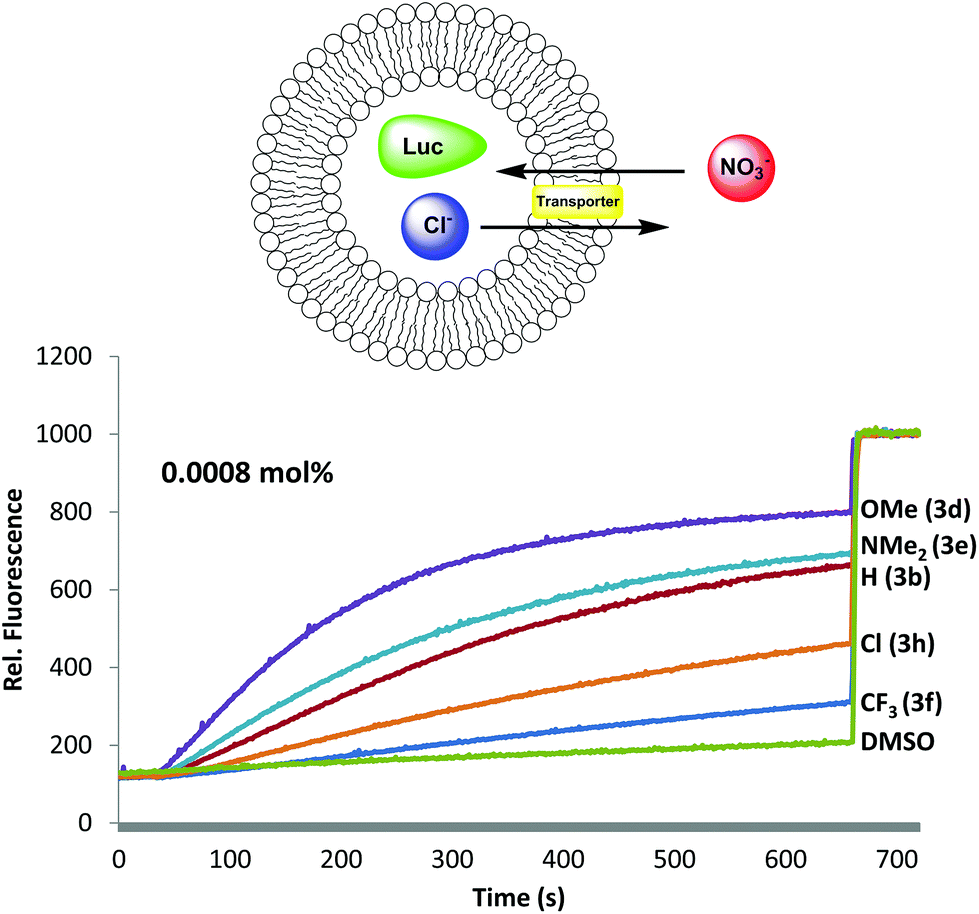
Having established that B-ring substitution influences the acid-base properties of –OAr analogs 3b, 3d–f and 3h, we next compared the ability of these synthetic prodigiosenes to catalyze the exchange of anions across phospholipid vesicles. Transmembrane transport was evaluated with a liposome model that uses the chloride-sensitive dye, lucigenin (LG), to monitor anion exchange.30 This assay works because LG fluorescence is quenched by Cl− but not by NO3−. Thus, egg-yolk phosphatidylcholine (EYPC) liposomes loaded with 1.0 mM LG and NaCl were suspended in a solution (pH 7.4) containing extravesicular NaNO3. Upon the addition of our synthetic prodigiosenes 3 the fluorescence of the intravesicular LG increases, indicating Cl− efflux and NO3− influx. Fig. 7 shows representative fluorescence curves, plotted as a function of time, after addition of analogs 3b, 3d–f and 3h. We quantified transport rates by (a) determining the initial rate of change of the relative fluorescence of the intravesicular LG and (b) estimating the half-life of the exchange reaction (Table 1). The data in Fig. 7 and Table 1 contain important findings. First, these synthetic prodigiosenes, especially 3d and 3e, are potent anion transporters and relatively low transporter loadings are needed to observe significant Cl− efflux from the liposomes. For example, we used only 0.0008 mol% of transporter relative to EYPC lipid (1 transporter for 125000 lipid molecules or about 2.5 transporters per liposome) to generate the data in Fig. 7. This is one of the lowest transporter loadings reported to date.5 Second, substitution of the B-ring with an –OAr group does not perturb the ability of these analogs to catalyze anion exchange. Indeed, the electron-rich 3d·H+ (R = Ph-pOMe) is qualitatively similar to the natural product prodigiosin 1·H+ in terms of the efficiency at which it exchanges Cl− and NO3− anions across the EYPC bilayer (see ESI Fig. S6†) Most importantly, compounds substituted with electron-donating groups (3d and 3e) are better anion transporters than analogs containing electron-withdrawing groups (3f and 3h). These results indicate that the catalytic efficiency of transmembrane anion exchange correlates with how much of the prodigiosene is in its protonated form 3·H+.31
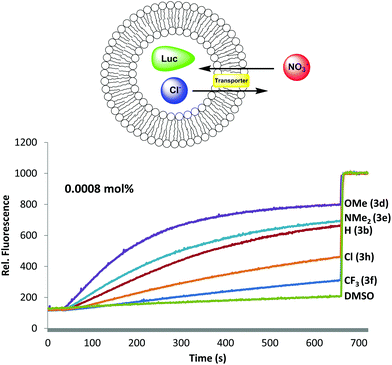 |
| | Fig. 7 Anion exchange assay for analogs 3b, 3d–f and 3h. Change in fluorescence of lucigenin (LG) observed in EYPC liposomes at 25 °C. The data was collected using 0.0008 mol% of transporter compounds relative to EYPC lipid. Liposomes containing 1 mM lucigenin, 20 mM HEPES buffer and 100 mM NaCl (pH 7.43) were suspend in 20 mM HEPES and 100 mM NaNO3 solution (pH 7.43); at t = 30 s, prodigiosene transporter was added and the fluorescence monitored (λex = 372 nm, λem = 504 nm); at t = 660 s, addition of triton-X; traces shown are an average of three trials. | |
In vitro anti-cancer properties
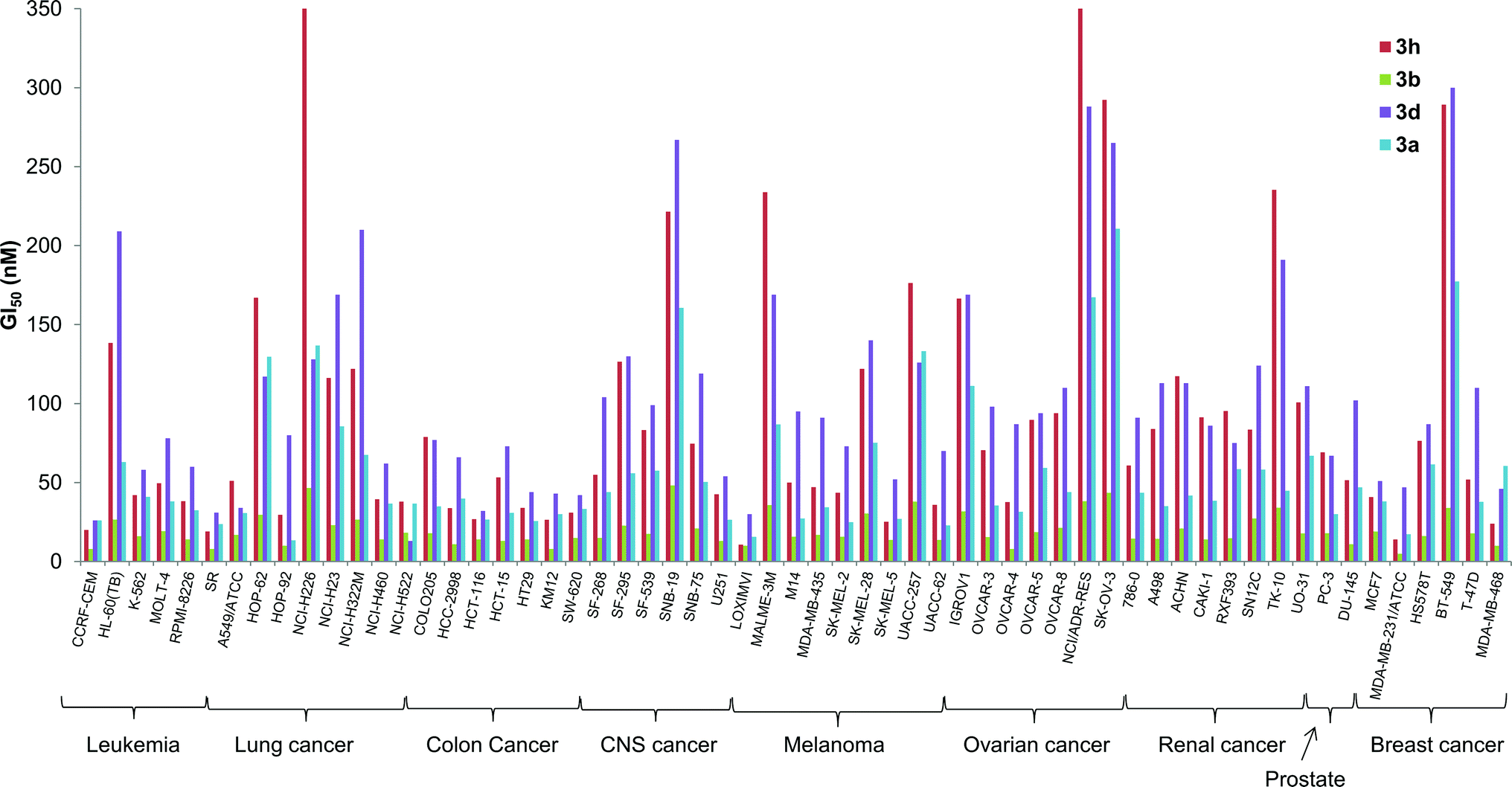
The in vitro activity of some of the B-ring analogs was evaluated at the National Cancer Institute (NCI, http://dtp.cancer.gov) against the standard NCI60 panel of 60 human cell lines.32 Prodigiosenes selected for testing were the benzyl (3a) and phenyl (3b) analogs and derivatives with an electron-donating (3d R = Ph-pOMe) and an electron-withdrawing group (3h R = Ph-pCl). Activities against the NCI60 panel are reported in Table 2 as mean values averaged over all 60 cell lines; these activities are (1) the half maximal growth inhibition concentration, GI50, (2) the total growth inhibition concentration, TGI, and (3) the half maximal lethal concentration, LC50. All four synthetic prodigiosenes exhibited GI50 values in the nM range, similar in magnitude to 1, indicating that replacement of the B-ring OMe by an O-aryl group is not universally detrimental to anti-cancer activity. They also show LC50 values in the μM range that could indicate a lower toxicity compared to the natural compound 1 (LC50 = 300 nM). The phenyl analog 3b was the most active of the derivatives with a mean GI50 value of 18 nM. A more detailed analysis in Fig. 8 shows that 3b was also typically the most active of the four synthetic prodigiosenes against all 60 cell lines.
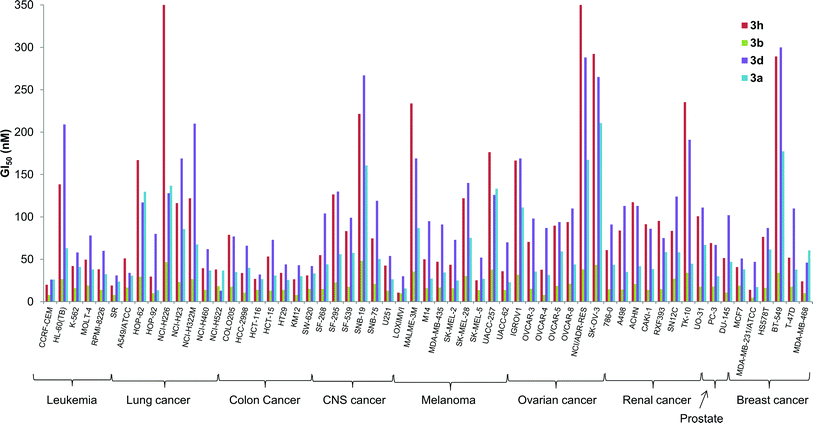 |
| | Fig. 8 GI50 concentrations (half maximal growth inhibition concentrations) of some synthetic prodigiosenes 3a, 3b, 3d and 3h against 60 human cancer cell lines representing 9 different cancer types; see http://http:dtp.cancer.gov. | |
Table 2
In vitro activity of some prodigiosenes over the NCI60 cancer cell lines. Values are a mean of two experiments (http://dtp.cancer.gov)
| |
1
|
3a (Bn) |
3b (Ph) |
3d (Ph-pOCH3) |
3h (Ph-pCl) |
|
GI50 = half maximal growth inhibition concentration.
TGI = total growth inhibition concentration.
LC50 = half maximal lethal concentration.
|
| GI50 (nM)a |
14 |
44 |
18 |
74 |
62 |
| TGI (μM)b |
2.1 |
0.6 |
0.2 |
0.8 |
1.9 |
| LC50 (μM)c |
0.3 |
4.8 |
2.5 |
7.2 |
14.4 |
Two questions that we sought to answer were would the prodigiosene basicity (1) correlate with its ability to catalyze transmembrane anion exchange and (2) correlate with its anti-cancer activity. The data presented herein suggest that the ease of protonation controls the transmembrane anion exchange. However, despite their significant in vitro anti-cancer activity, there is no evidence that those anti-cancer properties universally correlate with, or are caused by, the acid-base properties of the prodigiosene. Thus, the most basic prodigiosene 3d and the least basic prodigiosene 3h had similar mean GI50 values, 74 and 62 nM respectively, and both analogs were less potent than the phenyl analog 3b (GI50 = 18 nM).
Conclusions
Prodigiosenes, both naturally occurring and synthetic analogs, are outstanding catalysts for transport of anions across lipid membranes. Being able to control the efficiency of this transmembrane transport of anions would be valuable for various reasons, such as the potential development of therapeutics or anion sensors. In this study we synthesized some unique prodigiosene analogs with different –OAr substituents attached to the B-ring of the tripyrrolic skeleton. We measured the apparent pKa values of five B-ring analogs, namely the O-phenyl derivative (3b·H+), two O-aryl analogs with electron-donating groups and two O-aryl analogs with electron-withdrawing groups. These five derivatives were then tested for their ability to catalyze transmembrane exchange of Cl− and NO3− anions in EYPC liposomes. The data indicate that the efficiency of anion transport can indeed be modulated by the electronic nature of the O-aryl substituent on the B-ring. Thus, more basic analogs 3d (R = Ph-pOMe, pKa = 7.3) and 3e (R = Ph-pNMe2, pKa = 7.2) showed significantly greater Cl−/NO3− exchange rates than did the less basic analogs 3h (R = Ph-pCl, pKa = 6.8) and 3f (R = Ph-pCF3, pKa = 6.7). This suggests that the key factor controlling anion transport for this family of synthetic analogs, and presumably for the natural product prodigiosin, is the protonation of the prodigiosene to give an amphiphilic cation 3·H+. This cation extracts anions from water to give a neutral complex 3·HA that then diffuses rapidly across the lipid membranes. In addition to the synthetic and mechanistic work we also evaluated the anti-cancer activity of four synthetic prodigiosenes (NCI60 human tumour panel). All four analogs showed potent anti-cancer activities, with mean GI50 values in the nM range, indicating that substitution of the B-ring with OAr groups can give potent anti-cancer agents. Despite their promising in vitro anti-cancer activity there was no evidence that this activity is due to the ease of protonation of these synthetic prodigiosenes.
Acknowledgements
E. Marchal is supported by a trainee award from The Beatrice Hunter Cancer Research Institute with funds provided by Cancer Care Nova Scotia as part of The Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at CIHR. This work was supported by: a grant to JD by the Chemical Sciences, Geosciences and Biosciences Division, Basic Energy Sciences, US Department of Energy [DE-FG01-98ER14888]; and grants to AT from CIHR (133110), the Beatrice Hunter Cancer Research Institute and the Nova Scotia Health Research Foundation. We thank Dr Yiu-fai Lam, Director of the UMD NMR facility, for his help and advice concerning NMR experiments.
Notes and references
-
(a) A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348 RSC;
(b) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843 RSC.
-
(a) T. M. Fyles, Acc. Chem. Res., 2013, 46, 2847 CrossRef CAS PubMed;
(b) G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824 CrossRef CAS PubMed;
(c) H. Valkenier and A. P. Davis, Acc. Chem. Res., 2013, 46, 2898 CrossRef CAS PubMed;
(d) P. A. Gale, R. Pérez-Tomás and R. Quesada, Acc. Chem. Res., 2013, 46, 2801 CrossRef CAS PubMed.
-
(a) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed;
(b) A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791 CrossRef CAS PubMed.
-
(a) P. A. Gale, J. Garric, M. E. Light, B. A. McNally and B. D. Smith, Chem. Commun., 2007, 1736 RSC;
(b) P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sánchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886 CrossRef CAS PubMed.
- J. A. Cooper, S. T. G. Street and A. P. Davis, Angew. Chem., Int. Ed., 2014, 53, 5609 CrossRef CAS PubMed.
-
(a) V. Saggiomo, S. Otto, I. Marques, V. Felix, T. Torroba and R. Quesada, Chem. Commun., 2012, 48, 5274 RSC;
(b) H. Valkenier, C. J. E. Haynes, J. Herniman, P. A. Gale and A. P. Davis, Chem. Sci., 2014, 5, 1128 RSC.
-
(a) I. Alfonso and R. Quesada, Chem. Sci., 2013, 4, 3009 RSC;
(b) N. Busschaert and P. A. Gale, Angew. Chem., Int. Ed., 2013, 52, 1374 CrossRef CAS PubMed;
(c) B. Shen, X. Li, F. Wang, X. Yao and D. Yang, PLoS One, 2012, 7, e34694 CAS;
(d) C. Jiang, E. R. Lee, M. B. Lane, Y. F. Xiao, D. J. Harris and S. H. Cheng, Am. J. Physiol. Lung Cell. Mol. Physiol., 2001, 281, L1164 CAS.
-
(a) M. Oblatt-Montal, G. L. Reddy, T. Iwamoto, J. M. Tomich and M. Montal, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1495 CrossRef CAS;
(b) P. H. Schlesinger, R. Ferdani, J. Liu, J. Pajewska, R. Pajewski, M. Saito, H. Shabany and G. W. Gokel, J. Am. Chem. Soc., 2002, 124, 1848 CrossRef CAS PubMed.
-
(a) A. V. Koulov, T. N. Lambert, R. Shukla, M. Jain, J. M. Boon, B. D. Smith, H. Li, D. N. Sheppard, J.-B. Joos, J. P. Clare and A. P. Davis, Angew. Chem., Int. Ed., 2003, 42, 4931 CrossRef CAS PubMed;
(b) B. A. McNally, A. V. Koulov, T. N. Lambert, B. D. Smith, J.-B. Joos, A. L. Sisson, J. P. Clare, V. Sgarlata, L. W. Judd, G. Magro and A. P. Davis, Chem. – Eur. J., 2008, 14, 9599 CrossRef CAS PubMed.
-
(a) J. L. Sessler, L. R. Eller, W.-S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989 CrossRef CAS PubMed;
(b) D. R. I. Saez, J. Regourd, P. V. Santacroce, J. T. Davis, D. L. Jakeman and A. Thompson, Chem. Commun., 2007, 2701 Search PubMed;
(c) P. A. Gale, M. E. Light, B. McNally, K. Navakhun, K. E. Sliwinski and B. D. Smith, Chem. Commun., 2005, 3773 RSC;
(d) J. L. Seganish and J. T. Davis, Chem. Commun., 2005, 5781 RSC;
(e) J. T. Davis, P. A. Gale, O. A. Okunola, P. Prados, J. C. Iglesias-Sanchez, T. Torroba and R. Quesada, Nat. Chem., 2009, 1, 138 CrossRef CAS PubMed;
(f) S. Rastogi, E. Marchal, I. Uddin, B. Groves, J. Colpitts, S. A. McFarland, J. T. Davis and A. Thompson, Org. Biomol. Chem., 2013, 11, 3834 RSC.
-
(a) R. Pérez-Tomas and M. Vinas, Curr. Med. Chem., 2010, 17, 2222 CrossRef;
(b) N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. C. Salmond, Nat. Rev. Microbiol., 2006, 4, 887 CrossRef CAS PubMed;
(c) N. N. Gerber, J. Antibiot., 1975, 28, 194 CrossRef CAS;
(d) L. Mangione, M. E. Scoglio and V. Alonzo, Atti Soc. Peloritana Sci. Fis., Mat. Nat., 1976, 22, 149 CAS.
-
(a) T. Sato, H. Konno, Y. Tanaka, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biol. Chem., 1998, 273, 21455 CrossRef CAS PubMed;
(b) B. Montaner and R. Perez-Tomas, Life Sci., 2001, 68, 2025 CrossRef CAS.
- A. Füerstner, Angew. Chem., Int. Ed., 2003, 42, 3582 CrossRef PubMed.
-
(a) J. Regourd, A. A.-S. Ali and A. Thompson, J. Med. Chem., 2007, 50, 1528 CrossRef CAS PubMed;
(b) B. Díaz de Greñu, P. I. Hernández, M. Espona, D. Quiñonero, M. E. Light, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem. – Eur. J., 2011, 17, 14074 CrossRef PubMed;
(c) C. L. A. Hawco, E. Marchal, M. I. Uddin, A. E. G. Baker, D. P. Corkery, G. Dellaire and A. Thompson, Bioorg. Med. Chem., 2013, 21, 5995 CrossRef CAS PubMed;
(d) D. A. Smithen, A. M. Forrester, D. P. Corkery, G. Dellaire, J. Colpitts, S. A. McFarland, J. N. Berman and A. Thompson, Org. Biomol. Chem., 2013, 11, 62 RSC.
- R. D'Alessio, A. Bargiotti, O. Carlini, F. Colotta, M. Ferrari, P. Gnocchi, A. Isetta, N. Mongelli, P. Motta, A. Rossi, M. Rossi, M. Tibolla and E. Vanotti, J. Med. Chem., 2000, 43, 2557 CrossRef PubMed.
-
(a) K. Papireddy, M. Smilkstein, J. X. Kelly, S. M. Salem, M. Alhamadsheh, S. W. Haynes, G. L. Challis and K. A. Reynolds, J. Med. Chem., 2011, 54, 5296 CrossRef CAS PubMed;
(b) E. Marchal, D. A. Smithen, M. I. Uddin, A. W. Robertson, D. L. Jakeman, V. Mollard, C. D. Goodman, K. S. MacDougall, S. A. McFarland, G. I. McFadden and A. Thompson, Org. Biomol. Chem., 2014, 12, 4132 RSC.
- E. Marchal, M. I. Uddin, D. A. Smithen, C. L. A. Hawco, M. Lanteigne, D. P. Overy, R. G. Kerr and A. Thompson, RSC Adv., 2013, 3, 22967 RSC.
-
(a) C. Yamamoto, H. Takemoto, K. Kuno, D. Yamamoto, A. Tsubura, K. Kamata, H. Hirata, A. Yamamoto, H. Kano, T. Seki and K. Inoue, Hepatology, 1999, 30, 894 CrossRef CAS PubMed;
(b) S. Ohkuma, T. Sato, M. Okamoto, H. Matsuya, K. Arai, T. Kataoka, K. Nagai and H. H. Wasserman, Biochem. J., 1998, 334, 731 CAS.
- M. S. Melvin, J. T. Tomlinson, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 2000, 122, 6333 CrossRef CAS.
- M. Espona-Fiedler, V. Soto-Cerrato, A. Hosseini, J. M. Lizcano, V. Guallar, R. Quesada, T. Gao and R. Pérez-Tomás, Biochem. Pharmacol., 2012, 83, 489 CrossRef CAS PubMed.
- A. Hosseini, M. Espona-Fiedler, V. Soto-Cerrato, R. Quesada, R. Pérez-Tomás and V. Guallar, PLoS One, 2013, 8, e57562 CAS.
- M. S. Melvin, J. T. Tomlinson, G. Park, C. S. Day, G. R. Saluta, G. L. Kucera and R. A. Manderville, Chem. Res. Toxicol., 2002, 15, 734 CrossRef CAS PubMed.
-
(a) D. L. Boger and M. Patel, J. Org. Chem., 1988, 53, 1405 CrossRef CAS;
(b) V. Rizzo, A. Morelli, V. Pinciroli, D. Sciangula and R. D'Alessio, J. Pharm. Sci., 1999, 88, 73 CrossRef CAS PubMed;
(c) B. Jolicoeur and W. D. Lubell, Org. Lett., 2006, 8, 6107 CrossRef CAS PubMed;
(d) B. Jolicoeur and W. D. Lubell, Can. J. Chem., 2008, 86, 213 CrossRef CAS;
(e) C. Yu, L. Jiao, X. Tan, J. Wang, Y. Xu, Y. Wu, G. Yang, Z. Wang and E. Hao, Angew. Chem., Int. Ed., 2012, 51, 7688 CrossRef CAS PubMed;
(f) P. Kancharla and K. A. Reynolds, Tetrahedron, 2013, 69, 8375 CrossRef CAS PubMed.
-
T. Meul, in Eur. Pat. Appl, vol. 252363, Lonza A.-G., Switz. 1988, p. 4 Search PubMed.
-
R. D'Alessio, A. Rossi, M. Tibolla and L. Ceriani, in PCT Int. Appl, vol. WO 9730029, Pharmacia & Upjohn S.p.A., Italy, 1997, p. 47 Search PubMed.
- W.-R. Li, S. T. Lin, N.-M. Hsu and M.-S. Chern, J. Org. Chem., 2002, 67, 4702 CrossRef CAS PubMed.
- S. Martina, V. Enkelmann, G. Wegner and A. D. Schlueter, Synthesis, 1991, 613 CrossRef CAS PubMed.
- M. Garcia-Valverde, I. Alfonso, D. Quinonero and R. Quesada, J. Org. Chem., 2012, 77, 6538 CrossRef CAS PubMed.
-
(a) N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and W. A. Harrell Jr., Chem. Commun., 2010, 46, 6252 RSC;
(b) N. J. Andrews, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis, W. A. Harrell Jr. and P. A. Gale, Chem. Sci., 2011, 2, 256 RSC;
(c) N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426 CrossRef CAS PubMed;
(d) C. R. Yamnitz, S. Negin, I. A. Carasel, R. K. Winter and G. W. Gokel, Chem. Commun., 2010, 46, 2838 RSC;
(e) N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Felix, J. G. Frey and P. A. Gale, Chem. Sci., 2013, 4, 3036 RSC.
- B. A. McNally, A. V. Koulov, B. D. Smith, J.-B. Joos and A. P. Davis, Chem. Commun., 2005, 1087 RSC.
- Transmembrane anion transport by tambjamine alkaloids, relatives of the prodigiosenes, can be influenced by transporter lipophilicity (ref. 6a). Therefore we calculated values for logP, the logarithm of the octanol–water partition coefficient, for the synthetic transporters (see ESI†). Importantly, the analog with the highest Cl−/NO3− exchange rates, 3d·H+ (R = OMe, logP = 4.9) is significantly less lipophilic than the slowest transporter in this family 3h·H+ (R = CF3, logP = 5.8).
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01399a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a