 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective recognition of anionic cell membranes using targeted liposomes coated with zinc(II)-bis(dipicolylamine) affinity units†
Serhan
Turkyilmaz
ab,
Douglas R.
Rice
a,
Rachael
Palumbo
a and
Bradley D.
Smith
*a
aDepartment of Chemistry and Biochemistry, 236 Nieuwland Science Hall. and University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: smith.115@nd.edu
bFaculty of Pharmacy, Department of Pharmaceutical Chemistry, Istanbul University, 34116 Beyazit, Istanbul, Turkey
First published on 18th June 2014
Abstract
Zinc(II)-bis(dipicolylamine) (Zn2BDPA) coated liposomes are shown to have high recognition selectivity towards vesicle and cell membranes with anionic surfaces. Robust synthetic methods were developed to produce Zn2BDPA-PEG-lipid conjugates with varying PEG linker chain length. One conjugate (Zn2BDPA-PEG2000-DSPE) was used in liposome formulations doped with the lipophilic near-infrared fluorophore DiR. Fluorescence cell microscopy studies demonstrated that the multivalent liposomes selectively and efficiently target bacteria in the presence of healthy mammalian cells and cause bacterial cell agglutination. The liposomes also exhibited selective staining of the surfaces of dead or dying human cancer cells that had been treated with a chemotherapeutic agent.
Introduction
Zinc(II)-bis(dipicolylamine) (Zn2BDPA) coordination complexes are known to associate with phosphate polyanions in aqueous solution (Fig. 1A) and they have been developed for various supramolecular applications such as optical chemosensing, biomolecule labeling and catalytic hydrolysis.1–21 We have contributed to this effort by demonstrating that Zn2BDPA complexes have selective affinity for anionic cell membrane surfaces over the near-neutral membrane surfaces of healthy mammalian cells.22–28 This discovery has led to molecular imaging probes that can target two types of anionic cell systems with high biomedical significance, namely, bacterial cells and dead/dying mammalian cells. With bacterial cells, the surrounding envelope is anionic because it contains phosphorylated amphiphiles such as phosphatidylglycerol or lipotechoic acid in the case of Gram-positive bacteria, or lipid A in the case of Gram-negative bacteria. With dead/dying mammalian cells, the outer membrane surface becomes anionic due to the exposure of phosphatidylserine during the cell death process. We have shown that fluorescent Zn2BDPA probes can selectively stain these anionic cells even when they are within highly complex and heterogeneous biological environments such as cell culture and living animals.22–28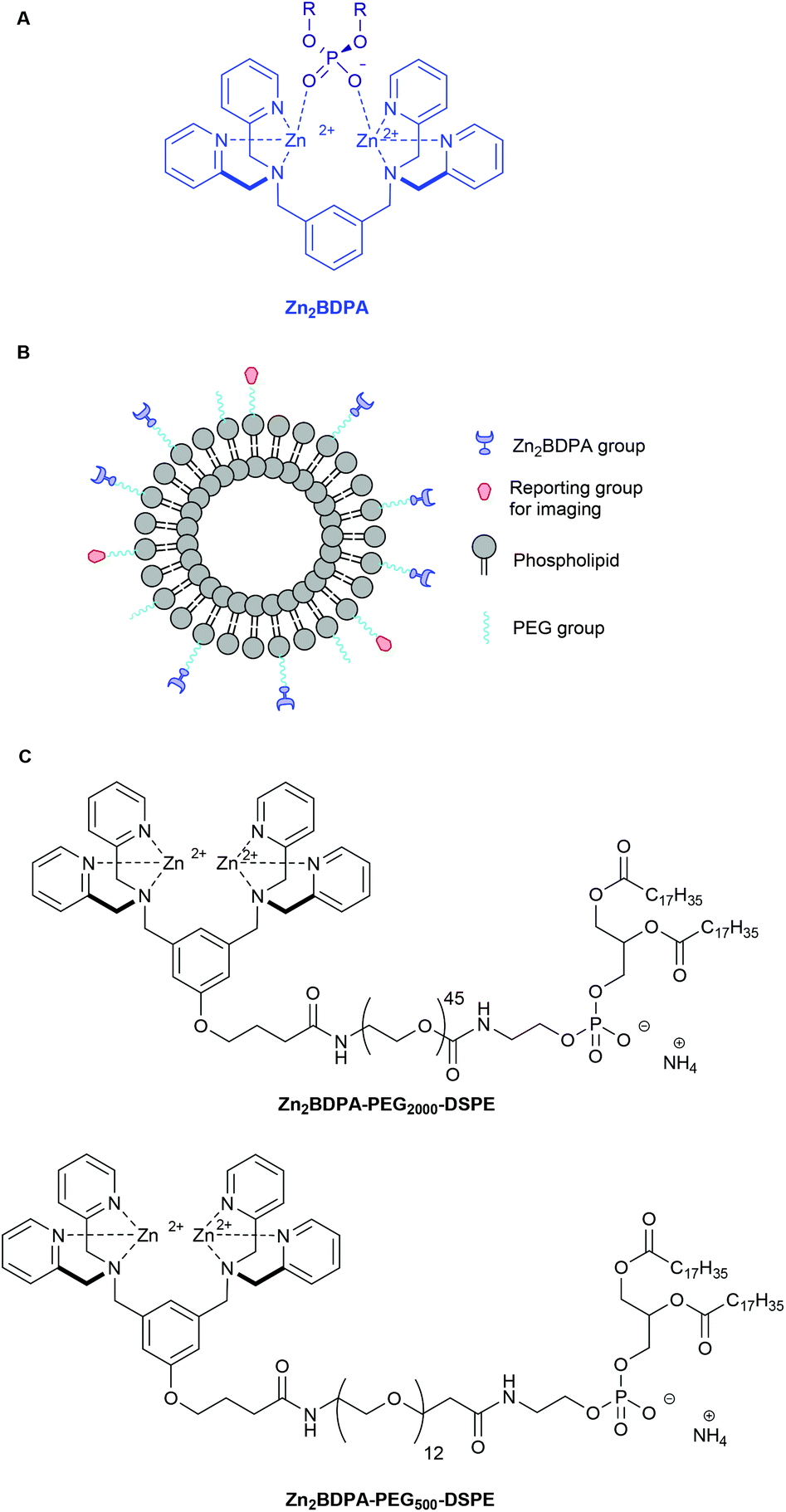 | ||
| Fig. 1 Association of a Zn2BDPA coordination complex with a phosphodiester (A), schematic picture of a Zn2BDPA coated liposome with PEG and Zn2BDPA groups on the inner leaflet omitted for clarity (B), structures of Zn2BDPA-lipid conjugates used in this study (C). | ||
Recently, we prepared and evaluated multivalent Zn2BDPA molecular probes that were equipped with four or more covalently attached Zn2BDPA targeting units and observed high selectivity for anionic membranes.29–31 Fluorescent versions of these multivalent Zn2BDPA probes were found to be effective optical imaging agents in cell culture and animal models of bacterial infection or cell death. A notable finding with the bacterial studies was the strong propensity of multivalent Zn2BDPA probes to selectively cross-link and agglutinate bacterial cells. The multivalent Zn2BDPA probes were not bactericidal, but the agglutination effect was universal regardless of Gram-type or cell morphology. These results suggest that multivalent Zn2BDPA molecular probes have promise as selective, broad spectrum bacterial agglutination agents for infection imaging and diagnostics. Similarly, they may also be incorporated into imaging and theranostic strategies that target mammalian cell death. For example, a hypothetical cancer treatment strategy might employ a multifunctional drug delivery vehicle to target the sites of endogenous cell death within a tumor. If the tumor is responsive to the delivered drug, more cell death occurs which, in principle, should amplify the drug delivery process during a subsequent treatment.32,33
A necessary requirement for each of these potential imaging and theranostic applications is the construction of effective multivalent Zn2BDPA imaging probes and drug delivery vehicles. We were attracted to liposomes as a biocompatible nanoparticle platform whose pharmacokinetic properties can be optimized by systematic modification of the amphiphilic building blocks. The ability of non-targeted stealth liposomes to exploit the enhanced permeation retention (EPR) effect and accumulate inside tumors is well known as one of the pioneering success stories of nanomedicine.34,35 In related fashion, stealth liposomes are known to collect in sites of bacterial infection within living subjects.36 In both of these cases, the liposome accumulation process is passive and does not involve direct molecular targeting. This present report, on the targeting ability of liposomes that are coated with multiple Zn2BDPA affinity units, is the first step in a process to develop targeted liposome systems that enhance the passive accumulation effects described above. There is a large and growing body of literature on the topic of targeted liposomes; that is, liposomes coated with targeting ligands having selective affinity for receptors or biomarkers of disease. Although the concept of targeted liposomes has alluring features, it also includes molecular design challenges that must simultaneously address conflicting requirements.37–42 Most notable is the need to coat the liposome surface with polymeric structures (e.g., PEG chains) that inhibit recognition and premature clearance of the liposomes by the reticuloendothelial system (RES). One potential solution is to attach the targeting ligands to the ends of the polymeric chains, but the current literature on this strategy does not make it clear if the targeting ligands will still be available for receptor binding, or if they will trigger undesired recognition by the RES. There is also the nontrivial synthetic chemistry challenge of fabricating the targeted liposomes. The most common approach is to append the targeting ligands to the ends of commercially available polyethyleneglycol-phospholipid conjugates such as PEG2000-DSPE. There are two limiting strategies. One is to react the targeting ligand with preformed liposomes containing a suitably reactive PEG2000-DSPE. The other approach is to first synthesize the functionalized PEG2000-DSPE conjugate and then assemble the liposomes. The latter method provides more opportunity to validate compositional purity of the liposome components, but there are still molecule characterization challenges due to polydispersity of the PEG chains in most commercial supplies of the starting materials. We believe that studies of targeted liposomes are best conducted using molecular building blocks with well-defined chemical structures and high compositional purity.
Here, we describe the preparation of targeted liposomes that are coated with multiple Zn2BDPA affinity units through the incorporation of a small fraction of amphiphilic Zn2BDPA-PEG-DSPE conjugates in the bilayer membranes (Fig. 1B). Specifically, we describe the synthesis of two conjugates, Zn2BDPA-PEG2000-DSPE and Zn2BDPA-PEG500-DSPE (Fig. 1C) and the in vitro molecular recognition properties of liposomes that incorporate these conjugates. We find that Zn2BDPA coated liposomes are able to rapidly cross-link a second population of anionic liposomes. Furthermore, we show that Zn2BDPA coated liposomes can selectively agglutinate bacterial cells in the presence of healthy mammalian cells and also selectively target the surfaces of dead/dying mammalian cancer cells. The results indicate that Zn2BDPA coated liposomes have great promise for wide range of targeted imaging and drug delivery applications.
Results and discussion
Synthesis and liposome preparation
Our strategy for the construction of Zn2BDPA-PEG-lipid conjugates was largely determined by the commercial availability of lipids and PEGylated lipids suitable for conjugation. Lipids terminating in amine groups such as DSPE and NH2-PEG2000-DSPE are commercially available. Furthermore, bis(dipicolylamine) (BDPA) conjugates of these lipids can be prepared through standard amide coupling chemistry. The BDPA carboxylic acid derivative 6, which would allow the preparation of these conjugates, was synthesized in a fashion similar to a BDPA amine derivative we reported earlier.43 Accordingly, dimethyl 5-hydroxyisophthalate (1) was reduced using LiAlH4 to give 3,5-bis(hydroxymethyl)phenol (2) in high yield,44 which was coupled with ethyl 4-bromobutanoate using K2CO3/Bu4NI in anhydrous DMF to give 3 in 31% yield. Numerous attempts at improving the yield for this alkylation proved fruitless. The diol 3 was efficiently converted to the dibromo derivative 4 using modified Appel conditions.45 The ligands for zinc were installed by reacting 4 with dipicolylamine using K2CO3 in anhydrous DMF to give 5 in good yield. Finally saponification of 5 afforded the desired BDPA carboxylic acid 6 quantitatively (Scheme 1).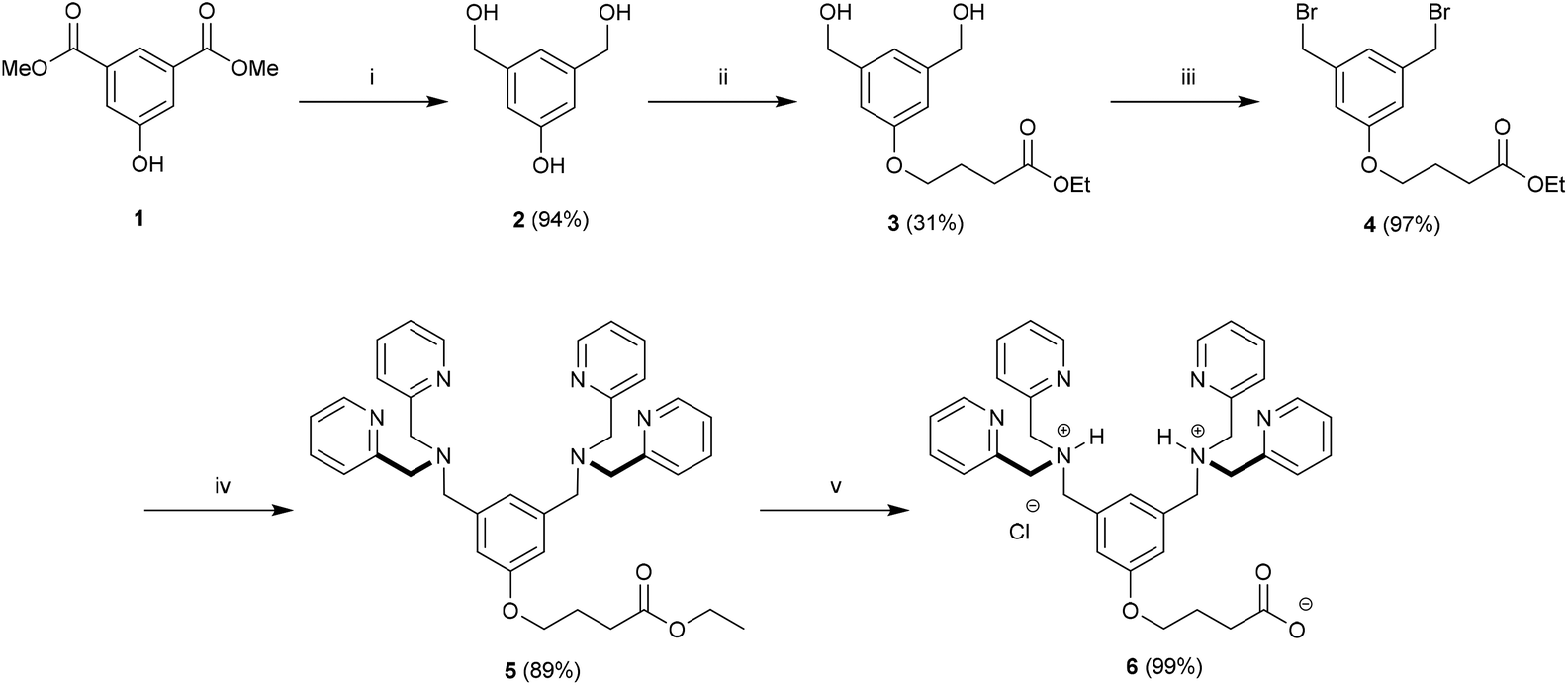 | ||
Scheme 1 Preparation of BDPA carboxylic acid 6. Conditions: (i) LiAlH4, THF, rt, 24 h; (ii) Ethyl 4-bromobutyrate, K2CO3, Bu4NI, 60 °C, 16 h; (iii) Ph3P, CBr4, DIPEA, THF, rt, 24 h; (iv) 2,2′-dipicolylamine, K2CO3, DMF, rt, 16 h; (v) 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 THF–H2O–MeOH, NaOH, reflux, 2 h. :1:1 THF–H2O–MeOH, NaOH, reflux, 2 h. | ||
With 6 in hand we turned our attention to the preparation of BDPA-PEG2000-DSPE (7). A multitude of methods can be considered for the amide bond formation between 6 and H2N-PEG2000-DSPE.46 We found that the EDC/DMAP mediated pentafluorophenol (PfpOH) activation of 6 followed by reaction with H2N-PEG2000-DSPE reliably afforded BDPA conjugate 7 (Scheme 2). Using this method it was possible to prepare tens of milligrams of 7 in one run and purification merely consisted of a relatively simple preparatory TLC procedure. It should be noted that commercially available H2N-PEG2000-DSPE is a polydisperse mixture of PEG chains with a mean degree of polymerization around 45 and this is reflected in the HRMS spectrum of 7 (ESI). To remove the mass spectral ambiguity regarding the identity of the product and to demonstrate the versatility of the synthetic methods we decided to prepare BDPA-PEG500-DSPE (10) from commercially available monodisperse FmocNH-PEG500-propionic acid (8). Reaction of PfpOH activated 8 with DSPE proved very sluggish in a wide variety of solvents due to the poor solubility of this phospholipid. This problem was overcome by refluxing the reaction. Removal of the Fmoc group using piperidine and reaction of the resulting H2N-PEG500-DSPE (9) with PfpOH activated 6 afforded the desired conjugate 10 in good yield (Scheme 3). It was possible to purify 10 using a relatively simple preparatory TLC procedure and the spectral data indicated monodispersity.
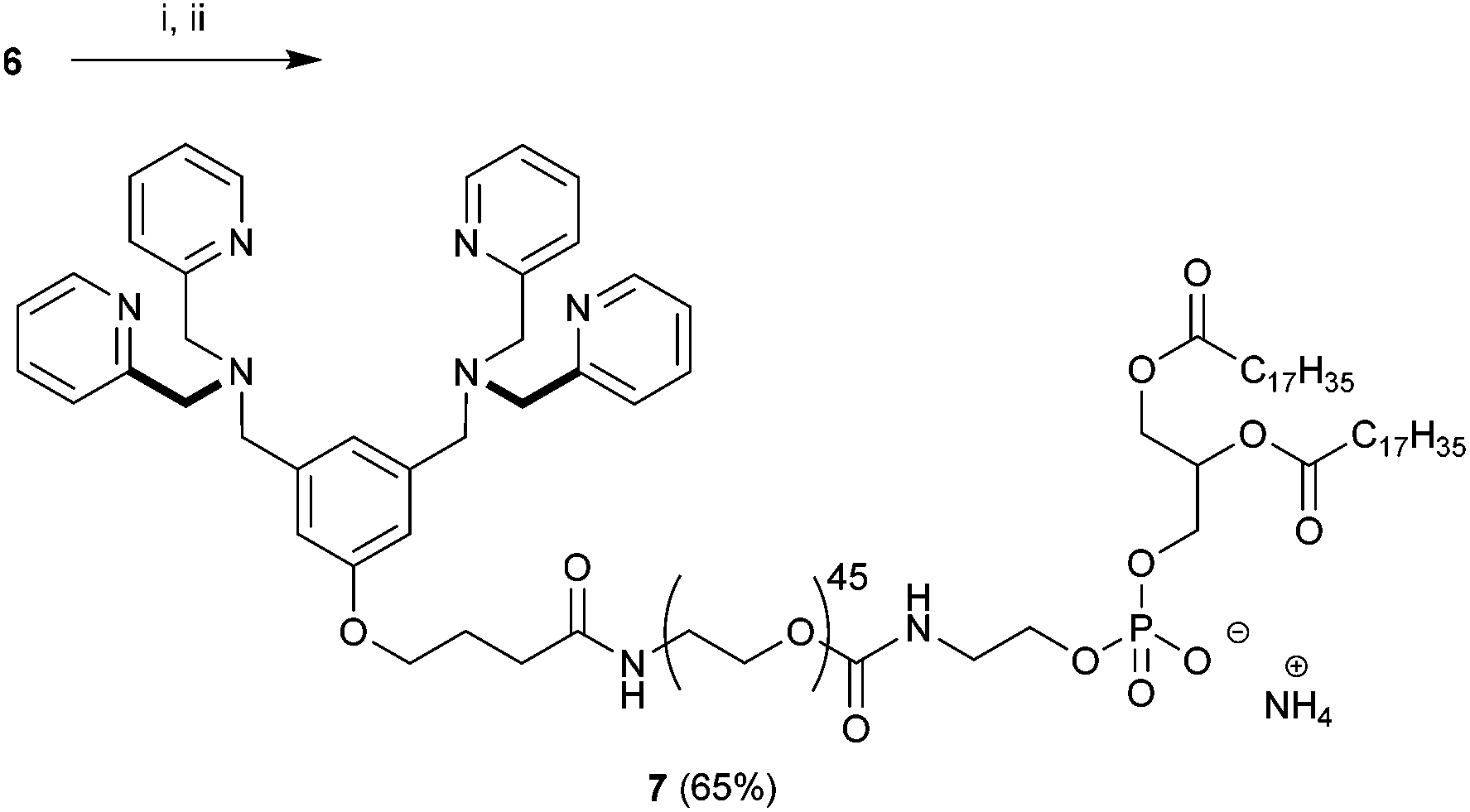 | ||
| Scheme 2 Preparation of BDPA-PEG2000-DSPE (7). Conditions: (i) PfpOH, EDC, DMAP, CHCl3, 0 °C to rt, 16 h followed by (ii) H2N-PEG2000-DSPE, DIPEA, CHCl3, rt, 24 h. | ||
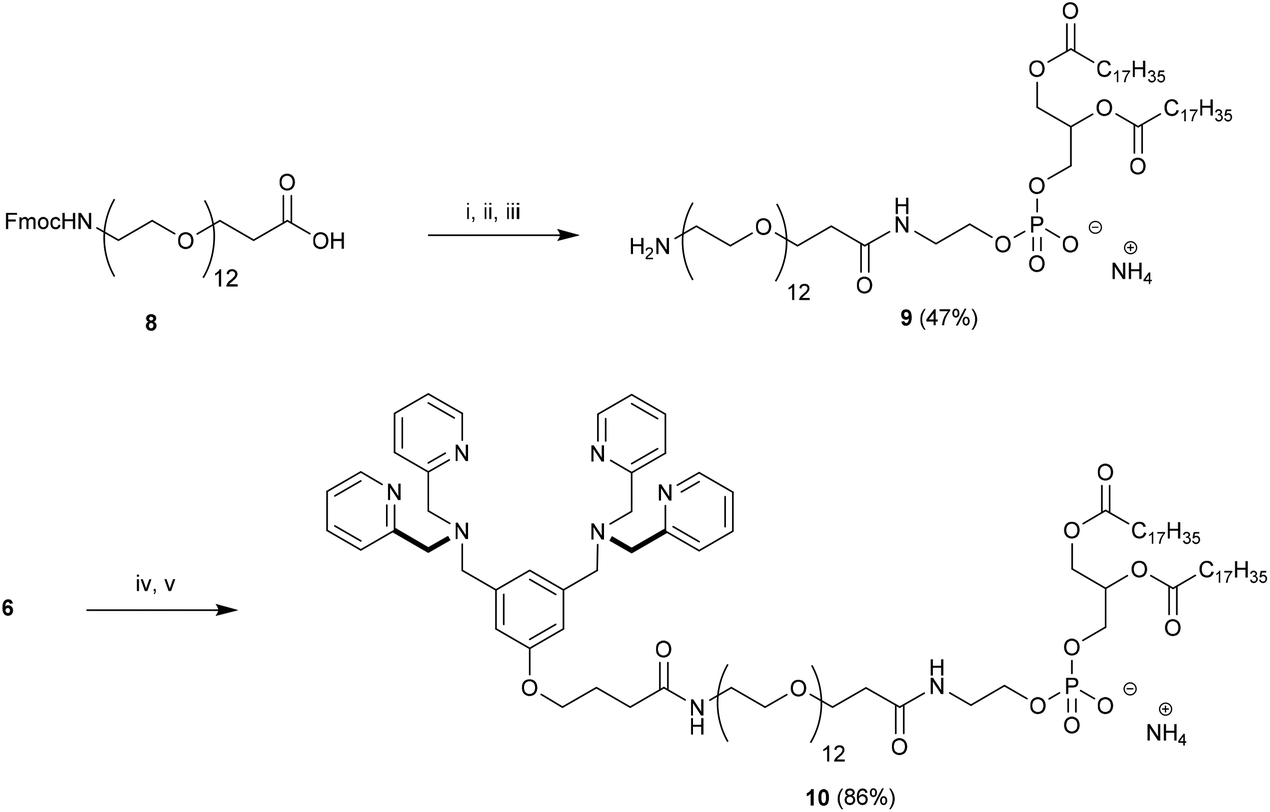 | ||
| Scheme 3 Preparation of BDPA-PEG500-DSPE (10). Conditions: (i) PfpOH, EDC, DMAP, CHCl3, 0 °C to rt, 16 h followed by (ii) DSPE, DIPEA, CHCl3, reflux, 24 h; (iii) piperidine–DMF 2:8, rt, 2 h; (iv) PfpOH, EDC, DMAP, CHCl3, 0 °C to rt, 16 h followed by (v) 9, DIPEA, CHCl3, rt, 24 h. | ||
For cuvette experiments, liposomal dispersions containing 2.5 mol% of the Zn2BDPA-PEG-DSPE amphiphiles were prepared using the film hydration/extrusion method.47 Lipid films composed of 7–cholesterol–POPC or 10–cholesterol–POPC (2.5:30:67.5 mol%, 3.32 μmol total lipid) were hydrated using HEPES buffer containing Zn(NO3)2. The Zn2+/BDPA molar ratio was 10 to ensure rapid and complete formation of the Zn2BDPA coordination complexes. In the absence of Zn2+, the dispersions were impossible to extrude, presumably due to bilayer self-aggregation caused by the lipophilic nature of the uncomplexed BDPA units. In the presence of Zn2+, the lipid films were readily dispersed into aqueous solution and easy to extrude through polycarbonate membranes with 200 nm diameter pores. Dynamic light scattering (DLS) analysis of the liposomes composed of Zn2BDPA-PEG2000-DSPE–cholesterol–POPC (2.5:30:67.5) indicated a monomodal size distribution with diameter = 154 ± 54 nm, PDI = 0.109, and ζ = 4.82 mV. The same analysis of liposomes composed of Zn2BDPA-PEG500-DSPE–cholesterol–POPC (2.5:30:67.5) indicated a bimodal size distribution with two diameters = 96 ± 15 nm, and 790 ± 140 nm, PDI = 1.000, and ζ = 4.00 mV. We infer that the larger diameter is due to liposome self-aggregation. Most likely, the shorter PEG chain in the Zn2BDPA-PEG500-DSPE is unable to fully block association of the terminal Zn2BDPA affinity units with the negatively charged phosphate diester groups on the opposing bilayer surface. Self-aggregation was not observed with the liposomes containing Zn2BDPA-PEG2000-DSPE, (four times longer PEG chain), therefore, all subsequent studies of Zn2BDPA coated liposomes used membrane compositions containing this conjugated amphiphile.
The selective affinity of Zn2BDPA coated liposomes for anionic membranes was vividly demonstrated by conducting experiments that mixed liposomes composed of Zn2BDPA-PEG2000-DSPE–cholesterol–POPC (2.5:30:67.5) with target liposomes of various compositions. As shown in Fig. 2, mixing with anionic liposomes composed of POPS–cholesterol–POPC (10:30:60) resulted in rapid and extensive precipitation. Mixing with anionic liposomes composed of DPPG–cholesterol–POPC (10:30:60) produced the same outcome (see ESI†). In contrast, mixing with uncharged liposomes composed of cholesterol–POPC (30:70) produced no precipitation. Control experiments showed that the anionic liposome systems do not form aggregates when exposed to Zn(NO3)2 alone, thus confirming that the Zn2BDPA affinity units are essential for the anionic membrane recognition process. These favorable liposome targeting results encouraged us to conduct cell recognition studies using both human and bacterial cells. The liposomes were doped with a small amount of the near-infrared fluorescent lipophilic dye, DiR, to enable effective visualization using fluorescence microscopy.
 | ||
| Fig. 2 Cross-linking of liposomes containing Zn2BDPA-PEG2000-DSPE and anionic target liposomes containing POPS. (A) Liposomes composed of Zn2BDPA-PEG2000-DSPE–cholesterol–POPC (2.5:30:67.5 mol%, 0.83 μmol total lipid) in zinc-containing HEPES buffer. (B) Liposomes composed of POPS–cholesterol–POPC (10:30:60 mol%, 0.83 μmol total lipid) in zinc-containing HEPES buffer. (C) Admixture of liposome samples A and B. | ||
Selective bacteria targeting using Zn2BDPA coated liposomes
The bacterial targeting of Zn2BDPA coated liposomes was evaluated by conducting imaging experiments using cultures of E. coli, P. aeruginosa, S. aureus, and K. pneumoniae. In each case, separate samples of bacteria (∼108 cells) were treated for 15 min with Zn2BDPA coated liposomes (Zn2BDPA-PEG2000-DSPE–DiR–cholesterol–POPC, 2:2:30:66) or untargeted liposomes (DiR–cholesterol–POPC, 2:30:68). The treated cells were pelleted by microcentrifugation and the tubes were imaged using a CCD camera. As shown in Fig. 3, the fluorescent Zn2BDPA coated liposomes were located primarily in the bacterial pellet, whereas, the fluorescent untargeted liposomes were primarily in the supernatant above the pellet. After pellet imaging, the bacterial cells were rinsed twice, dispersed into solution by vortexing, and subjected to fluorescence microscopy. Shown in Fig. 4 are typical micrographs of cross-linked bacteria/liposome aggregates. Analogous micrographs of the pelleted bacteria that had been treated with fluorescent untargeted liposomes showed no evidence for bacterial cross-linking (Fig. ESI-6†).
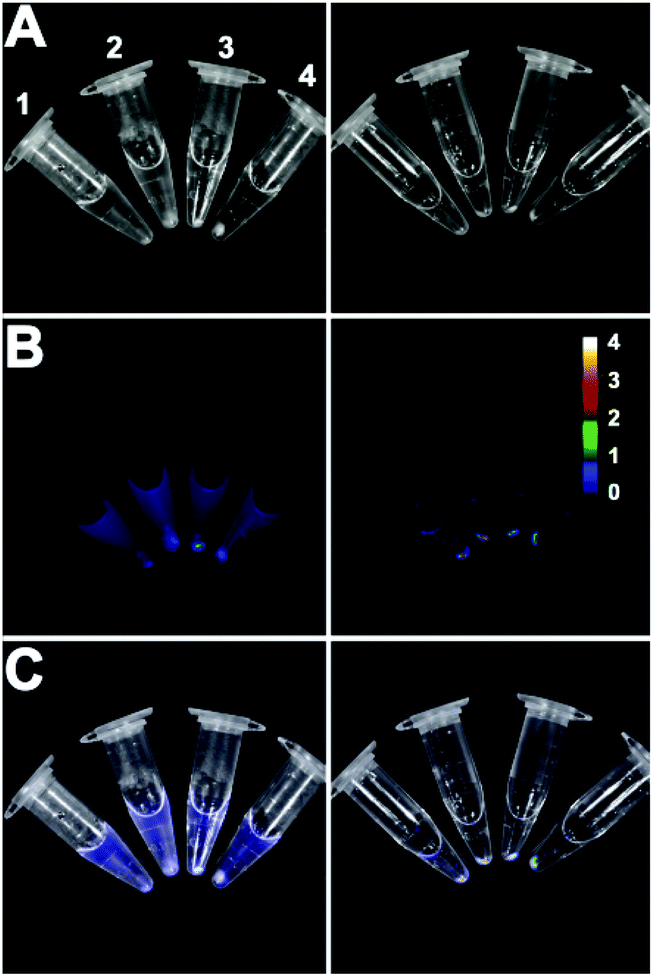 | ||
| Fig. 3 Four strains of pelleted bacteria after treatment with fluorescent untargeted liposomes (left column) or fluorescent Zn2BDPA coated liposomes (right column) and imaged using a CCD camera. (A) Brightfield; (B) near-infrared fluorescence; (C) merge. 1 = E. coli, 2 = P. aeruginosa, 3 = S. aureus and 4 = K. pneumoniae. Scale bar indicates near-infrared emission intensity for both fluorescence images in row B. | ||
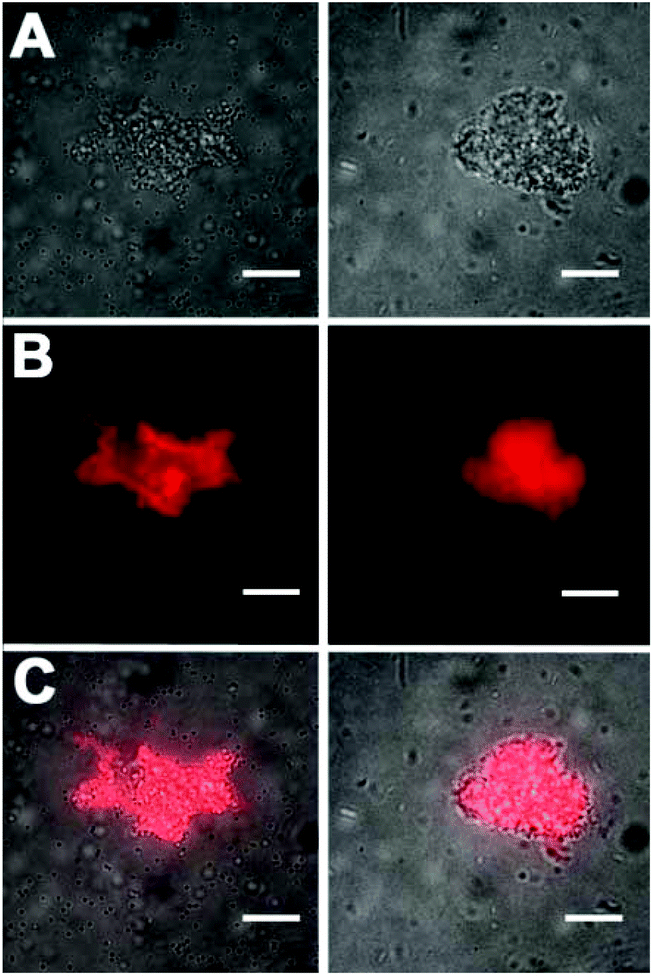 | ||
| Fig. 4 Representative fluorescence microscopy images of bacteria after treatment with fluorescent Zn2BDPA coated liposomes. (left column) Gram-positive S. Aureus bacteria, (right column) Gram-negative E. coli. (Top row) Brightfield image of cross-linked bacteria/liposome aggregate; (Middle row) Near-infrared fluorescence emission from Zn2BDPA liposomes bound to the bacteria; (Bottom row) Overlay of brightfield and fluorescence images. No fluorescence staining of bacterial cells was observed with fluorescent untargeted liposomes. Scale bar = 30 μm. | ||
Additional cell microscopy experiments demonstrated the selectivity of the Zn2BDPA coated liposomes for bacterial cells over healthy mammalian cells. Mixtures of near-infrared fluorescent liposomes (coated with Zn2BDPA or untargeted), MDA-MB-211 human breast cancer cells and GFP-expressing P. aeruginosa bacteria were incubated for 15 min. As shown in Fig. 5, only the mixtures with Zn2BDPA coated liposomes resulted in bacterial agglutination. Fig. 5C and 5D show strong co-localization of the near-infrared Zn2BDPA coated liposomes and the green bacterial fluorescence, and no association of the Zn2BDPA coated liposomes with the mammalian cell surfaces. The untargeted liposomes did not interact with the bacteria which remained widely dispersed across the entire micrograph. These results are consistent with our previous observations using multivalent molecular probes with four or more covalently attached Zn2BDPA targeting units, and the independent work of others who have reported that magnetic Zn2BDPA nanoparticles can be used to separate bacterial cells from mammalian blood cells.48
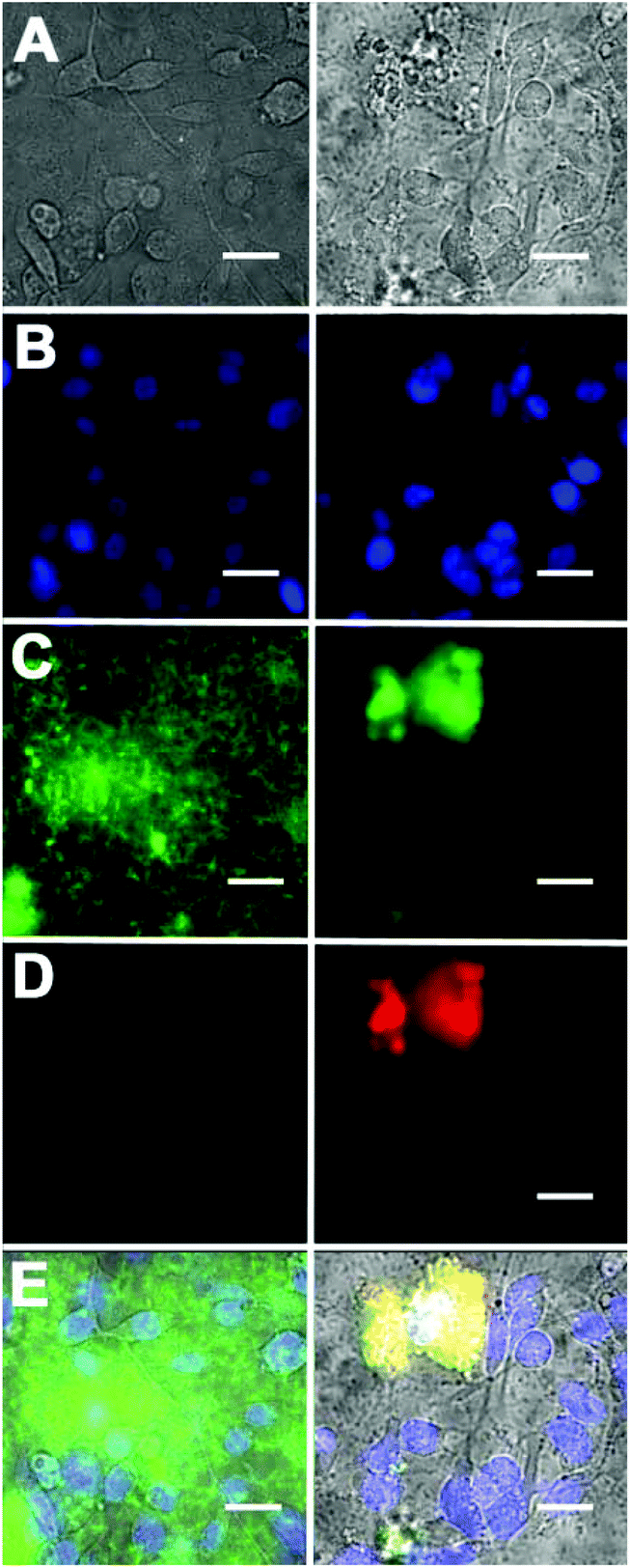 | ||
| Fig. 5 Fluorescence micrographs of a mixture of DAPI stained MDA-MB-211 human breast cancer cells, GFP-expressing P. aeruginosa, and either near-infrared fluorescent untargeted liposomes (left column) or near-infrared fluorescent Zn2BDPA coated liposomes (right column). (row A) Brightfield images; (row B) Human cell nuclei stained with blue-emitting DAPI; (row C) green-emitting P. aeruginosa; (row D) near-infrared fluorescence from Zn2BDPA coated liposomes; (row E) Overlay of A, B, C and D. Scale bar = 30 μm. | ||
Mammalian cell death targeting using Zn2BDPA coated liposomes
Standard MTT cell vitality assays showed that the Zn2BDPA coated liposomes were not toxic to MDA-MB-231 human breast cancer cells (Fig. ESI-7†). Furthermore, fluorescence microscopy studies showed that fluorescently labeled Zn2BDPA coated liposomes did not stain healthy MDA-MB-231 cells, but they did stain dead and dying cells that had been treated with a cytotoxic agent. Specifically, MDA-MB-211 cells were incubated with etoposide (10 μM) followed by fluorescent liposomes and PSVue480, a commercially available Zn2BDPA-fluorescein molecular conjugate that has been validated as a green-emitting fluorescent probe that targets exposed PS on the exterior of dead and dying cells. As shown in Fig. 6, the near-infrared Zn2BDPA coated liposomes co-localized with PSVue480. Untargeted liposomes had no affinity for the dead and dying cell population.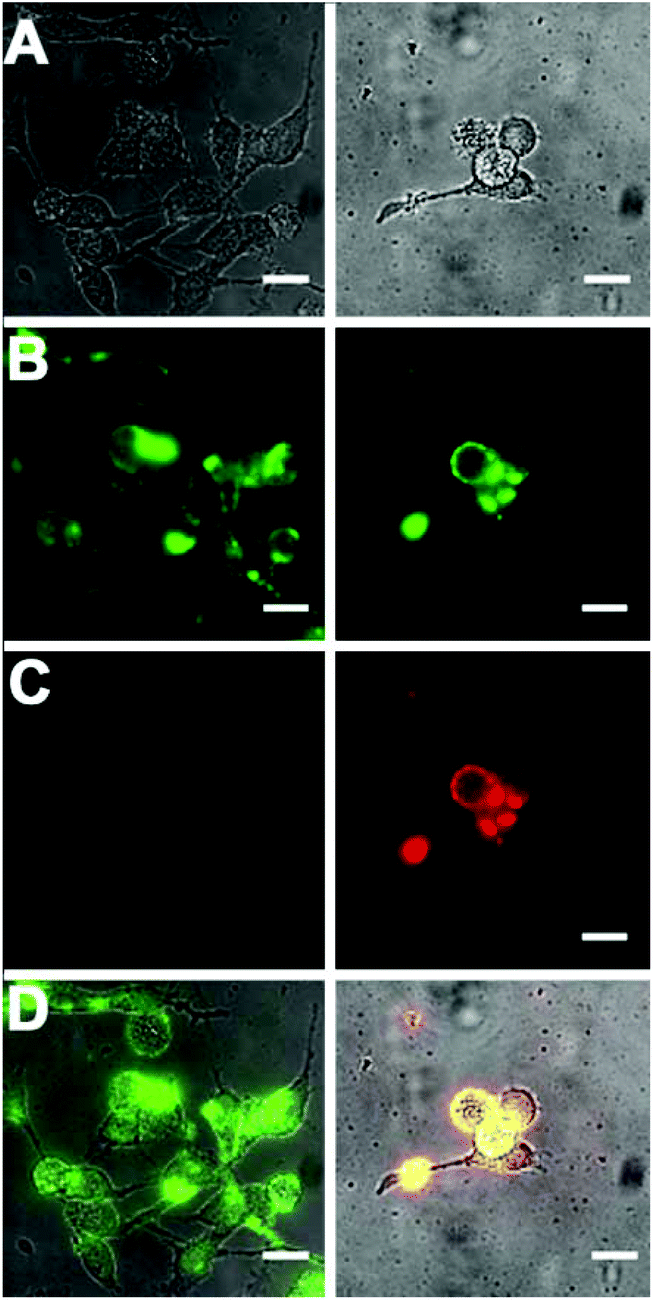 | ||
| Fig. 6 Micrographs of human MDA-MB-211 cells treated with cytotoxic etoposide (10 μM) for 16 h and stained with green-emitting PSVue480 (10 μM), and either near-infrared fluorescent untargeted (left column) or Zn2BDPA coated liposomes (right column). (row A) Brightfield; (row B) Green-emission from cells stained with PSVue480; (row C) Near-infrared emission from cells stained with liposomes; (row D) Overlay of A, B, and C. Scale bar = 30 μm. | ||
Conclusion
The two amphiphilic conjugates, Zn2BDPA-PEG2000-DSPE and Zn2BDPA-PEG500-DSPE (Fig. 1C), were prepared using reliable synthetic methods and incorporated into liposomes at low molar fractions. Liposomes incorporating the shorter conjugate exhibited self-aggregation, whereas Zn2BDPA coated liposomes incorporating the longer conjugate remained highly dispersed in aqueous solution. The Zn2BDPA coated liposomes formed cross-linked precipitates with target liposomes containing anionic phospholipids, and did not interact with the liposomes composed of uncharged phospholipids that were mimics of the surfaces of healthy mammalian cells. The Zn2BDPA coated liposomes selectively agglutinated bacterial cells in the presence of healthy human cells. The selective agglutination effect is most likely universal for both Gram-positive and Gram-negative bacteria since it targets the anionic amphiphiles in the bacterial envelope,49 and is complementary to the highly specific cell recognition exhibited by antibodies which typically target a specific antigenic protein. The Zn2BDPA affinity unit is not bactericidal but multivalent Zn2BDPA coated liposomes may have value as immobilization agents that sequester an infection within an organism. Alternatively Zn2BDPA coated liposomes may have potential as antibiotic delivery vehicles.50,51 The selectivity for anionic cell surfaces was further demonstrated by selective staining of dead or dying mammalian cells in the presence of healthy cells, an experimental outcome that is consistent with the targeted liposome results of others.52–54 While Zn2BDPA coated liposomes could simply be employed as dead cell stains for in vitro assays, we envision a more ambitious in vivo application as a cell death “theranostic” platform that combines therapeutic and diagnostic capabilities within a single nanoparticle. Moreover, a liposomal system with both cell death targeting and cytotoxic delivery capability has the potential of producing amplified drug delivery to the dead and dying cells within cancerous tissue.32,33Experimental
Materials
FmocNH-PEG500-propionic acid (8) was obtained from Polypure (Oslo, Norway). H2N-PEG2000-DSPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), 1,2-dipalmitoyl-sn-glycero-3-phosphatidylglycerol (DPPG), were obtained from Avanti Polar Lipids (Alabaster, Alabama, USA). The following fluorescent molecular probes were purchased and used as supplied PSVue480 (Molecular Targeting Technologies Inc.), DAPI and DiR (Life Technologies).Synthesis
Proton (1H-NMR) and carbon (13C-NMR) nuclear resonance spectra were recorded on Varian UnityPlus 300 (300 MHz for 1H, 75 MHz for 13C), Varian INOVA 500 (500 MHz for 1H, 125 MHz for 13C), and Bruker Ascend 500 III HD (500 MHz for 1H, 125 MHz for 13C) NMR spectrometers. High resolution mass spectra were recorded on a Bruker micrOTOF II mass spectrometer using electrospray ionization (ESI). Analytical TLC was performed on EMD aluminum-backed 250 μm silica gel 60 F254 and on EMD aluminum-backed 250 μm neutral aluminum oxide 60 F254 plates. Analytical TLC visualization was done under UV light (254 nm) or using conventional staining methods (I2, CAM, ninhydrin, sulfuric acid charring, and so on). Preparatory scale TLC (P-TLC) was done using SiliCycle glass-backed 1000 and 2000 μm silica gel 60 F254 plates. Flash column chromatography (FCC) was done by using either SiliCycle SiliaFlash P60 silica gel (40–63 μm, mesh 230–400) or Aldrich neutral aluminum oxide (Brockman I, 150 mesh, 58 Å). Anhydrous solvents for reactions were procured from commercial sources except for THF (purified using an Innovative Technologies SPS-100-2 solvent purification system) and CHCl3 (distilled from P2O5).:1 EtOAc–Hex) to give 241 mg (97% yield) of product as a pale yellow oil. Rf = 0.48 (SiO2, 1:1 EtOAc–Hex). 1H NMR (300 MHz, CDCl3) δ 1.27 (t, J = 7.18 Hz, 3H), 2.12 (quin, J = 6.70 Hz, 2H), 2.53 (t, J = 7.29 Hz, 2H), 4.03 (t, J = 6.10 Hz, 2H), 4.16 (q, J = 7.18 Hz, 2H), 4.43 (s, 4H), 6.86 (d, J = 1.20 Hz, 2H), 7.00 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 14.50, 24.77, 30.92, 33.13, 60.75, 67.13, 115.39, 122.10, 139.84, 159.46, 173.37. HRMS (ESI+) calculated for C14H18Br2O3 ([M + H]+) 392.9695; found 392.9718.
:2) and 1.05 g of product (96% yield) was obtained as a dark orange/brown oil. Rf = 0.77 (Al2O3, CHCl3–MeOH 98:2). 1H NMR (300 MHz, CDCl3) δ 1.26 (t, J = 7.18 Hz, 3H), 2.11 (quin., J = 6.70 Hz, 2H) 2.53 (t, J = 6 Hz, 2H), 3.68 (s, 4H), 3.84 (s, 8H), 4.01 (t, J = 6.10 Hz, 2H), 4.15 (q, J = 7.18 Hz, 2H), 6.88 (s, 2H), 7.08 (s, 1H), 7.15 (ddd, J1 = 6.76 Hz, J2 = 4.96 Hz, J3 = 1.91 Hz, 4H), 7.57–7.68 (m, 8H), 8.52 (d, J = 4.78 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 14.47, 24.92, 31.10, 58.75, 60.22, 60.65, 66.89, 113.75, 121.78, 122.23, 123.00, 136.69, 140.70, 149.17, 159.24, 159.78, 179.49. HRMS (ESI+) calculated for C32H42N6O3 ([M + H]+) 631.3391, found 631.3423.
:1:1) was added 60 μl (0.34 mmol) 20% (w/w) NaOH (aq.). The reaction was refluxed for 2 h, at which time TLC (Al2O3, CHCl3–MeOH 98:2) indicated complete conversion of the starting material. The pH of the reaction was adjusted to ∼7 by addition of ∼300 μl 1 M HCl. The solvents were removed under reduced pressure and the remaining solids were washed 3 times with 10 mL CHCl3. The combined organic layers were dried with MgSO4. Filtration and removal of solvent afforded ∼50 mg (∼99% yield) of the desired compound (BDPA-acid) as a pale yellow oil. Rf = 0.46 (Al2O3, CHCl3–MeOH–H2O 65:30:5); 0.27 (SiO2, CHCl3–MeOH–NH4OH 8:2:0.2). 1H NMR (500 MHz, CD3OD) δ 2.05 (quin., 2H, J = 6.72 Hz), 2.47 (t, 2H, J = 7.21 Hz), 3.63 (s, 4H), 3.77 (s, 8H), 4.00 (t, J = 6.36 Hz, 2H), 6.83 (s, 2H), 6.99 (s, 1H), 7.21–7.28 (m, 4H), 7.64 (d, J = 7.83 Hz, 4H), 7.76 (td, J = 7.70 Hz, J = 1.71 Hz, 4H), 8.31–8.46 (m, 4H). 13C NMR (125 MHz, CD3OD) δ 24.80, 30.56, 58.69, 59.70, 66.94, 114.01, 121.77, 122.61, 123.54, 137.49, 140.16, 148.22, 159.22, 159.37, 176.07. HRMS (ESI+) calculated for C36H39N6O3 ([M + H]+) 603.3078; found 603.3067.
:2:0.2 CHCl3–MeOH–NH4OH as the eluent followed by a second P-TLC run on SiO2 using 8:1:0.1 CHCl3–MeOH–NH4OH as the eluent to give 24 mg (∼65% yield) of product as a yellow/orange solid film. Rf = 0.25 (SiO2, 8:1:0.1 CHCl3–MeOH–NH4OH). 1H NMR (300 MHz, CDCl3, CD3OD) δ 0.84 (t, J = 6 Hz, 6H), 1.21 (m, ∼65H), 1.54 (m, 4H), 2.07 (m, 2H), 2.24 (td, J1 = 7.53 Hz, J2 = 2.39 Hz, 4H), 2.36 (m, 2H), 3.38 (m, 4H), 3.44–3.76 (m, ∼216H), 3.85 (m, 10H), 3.95 (m, 6H), 4.07–4.22 (m, 4H), 4.34 (dd, J1 = 12.08 Hz, J2 = 3.23 Hz, 1H), 5.17 (m, 1H), 6.85 (s, 2H), 7.03 (s, 1H), 7.13–7.20 (m, 4H), 7.54–7.71 (m, 8H), 8.45 (d, J = 4.54 Hz, 4H). 13C NMR (125 MHz, CDCl3, CD3OD) δ 14.25, 22.84, 25.04, 25.07, 25.50, 29.32, 29.51, 29.81, 29.86, 32.08, 32.87, 34.27, 34.44, 39.32, 42.41, 58.72, 59.69, 62.81, 63.57, 63.68, 64.42, 67.27, 69.89, 70.02, 70.23, 70.38, 70.63, 70.69, 114.18, 121.89, 122.55, 123.34, 137.23, 139.96, 148.77, 158.83, 159.39, 173.32, 173.72. HRMS (ESI+) calculated for C168H298N8O56P− ([(M + 3H)/2]+) 1679.0339, found 1679.5384.
:2:0.2 CHCl3–MeOH–H2O. Enriched fractions were pooled, the solvent was removed under reduced pressure, the residue was dissolved minimal CHCl3, and was applied to a silica preparatory TLC plate which was developed using 8:2:0.2 CHCl3–MeOH–H2O to give 178 mg pure product. To remove ambiguity regarding the counterion 150 mg of product was dissolved in 50 mL CHCl3 and washed 2 times with 50 mL saturated NaCl which was acidified to pH 4. The organic phase was dried using Na2SO4 and removal of solvent in vacuo yielded 146 mg (∼65% yield) of FmocNH-PEG500-DSPE. Rf = 0.46 (SiO2, CHCl3–MeOH–H2O 8:2:0.2). 1H NMR (300 MHz, CDCl3) δ 0.88 (t, 3H), 1.19–1.39 (m, ∼60H), 1.58 (m, 5H), 2.28 (td, J = 7.53, 2.63 Hz, 4H), 2.5 (t, J = 5.50 Hz, 2H), 3.26–3.50 (m, 5H), 3.50–3.82 (m, ∼56H), 3.88 (t, J = 6.22 Hz, 1H), 4.00 (t, J = 5.86 Hz, 4H), 4.08–4.30 (m, 3H), 4.31–4.50 (m, 4H), 5.17–5.27 (m, 1H), 5.53 (m, 1H), 7.28–7.35 (m, 2H), 7.41 (t, J = 7.18 Hz, 2H), 7.61 (d, J = 7.41 Hz, 2H), 7.77 (d, J = 7.41 Hz). 13C NMR (75 MHz, CDCl3) δ 14.36, 15.34, 22.92, 25.08, 29.36, 29.59, 29.94, 32.14, 34.26, 34.41, 35.43, 36.56, 41.13, 42.76, 47.46, 62.41, 66.75, 67.43, 70.05, 70.26, 70.37, 70.75, 120.16, 125.28, 127.25, 127.86, 141.50, 144.19, 172.59, 173.20, 173.57, 173.62. HRMS (ESI+) calculated for C83H146N2O23P− ((M + 2H)+) 1569.0054, found 1568.9970. 146 mg (91.8 μmol) of FmocNH-PEG500-DSPE was dissolved in 5 mL 20% piperidine in DMF. The reaction was allowed to proceed for 2 h at room temperature, after which the solvent was removed in vacuo and the residue was subjected to flash column chromatography (SiO2, CHCl3–MeOH–water 8:2:0.2) 4 times to yield enriched product. This material was purified after two runs on preparatory TLC (SiO2, CHCl3–MeOH–H2O 8:2:0.2). 90 mg (∼73% yield) of product was obtained as a white solid. Rf = 0.26 (SiO2, CHCl3–MeOH–H2O 8:2:0.2). 1H NMR (300 MHz, CDCl3) δ 0.88 (t, 6H), 1.26 (m, ∼58H), 1.59 (m, 4H), 2.29 (td, J = 7.59, 3.95 Hz, 5H), 2.50 (t, J = 6.10 Hz, 2H), 3.19 (m, 2H), 3.38–3.52 (m, 3H), 3.53–3.86 (m, ∼51H), 3.94–4.09 (m, 2H), 4.17 (dd, J = 12.08, 6.58 Hz, 1H), 4.40 (dd, J = 11.96, 3.35, 1H), 5.22 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 13.69, 22.51, 24.74, 24.77, 29.00, 29.18, 29.22, 29.51, 29.55, 31.79, 33.91, 34.04, 36.29, 39.42, 62.18, 64.18, 64.88, 66.64, 66.99, 69.52, 69.66, 69.73, 69.83, 69.90, 69.97, 70.09, 70.17, 70.23, 173.35, 173.75. HRMS (ESI+) calculated for C68H134N2O21P− ((M + 2H)+) 1347.9368, found 1347.9354; MS (ESI+) calculated for C68H134N2O21P− ((M + H + Na)+) 1369.9187, found 1369.9177.
:2:0.2 CHCl3–MeOH–NH4OH as the eluent followed by a second P-TLC run on SiO2 using 8:1:0.1 CHCl3–MeOH–NH4OH as the eluent to give 100 mg (∼86% yield) of product as an orange/brown solid film. Rf = 0.50 (SiO2, 8:2:0.2 CHCl3–MeOH–NH4OH). 1H NMR (300 MHz, CDCl3) δ 0.87 (t, J = 6 Hz, 6H), 1.25 (m, 60H), 1.57 (m, 4H), 2.11 (m, 2H), 2.27 (m, 4H), 2.41 (t, J = 6 Hz, 2H), 2.48 (t, J = 6 Hz, 2H), 3.45 (m, 4H), 3.50–3.79 (m, 59H), 3.86 (s, 8H), 3.99 (m, 6H), 4.17 (dd, J1 = 11.96 Hz, J2 = 6.70 Hz, 1H), 4.39 (dd, J1 = 12.20 Hz, J2 = 3.11 Hz, 1H), 5.23 (m, 1H), 6.88 (s, 2H), 7.06 (s, 1H), 7.17 (m, 4H), 7.58–7.71 (m, 8H), 8.53 (d, J = 5.02 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 14.10, 22.66, 24.85, 24.89, 25.28, 29.11, 29.30, 29.33, 29.63, 29.68, 31.89, 32.76, 34.07, 34.25, 36.74, 39.15, 58.48, 59.50, 62.65, 66.98, 67.45, 69.84, 70.07, 70.10, 70.43, 113.72, 122.11, 122.95, 136.11, 148.72, 159.07, 171.36, 172.48, 172.99, 173.37. HRMS (ESI−) calculated for C104H170N8O23P− (M−) 1931.2155, found 1931.2042.
Liposome studies
:30:67.5) and 250 μL of target liposomes with various compositions. The results were photographed using a digital camera.
:2:30:66) or untargeted liposomes (DiR–cholesterol–POPC, 2:30:68). The treated cells were pelleted by microcentrifugation at 5000 rpm for 5 min and the tubes were imaged using a CCD camera within a Xenogen imaging station. A region of interest analysis of the fluorescence images indicated that the pellets contained 98% and 6% of the Zn2BDPA coated liposomes and untargeted liposomes, respectively. The bacterial cells were washed twice with buffer to reduce background fluorescence, dispersed into solution then onto slides, and micrographs were acquired using a Nikon Eclipse TE2000-U epifluorescence microscope with a 60× objective and a Photometrics Cascade 512B CCD. Near infrared fluorescence images were captured using a Cy7 filter set (Exciter HQ710/75x, Dichroic Q750LP, Emitter HQ810/90m).
Fluorescence microscopy was used to demonstrate the selectivity of the Zn2BDPA coated liposomes for bacterial cells over healthy mammalian cells. The nuclei of confluent MDA-MB-231 human breast cancer cells were stained by incubating with DAPI (1 μg mL−1 DAPI in PBS) for 30 minutes. Separate samples of the DAPI stained MDA-MB-231 cells were washed with HEPES buffer then treated with near-infrared fluorescent Zn2BDPA coated liposomes or untargeted liposomes in HEPES buffer. An aliquot of GFP-expressing P. aeruginosa (20 μL from a broth grown to an OD600 = 0.2) was added to the samples, which were incubated for 15 minutes then imaged using fluorescence microscopy. Fluorescence images were captured using UV (ex: 340/80, em: 435/85), GFP (ex: 450/90, em: 500/50), or Cy7 filter sets.
Mammalian cell toxicity
Toxicity of the liposomal formulations was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) cell vitality assay. MDA-MB-231 human breast cancer cells (ATCC) were seeded into 96-microwell plates, and grown to a confluency of 85% in RPMI media with 10% fetal bovine serum, and 1% streptavidin L-glutamate at 37 °C and 5% CO2. The Vybrant MTT Cell Proliferation Assay Kit (Invitrogen, Eugene, USA) was performed according to the manufacturer's protocol and validated using 10 μM etoposide as a positive control for high toxicity. The cells were treated with untargeted liposomes (cholesterol–POPC, 30:70) and Zn2BDPA coated liposomes (Zn2BDPA-PEG2000-DSPE–cholesterol–POPC, 2:30:68) and incubated for 18 h at 37 °C. The medium was removed and replaced with 100 μL of RPMI media containing MTT (1.2 mM). An SDS detergent solution was added and incubated at 37 °C and 5% CO2 for an additional 4 hours. The absorbance of each well was read at 570 nm and the normalized data (measured in triplicate) are shown in Fig. ESI-7.† The results indicate that the untargeted and Zn2BDPA coated liposomes induce negligible amounts of cell death at concentrations <10 μM and <25 μM respectively.
Mammalian cell death imaging using fluorescent liposomes
Confluent MDA-MB-211 cells were treated with etoposide (10 μM) for 16 hours to induce apoptotic cell death. Separate populations of apoptotic MDA cells were treated with near-infrared fluorescent Zn2BDPA coated liposomes or untargeted liposomes in HEPES buffer and allowed to incubate for 15 minutes. The cells were then washed three times followed by treatment with green-emitting cell death probe PSVue480 (10 μM) for 15 minutes and three subsequent wash steps. Fluorescence micrographs were captured using the microscope described above and a GFP or Cy7 filter set.Acknowledgements
This study was supported by the NIH (RO1GM059078 to B.D.S. and T32GM075762 to D.R.R.), Walther Cancer Research Foundation, the University of Notre Dame, the Notre Dame Integrated Imaging Facility, and the Freimann Life Sciences Center. S.T. gratefully acknowledges support from the Scientific and Technological Research Council of Turkey (Grant number 114C041).Notes and references
- T. Sakamoto, A. Ojida and I. Hamachi, Chem. Commun., 2009, 141–152 RSC.
- M. Kruppa and B. Konig, Chem. Rev., 2006, 106, 3520–3560 CrossRef CAS PubMed.
- E. J. O'Neil and B. D. Smith, Coord. Chem. Rev., 2006, 250, 3068–3080 CrossRef PubMed.
- S. K. Kim, D. H. Lee, J.-I. Hong and J. Yoon, Acc. Chem. Res., 2008, 42, 23–31 CrossRef PubMed.
- Y. Zhou, Z. Xu and J. Yoon, Chem. Soc. Rev., 2011, 40, 2222–2235 RSC.
- H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC.
- M. S. Han and D. H. Kim, Angew. Chem., Int. Ed., 2002, 41, 3809–3811 CrossRef CAS.
- D. H. Lee, J. H. Im, S. U. Son, Y. K. Chung and J. I. Hong, J. Am. Chem. Soc., 2003, 125, 7752–7753 CrossRef CAS PubMed.
- D. H. Lee, S. Y. Kim and J. I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777–4780 CrossRef CAS PubMed.
- J. H. Lee, J. Park, M. S. Lah, A. Chin and J. I. Hong, Org. Lett., 2007, 9, 3729–3731 CrossRef CAS PubMed.
- S. W. Bae, M. S. Cho, A. R. Jeong, B. R. Choi, D. E. Kim, W. S. Yeo and J. I. Hong, Small, 2010, 6, 1499–1503 CrossRef CAS PubMed.
- D. J. Oh, K. M. Kim and K. H. Ahn, Chem. – Asian J., 2011, 6, 2033–2038 CrossRef PubMed.
- G. Liu, K. Y. Choi, A. Bhirde, M. Swierczewska, J. Yin, S. W. Lee, J. H. Park, J. I. Hong, J. Xie, G. Niu, D. O. Kiesewetter, S. Lee and X. Chen, Angew. Chem., Int. Ed., 2012, 51, 445–449 CrossRef CAS PubMed.
- K. Honda, S. H. Fujishima, A. Ojida and I. Hamachi, ChemBioChem, 2007, 8, 1370–1372 CrossRef CAS PubMed.
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452–10459 CrossRef CAS PubMed.
- K. Honda, E. Nakata, A. Ojida and I. Hamachi, Chem. Commun., 2006, 4024–4026 RSC.
- J. A. Drewry, S. Burger, A. Mazouchi, E. Duodu, P. Ayers, C. C. Gradinaru and P. T. Gunning, MedChemComm, 2012, 3, 763–770 RSC.
- J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192–200 CrossRef CAS PubMed.
- V. Ganesh, K. Bodewits, S. J. Bartholdson, D. Natale, D. J. Campopiano and J. C. Mareque-Rivas, Angew. Chem., Int. Ed., 2009, 48, 356–360 CrossRef CAS PubMed.
- E. Kinoshita, M. Takahashi, H. Takeda, M. Shiro and T. Koike, Dalton Trans., 2004, 1189–1193, 10.1039/b400269e.
- L. Ciavatta, J. C. Mareque, D. Natale and F. Salvatore, Ann. Chim., 2006, 96, 317–325 CrossRef CAS.
- A. V. Koulov, K. A. Stucker, C. Lakshmi, J. P. Robinson and B. D. Smith, Cell Death Differ., 2003, 10, 1357–1359 CrossRef CAS PubMed.
- A. V. Koulov, R. G. Hanshaw, K. A. Stucker, C. Lakshmi and B. D. Smith, Isr. J. Chem., 2005, 45, 373–379 CrossRef CAS.
- R. G. Hanshaw, C. Lakshmi, T. N. Lambert, J. R. Johnson and B. D. Smith, ChemBioChem, 2005, 6, 2214–2220 CrossRef CAS PubMed.
- W. M. Leevy, S. T. Gammon, J. R. Johnson, A. J. Lampkins, H. Jiang, M. Marquez, D. Piwnica-Worms, M. A. Suckow and B. D. Smith, Bioconjugate Chem., 2008, 19, 686–692 CrossRef CAS PubMed.
- B. A. Smith, W. J. Akers, W. M. Leevy, A. J. Lampkins, S. Xiao, W. Wolter, M. A. Suckow, S. Achilefu and B. D. Smith, J. Am. Chem. Soc., 2010, 132, 67–69 CrossRef CAS PubMed.
- B. A. Smith, S. Xiao, W. Wolter, J. Wheeler, M. A. Suckow and B. D. Smith, Apoptosis, 2011, 16, 722–731 CrossRef PubMed.
- B. A. Smith, B. W. Xie, E. R. van Beek, I. Que, V. Blankevoort, S. Xiao, E. L. Cole, M. Hoehn, E. L. Kaijzel, C. W. Lowik and B. D. Smith, ACS Chem. Neurosci., 2012, 3, 530–537 CrossRef CAS PubMed.
- S. Xiao, S. Turkyilmaz and B. D. Smith, Tetrahedron Lett., 2013, 54, 861–864 CrossRef CAS PubMed.
- S. Xiao, L. Abu-Esba, S. Turkyilmaz, A. G. White and B. D. Smith, Theranostics, 2013, 3, 658–666 CrossRef CAS PubMed.
- B. A. Smith, K. M. Harmatys, S. Xiao, E. L. Cole, A. J. Plaunt, W. Wolter, M. A. Suckow and B. D. Smith, Mol. Pharmaceutics, 2013, 10, 3296–3303 CrossRef CAS PubMed.
- X. He, N. Bonaparte, S. Kim, B. Acharya, J. Y. Lee, L. Chi, H. J. Lee, Y. K. Paik, P. G. Moon, M. C. Baek, E. K. Lee, J. H. Kim, I. S. Kim and B. H. Lee, J. Controlled Release, 2012, 162, 521–528 CrossRef CAS PubMed.
- K. Wang, M. H. Na, A. S. Hoffman, G. Shim, S. E. Han, Y. K. Oh, I. C. Kwon, I. S. Kim and B. H. Lee, J. Controlled Release, 2011, 154, 214–217 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- E. A. Azzopardi, E. L. Ferguson and D. W. Thomas, J. Antimicrob. Chemother., 2013, 68, 257–274 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- P. Ruenraroengsak, J. M. Cook and A. T. Florence, J. Controlled Release, 2010, 141, 265–276 CrossRef CAS PubMed.
- N. Bertrand and J. C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef CAS PubMed.
- I. K. Kwon, S. C. Lee, B. Han and K. Park, J. Controlled Release, 2012, 164, 108–114 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and T. L. Andresen, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 481–503 CrossRef CAS PubMed.
- C. Lakshmi, R. G. Hanshaw and B. D. Smith, Tetrahedron, 2004, 60, 11307–11315 CrossRef CAS PubMed.
- P. R. Ashton, D. W. Anderson, C. L. Brown, A. N. Shipway, J. F. Stoddart and M. S. Tolley, Chem. – Eur. J., 1998, 4, 781–795 CrossRef CAS.
- R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801–811 CrossRef.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- A. Samad, Y. Sultana and M. Aqil, Curr. Drug Delivery, 2007, 4, 297–305 CrossRef CAS.
- J. J. Lee, K. J. Jeong, M. Hashimoto, A. H. Kwon, A. Rwei, S. A. Shankarappa, J. H. Tsui and D. S. Kohane, Nano Lett., 2014, 14, 1–5 CrossRef CAS PubMed.
- C. Ratledge and S. G. Wilkinson, Microbial Lipids, Academic Press, London, 1988 Search PubMed.
- C. M. Huang, C. H. Chen, D. Pornpattananangkul, L. Zhang, M. Chan, M. F. Hsieh and L. F. Zhang, Biomaterials, 2011, 32, 214–221 CrossRef CAS PubMed.
- K. W. Yang, B. Gitter, R. Ruger, G. D. Wieland, M. Chen, X. L. Liu, V. Albrecht and A. Fahr, Photochem. Photobiol. Sci., 2011, 10, 1593–1601 CAS.
- Y. S. Cho, K. M. Kim, D. Lee, W. J. Kim and K. H. Ahn, Chem. – Asian. J., 2013, 8, 755–759 CrossRef CAS PubMed.
- B. Garnier, A. Bouter, C. Gounou, K. G. Petry and A. R. Brisson, Bioconjugate Chem., 2009, 20, 2114–2122 CrossRef CAS PubMed.
- For a general review regarding liposomes coated with metal complexes, including those with targeting capability, see: B. Gruber and B. Konig, Chem. – Eur. J., 2013, 19, 438–448 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, liposome characterization and cell toxicity data. See DOI: 10.1039/c4ob00924j |
| This journal is © The Royal Society of Chemistry 2014 |