Direct functionalization of benzylic C–Hs with vinyl acetates via Fe-catalysis†‡
Chun-Xiao
Song
a,
Gui-Xin
Cai
a,
Thomas R.
Farrell
b,
Zhong-Ping
Jiang
a,
Hu
Li
a,
Liang-Bing
Gan
*a and
Zhang-Jie
Shi
*ac
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Peking University, Beijing, China. E-mail: zshi@pku.edu.cn; Fax: +86 010-62760890; Tel: +86 010-62760890
bDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA
cState Key Laboratory of Organometallic Chemistry Chinese Academy of Sciences, Shanghai 200032, China
First published on 7th September 2009
Abstract
Direct cross-coupling to construct sp3 C–sp3C bonds viaFe-catalyzed benzylic C–H activation with 1-aryl vinyl acetate was developed.
Transition metal-catalyzed cross-coupling reactions, such as the Heck reaction, have been well developed and become some of the most powerful methods to construct C–C bonds.1 Other developments have included coupling aliphatic halides, instead of aryl/alkenyl halides, with the functionalized olefins.2 Recent advances in approaching this goal have indicated that aryl C–H bonds could be applied to C–C bond formation by coupling with functionalized alkenes via Pd(II) catalysis under oxidative conditions, avoiding the use of aryl halides.3 Undoubtedly, direct olefination of sp3 C–H bonds is more difficult due to their intrinsic properties. To the best of our knowledge, only two examples have been reported so far. Sames et al. first reported intramolecular C–C formation of the N-adjacent sp3 C–H bond of pyrroline, followed by isomerization with a tethered olefin, catalyzed by Ir complexes.4 The other example, reported by Liet al., showed direct olefination of sp3 C–H bonds, which was activated by both an adjacent N atom and an aryl group, presumably through an oxidation–Baylis–Hillman sequence.5
Compared to late transition metal-catalyzed processes, early transition metal-catalyzed transformations have unique features due to their cost effectiveness and different mechanistic insights.6 Among all the first row transition metals, iron draws much attention due to its high abundance, low price and low toxicity.7 Recently, Fe catalysis has been widely expanded,8 and in particular, iron catalysts have led to great advances in the direct oxidation8n,9 of C–H bonds, as well as in C–C bond formation.10 Herein, our developments offer a novel and useful method to construct C–C bonds viairon-catalyzed direct benzylic C–H transformations with functionalized olefins.
We originally planned to explore direct olefination through a Heck-type process via sp3 C–H activation (Scheme 1). The initial efforts were made to search for the solution of this design from diphenyl methane 1a, which showed good activity in various transformations.11 Different substituted olefins were tested in the presence of various transition metal catalysts and oxidants. Under different conditions, acrylate derivatives, which showed high reactivity in traditional Heck reactions,1 were not efficient. Electron-rich olefins, such as 1-hexene,12n-butyl vinyl ether13 and 3,4-dihydropyran,14 also failed.
 | ||
| Scheme 1 Original design of the cross-coupling via sp3 C–H activation with alkenes. | ||
To our delight, when styrene was used in the presence of FeCl2 as a catalyst and di-tert-butyl peroxide (DTBP) as an oxidant, the coupling product was observed by GC-MS and confirmed by crude 1H NMR, accompanied by other undetermined byproducts (Scheme 2). Unfortunately, the efficiency could not be significantly improved, although various reaction parameters were screened. To our surprise, when other substituted styrenes were surveyed, the reaction was highly inhibited. Although this reaction demonstrated the first example of direct olefination of sp3 C–H bonds as a Heck-type transformation, the low efficiency and limited substrate scope hardly made this transformation applicable.
 | ||
| Scheme 2 Direct olefination of benzylic C–H bonds with styrene. | ||
Further tests opened a new channel to approach our original design. Interestingly, when 1-phenyl vinyl acetate 2a was submitted to the aforementioned conditions, a new product 3aa was observed. To optimize this transformation, different conditions were screened (ESI, Table S1‡) and we found that in the absence of either FeCl2 or DTPE, the reaction was terminated. The results also indicated that other common metal salts, such as Co, Cu, Mn and Pd, were not suitable for this transformation. Due to its availability, low price, catalytic efficiency and tolerance of environments, FeCl2 was determined to be the most effective catalyst. DTPE was found as the best oxidant and the desired product was isolated in 74% yield (ESI, Table S1, entry 7‡). Other peroxides, such as TBHP and tBuOOCOPh could also promote the reaction, but with lower efficiencies. Notably, stable poly-tert-butylperoxylated fullerene was first observed to show partial reactivity.15
Different benzylic substrates were explored (Table 1), and we found that various derivatives of diphenylmethane were suitable. Electronic effects seemingly played an important role. Electron-withdrawing groups partially promoted the yields (entries 2–4), but in contrast, electron-donating groups decreased the efficiency. For example, the presence of a methoxyl group on the phenyl ring decreased the yield obviously (entry 6). Steric effects in ortho-substituted substrate 1g also dramatically lowered the yield (entry 7). Aside from substituted phenyls, naphthyl and seven-membered cyclic diphenyl methane derivatives were suitable substrates, and the desired products were isolated in good yields (entries 5, 8 and 9). The reaction could be extended to the isochroman and the desired product 3ja was obtained in 59% yield, with functionalization at the highly reactive benzylic position adjacent to the O-atom (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Ar1 | R | 1 | Yield (%) |
| a The reaction was carried out in 4.0 mmol of 1 and 0.5 mmol of 2a, in the presence of 0.6 mmol of DTBP and 0.05 mmol of FeCl2 under N2 at 100 °C for 24 h and isolated yields were obtained after column chromatography if without further note. | ||||
| 1 | Ar1 = R = Ph | 1a | 3aa (74) | |
| 2 | Ar1 = R = 4-FPh | 1b | 3ba (62) | |
| 3 | 4-FPh | Ph | 1c | 3ca (73) |
| 4 | 4-CIPh | Ph | 1d | 3da (63) |
| 5 | 4-PhPh | Ph | 1e | 3ea (77) |
| 6 | 4-MeOPh | Ph | 1f | 3fa (45) |
| 7 | 2-MeOPh | Ph | 1g | 3ga (13) |
| 8 | 2-Nap | Ph | 1h | 3ha (69) |
| 9 |

|
1i | 3ia (77) | |
| 10 |

|
1j | 3ja (59) | |

Further exploration extended the substrate scope to the benzylic C–H of substituted toluene. When toluene 1k was submitted, the desired product was obtained in a moderate yield (eqn (1), Scheme 3). The benzylic methyl group of 1l showed excellent reactivity, which may arise from stabilization of the proposed radical intermediate (eqn (2), Scheme 3).16
 | ||
| Scheme 3 Fe-catalyzed direct olefination of toluene 1k and derivatives 1l with 1-phenyl vinyl acetate 2a. | ||
Different substituted 1-aryl vinyl acetates were also explored (Table 2). We found that steric effects affected the efficiency: ortho-tolyl substitution lowered the yield to 52% from 65% and 68% with para- and meta-tolyl substitutions, respectively (cf. entry 4 with entries 2 and 3). Electronic effects similarly favored the electron withdrawing groups (cf. entry 5 with entries 6 and 7). It is noteworthy that C–X (X = Br or F) and ester groups tolerated the conditions well, and these groups could be transformed into different functionalities (3af, 3ag and 3ah, entries 6–8). Unfortunately, 2-propenyl acetate is not suitable and only a small amount of desired product was observed by GC-MS.
|
|
|||
|---|---|---|---|
| Entry | R | 3 | Yield (%) |
| a The reaction was carried out in the 4.0 mmol of 1 and 0.5 mmol of 2 in the presence of 0.6 mmol of DTBP and 0.05 mmol of FeCl2 under N2 at 80 °C for 24 h and isolated yields were obtained after column chromatography if without further note. | |||
| 1 |

|

|
3aa (74) |
| 2 |
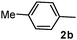
|

|
3ab (65) |
| 3 |

|

|
3ac (68) |
| 4 |
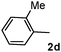
|
|
3ad (52) |
| 5 |

|

|
3ae (52) |
| 6 |

|
|
3af (69) |
| 7 |

|

|
3ag (70) |
| 8 |

|

|
3ah (59) |

Considering the safety problem arising from the possible explosion of peroxide in the presence of transition metal species, we further experimentally modified this transformation (Scheme 4). When both vinyl acetate 2a and DTBP was mixed and added dropwise in a half hour, 3aa was isolated in 61% yield. To our delight, a better efficiency was obtained with the addition of only DTBP in 0.5 h. The length of the adding time to 10 h did not affect the yield significantly. Thus, this operation reduced the hazard by the decrease of the DTBP concentration.
 | ||
| Scheme 4 Promotion the safty of Fe-catalyzed alkylation with 1a and 2a by slow addition of DTBP and/or 2a. | ||
The radical process was highly preferred to facilitate their transformations like Fenton’s process.9g,10a,b The chemistry demonstrated here may also go through the radical process as shown in Scheme 5, path A. The occasional observation of tert-butyl ether and dimerization of diphenyl methane support this hypothesis. An intermolecular isotopic competitive study (KH/D = 2.4) indicated that the proton abstraction process may be involved in the rate determining step. Another possibility for the mechanism is the cationic pathway (Scheme 5, path B). In this assumption, a radical species, generated from the first step with DTBP, might be further oxidized to a benzyl cation, which undergoes electrophilic attack to produce the desired product. However, the observed electronic feature of diarylmethane substrates does not seem to support this hypothesis.
 | ||
| Scheme 5 Proposed mechanism on direct C–C formation viaFe-catalyzed benzylic C–H activationvia either radical (path A) or cationic (path B) process. I and II were observed as major byproducts. | ||
In summary, we have described a novel method to construct sp3 C–C bonds viaFe-catalyzed benzylic C–H activation under mild conditions. Functionalized olefins, such as vinyl acetate and styrene, were first applied as a partner to direct coupling with C–H bonds. To the best of our knowledge, this is the first example that shows Heck-type cross coupling through sp3 C–H activationvia a transition metal-catalyzed transformation. Further studies are under way to clearly understand the mechanistic pathway and to apply this chemistry to organic synthesis.
Notes and references
- (a) R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518 CrossRef CAS; (b) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CAS; (c) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (d) R. F. Heck, Palladium Reagents in Organic Synthese, Academic Press, New York, 1985 Search PubMed.
- (a) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514 CrossRef CAS; (b) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068 CrossRef CAS; (c) L. Firmansjah and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 11340 CrossRef CAS.
- (a) Y. Fujiwara, I. Moritani, S. Danno and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166 CrossRef CAS; (b) I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119 CrossRef; (c) Y. Fujiwara, R. Asano, I. Moritani and S. Teranishi, J. Org. Chem., 1976, 41, 1681 CrossRef CAS; (d) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (e) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. De Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS; (f) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS; (g) G. Dyker, Handbook of C–H Transformations. Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005, and references therein Search PubMed.
- B. DeBoef, S. J. Pastine and D. Sames, J. Am. Chem. Soc., 2004, 126, 6556 CrossRef CAS.
- Z. Li, D. S. Bohle and C. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS.
- (a) A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS; (b) A. Fürstner and R. Martin, Chem. Lett., 2005, 34, 624 CrossRef.
- (a) T. Nagano and T. Hayashi, Org. Lett., 2004, 6, 1297 CrossRef CAS; (b) A. Fürstner, A. Leitner, M. Mendez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856 CrossRef; (c) M. Nakamura, K. Matsuo, S. Ito and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 3686 CrossRef CAS; (d) I. Sapountzis, W. Lin, C. C. Kofink, C. Despotopoulou and P. Knochel, Angew. Chem., Int. Ed., 2005, 44, 1654 CrossRef CAS; (e) K. Itami, S. Higashi, M. Mineno and J. Yoshida, Org. Lett., 2005, 7, 1219 CrossRef CAS; (f) L. K. Ottesen, F. Ek and R. Olsson, Org. Lett., 2006, 8, 1771 CrossRef CAS; (g) K. Bica and P. Gaertner, Org. Lett., 2006, 8, 733 CrossRef CAS; (h) J. Kischel, K. Mertins, D. Michalik, A. Zapf and M. Beller, Adv. Synth. Catal., 2007, 349, 865 CrossRef CAS; (i) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788 CrossRef CAS; (j) T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2007, 129, 9844 CrossRef CAS; (k) C. Y. Li, X. B. Wang, X. L. Sun, Y. Tang, J. C. Zheng, Z. H. Xu, Y. G. Zhou and L. X. Dai, J. Am. Chem. Soc., 2007, 129, 1494 CrossRef CAS; (l) A. Fürstner, K. Majima, R. Martin, H. Krause, E. Kattnig, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 1992 CrossRef; (m) A. Guérinot, S. Reymond and J. Cossy, Angew. Chem., Int. Ed., 2007, 46, 6521 CrossRef CAS; (n) A. Correa, O. G. Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (o) A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2007, 46, 8862 CrossRef CAS; (p) O. Bistri, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 586 CrossRef CAS; (q) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef; (r) M. Carril, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 4862 CrossRef CAS.
- (a) P. Stavropoulos, R. Celenligil-Cetin and A. E. Tapper, Acc. Chem. Res., 2001, 34, 745 CrossRef CAS; (b) C. Walling, Acc. Chem. Res., 1998, 31, 155 CrossRef; (c) P. A. MacFaul, D. D. M. Wayner and K. U. Ingold, Acc. Chem. Res., 1998, 31, 159 CrossRef CAS; (d) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939 CrossRef CAS; (e) M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 2740 CrossRef; (f) K. Suzuki, P. D. Oldenburg and L. Que Jr, Angew. Chem., Int. Ed., 2008, 47, 1887 CrossRef CAS; (g) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS; (h) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124 CrossRef CAS.
- (a) Z. Li, L. Cao and C. Li, Angew. Chem., Int. Ed., 2007, 46, 6505 CrossRef CAS; (b) Y. Zhang and C. Li, Eur. J. Org. Chem., 2007, 4654 CrossRef CAS; (c) Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS; (d) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and S.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS.
- (a) B. M. Choudary, A. D. Prasad, V. Bhuma and V. Swapna, J. Org. Chem., 1992, 57, 5841 CrossRef; (b) F. G. Bordwell, J. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229 CrossRef CAS.
- F. G. Bordwell and M. L. Peterson, J. Am. Chem. Soc., 1954, 76, 3952 CrossRef CAS.
- J. Furukawa, A. Onishi and T. Tsuruta, J. Org. Chem., 1958, 23, 672 CrossRef CAS.
- F. M. Menger and C. H. Chu, J. Org. Chem., 1981, 46, 5044 CrossRef CAS.
- L. Gan, S. Huang, X. Zhang, A. Zhang, B. Cheng, H. Cheng, X. Li and G. Shang, J. Am. Chem. Soc., 2002, 124, 13384 CrossRef CAS.
- C. D. Cook, J. Org. Chem., 1953, 18, 261 CrossRef CAS.
Footnotes |
| † This article is part of a ChemComm ‘Catalysis in Organic Synthesis’ web-theme issue showcasing high quality research in organic chemistry. Please see our website (http://www.rsc.org/chemcomm/organicwebtheme2009) to access the other papers in this issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, analytical and spectral data, and NMR spectra of compounds. See DOI: 10.1039/b911031c |
| This journal is © The Royal Society of Chemistry 2009 |