Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines?
Örn
Almarsson
*a and
Michael J.
Zaworotko
*b
aTransForm Pharmaceuticals, Inc., 29 Hartwell Avenue, Lexington, MA 02421, USA. E-mail: almarsson@transformpharma.com; Fax: +781 863 6519; Tel: +781 674 7894
bDepartment of Chemistry, University of South Florida, SCA400, 4202 E. Fowler Avenue, Tampa, FL 33620, USA. E-mail: xtal@usf.edu; Fax: +813 974 3203; Tel: +813 974 4129
First published on 5th August 2004
Abstract
The evolution of crystal engineering into a form of supramolecular synthesis is discussed in the context of problems and opportunities in the pharmaceutical industry. Specifically, it has become clear that a wide array of multiple component pharmaceutical phases, so called pharmaceutical co-crystals, can be rationally designed using crystal engineering, and the strategy affords new intellectual property and enhanced properties for pharmaceutical substances.
Dr Örn Almarsson is senior director of Pharmaceutical Chemistry at TransForm Pharmaceuticals, Inc. Prior to joining TransForm in December of 2000, he was a research fellow in the department of Pharmaceutical Research and Development at Merck Research Laboratories in West Point, Pennsylvania. He received his BS degree in chemistry from the University of Iceland in 1988 and his PhD in physical-organic chemistry from the University of California at Santa Barbara in 1994. His PhD thesis work, performed under Prof. Thomas C. Bruice, is in the field of bio-organic mechanisms of nicotinamide co-factor redox reactions in dehydrogenases and the mechanisms of O–O bond cleavage in peroxidase model systems. At TransForm, Dr Almarsson has focused on development and application of high-throughput crystallization technologies for pharmaceutical compounds. His current work involves optimizing crystal forms for formulation of drug development candidates with the aim of enhancing biopharmaceutical performance and drug delivery properties. |
Dr Mike Zaworotko is Professor and Chair of the Department of Chemistry at the University of South Florida, USF. He was born in Wales in 1956 and received his BSc and PhD degrees from Imperial College (1977) and the University of Alabama (1982), respectively. He joined USF in 1999. Current research interests include the following: crystal engineering, nanotechnology, supramolecular chemistry, X-ray crystallography. Particular emphasis is currently placed upon applications of crystal engineering in the context of nanoscale structures, magnetism, porosity and compositions of active pharmaceutical ingredients. Dr Zaworotko has published over 210 peer reviewed papers and he currently serves on the editorial boards of J. Chemical Crystallography and Crystal Growth & Design. |
1 Introduction
“Benzoic acid and other carboxylic acids have been shown to be associated to double molecules in solution in certain solvents, such as benzene, chloroform, carbon tetrachloride and carbon disulfide...Benzoic acid exists in the monomeric form in solution in acetone, acetic acid, ethyl ether, ethyl alcohol, ethyl acetate and phenol; in these solutions the single molecules are stabilized by hydrogen bond formation with the solvent.” (Linus Pauling in The Nature of the Chemical Bond, 2nd edition, Cornell University Press, 1948.)In terms of intrinsic value, active pharmaceutical ingredients (API's) are among the most valuable materials on the planet. It is therefore surprising that the growing field of crystal engineering1–3 and its ability to produce new and potentially valuable materials has only addressed API's within the last two years.4–9 Pharmaceuticals are generally comprised of an API, a formulation containing inactive ingredients as a carrier system, and a package for market performance and appeal. The vast majority of API's occur as solids. Crystalline API's are strongly preferred due to their relative ease of isolation, the rejection of impurities inherent to the crystallization process and the physico-chemical stability that the crystalline solid state affords. The problems that arise with the use of crystalline material are usually related to poor solubility properties and the existence of more than one crystalline form of an API. In terms of regulatory approval crystalline forms of an API have traditionally been limited to polymorphs, salts and stoichiometric solvates (pseudopolymorphs).10 However, crystal engineering affords a paradigm for rapid development of a fourth class of API's, that of pharmaceutical co-crystals.
Crystal engineering can be defined as application of the concepts of supramolecular chemistry to the solid state with particular emphasis upon the idea that crystalline solids are de facto manifestations of self-assembly. Crystal structures can therefore be regarded as the result of a series of weak but directional molecular recognition events. With understanding comes the possibility of design and it is the advent of supramolecular synthesis1–3 that facilitates the rational design of new structures and compositions. The roots of crystal engineering can be traced at least as far back as the 1930's, when Pauling defined the chemical bond in both covalent and noncovalent terms.11 The term “crystal engineering” was coined by Pepinsky in 195512 but was not implemented until Schmidt studied a series of solid state reactions in crystalline solids.13 Indeed, solvent free synthesis continues to represent an active area of research in the context of crystal engineering.14,15 Based upon literature citations,† it is apparent that crystal engineering enjoyed rapid growth during the 1990's, especially in terms of organic solids and metal-organic solids but also in terms of organometallic16 and inorganic structures.17
What are pharmaceutical co-crystals? Herein we define pharmaceutical co-crystals as being a subset of a broader group of multi-component crystals that also includes salts, solvates (pseudopolymorphs), clathrates, inclusion crystals and hydrates. In a supramolecular context, solvates and pharmaceutical co-crystals are related to one another in that at least two components of the crystal interact by hydrogen bonding and, possibly, other noncovalent interactions rather than by ion-pairing. Neutral compounds and salt forms alike have the potential to be solvated (i.e. interact with solvent molecules) or co-crystallized (i.e. interact with a co-crystal former). Solvate molecules and co-crystal formers can include organic acids or bases that remain in their neutral form within the multi-component crystal. The primary difference is the physical state of the isolated pure components: if one component is a liquid at room temperature, the crystals are referred to as solvates; if both components are solids at room temperature, the products are referred to as co-crystals. While at first glance these differences may seem inconsequential, they have profound impact on the preparation, stability, and ultimately on developability of products. Furthermore, whereas solvates are commonplace because they often occur as a serendipitous result of crystallization from solution, co-crystals, especially pharmaceutical co-crystals, represent a relatively unexplored class of compounds. On the other hand, as will become clear herein, pharmaceutical co-crystals can be rationally designed and there are many more potential co-crystal formers than there are solvents or counterions.
The complex nature of API structures means that they inherently contain exterior functional groups that engage in molecular recognition events. Indeed, it is the very presence of these functional groups that affords biological activity but also provides an ability to engage in more than one supramolecular event with itself, a solvent molecule or co-crystal former, thereby forming polymorphs, solvates or co-crystals, respectively. It is important to note that there are two basic types of molecular recognition that facilitate the formation of polymorphs, solvates and co-crystals. Functional groups that are self-complementary are capable of forming supramolecular homosynthons. For example, as revealed by Scheme 1a, carboxylic acid moieties and amide moieties can form homodimers via a two-point donor-acceptor molecular recognition path. However, it is also possible for functional groups to engage with a different but complementary functional group, as noted by Pauling. Indeed, carboxylic acids and amides are complementary with each other and can interact through formation of a supramolecular heterosynthon (Scheme 1b). This particular motif has been studied for some time in the context of co-crystals.18
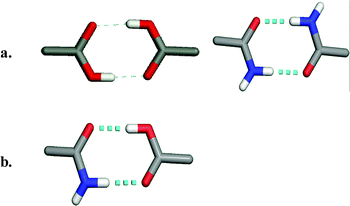 | ||
| Scheme 1 The formation of supramolecular synthons between acids and amides: (a) supramolecular homosynthons as exhibited by acid–acid and amide–amide dimers; (b) supramolecular heterosynthons as exhibited by acid–amide dimers. | ||
In this contribution we detail the current and potential impact of crystal engineering on our understanding of polymorphs, solvates and co-crystals with particular emphasis upon API's. Carboxylic acid and amide moieties are widely encountered in API's and studied in model compounds. They will therefore be used extensively in this contribution even though it should be remembered that they represent just a microcosm of the functional group diversity that exists in API's.
2 Crystal engineering in the context of polymorphs
“A solid crystalline phase of a given compound resulting from the possibility of at least two different arrangements of the molecules of that compound in the solid state” (W.C. McCrone in Physics and Chemistry of the Organic Solid State, Vol II, Wiley Interscience, New York, 725–726, 1965.)McCrone's definition of a polymorph as presented above is particularly appropriate in the context of drugs, since the existence of highly functional API's invites multiple modes of self-organization and amounts to promiscuity in self-assembly. It is this feature and conformational flexibility that are the primary driving forces for the existence of crystal polymorphism. It is therefore not surprising that it is well and long documented that API's can exist in several polymorphic, solvated and/or hydrated forms.10,18 This tendency for polymorphism represents both a problem and an opportunity in pharmaceutical research. Lack of reliability of manufacturing and physical (and sometimes chemical) instability of a given polymorph can be an issue for a drug developer, while a novel polymorph in the hands of a competitor can provide options for generic pharmaceutical competition.
We shall focus upon polymorphism from a supramolecular perspective with emphasis upon two functional groups that are commonly encountered in API's: carboxylic acids and amides.
2.1 Structures in which carboxylic acids are involved in self-organization.
Carboxylic acid moieties represent perhaps the longest and most widely studied functional group in terms of our understanding of hydrogen bonding in both solution and the solid state.11 In the context of crystal structures, carboxylic acids exhibit a remarkable range of diversity in their supramolecular chemistry and this in turn leads to observation of polymorphs in even the most simple of chemical structures. There are two primary modes for carboxylic acids to self-organize in the form of supramolecular homosynthons: the dimer and the catemer. Such “supramolecular isomerism” is the origin of polymorphism exhibited by the two polymorphs of chloroacetic acid (Fig. 1). Fig. 1a illustrates the dimer motif which occurs in one polymorph19 whereas Fig. 1b presents the second form, in which a catemer supramolecular synthon results in the formation of a tetrameric supramolecular assembly.20 It should be noted that carboxylic acid polymorphs are not always a consequence of isomerism in supramolecular homosynthons. For example, they can result from factors such as different crystal packing arrangements of dimer motifs or, if appropriate, torsional flexibility, which can afford conformational polymorphism.21 Nevertheless, there are other simple carboxylic acids that exhibit polymorphism because of dimer/catemer supramolecular isomerism (e.g. hydroxybenzoic acid,22 oxalic acid23 and tetrolic acid24).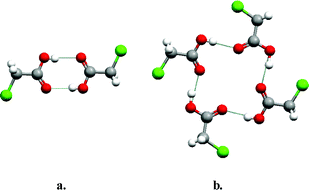 | ||
| Fig. 1 The self-organization modes seen in the two reported polymorphs of chloroacetic acid: (a) centrosymmetric dimer; (b) catemer motif, which leads to a tetrameric assembly. | ||
The story does not end there: whereas there are over 4000 entries in the Cambridge Structural Database25 (CSD) of crystal structures in which at least one carboxylic acid moiety is present, 1179 exhibit the dimer motif (29.4%) and only 86 exhibit the catemer motif (2.1%). In other words, the formation of supramolecular homosynthons is not the dominant supramolecular event in the solid state even if it might be in solution. An analysis of the remaining carboxylic acid containing crystal structures reveals that they typically form supramolecular structures that involve a carboxylic acid and a different functional group, i.e. they form supramolecular heterosynthons. The ability of a molecule to engage in either supramolecular homosynthons or supramolecular heterosynthons represents another avenue for the existence of polymorphism.
Polymorphism in molecules which contain multiple functional groups is exemplified by Fig. 2, which presents the monoclinic and triclinic forms of 2-(2-methyl-3-chloroanilino)-nicotinic acid,26 a molecule that exhibits analgesic and anti-inflammatory properties. Fig. 2 reveals that 2-(2-methyl-3-chloroanilino)-nicotinic acid can self-organize via either supramolecular homosynthons or supramolecular heterosynthons: (a) generation of head-to-tail chains sustained by a carboxylic acid–pyridine supramolecular heterosynthon; (b) formation of centrosymmetric dimers sustained by the carboxylic acid supramolecular homosynthon.
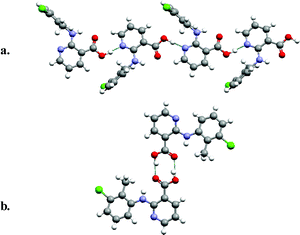 | ||
| Fig. 2 The monoclinic (a) and triclinic (b) forms of 2-(2-methyl-3-chloroanilino)-nicotinic acid, an analgesic/anti-inflammatory molecule. | ||
It is important to emphasize the distinction between supramolecular homosynthons and supramolecular heterosynthons since the latter represent a possible entry into the realm of multiple-component crystals and a diverse range of compositions of matter and physical properties. That carboxylic acids represent such a large subset of the CSD makes it possible to ask an important question: are supramolecular heterosynthons not just rational but also predictable? In the context of the pyridine–carboxylic supramolecular heterosynthon the CSD reveals that there are 424 compounds that contain both a carboxylic acid and an aromatic nitrogen base. 198 of these compounds (46.7%) exhibit the supramolecular heterosynthon rather than one of the carboxylic acid supramolecular homosynthons (Scheme 2). When one considers that many of the compounds in this dataset contain multiple functional groups this is a remarkably high rate of occurence.
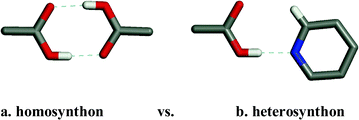 | ||
| Scheme 2 The homosynthon vs. heterosynthon motifs observed in crystal structures of compounds in which both carboxylic acids and pyridine moieties are present. The heterosynthon dominates, occurring in 119/245 crystal structures whereas the homosynthon occurs in only 10 crystal structures. | ||
2.2 Structures in which primary amides are involved in self-organization
Primary amides are also well represented in the CSD, with 1152 entries. The dominant supramolecular homosynthon is the centrosymmetric dimer as presented in Scheme 1. This homosynthon contains complementary hydrogen bond donors and acceptors and is capable of further self-assembly, thereby generating supramolecular tapes or sheets. Fig. 3a illustrates how chloroacetamide forms a tape network based upon self-organization of homodimers.27–30 Interestingly, chloroacetamide also exhibits polymorphism and for the same fundamental reason as chloroacetic acid: it exhibits a catemer structure as well as a homodimer structure.31 The polymorphic form of chloroacetamide that is the result of catemer motifs is illustrated in Fig. 3b. It reveals that the superstructure is also that of a tape. The two forms of chloroacetamide crystallize in the same space group with almost identical cell parameters. This is an extremely unusual situation and is presumably related to the fact that the two tapes are similar in terms of dimensions and exterior features.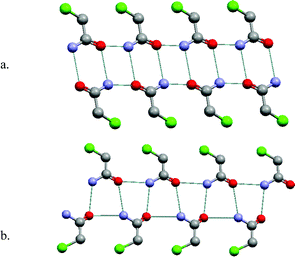 | ||
| Fig. 3 The self-organization modes seen in two polymorphs of chloroacetamide: (a) centrosymmetric dimer that self-assembles as 1-D tapes; (b) catemer motif, which also forms 1-D tapes. | ||
Chloroacetic acid and chloroacetamide serve as illustrations of how even small molecules with only one hydrogen bonding group can generate polymorphs based upon supramolecular isomerism. A similar analogy can be found in API's that contain acid and amide moieties. Piracetam, a learning process drug, is an amide-containing API that exemplifies the type of polymorphism that occurs when supramolecular isomerism occurs in supramolecular homosynthons. There are three forms of Piracetam reported in the CSD.32,33 Two of these forms exist as tapes that are sustained by the amide homodimer and NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(carboxamide) hydrogen bonds (Fig. 4a).32 The third form is sustained by catemer chains that are crosslinked by N–H⋯OC(carboxamide) hydrogen bonds (Fig. 4b).33 The superstructure can therefore be described as hydrogen bonded sheets.
C(carboxamide) hydrogen bonds (Fig. 4a).32 The third form is sustained by catemer chains that are crosslinked by N–H⋯OC(carboxamide) hydrogen bonds (Fig. 4b).33 The superstructure can therefore be described as hydrogen bonded sheets.
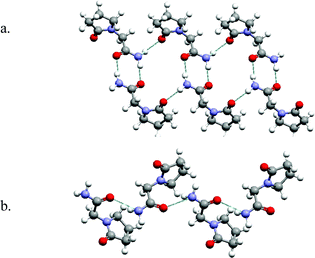 | ||
| Fig. 4 The network structures formed by Piracetam: (a) homodimers form supramolecular tapes two forms; (b) 1-D chains sustained by the catemer motif are found in the third form. | ||
To summarize the points made thus far:
• Single component crystals that contain carboxylic acid or amide moieties are prone to polymorphism even if only one hydrogen bonding moiety is present and supramolecular homosynthons are the primary molecular recognition events.
• In the case of API's, the situation is further complicated by the presence of additional hydrogen bonding moieties, which can lead to the formation of supramolecular heterosynthons.
• Carboxylic acid and amide groups were chosen as examples, because they are prevalent in the CSD and in API's. However, the points made thus far can be regarded as being generally relevant. For example, we recently reported34 how alcohol–ether heterosynthons can afford polymorphic forms of butylated hydroxy anisole, an antioxidant that is commonly used in solid dosage forms of API's.35,36 The difference between the two forms is striking: form I exists as the result of 4-fold helical chains: form II contains discrete hexamers.
How one might exploit supramolecular heterosynthons for the crystal engineering of new compositions of matter will form the basis of the remainder of this contribution.
3 Crystal engineering in the context of co-crystals
“Supramolecular synthons are structural units within supermolecules that can be formed and/or assembled by known or conceivable synthetic operations involving intermolecular interactions”. (Gautam R. Desiraju Angew. Chem. Int. Ed. Engl., 34, 2311, 1995.)How does one develop a strategy for the preparation of co-crystals? Solvates are frequently encounted but are typically the result of serendipity rather than design and are often found as by-products of polymorph and salt screens. Co-crystals, on the other hand, are less ubiquitous but are more prone to rational design. Co-crystals have been prepared by melt-crystallization, by grinding37 and by recrystallization from solvents.14,15 Pharmaceutical co-crystals have the potential to be much more useful in pharmaceutical products than solvates or hydrates. First, the number of pharmaceutically acceptable solvents is very small. Secondly, solvents tend to be more mobile and have higher vapour pressures than small molecule co-crystal formers. It is not unusual to observe dehydration/desolvation of hydrates/solvates in solid dosage forms, depending on storage conditions. Solvent loss frequently leads to amorphous compounds, which are generally less chemically stable and can crystallize into less soluble forms. In contrast to solvents, most co-crystal formers are unlikely to evaporate from solid dosage forms, making phase separation less likely.
3.1 Co-crystals based upon acids or amides
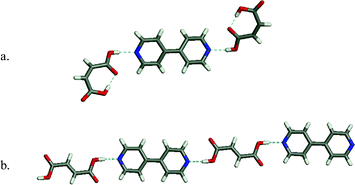
As suggested earlier, an effective approach to understanding and designing co-crystals is to apply the paradigm of supramolecular synthesis, in particular exploitation of supramolecular heterosynthons. The ubiquity of acids and amides in the CSD makes them appropriate foci for design and synthesis. Indeed, the acid–amide supramolecular heterosynthon illustrated in Scheme 1a has been exploited by several groups for the generation of co-crystals18,38–41 and the CSD reveals that there are 118 crystal structures in which both an acid and an amide moiety are present. Remarkably, 58 of these structures exhibit the acid–amide supramolecular heterosynthon whereas only 11 structures exhibit the acid homodimer and only 28 exhibit the amide homodimer. Fig. 5 presents two prototypal examples of co-crystals that are sustained by the acid–amide supramolecular heterosynthon: succinic acid : benzamide18 and urea : glutaric acid.38 Acid–amide supramolecular heterosynthons are not the only examples of robust heterosynthons that are favored over the parent homosynthons. Acid–pyridine supramolecular heterosynthons, a subset of the acid–aromatic amine set described earlier, occur in 119 of the 245 crystal structures that contain both functional groups. Remarkably, only 10 of these 245 structures contain acid–acid homosynthons (Scheme 2).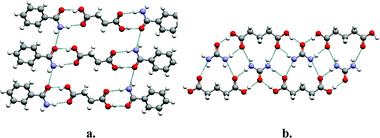 | ||
| Fig. 5 Two examples of co-crystals that are sustained by the acid–amide supramolecular heterosynthon: (a) succinic acid : benzamide (1:2); (b) urea : glutaric acid (1:1). | ||
Representative examples of co-crystals that are sustained by the pyridine–carboxylic acid supramolecular synthon are presented in Fig. 6. Maleic acid : 4,4′-bipyridine forms a discrete 2:1 adduct42 whereas fumaric acid : 4,4′-bipyridine forms in 1:1 stoichiometry and thereby generates a 1-D chain.42
 | ||
| Fig. 6 Two examples of co-crystal structures formed by the acid–pyridine supramolecular heterosynthon: (a) maleic acid: 4,4′-bipyridine; (b) fumaric acid : 4,4′-bipyridine. | ||
3.2 Functional co-crystals
Examples of co-crystals have existed in conductive organic crystals, non-linear optical crystals, dyes, pigments and agrochemicals for some time43 but have only recently been applied to API's. Several recent papers emphasize the importance of understanding supramolecular heterosynthons in the synthesis of pharmaceutical co-crystals. For example, the ability to insert 4,4′-bipyridine and related molecules between the carboxylic acid dimers of aspirin, rac-ibuprofen, and rac-flurbiprofen was recently reported.6Fig. 7 illustrates two of these structures, which further demonstrate the ability of the pyridine–carboxylic acid heterosynthon to compete with a carboxylic acid dimer homosynthon (Scheme 2).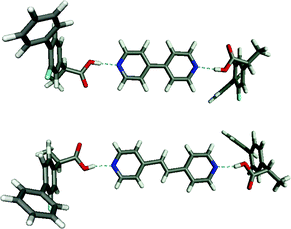 | ||
| Fig. 7 The 2:1 supramolecular adducts formed by flurbiprofen and 4,4′-bipyridine (top) and 4,4′-dipyridylethane (bottom). Similar structures occur for ibuprofen and aspirin. | ||
A second study focused on finding multiple solvates and co-crystals of carbamazepine.5 Carbamazepine represents an excellent test case since four polymorphs and two solvates of carbamazepine have been reported in the literature. In all of the compounds for which structural data is available, carbamazepine molecules crystallize as amide dimers (Fig. 8). The crystal structures illustrate that each dimer contains a peripheral H-bond donor and acceptor pair that is unsatisfied due to geometric constraints imposed by the drug molecule. Simple H-bond acceptor solvents like acetone and DMSO insert themselves to fill voids between the adjacent pairs of dimers. Multiple co-crystal formers having hydrogen bonding groups likewise insert themselves into the void, including saccharin and nicotinamide. The amide homosynthon can also be broken to form heterosynthon Ib. This was achieved to form solvates with acetic, formic, and butyric acids and co-crystals with trimesic and nitroisophthalic acid. The crystal structures of the carbamazepine : saccharin co-crystal and the formic acid solvate are illustrated in Fig. 9.
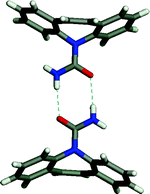 | ||
| Fig. 8 The carbamazepine dimers that exist in all previously reported solvates and polymorphs of carbamazepine. | ||
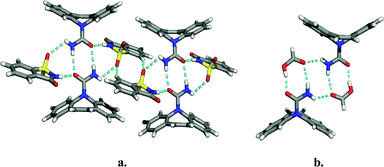 | ||
| Fig. 9 Examples of the supramolecular adducts formed in the crystal structures of co-crystals and solvates of carbamazepine: (a) saccharin co-crystal; (b) carbamazepine:formic acid solvate. | ||
A study of adducts of acetaminophen (paracetamol) with ethers and amines provides additional examples of supramolecular synthons for co-crystal formation (Scheme 3).9 While supramolecular homosynthon IIIa could have formed, both known forms of the pure material consist of linear head-to-tail chains held together through motif IIIb; the chains are cross-linked through synthon IIIc. The linear chain structure is preserved in co-crystals with 4,4′-bipyridine, but the cross-linking interaction IIIc is replaced by IIId, in which the 4,4′-bipyridine is hydrogen bonded to the amide hydrogen. The chains remain cross-linked but only through pi-stacking interactions between 4,4′-bipyridine pairs on neighboring chains. In co-crystals with piperazine, the acetaminophen forms head-to-head chains through IIIe. Each chain is joined to the next through a layer of piperazine molecules that interact through heterosynthons IIIf and IIIg. The paper also includes many solvates that will not be reviewed here, but their supramolecular synthons should also be applicable in the context of co-crystal design and formation.
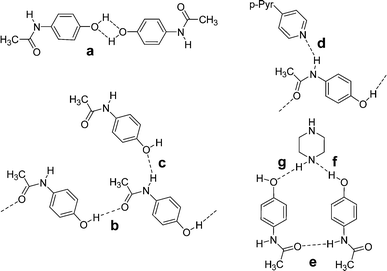 | ||
| Scheme 3 The supramolecular synthons observed in co-crystals of acetaminophen (paracetamol): IIIa–c occur in polymorphs whereas IIId and IIIe occur in co-crystals. | ||
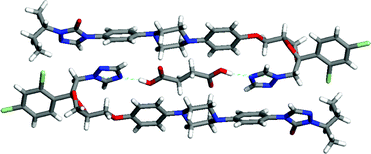
The analysis of molecules for complementarity of supramolecular synthons represents a valuable approach to screening that a knowledgeable scientist can exploit to narrow the search for co-crystals. However, an early study of 1:1 molecular complexes between the antibacterial agents trimethoprim (TMP) and sulfamethoxypyridazine (SMP) highlights the need to explore the space beyond those leading to expected interactions.44 Each complex contains an 8-membered, hydrogen-bonded ring joining the two molecules as shown in Fig. 10. The specific ring structures formed are not those that might have been predicted by inspection of the structures of the neutral molecules. Instead, the synthons are derived from the 2-aminopyridine of TMP and the zwitterionic form of SMP involving the sulfonamide (pKa ∼ 7) and pyridazine (pKa ∼ 2). The zwitterion is a thermodynamically unfavorable form of SMP in aqueous solution. This example of assembly through an unstable intermediate underscores the limitation of the approach of analyzing co-crystal formation solely on the basis of pKa arguments. A more comprehensive approach is needed. HT crystallization offers the possibility to uncover unexpected interactions by screening against a full library of pharmaceutically acceptable molecules instead of limiting the studies to co-crystal formers with perceived complementarity.
 | ||
| Fig. 10 The 8-membered hydrogen-bonded ring that links antibacterial agents trimethoprim (TMP) and sulfamethoxypyridazine (SMP). | ||
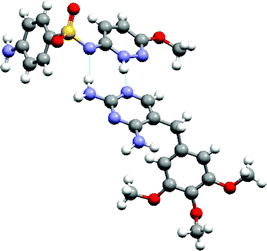
The more comprehensive approach to study expected and unexpected co-crystal formation events is high-throughput (HT) crystallization. The discovery of pharmaceutically acceptable co-crystals consisting of hydrogen-bonded trimers of two molecules of cis-itraconazole, a triazole anti-fungal agent, and a molecule of a 1,4-dicarboxylic acid resulting from a HT crystallization screen was recently reported.8 The crystal structure of the succinic acid co-crystal (Fig. 11) reveals a supramolecular heterosynthon between the triazole of each pair of drug molecule and carboxylic acid moieties on a single diacid molecule. The extended succinic acid molecule fills a pocket, while bridging the triazole groups. The interaction between the 1,4-diacid and the strongest base on itraconazole (piperazine) is not observed in the co-crystal structure. Other 1,4-diacids capable of extended (anti-) conformations also yielded co-crystals with itraconazole, while co-crystals could not be made from maleic acid with Z-regiochemistry, or from 1,3- or 1,5-dicarboxylic acids. Hence, structural fit appears to be far more important than acid–base strength complementarity for co-crystallization of itraconazole with 1,4-dicarboxylic acids.
 | ||
| Fig. 11 The 2:1 supramolecular adduct formed by itraconazole and succinic acid. | ||
The structures presented herein demonstrate that pharmaceutical co-crystals represent an interesting and emerging class of pharmaceutical materials in terms of rational design, projected diversity and applicability. Furthermore, the study of pharmaceutical co-crystals, along with polymorphs, solvates, salts and hydrates, is perfectly suited to HT crystallization experimentation and could in the future be considered an integral part of form selection processes in pharmaceutical research and development.
4 Conclusions and future directions
“What would the properties of materials be if we could really arrange the atoms the way we want them?…we will get an enormously greater range of possible properties that substances can have, and of different things that we can do.” (Richard P. Feynman, December 29, 1959).In the world of pharmaceuticals, the opportunity presented by co-crystals appears to be significant. Published examples of pharmaceutical co-crystals are few as yet, but we now believe the approach can be applied broadly to API's. The design and selection of optimal pharmaceutical materials based on supramolecular synthesis is a relatively low-risk strategy, because the approach employs principles of molecular recognition and self-assembly rather than creating covalent bonds. Therefore, there are no covalent modifications of the API in question. Nevertheless, some big questions remain:
4.1 How large is the space of pharmaceutical co-crystals?
Compared with the space of salt forms, solvates and polymorphs, how large is the space of pharmaceutical co-crystals? Polymorphism tendency of pharmaceutical substances varies greatly, but the general observation is that most compounds are at some time or another going to display polymorphic behavior. Typically, the extent of polymorphism of pharmaceuticals is limited to a handful of different crystal forms. A recent review classifies highly polymorphic materials as having 4 or more forms.45 Solvates (including hydrates) can be more numerous, and in certain cases very large numbers of solvates can be observed. Indeed, one study suggests that sulfathiazole is inordinately promiscuous in terms of solvate formation, with over one hundred solvates found.46 Salt forms can be numerous as well, with over 90 acids and 30 bases being considered suitable for pharmaceutical salt selection.47 Examples of compounds possessing a dozen or more crystalline salt forms have been published.48,49 It is important to remember that salt formation is generally directed at one acidic or basic functional group. In contrast, co-crystals can simultaneously address multiple functional groups (synthons) in a single drug molecule. In addition, the space is not limited to binary combinations (such as acid–base pairs) since tertiary and quaternary co-crystals are realistic possibilities. Co-crystal formers for pharmaceutical use remain to be enumerated fully, but we argue that well over a hundred solid materials with GRAS status (including food additives and other well-accepted substances) can be employed. Even more provocatively, one might consider using sub-therapeutic amounts of eminently safe drug substances, such as aspirin and acetaminophen, as legitimate co-crystal formers, thus expanding the arsenal even further. Taken together with the high dimensionality and resulting combinatorial nature of supramolecular assembly, the space of pharmaceutical co-crystals would appear to be extremely large: one can easily envision thousands of possibilities for any given drug with at least two synthons present in the molecule. Such diversity will probably be best addressed with combinatorial methodologies, such as high-throughput crystallization.4.2 Can there be rational, directed design of pharmaceutical co-crystal phases?
This is another question which relates to the prospect for design. Crystal structures are inherently unpredictable, but the interactions that occur prior to a crystal forming or growing are predictable. An analogy can be drawn to salt selection,47,50 in which pKa arguments are used to select acid–base pairs that can be converted to salt compounds. The prediction of the proton transfer event is based on solution data, but the occurrence of a crystalline salt form cannot be predicted a priori. Based on the examples of rational synthon selection presented here, it follows that strategies of rational design of co-crystal experimentation are viable.4.3 Are pharmaceutical co-crystals more or less prone to polymorphism than other pharmaceutical phases?
This question will not have a direct answer, because to prove the absence of polymorphism is tantamount to “proving the negative”. But if one considers the argument that compounds have a lower degree of self-complementarity than complementarity to a rationally selected co-crystal former, one might suspect that a compound polymorphic in the pure state could display a decreased tendency to polymorphism as a co-crystal relative to the pure phase. Support for or defeat of this argument will involve significant research. Initial indications are that polymorphic substances may provide good candidates for co-crystal formation.39a As an example, carbamazepine can exist as four well characterized polymorphs51 and a dihydrate.52 This drug was recently converted to many co-crystals.5 In terms of assessing polymorphism, one co-crystal of carbamazepine and saccharin has only displayed one packing arrangement, despite testing via HT crystallization in over 2000 experiments.53 In contrast, two co-crystal structures of a N,N′-bis(para-bromophenyl)melamine-diethylbarbital demonstrate how a specific heterosynthon between the two molecules is robust, but packing of the tapes into a crystalline arrangement can lead to two discrete polymorphs.54 Hence, there may be opportunity to reduce the practical extent of polymorphism of drug compounds specifically by co-crystal formation although there may be exceptions.4.4 What opportunities exist for tuning physico-chemical properties by pharmaceutical co-crystal formation?
This is perhaps the most important question, because it is after all the complex interplay of form, function and performance attributes that determine success (or failure) of a particular pharmaceutical formulation.55 Issues ranging from poor solubility and inadequate dissolution properties to lack of crystallinity and attendant instability plague the industry.56 Poor aqueous solubility is a growing problem in the industry and it is having an impact on the productivity of drug research. The solubility issue hampers pre-clinical study of a new drug candidate, and can limit dosing and bioavailability. New strategies to deal with these problems are badly needed. Why are API's increasingly found to be of low solublility? There are varying ways to speculate around this question, but some would point to the methodologies that are now employed to discover pharmacologically active compounds. In vitro assays have largely replaced in vivo animal screens as means to discover active compounds. The challenge of drug delivery is not addressed until there is a real desire to advance a compound into the development process. In addition, combinatorial chemistry and application of genomics have generated molecular targets for which the most potent lead compounds are inherently poorly soluble. Attempts to engineer compounds via medicinal chemistry avenues often lead to frustration, as the in vitro activity is frequently lost with increased water solubility. In the end, a discovery project may end up advancing a compound for which the stable crystal form exhibits inadequate aqueous solubility or dissolution rate that leads to poor oral absorption or inability to deliver by other routes (e.g. injection or inhalation). The most common strategy currently employed for improving bioavailability and optimizing drug delivery is to prepare salt forms of ionizable compounds, using pharmaceutically acceptable acids and bases. However, in the case of compounds that cannot form stable salts in aqueous medium, pharmaceutical scientists are left with few good options for material design, and must resort to particle size reduction to the nanometer range, deliberate amorphization, or solubilization in non-aqueous vehicles. These processes lead to formulations that have more physico-chemical problems than crystalline preparation.55 In terms of addressing the stability issue, one of the most important challenges presently is crystallization of compounds that are amorphous. In general, amorphs are undesirable forms due to physical instability (at the very least, there is the theoretical possibility of the material crystallizing at some point). Chemical reactivity can be significantly increased in amorphous states relative to crystalline forms.57 In addition, amorphous forms tend to be hygroscopic and have low powder densities, giving rise to significant processing challenges. One reason for the resistance to crystallization is undoubtedly the mismatch that can occur in the number of hydrogen bond donors and acceptors in a molecule. In such cases, a solvate (or series of solvates) that leads to more satisfied hydrogen bond arrangements may be produced, while the pure, desolvated substance remains amorphous. An example of this situation is the HCl salt of the ACE inhibitor quinapril HCl.57b The opportunity exists to use co-crystallization to replace the solvate, while taking advantage of the supramolecular synthons that are suggested by the solvate structures. Given a co-crystal form thus obtained, one can expect the crystallinity of the material to result in greater stability and other desirable properties as compared with the amorphous form. In terms of solubility, amorphous compounds can have significant advantage over crystalline forms.58,59 Though this advantage could find use in isolated cases, the lack of a crystalline form and concern over phase changes make the use of amorphous drugs in market formulations undesirable. When a crystalline form of a pure phase exhibits poor solubility or slow dissolution rate in aqueous media that translates to inadequate bioperformance, the strategies of salt selection and co-crystal formation should be considered. While salts can be made of acidic and basic drugs, the large space of non-ionizable compounds are generally candidates for co-crystal exploration. The in vitro dissolution profile of carbamazepine-saccharin co-crystal60 illustrates the superior dissolution of the drug molecule in that context as compared with one of the pure anhydrous polymorphs. While the polymorph transiently supersaturates in the aqueous medium and subsequently precipitates to eventually form the known dihydrate, the co-crystal supersaturates to a sustained two-fold equilibrium solubility of the dihydrate. Such supersaturation behavior has been found to influence the bioavailability of carbamazepine.61 While equilibrium solubility of drug compounds in co-crystals may be less affected than in the context of salt forms, the kinetic aspects of solubilization provide the key to many successful formulation strategies, such as oral immediate or controlled release. Co-crystals clearly open up a vast space of possibilities for exploring the range of dissolution characteristics, and facilitate co-optimization with other parameters, such as stability and processability.To summarize, despite the need for a greater understanding and control of the crystalline phases for pharmaceutical development, the concepts of supramolecular synthesis and crystal engineering have remained underexploited in the world of drugs. As presented herein, applying the concept of supramolecular synthesis to the development of pharmaceutical co-crystals would seem to represent a new paradigm that would address both intellectual and property issues related to drug development and delivery, especially when supramolecular synthesis is coupled with HT screening technologies.
Notes and references
- (a) G. R. Desiraju, Crystal Engineering—The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed; (b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- M. C. Etter, J. Chem. Phys., 1991, 95, 4601–4610 CAS; M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- Ö. Almarsson, M. B. Hickey, M. L. Peterson, S. L. Morissette, C. McNulty, S. Soukasene, M. Tawa, M. MacPhee and J. F. Remenar, Cryst. Growth Des., 2003, 3, 927–933 CrossRef CAS.
- S. G. Fleischman, S. S. Kuduva, J. A. McMahon, B. Moulton, R. B. Walsh, N. Rodriguez-Hornedo and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 909–919 CrossRef CAS.
- S. G. Fleischman, L. Morales, B. Moulton, N. Rodríguez-Hornedo, R. B. Walsh and M. J. Zaworotko, Chem. Commun., 2003, 186–187 RSC.
- S. Datta and D. J. W. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42–57 Search PubMed.
- J. F. Remenar, S. L Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzman and Ö. Almarsson, J. Am. Chem. Soc., 2003, 125, 8456–8457 CrossRef CAS.
- I. D. H. Oswald, D. R. Allen, P. A. McGregor, W. D. S. Motherwell, S. Parsons and C. R. Pulham, Acta Crystallogr., 2002, B58, 1057–1066 CAS.
- (a) J. K. Haleblian, J. Pharm. Sci., 1975, 64, 1269 CAS; (b) J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 2nd edn., 1948 Search PubMed.
- R. Pepinsky, Phys. Rev., 1955, 100, 971 CAS.
- G. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS.
- F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS.
- T. Friscic and L. R. MacGillivray, Chem. Commun., 2003, 1306–1307 RSC.
- D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1405 CrossRef CAS.
- R. C. Finn, J. Zubieta and R. C. Haushalter, Prog. Inorg. Chem., 2003, 51, 421–601 CAS.
- (a) L. Leiserowitz and F. Nader, Angew. Chem., 1972, 84, 536; (b) C. M. Huang, L. Leiserowitz and G. M. J. Schmidt, J. Chem. Soc. Perkins Trans. 2, 1973, 5, 503 RSC; (c) Z. Berkovitch-Yellin, L. Leiserowitz and F. Nader, Acta Cryst., 1977, B33, 3670 CAS.
- J. A. Kanters, G. Roelofsen and T. Feenstra, Acta. Cryst. B., 1976, 32, 3331 CrossRef.
- J. A. Kanters and G. Roelofsen, Acta. Cryst. B., 1976, 32, 3328 CrossRef.
- J. Bernstein and A. T. Hagler, J. Am. Chem. Soc., 1978, 100, 673–681 CrossRef CAS.
- G. V. Gridunova, Kristallografiya, 1982, 27, 267 Search PubMed.
- J. L. Derissen and P. H. Smit, Acta Cryst. B, 1974, 30, 2240 CrossRef.
- V. Benghiat and L. Leiserowitz, J. Am. Chem. Soc. Perkins Trans. 2, 1972, 12, 1763 RSC.
- F. H. Allen and O. Kennard, Chem. Des. Automation News, 1993, 8, 31 Search PubMed.
- M. Takasuka, H. Nakai and M. Shiro, J. Chem. Soc. Perkins Trans. 2, 1982, 9, 1061 RSC.
- J. Dejace, Acta. Cryst., 1955, 8, 851 CrossRef CAS.
- B. R. Penfold and W. S. Simpson, Acta. Cryst., 1956, 9, 831 CrossRef CAS.
- B. Kalyanaraman, L. D. Kispert and J. L. Atwood, J. Cryst. Mol. Struct., 1978, 8, 175 Search PubMed.
- Y. Kato and N. Oguro, Mem. Osaka Kyoiku Univ. Ser. 3, 1982, 30, 155 Search PubMed.
- M. Katayama, Acta. Cryst., 1956, 9, 986 CrossRef CAS.
- G. Admiraal, J. C. Eikelenboom and A. Vos, Acta. Cryst. B., 1982, 38, 2600 CrossRef.
- D. Louer, M. Louer, V. A. Dzyabchenko, V. Agafonov and R. Ceolin, Acta. Cryst. B., 1995, 51, 182 CrossRef.
- J. A. McMahon, M. J. Zaworotko and J. F. Remenar, Chem. Commun., 2004, 278 RSC.
- H. Verhagen, P. A. E. L. Schilderman and J. C. S Kleinjans, Chem-Biol. Interactions, 1991, 80, 109 Search PubMed.
- J. W. Finley and P. Given Jr, Food Chem. Toxicol., 1986, 24, 999 CrossRef CAS.
- A. V. Trask, S. W. D. Motherwell and W. Jones, Chem. Commun., 2004, 890–891 RSC.
- V. Videnova-Adrabinska and M. C. Etter, J. Chem. Cryst., 1995, 25, 823 CAS.
- (a) C. B. Aakeroy, A. M. Beatty and B. A. Helfrich, Angew. Chem. Int. Ed. Engl., 2001, 40, 3240 CrossRef CAS; (b) C. B. Aakeroy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef.
- (a) S. Ohba, H. Hosomi and Y. Ito, J. Am. Chem. Soc., 2001, 123, 6349 CrossRef CAS; (b) Y. Ito, H. Hosomi and S. Ohba, Tetrahedron, 2000, 56, 6833 CrossRef CAS; (c) H. Hosomi, S. Ohba and Y. Ito, Acta. Cryst. C., 2000, 56, 507.
- (a) G. Smith, K. E. Baldry, K. A. Byriel and C. H. L. Kennard, Aust. J. Chem., 1997, 50, 727 CrossRef CAS; (b) R. C. Bott, G. Smith, V. D. Wermuth and N. C. Dwyer, Aust. J. Chem., 2000, 53, 767 CrossRef CAS; (c) G. Smith, M. G. Coyne and J. M. White, Aust. J. Chem., 2000, 53, 203 CrossRef CAS; (d) T. J. M. Luo and G. T. R. Palmore, Cryst. Growth Des., 2002, 2, 337 CrossRef CAS; (e) P. Vishweshwar, A. Nangia and V. M. Lynch, Cryst. Growth Des., 2003, 3, 783–790 CrossRef CAS; (f) L. S. Reddy, A. Nangia and V. M. Lynch, Cryst. Growth Des., 2004, 4, 89–94 CrossRef CAS.
- S. Chatterjee, V. R. Pedireddi and C. N. R. Rao, Tetrahedron Lett., 1998, 39, 2843 CrossRef CAS.
- (a) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS; (b) G. de la Torre, P. Vazquez, F. Agullo-Lopez and T. Torres, J. Mater. Chem., 1998, 8, 1671 RSC.
- G. Bettinetti, M. R. Caira, A. Callegari, M. Marcello, M. Sorrenti and C. Tadini, J. Pharm. Sci., 2000, 89, 478–489 CrossRef CAS.
- S. Morissette, Ö. Almarsson, M. Peterson, J. Remenar, M. Read, A. Lemmo, S. Ellis, M. Cima and C. Gardner, Adv. Drug Deliv. Rev., 2004, 56(3), 275–300 CrossRef.
- A. L. Bingham, D. S. Hughes, M. B. Hursthouse, R. W. Lancaster, S. Tavener and T. L. Threlfall, Chem. Comm., 2001, 603 RSC.
- P. H. Stahl and M. Nakano, Pharmaceutical Aspects of the Drug Salt Form, in Handbook of Pharmaceutical Salts: Properties, Selection, and Use, ed. P. H. Stahl and C.G. Wermuth, Wiley-VCH/VCHA, New York, 2002 Search PubMed.
- S. M. Berge, L. D. Bighley and D. C. Monkhouse, J. Pharm. Sci., 1977, 66, 1–19 CAS.
- J. F. Remenar, J. M. MacPhee, B. K. Larson, V. A. Tyagi, J. H. Ho, D. A. McIlroy, M. B. Hickey, P. B. Shaw and O. Almarsson, Org. Process. Res. Dev., 2003, 7, 990–996 Search PubMed and references therein.
- P. L. Gould, Int. J. Pharm., 1986, 33, 201 CrossRef CAS.
- A. L. Grzesiak, M. Lang, K. Kim and A. J. Matzger, J. Pharm. Sci., 2003, 92, 2260 CrossRef CAS.
- F. U. Krahn and J. B. Mielck, Pharm. Acta Helv., 1987, 62, 247 CAS.
- M. B. Hickey, personal communication.
- J. A. Zerkowski, J. C. MacDonald and G. M. Whitesides, Chem. Mater., 1997, 9, 1933 CrossRef CAS.
- S.-D. Clas, Curr. Opin. Drug Disc. Dev., 2003, 6, 550 Search PubMed.
- Ö. Almarsson and C. R. Gardner, Current Drug Discovery, 2003, pp. 21–26 Search PubMed.
- (a) S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid-State Chemistry of Drugs, SSCI, Inc., West Lafayette, IN, 1999 Search PubMed; (b) S. R. Byrn, A. W. Newman and W. Xu, Adv. Drug Deliv. Rev., 2000, 48, 115 CrossRef.
- B. C. Hancock and M. Parks, Pharm. Res., 2000, 17, 397–404 CrossRef CAS.
- S.-D. Clas, M. Cotton, E. Moran, S. Spagnoli, G. Zografi and E. B. Vadas, Thermochim. Acta., 1996, 288, 83 CrossRef CAS.
- N. Rodriguez-Hornedo, personal communication.
- Y. Kobayashi, S. Ito, S. Itai and K. Yamamoto, Int. J. Pharm., 2000, 193, 137 CrossRef CAS.
Footnote |
| † There has been steady growth in occurrence of the term “crystal engineering” in titles, keywords or abstracts as follows for 1989–2003, respectively: 4, 6, 6, 14, 26, 22, 19, 45, 59, 67, 96, 93, 129, 129, 133. The actual number of papers addressing the general subject of crystal engineering is much larger. |
| This journal is © The Royal Society of Chemistry 2004 |