Vinyl fluoride as an isoelectronic replacement for an enolate anion: Inhibition of type II dehydroquinases
Martyn
Frederickson
a,
John R.
Coggins
b and
Chris
Abell
*a
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk
bDivision of Biochemistry and Molecular Biology, IBLS, Glasgow University, Glasgow, UK G12 8QQ
First published on 26th July 2002
Abstract
A vinyl fluoride analogue of the intermediate in the reaction catalysed by type II dehydroquinase enzymes has been synthesized over seven steps from (−)-quinic acid and shown to be a potent enzyme inhibitor.
The shikimate1,2 and quinate3 pathways (the enzymatic routes from acyclic carbohydrates to essential aromatics utilized by micro-organisms, fungi and the higher plants) continue to elicit major research efforts especially since the recent discovery4 of the shikimate pathway in apicomplexan parasites including the most virulent of the malarial parasites (Plasmodium falciparum).
Dehydroquinase [3-dehydroquinate dehydratase; E.C. 4.2.1.10] which catalyses the reversible conversion of 3-dehydroquinate (DHQ) 1 to 3-dehydroshikimate (DHS) 3 is uniquely common to both pathways. It occurs in two distinct forms each with differing chemical and biochemical properties. Type I enzymes are heat labile protein dimers5 that catalyse a syn-elimination6 of water through Schiff's base intermediates7 involving a conserved lysine. Type II dehydroquinases are more thermally robust dodecameric species8 that effect an anti-dehydration via an enzyme stabilized enolate 29 (Scheme 1).
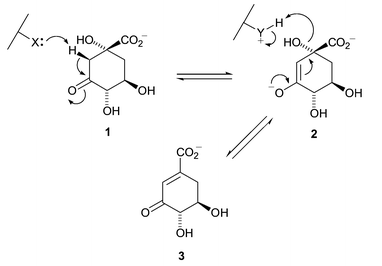 | ||
| Scheme 1 | ||
The mechanistic differences of the two enzyme types led us to design inhibitors that were selective for the type II proteins which are found in a number of clinically important bacterial strains including Mycobacterium tuberculosis. Based upon the idea that compounds which mimicked the enolate intermediate 2 were likely to show such selectivity we prepared derivatives which, when compared to DHQ, possessed either increased sp2 character at C(2) and C(3) (alkene 4) or had superior hydrogen bonding potential at C(3) (oxime 5) (Fig. 1).
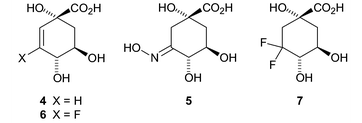 | ||
| Fig. 1 | ||
We were thus able to demonstrate10 that 4 and 5 were potent and highly selective (up to 1000 fold) competitive reversible inhibitors of type II dehydroquinases. Recent crystallographic studies11 on the complex formed between alkene 4 and the type II dehydroquinase from Streptomyces coelicolor (PDB accession code : 1GU1) has allowed its mode of action to be fully determined.
Our search for even more effective inhibitors focused on derivatives in which the true geometric and electronic features of enolate 2 were more accurately mimicked, particularly the electron distribution and high electron density generated at C(3). Despite the relative rarity of the vinyl fluoride functionality we chose to replace the enolate oxyanion with its closest chemical equivalent, the isosteric and isoelectronic fluorine atom. Herein we describe the use of the vinyl fluoride moiety as an isosteric and isoelectronic replacement for an enolate anion and report the syntheses of fluorovinyl 6 and difluoro 712 acids.
Acids 6 and 7 were prepared over seven steps from (−)-quinic acid 8 (Scheme 2). Esterification of 8 was accomplished concomitantly with protection of the 4,5-trans-diol as the cyclic bis-ketal 9 as described previously.13 Ruthenium(III) catalysed periodate oxidation to the C(3) ketone 10 followed by MOM protection of the tertiary alcohol at C(1) afforded the fully protected 3-dehydroquinate 11.14 Incorporation of fluorine at C(3) proved to be somewhat capricious. It was ultimately reproducibly achieved15 upon treatment of ketone 11 in boiling anhydrous DME under argon with multiple portions of DAST to afford a mixture of two chromatographically separable products 12 and 13. Extensive NMR analysis (1H, 13C and 19F in deuteriochloroform) showed the less polar compound to be the desired vinyl fluoride 12 (δF −115.9 coupled to δC 159.6). Determination of the structure and relative stereochemistry of the more polar, crystalline product 13 (δF −113.3 and −103.3 coupled to δC 119.7) proved possible by X-ray crystallographic analysis16,17 (Fig. 2). The product was thereby unequivocally shown to be the corresponding C(3) gem-difluoride.
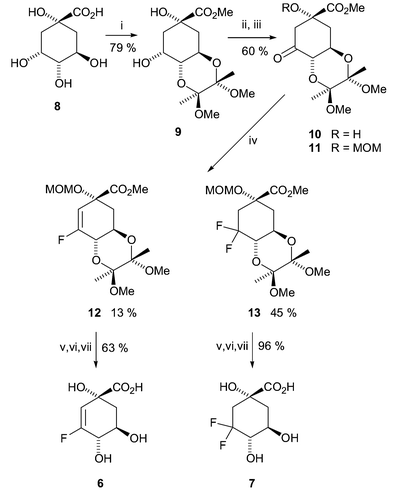 | ||
| Scheme 2 Reagents and conditions: i, MeC(OMe)2C(OMe)2Me, HC(OMe)3, CSA, MeOH, 65 °C, Ar, 16 h; ii, RuCl3, KIO4, K2CO3, H2O, CHCl3, 20 °C, 48 h; iii, CH2(OMe)2, P2O5, CHCl3, 20 °C, 16 h; iv, DAST, DME, 80 °C, Ar, 6 h; v, TFA, H2O, 60 °C, 2 h; vi, NaOH, H2O, 20 °C, 30 min; vii, Amberlite IR-120(H), H2O, 20 °C, 10 min. | ||
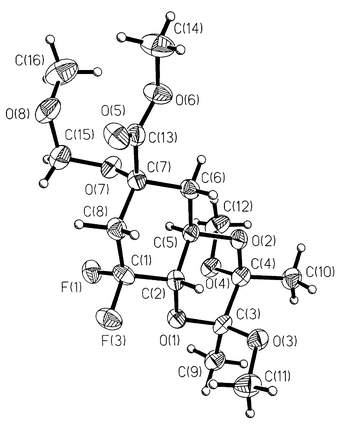 | ||
| Fig. 2 Single crystal X-ray structure of 13. | ||
The desired product 12 was the minor of the two (12 13%, 13 45%). Attempted dehydrofluorination of 13 to afford the required fluoride 12 proved to be unsuccessful under a variety of basic conditions both with and without the addition of silver salts (added to encourage fluoride precipitation). Nonetheless, three step deprotection of 12 and 13 (acid catalysed bis-ketal and MOM deprotection, saponification and ion-exchange) afforded the fluorovinyl 6 and difluoro 7 acids (63% and 96% respectively).
The fluorovinyl acid 6 was assayed against both type I and type II dehydroquinases and was shown to be highly selective for the type II proteins. Specifically 6 has a Ki of 10 μM against the dehydroquinase from Mycobacterium tuberculosis, making it the most potent inhibitor reported for this enzyme. Significantly, 4 which has a hydrogen instead of a fluorine at C(3), has a Ki of 200 μM against the same enzyme. Full biochemical results together with details of X-ray crystallographic studies of complexes of 6 with type II enzymes will be reported elsewhere.
Fluorine has been widely utilized to alter both the chemical and biochemical properties of many compounds, chiefly as a replacement for H, OH or neutral oxygen atoms (as part of a CHF of CF2 group); those pertinent to the shikimate pathway include 2-fluoro,18 6-fluoro19 (both H replacements), 3-fluoro20 and 3,5-difluoroshikimates21 (OH groups). This is the first report of fluorine as a replacement for an oxyanion, which it resembles more closely than a hydroxy group, by being both isosteric and isoelectronic.
We thank Neil Feeder for the X-ray crystallographic analysis of difluoride 13, John Greene and David Robinson for protein purification, and the BBSRC for postdoctoral support (MF).
Notes and references
- E. Haslam, Shikimic acid: Metabolism and Metabolites, John Wiley and Sons, Chichester, 1993 Search PubMed.
- C. Abell, in Comprehensive Natural Products Chemistry, ed. U. Sankawa, Elsevier, Amsterdam, 1999, vol. 1, p. 573 Search PubMed.
- N. H. Giles, M. E. Case, J. A. Baum, R. F. Geever, L. Huiet, V. B. Patel and B. M. Tyler, Microbiol. Rev., 1985, 49, 338 Search PubMed.
- F. Roberts, C. W. Roberts, J. J. Johnson, D. E. Kyle, T. Krell, J. R. Coggins, G. H. Coombs, W. K. Milhous, S. Tzipori, D. J. P. Ferguson, D. Chakrabarti and R. McLeod, Nature, 1998, 393, 801 CrossRef CAS.
- S. Chaudhuri, K. Duncan, L. D. Graham and J. R. Coggins, Biochem. J., 1991, 275, 1 CAS.
- B. W. Smith, M. J. Turner and E. Haslam, J. Chem. Soc., Chem. Commun., 1970, 842 Search PubMed.
- A. Schneier, C. Kleanthous, R. Deka, J. R. Coggins and C. Abell, J. Am. Chem. Soc., 1991, 113, 9416 CrossRef.
- A. R. Hawkins, W. R. Reinhert and N. H. Giles, Biochem. J., 1982, 203, 769 CAS.
- J. M. Harris, C. Gonzalez-Bello, C. Kleanthous, A. R. Hawkins, J. R. Coggins and C. Abell, Biochem. J., 1996, 319, 333 CAS.
- M. Frederickson, E. J. Parker, A. R. Hawkins, J. R. Coggins and C. Abell, J. Org. Chem., 1999, 64, 2612 CrossRef CAS.
- A. W. Roskaz, D. A. Robinson, T. Krell, I. S. Hunter, M. Frederickson, C. Abell, J. R. Coggins and A. J. Lapthorn, Structure, 2002, 10, 493 CrossRef.
- The corresponding 3,3-difluoro derivative of DHS has been described previously: S. Jiang, G. Singh, D. J. Boam and J. R. Coggins, Tetrahedron Asym., 1999, 10, 4087 Search PubMed.
- J.-L. Montchamp, F. Tian, M. E. Hart and J. W. Frost, J. Org. Chem., 1996, 61, 3897 CrossRef CAS.
- F. Tian, J.-L. Montchamp and J. W. Frost, J. Org. Chem., 1996, 61, 7373 CrossRef CAS.
- For optimal results, recrystallized ketone 11 [from hexane and diethyl ether] was used with fresh samples of DAST and anhydrous DME.
- Crystal data for 13: C16H26F2O8, M = 384.37, tetragonal, a = b = 19.184 (3), c = 10.165 (4) Å, U = 3740.9 (17) Å3, T = 180 (2) K, space group P41212 (no. 92), Z = 8, μ (Mo-Kα) = 0.12 mm−1, 3845 reflections measured, 3296 unique (Rint = 0.028) which were used in all calculations. The final wR(F2) were 0.047 [I > 2σ(I)] and 0.074 (all data).
- Full crystallographic data including lists of bond length, bond angles, hydrogen coordinates and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre, CCDC 184932. See http://www.rsc.org/suppdata/cc/b2/b205105m/ for crystallographic files in .cif or other electronic format.
- R. H. Rich and P. A. Bartlett, J. Org. Chem., 1996, 61, 3916 CrossRef CAS.
- J. K. Sutherland, W. J. Watkins, J. P. Bailey, A. K. Chapman and G. M. Davies, J. Chem. Soc., Chem. Commun., 1989, 1386 RSC.
- R. Brettle, R. Cross, M. Frederickson, E. Haslam, F. S. MacBeath and G. M. Davies, Bioorg. Med. Chem. Lett., 1996, 6, 1275 CrossRef CAS.
- F. J. Weigert and A. Shenvi, J. Fluor. Chem., 1994, 66, 19 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |