DOI:
10.1039/D4CE01018C
(Paper)
CrystEngComm, 2025,
27, 1707-1721
Structural analysis, in vitro anti-tubercular activities, and in silico ADMET evaluation of ethyl 7-methoxy-3-(4-substituted benzoyl)indolizine-1-carboxylates†
Received
5th October 2024
, Accepted 10th December 2024
First published on 9th January 2025
Abstract
Within the context of crucial exploration for innovative tuberculosis drugs, especially to counteract emerging drug resistance, we present a newly developed series of ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate analogues with various substituents (5a–p) to analyze their anti-tubercular efficacy. The synthesized indolizine derivatives were characterized by several spectroscopic methods such as FT-IR, 1H-NMR, and 13C-NMR. The structural aspects of these derivatives (5a, 5b, 5d, 5h, 5l, 5n, and 5p) were elucidated using single-crystal X-ray diffraction. Hirshfeld surface analysis and 2D fingerprint plots aided in investigating intermolecular hydrogen bonding and non-covalent interactions, and their impact on crystal packing. Moreover, computational analyses such as MEP, and energy framework calculations have identified the compound's interaction sites and inter-molecular interaction energies. FMO analysis was carried out to determine the global reactivity parameters of the synthesized compounds. NCI and QTAIM studies were conducted to evaluate the nature as well as qualitative and quantitative descriptions of intra and inter-molecular hydrogen bonding interactions present in these molecules and their crystal packing. The in vitro whole-cell anti-tubercular activity of the indolizine derivatives (5a–p) was evaluated against Mycobacterium tuberculosis H37Rv (susceptible), multidrug-resistant, and extensively drug-resistant strains (ATCC 27294 – American type cell culture). It is worth mentioning that some of these molecules (5a–5h, 5l, 5n, and 5p) exhibited potent activity against H37Rv and MDR-MTB strains, with minimum inhibitory concentrations ranging from 4–32 μg mL−1. In the case of the XDR-MTB strain, only compounds 5d, 5e, 5f, 5h, 5l, and 5n, showed moderate to good activities with MIC values ranging from 16–128 μg mL−1. Additionally, ADMET (in silico) studies of the potent compounds emphasized their drug-likeness, reinforcing their potential as promising compounds for further development as anti-TB agents.
1. Introduction
Tuberculosis (TB) is a highly contagious disease and a significant cause of illness and death, especially among low socioeconomic groups and marginalized sections of the community. This infectious disease is primarily caused by Mycobacterium tuberculosis (MTB), with a predominant impact on the lungs (pulmonary); however, it can disseminate to other organs in the body (extrapulmonary).1 TB continues to be a major threat to the human population. According to the World Health Organization (WHO), TB is the 13th leading cause of mortality and the second most common infectious disease-related cause of death, after COVID-19.2 In 2022, TB resulted in nearly 1.4 million deaths, including 208![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths among HIV/AIDS-positive patients.3 HIV-positive patients are 19 times more likely to develop tuberculosis than HIV-negative individuals. The present threat of TB to global public health is a consequence of the rising number of cases.4
000 deaths among HIV/AIDS-positive patients.3 HIV-positive patients are 19 times more likely to develop tuberculosis than HIV-negative individuals. The present threat of TB to global public health is a consequence of the rising number of cases.4
Several factors have contributed to the ongoing global health threat of TB, including drug resistance such as multidrug-resistant tuberculosis (MDR-TB), extensively drug-resistant tuberculosis (XDR-TB), and totally drug-resistant tuberculosis (TDR-TB) as well as co-morbidities with acquired immunodeficiency syndrome (AIDS) and the risk factors associated with developing diabetes mellitus in TB patients.5 The US Food and Drug Administration (US-FDA) has approved a few medications to advance TB care. When combined with first- or second-line anti-TB drugs, bedaquiline (approved in late 2012 under the FDA's accelerated review program) was found to have strong anti-TB effects and promising activity against MDR and XDR-TB, resulting in reduced treatment duration.6,7 However, nausea, elongation of the QT interval, and drug interactions with cytochrome P3A4 inducers and inhibitors have been reported as side effects of bedaquiline.8,9 In the latter part of 2013, the European Medicines Agency approved delamanid as the second anti-TB drug.10 However, there have been recent cases where strains of MTB have acquired resistance to both bedaquiline and delamanid due to prolonged treatment. Pretomanid, the most recent drug candidate from the TB therapeutic pathway, was approved by the FDA in 2019 for the treatment of MDR and XDR TB.11 Despite the availability of drugs, which are often cytotoxic and have reduced efficacy due to the evolution of resistant bacterial strains, there has not been significant progress in anti-TB drug discovery. A recent push for the development of new therapeutic agents to combat MDR, XDR, and TDR-TB has led to the identification of several promising anti-TB candidates.12,13 Among these, TBA-7371 (an azaindole) and Q203, or telacebec (an imidazopyridine amide), are particularly promising therapeutic candidates for the prospective treatment of MDR and XDR TB (Fig. S1†).9,14
Nevertheless, there is a growing demand for the development of new drug targets specific to mycobacteria that exhibit efficacy similar to existing anti-TB treatments. One promising approach in combating multidrug-resistant TB (MDR-TB) is the investigation of multi-targeting drugs, which involve a single molecule binding to multiple biological targets.15 This approach, known as polypharmacology, has shown promising results in improving effectiveness, synergistic effects, and adverse event profiles, while also reducing drug–drug interactions and the risk of resistance development. For example, SQ109, a therapeutic candidate in the TB drug pipeline, is a known MmpL3 inhibitor with good activity against MTB enzymes MenA and MenG.16 Additionally, numerous natural products have recently been identified as effective molecules against clinical MDR strains of MTB.17
Consequently, designing new chemical entities with various mechanisms of action plays a crucial role in treating H37Rv (susceptible), MDR, XDR, and TDR-TB infections.8,9 Considering the urgent need to combat emerging drug resistance, there is a strong necessity for novel drug development in tuberculosis treatment. This study continues our research on the design and synthesis of nitrogen-containing heterocyclic compounds, specifically “indolizine”, as anti-TB agents. In synthetic chemistry,18–22 indolizines have garnered significant interest due to their importance in various medicinal applications, including treatment for tuberculosis,23–27 antioxidant28 properties, anticancer,29 antimicrobial,30,31 anti-inflammatory,32–35 and larvicidal activity,36,37 and as NmeNANAS38 inhibitors. The above evidence propelled us to design, characterize, and evaluate the structure–activity relationship (SAR) by examining the influence of different R1 and R2 substitutions on the indolizine scaffold for their anti-tubercular potential against three MTB strains: H37Rv, rifampicin/isoniazid-resistant (MDR), and XDR (Scheme 1).
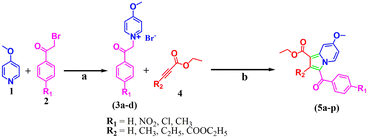 |
| | Scheme 1 The synthetic scheme involving the development of ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate (5a–p); (a) dry acetone, RT, 24 h. (b) K2CO3, DMF, RT, 30 min. | |
2. Experimental section
2.1 General chemical synthesis and characterization
All the chemicals for synthesizing the title compounds (5a–p) were purchased from Sigma-Aldrich, and/or Tokyo Chemical Industry (TCI) Company, Ltd. The solvents required for synthesis and crystallization were acquired from Thermo Fisher Scientific Company. The compounds were purified by column chromatography using Merck silica gel. Reaction progress was monitored by thin-layer chromatography (TLC) on 0.25 mm Merck 60 F254 silica gel plates, using a petroleum ether and ethyl acetate solvent system. UV light (254 nm and 365 nm) was used for product visualization, and the product was extracted by evaporating the solvent with a Heidolph rotary evaporator. The Bio-Technique vacuum pump and oven were used to dry the compounds. A DBK programmable melting point apparatus was used to determine the melting point of the synthesized compounds (5a–p). The Fourier transform infrared spectra (FT-IR) for indolizine derivatives were recorded on the IR affinity-1 spectrophotometer (Shimadzu) using the KBr diffuse reflectance system (DRS). In addition, NMR (1H and 13C) spectra were collected on Bruker ADVANCE III 400 MHz instruments in DMSO-d6 as a solvent with TMS set as an internal standard. The chemical shift (δ) values were expressed in ppm relative to TMS, while the coupling constant (J) values were determined in units of hertz (Hz). The splitting pattern is abbreviated as s, singlet, d, doublet, t, triplet, m, multiplet.
2.2 General synthetic procedure for the preparation of intermediates 4-methoxy-1-(2-(4-substituted phenyl)-2-oxoethyl)pyridin-1-ium bromide (3a–d)
To a stirred solution of 4-methoxy pyridine (0.0107 mol) in 25 mL of dry acetone, 4-substituted phenacyl bromide (0.0107 mol) was added in a 100 mL round-bottom (RB) flask and the reaction mixture was stirred at room temperature for 24 h. The completion of the reaction mixture was monitored using TLC. The solid formed was filtered and dried under vacuum to obtain the intermediate product 4-methoxy-1-(2-(4-substituted phenyl)-2-oxoethyl)pyridin-1-ium bromide (3a–d), with a yield of 96–99%.
2.3 General procedure for the preparation of ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylates (5a–p)
To a stirred solution of intermediate 4-methoxy-1-(2-(4-substituted phenyl)-2-oxoethyl)pyridin-1-ium bromide (3a–d) (0.0024 mol) and anhydrous K2CO3 (0.0048 mol) in 10 mL of dry DMF solvent in a 50 mL RB flask, substituted alkyne carboxylate (0.0024 mol) was added and stirred for 30 min at room temperature. The completion of the reaction mixture was monitored using TLC. After the reaction was completed, the reaction mixture was poured into cold water and extracted with ethyl acetate (50 ml), and then evaporated under reduced pressure with the help of a Heidolph rotary evaporator. Further, the obtained solid was purified through column chromatography using 60–120 mesh silica gel with petroleum ether (60–80), and ethyl acetate was used as an eluent solvent in the ratio (7:3) to obtain the pure ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate (5a–p). The product yield is in the range of 60–82% (Table 1), and the plausible mechanism route for the synthesized compounds (5a–p) is mentioned in Fig. S2.† The physicochemical constants of ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate derivatives are tabulated in Table 1, and the characterization details are reported in the ESI† (Fig. S3–S35).
Table 1 Physicochemical characteristics of ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate analogues (5a–p)
| Code |
Molecular formula |
R1 |
R2 |
Yielda,b (%) |
M.P. (°C) |
clogPc |
|
All the synthetic compounds were characterized by physical and spectral data.
Yield was calculated after column chromatography purification and confirmation.
ChemBioDraw Ultra 16.0v program was used to calculate clogP.
|
|
5a
|
C19H17NO4 |
H |
H |
79 |
132–134 |
4.3781 |
|
5b
|
C20H19NO4 |
H |
CH3 |
65 |
125–127 |
4.8771 |
|
5c
|
C21H21NO4 |
H |
C2H5 |
68 |
111–113 |
5.4061 |
|
5d
|
C22H21NO6 |
H |
CO2CH2CH3 |
70 |
126–128 |
3.8741 |
|
5e
|
C19H16N2O6 |
NO2 |
H |
64 |
169–172 |
4.2535 |
|
5f
|
C20H18N2O6 |
NO2 |
CH3 |
72 |
160–162 |
4.7525 |
|
5g
|
C21H20N2O6 |
NO2 |
C2H5 |
66 |
145–147 |
5.2815 |
|
5h
|
C22H20N2O8 |
NO2 |
CO2CH2CH3 |
62 |
184–186 |
3.7419 |
|
5i
|
C19H16ClNO4 |
Cl |
H |
75 |
164–166 |
5.0993 |
|
5j
|
C20H18ClNO4 |
Cl |
CH3 |
61 |
122–124 |
5.5983 |
|
5k
|
C21H20ClNO4 |
Cl |
C2H5 |
67 |
108–110 |
6.1273 |
|
5l
|
C22H20ClNO6 |
Cl |
CO2CH2CH3 |
82 |
148–150 |
4.5899 |
|
5m
|
C20H19NO7 |
CH3 |
H |
68 |
150–152 |
4.8771 |
|
5n
|
C21H21NO4 |
CH3 |
CH3 |
63 |
112–114 |
5.3761 |
|
5o
|
C22H23NO4 |
CH3 |
C2H5 |
60 |
110–112 |
5.9051 |
|
5p
|
C23H23NO6 |
CH3 |
CO2CH2CH3 |
78 |
162–164 |
4.3731 |
2.4 Methods for the growth of single crystals
A minimum 5 mg amount of compound (purified by column chromatography) was taken in a 5 ml glass vial and dissolved in a small amount (1–2 ml) of ethanol solvent to obtain a single crystal. After the proper dissolution of compounds (5a–p), it was covered with parafilm, and 3 to 4 small holes were made on the top. The glass vial was then kept for slow evaporation at low temperatures under dust-free and vibration-free conditions to obtain single crystals (Fig. S36†), and a Nikon (H600L) polarized optical microscope (POM) was used to select a good quality single crystal for data collection.
2.5 Single crystal X-ray data collection and structure refinement
Crystals of the synthesized compounds with different shapes, which were shiny and colorless, were acquired through the slow evaporation technique at low temperatures using ethanol as the solvent. The good single-crystals of 5a, 5b, 5d, 5h, 5l, 5n, and 5p were obtained and collected at the X-ray diffraction beamline (XRD1) of the Elettra Synchrotron (Trieste, Italy).39 The crystal was immersed in NHV oil (Jena Bioscience, Jena, Germany) and subsequently fixed to the goniometer head with nylon loops (MiTeGen, Ithaca, USA). The complete dataset was collected at 100 K using a nitrogen stream provided by an Oxford Cryostream 700. Data were collected using a Pilatus 2M hybrid-pixel area detector (DECTRIS Ltd., Baden-Daettwil, Switzerland) with a monochromatic wavelength of 0.700 Å and employing the rotating crystal method. The diffraction data were indexed and integrated using the XDS software.40 The structure was solved using Olex2 software,41 employing the intrinsic phasing SHELXT program42 for structure solution, followed by refinement using the SHELXL package,43 which utilizes least-squares minimization. Mercury 4.2.0 software and the POV-Ray program were utilized to generate the molecular graphics and images.44,45 Geometric calculations and the generation of the hydrogen bond table were performed using the PLATON program.46 Hydrogen atoms in all 5a, 5b, 5d, 5h, 5l, 5n, and 5p compounds were located from the diffraction maps. The C–H bond distances were constrained to 0.95 Å for CH groups, 0.99 Å for CH2 groups, and 0.98 Å for CH3 groups. The structure was refined to R factors of 0.0423, 0.0359, 0.0368, 0.0470, 0.035, 0.0429, and 0.0374 for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p respectively. Refinement factors and crystallographic data for the compound discussed, along with CIF files deposited at the Cambridge Crystallographic Data Centre (CCDC), are mentioned in Table S1.†
2.6 Hirshfeld surface analysis
The Hirshfeld surface at the molecular level is built around a spherical atom using electron density distribution.47,48 The uniqueness of the Hirshfeld surface lies in its association with a specific crystal structure and a defined set of spherical atomic electron densities. This characteristic suggests the possibility of gaining additional insight into the intermolecular interactions in molecular crystals.49 On Hirshfeld surfaces, the distances de and di represent the distances from the surface to the external nucleus and from the surface to the internal nucleus, respectively. The 2D fingerprint plot, formed by combining de and di, summarizes intermolecular contacts within the crystal.50 The Hirshfeld surfaces, including dnorm, di, de, shape index, curvedness, and two-dimensional (2D) fingerprint plots, were generated using the Crystal Explorer 21.5 software to visualize the intermolecular interactions within these molecules.51,52
2.7 Energy framework analysis
An energy framework analysis was employed to graphically depict the nature of the energy associated with the short interaction within the crystal structure. The energy framework provides a qualitative description of the three-dimensional topology of the associated interaction present within the crystal structure, which was carried out using Crystal Explorer 21.5.52,53 This approach provides an understanding of intermolecular interaction energy and their corresponding partitioned energy through 3D-graphical representations. The energy framework analysis was performed using the B3LYP method with the 6-31G(d,p) basis set.
2.8 Computational analysis
Density functional theory (DFT) calculations were performed using the GAUSSIAN 09 program for the synthesized compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p.54 The molecular structures of the compounds were derived from single-crystal X-ray crystallographic data. The single point energy calculations were executed using the B3LYP method with the 6-311G(d,p) basis set.55 The results were visualized using the GAUSS VIEW 6.0 program. The reactivity parameters for the mentioned compounds were derived from the FMO analysis. Additionally, MEP surfaces were generated to identify potential active sites involved in non-covalent interaction formation.56 Furthermore, essential tools such as NCI and QTAIM analyses were utilized to assess the strength and qualitative and quantitative nature of HBs present within the molecular assembly of these compounds using the Multiwfn 3.6 package,57 with results visualized using VMD.58
2.9 Anti-tubercular activity
The designed compounds (5a–p) were assessed for their ability to combat tuberculosis by testing their activity against three different types of MTB strains, specifically H37Rv, MDR, and XDR strains. This evaluation was conducted using the resazurin microplate assay (REMA) method, which utilizes colorimetric measurements.59 A 100 μL portion of Middelbrook 7H9 broth prepared and administered aseptically was placed in each 96-well flat-bottomed microtiter plate with covers (Lasec, South Africa). The test compounds were precisely measured, dissolved in a suitable solvent, and subjected to filter sterilization with a 0.2-micron polycarbonate filter to ensure accuracy. The test samples were divided into smaller portions and placed in cryovials before storing at −20 °C. Then, 100 μL of each test sample was introduced into the wells containing Middlebrook 7H9 broth. The broth was enriched with 0.1% casitone, 0.5% glycerol, and 10% OADC, which stands for oleic acid, albumin, dextrose, and catalase. The test samples were diluted stepwise, with each dilution being two times weaker than the previous one, directly in the microtiter plate's broth. This process resulted in a range of desired concentrations from 40–0.625 μg mL−1.
Fresh inoculums were prepared from clinical isolates obtained from Middlebrook 7H11 agar plates. The process involved scraping a loopful of colonies and re-suspending them into Middlebrook 7H9 broth containing glass beads. The initial density of the inoculum was calibrated to match the McFarland number 1 standard. Afterwards, it was further diluted in M7H9 broth at a ratio of 1:10. Subsequently, 100 μL of this diluted inoculum was added to both the test samples and wells without any drugs. Each isolate was accompanied by a growth control and a sterile control. To prevent evaporation during incubation, the sterile M7H9 broth was added to the perimeter wells. The plate was securely covered and placed inside a plastic bag before being placed in an incubator at 37 °C. Following an incubation period of 8 days, a volume of 30 μL from a 0.02% working solution of resazurin salt was introduced into each microtiter well. After incubating the plates overnight, they were examined the next day. A color change from blue to pink indicated a positive reaction, indicating the reduction of resazurin to resorufin. This confirmed the viability and growth of MTB cells and, consequently, their drug resistance. The MICs were determined by identifying the lowest concentration of the drug that prevented the organism's growth without observing any color changes in the well.
2.10 Safety studies (in vitro)
The safety of the tested indolizines was evaluated using an MTT assay. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cytotoxicity assay was used to evaluate the cytotoxic effect of the most promising compounds against peripheral blood mononuclear cells (PBMCs) according to the described protocol.60 Cells were pipetted (90 μL of cell culture, 1 × 105 cells per mL) into each well of 96-well microtiter plates, and the outer wells were filled with PBS (phosphate buffered saline) to prevent the medium from evaporation during incubation. After that, plates were incubated at 37 °C for 24 h. Each well of the plate was then treated with 10 mL of the compounds (1000–5 mg mL−1). In the control wells, the negative control DMSO (dimethyl sulfoxide) and media were added. The plates were incubated for 2 days at 37 °C in a humidified incubator that contained a 5% CO2 atmosphere. After the incubation, 20 mL of MTT reagent (5 mg mL−1) was added to the individual well. The plate was then incubated for 4 h at 37 °C (5% CO2 incubator). The media was then removed, and an aliquot of 100 μL DMSO was added to each well to dissolve the formazan crystals formed in metabolically active cells. After that, the plates were incubated for an extra hour. The absorbance of the formazan was evaluated at 590 nm using an ELISA plate reader (Thermo Scientific Multiskan GO).
3. Results and discussion
3.1 Spectroscopic data
The FT-IR spectra of the ethyl 3-(4-substituted benzoyl)-7-methoxyindolizine-1-carboxylate compounds showed the aromatic C–H stretching frequency ∼3150–3000 cm−1. The C–H aliphatic stretching frequency of the methyl and methylene groups was observed at the regions of 2990–2950 cm−1 and 2870–2840 cm−1, respectively. Also, the carbonyl functional stretching frequency of the ester group was observed at 1720–1700 cm−1, and the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequency was observed in the 1340–1320 cm−1 region. para-Substituted compounds showed their stretching frequency at 810–790 cm−1 (Fig. S3†). In the 1H-NMR spectrum, the fused heteroaromatic ring protons of the indolizine ring have appeared at 7.55 to 7.52 ppm, and the ester CH2 protons have appeared at 4.29 to 4.27 ppm. Further, the CH3 protons attached to the heteroatom oxygen, which is a part of the indolizine moiety, appeared at 3.93 to 3.89 ppm, and ester CH3 protons appeared at 1.36 to 1.33 ppm. The 1H and 13C-NMR spectra of respective indolizine derivatives are mentioned in the ESI† (Fig. S4–S35).
N stretching frequency was observed in the 1340–1320 cm−1 region. para-Substituted compounds showed their stretching frequency at 810–790 cm−1 (Fig. S3†). In the 1H-NMR spectrum, the fused heteroaromatic ring protons of the indolizine ring have appeared at 7.55 to 7.52 ppm, and the ester CH2 protons have appeared at 4.29 to 4.27 ppm. Further, the CH3 protons attached to the heteroatom oxygen, which is a part of the indolizine moiety, appeared at 3.93 to 3.89 ppm, and ester CH3 protons appeared at 1.36 to 1.33 ppm. The 1H and 13C-NMR spectra of respective indolizine derivatives are mentioned in the ESI† (Fig. S4–S35).
3.2 Single crystal structure X-ray diffraction analysis
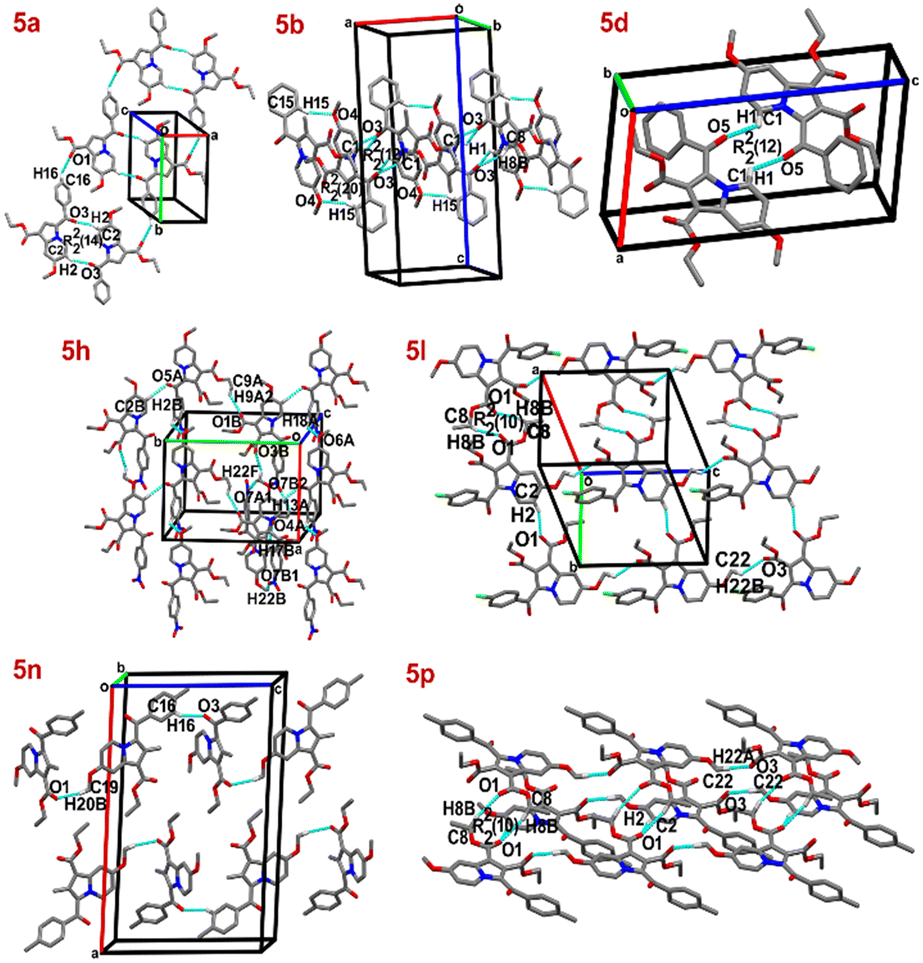
The synthesized compounds 5a, 5d, 5l, and 5p crystallize in the triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) whereas 5b, 5h, and 5n crystallize in the monoclinic crystal system with the space group P21/c. The respective lattice parameters and other crystallographic information for the mentioned compounds are provided in Table S1.† The molecular structures of the seven crystals are depicted in Fig. 1. The molecular packing of these compounds in the solid state is mostly influenced by the weak C–H⋯O hydrogen bonding along with the additional C–H⋯π hydrogen bonding interactions (Fig. 2 and S37 and Table S2†). Prominent intra- and inter-molecular H-bonding interactions associated with these reported compounds are depicted in Table 2.
whereas 5b, 5h, and 5n crystallize in the monoclinic crystal system with the space group P21/c. The respective lattice parameters and other crystallographic information for the mentioned compounds are provided in Table S1.† The molecular structures of the seven crystals are depicted in Fig. 1. The molecular packing of these compounds in the solid state is mostly influenced by the weak C–H⋯O hydrogen bonding along with the additional C–H⋯π hydrogen bonding interactions (Fig. 2 and S37 and Table S2†). Prominent intra- and inter-molecular H-bonding interactions associated with these reported compounds are depicted in Table 2.
 |
| | Fig. 1 Molecular structures of 5a, 5b, 5d, 5h, 5l, 5n, and 5p with anisotropic displacement ellipsoids drawn at the 50% probability level. Intramolecular hydrogen bonds are drawn as dotted lines, and H-atoms not involved in interactions are omitted for clarity. | |
 |
| | Fig. 2 The molecular assembly of 5a, 5b, 5d, 5h, 5l, 5n, and 5p through dimer motifs and other weak C–H⋯O interactions. | |
Table 2 The prominent intra and inter-molecular H-bonding interactions are associated with the compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p
| Code |
Intra and inter-molecular H-bonding interactions |
| D–X⋯A |
D–X (Å) |
X⋯A (Å) |
D⋯A (Å) |
<D–X⋯A (°) |
|
5a
|
C1–H1⋯O3 |
0.917(1) |
2.246(2) |
2.884(1) |
126.2(1) |
| C4–H4⋯O1 |
0.966(1) |
2.501(1) |
3.064(1) |
117.1(1) |
| C2–H2⋯O3i |
0.978(2) |
2.281(2) |
3.199(1) |
156.1(1) |
| C16–H16⋯O1ii |
0.977(2) |
2.586(2) |
3.326(1) |
132.8(1) |
| Symmetry codes: (i) 2 − x, 1 − y, −z; (ii) 1 + x, −1 + y, z |
|
5b
|
C1–H1⋯O3 |
0.939(1) |
2.179(1) |
2.829(1) |
125.5(1) |
| C4–H4⋯O2 |
0.966(1) |
2.359(1) |
2.922(1) |
116.7(8) |
| C11–H11B⋯O1 |
0.991(1) |
2.467(1) |
2.926(1) |
107.8(9) |
| C1–H1⋯O3i |
0.939(1) |
2.478(1) |
3.130(1) |
126.7(1) |
| C8–H8B⋯O3ii |
0.963(1) |
2.398(1) |
3.288(1) |
153.5(1) |
| C15–H15⋯O4iii |
0.989(1) |
2.495(1) |
3.420(1) |
155.7(1) |
| Symmetry codes: (i) −x, 2 − y, −z; (ii) 1 + x, y, z; (iii) 1 − x, 2 − y, −z |
|
5d
|
C1–H1⋯O5 |
0.940(1) |
2.267(1) |
2.878(1) |
121.0(9) |
| C4–H4⋯O2 |
0.931(1) |
2.399(1) |
2.945(1) |
116.5(9) |
| C1–H1⋯O5i |
0.957(1) |
2.390(1) |
3.068(1) |
127.5(9) |
| Symmetry codes: (i) −1 − x, 1 − y, 1 − z |
|
5h
|
C1B–H1B⋯O5B |
0.981(2) |
2.269(2) |
2.861(2) |
122.7(1) |
| C4B–H4B⋯O1B |
0.977(2) |
2.497(2) |
3.029(2) |
117.8(1) |
| C2B–H2B⋯O5Ai |
0.98(2) |
2.519(2) |
3.459(3) |
169.0(2) |
| C9A–H9A2⋯O1Bii |
0.98(2) |
2.573(2) |
3.484(4) |
154.0(2) |
| C17B–H17B⋯O4Aiii |
0.956(2) |
2.600(2) |
3.484(2) |
158.6(2) |
| C18A–H18A⋯O6Aiv |
1.006(2) |
2.533(2) |
3.115(2) |
117.8(1) |
| C22A–H22B⋯O7B1v |
1.008(2) |
2.489(2) |
3.453(2) |
164.1(2) |
| C22A–H22B⋯O3Bvi |
0.99(2) |
2.540(2) |
3.478(2) |
159.1(2) |
| C22B–H22F⋯O3Bvii |
1.02(2) |
2.521(2) |
3.363(2) |
140.2(2) |
| C22B–H22E⋯O7A1viii |
0.98(2) |
2.524(2) |
3.486(2) |
163.3(2) |
| Symmetry codes: (i) 1/2 − x, −1/2 + y, 1/2 − z; (ii) −1/2 + x, 1/2 − y, 1/2 + z; (iii) 1 + x, y, z; (iv) −1 + x, y, z; (v) −1 + x, y, z; (vi) −1/2 + x, 1/2 − y, 1/2 + z; (vii) −1 + x, y, z; (viii) 1 + x, y, z |
|
5l
|
C1–H1⋯O5 |
0.939(2) |
2.244(1) |
2.896(1) |
126.0(1) |
| C4–H4⋯O2 |
0.933(1) |
2.355(2) |
2.904(1) |
117.4(1) |
| C2–H2⋯O1i |
0.970(2) |
2.282(2) |
3.194(2) |
156.5(1) |
| C8–H8B⋯O1ii |
0.952(1) |
2.537(1) |
3.462(1) |
164.0(1) |
| C22–H22B⋯O3iii |
0.963(2) |
2.574(2) |
3.222(1) |
124.9(1) |
| Symmetry codes: (i) −1 + x, y, z; (ii) 1 − x, −y, 1 − z; (iii) x, y, 1 + z |
|
5n
|
C1–H1⋯O3 |
0.952(1) |
2.262(1) |
2.832(1) |
117.7(1) |
| C4–H4⋯O2 |
0.969(2) |
2.337(1) |
2.889(1) |
115.4(1) |
| C11–H11B⋯O1 |
0.987(1) |
2.457(2) |
2.943(1) |
110.0(9) |
| C16–H16⋯O3i |
0.973(2) |
2.524(2) |
3.420(1) |
153.1(1) |
| C19–H20B⋯O1ii |
0.979(2) |
2.473(1) |
3.4425(2) |
170.5(1) |
| Symmetry codes: (i) x, 1/2y, 1/2 + z; (ii) x, −1/2 − y, −1/2 + z |
|
5p
|
C1–H1⋯O5 |
0.951(1) |
2.262(1) |
2.895(1) |
123.3(1) |
| C4–H4⋯O2 |
0.941(1) |
2.361(1) |
2.913(13) |
117.2(1) |
| C2–H2⋯O1i |
0.977(2) |
2.284(1) |
3.208(14) |
157.5(1) |
| C8–H8B⋯O1ii |
0.972(1) |
2.495(1) |
3.443(12) |
165.1(1) |
| Symmetry codes: (i) −1 + x, y, z; (ii) 1 − x, −y, 1 − z |
Crystal structure and packing of 5a.
The compound 5a crystallizes in the triclinic space group P with two molecules in the unit cell (Z = 2) and one in the asymmetric unit (Z′ = 1). It forms a ring dimer with a R22(14) graph-set motif via C2–H2⋯O3 [2.501(1) Å, 156.1(1)°] hydrogen bonding interactions. Meanwhile, other C–H⋯O interactions [2.586(2) Å, 132.8(1)°] propagate along the ab-plane in the crystal structure (Fig. 2, Table 2). Additionally, C–H⋯π interactions [π = Cg, C8–H8B⋯Cg1, 2.862(1) Å, 160.7(1)°] between the methylene group of the ester and the indolizine moiety (Fig. S37, Table S2†), as well as π⋯π (CgCg) interactions [Cg2⋯Cg2, 3.621(9) Å], stabilize the molecular assembly (Fig. S38, Table S3†).
Crystal structure and packing of 5b.
The compound 5b crystallizes in the monoclinic space group P21/c with four molecules in the unit cell (Z = 4) and one in the asymmetric unit (Z′ = 1). Its molecular assembly prefers the formation of two dimers with R22(12) and R22(20) graph-set motifs by C–H⋯O [C1–H1⋯O3, 2.478(1) Å, 126.7(1)°; C15–H15⋯O4, 2.495(1) Å, 155.7(1)°] hydrogen bonding interactions (Fig. 2, Table 2). Further the methylene group of the ester engages C–H⋯π interaction with the indolizine and benzoyl moieties which stabilized the crystal packing [C8–H8A⋯Cg1, 2.954(1) Å, 128.3(8)°, C20–H20A⋯Cg3, 2.973(1) Å, 143.7(9)°] (Fig. S37, Table S2†). In addition, the π⋯π interaction [Cg1⋯Cg2, 3.778(9) Å] stabilizes the molecular assembly (Fig. S38, Table S3†).
Crystal structure and packing of 5d.
The compound 5d crystallizes in the triclinic space group P with two molecules in the unit cell (Z = 2) and one molecule in the asymmetric unit (Z′ = 1). The ring dimer with a R22(10) graph-set motif is stabilized by C1–H1⋯O5 [2.390(1) Å, 127.5(9)°] hydrogen bonding interactions (Fig. 2, Table 2). The methylene group of the ester forms C–H⋯π interaction with both the indolizine and benzoyl moieties to stabilize this molecular assembly [C9–H9A⋯Cg3, 2.927(1) Å, 155.2(1); C9–H9C⋯Cg1, 2.882(1) Å, 166.9(1); C13–H13B⋯Cg3 2.788(1) Å, 132.0(1), (Fig. S37, Table S2†). Moreover, the crystal packing is stabilized by π⋯π interaction [Cg2⋯Cg2, 3.559(7) Å] (Fig. S38, Table S3†).
Crystal structure and packing of 5h.
The compound 5h crystallizes in the monoclinic space group P21/c with eight molecules in the unit cell and two molecules in the asymmetric unit cell (Z = 8, Z′ = 2). The molecule exhibits several weak C–H⋯O hydrogen bonds, namely C2B–H2B⋯O5A [2.519(2) Å, 169.0(2)°], C9A–H9A2⋯O1B [2.573(2) Å, 154.0(2)°] and C22B–H22F⋯O3B [2.521(2) Å, 140.2(2)°] in their crystal packing (Fig. 2, Table 2). Unlike other crystal structures, none of these C–H⋯O interactions form dimers. However, the methylene group of the ester forms C–H⋯π interactions with the indolizine moiety, specifically C12A–H12A⋯Cg1 [2.876(2) Å, 146.6(1)°], C12A–H12A⋯Cg2 [2.901(2) Å, 151.7(1)°], and C13B–H13F⋯Cg8 [2.961(3) Å, 170(2)°] interactions (Fig. S37, Table S2†). Additionally, a π⋯π interaction [Cg1⋯Cg2, 3.586(1) Å] contributes to the stabilization of the molecular assembly (Fig. S38, Table S3†).
Crystal structure and packing of 5l.
The compound 5l crystallizes in the triclinic space group P with two molecules in the unit cell and one molecule in the asymmetric unit cell (Z = 2, Z′ = 1). The crystal structure shows a ring dimer with a R22(10) graph-set motif self-assembled by C8–H8B⋯O1 [2.537(1) Å, 164.0(1)°] hydrogen bonds along with other C–H⋯O interactions (Fig. 2, Table 2). In addition, the crystal packing is consolidated by C–H⋯π interaction through the methylene group of the ester with the indolizine moiety such as C13–H13C⋯Cg1 [2.787(2) Å, 154.0(1)°] and C13–H13C⋯Cg2 [2.970(2) Å, 130.0(2)°] (Fig. S37, Table S2†). Furthermore, the molecular assembly is stabilized through π⋯π interactions [Cg1⋯Cg2, 3.790(9) Å; Cg2⋯Cg2, 3.842(9) Å] (Fig. S38, Table S3†).
Crystal structure and packing of 5n.
The compound 5n crystallizes in the monoclinic space group P21/c with two molecules in the unit cell and one in the asymmetric unit (Z = 2, Z′ = 1). The crystal structure is stabilized by C–H⋯O interactions, without the formation of dimers as seen in other structures [C16–H16⋯O3, 2.524(2) Å, 153.1(1)°] and [C19–H20B⋯O1, 2.473(2) Å, 170.5(1)°] (Fig. 2, Table 2). In addition to this, C–H⋯π interactions [C11–H11B⋯Cg1, 2.915(1) Å, 113.4(1)°] between the methylene group of the ester and the indolizine moiety (Fig. S37, Table S2†), along with a π⋯π interaction [Cg1⋯Cg2, 3.483(9) Å], stabilize the molecular assembly (Fig. S38, Table S3†).
Crystal structure and packing of 5p.
The synthesized compound 5p crystallizes in the triclinic space group P with two molecules in the unit cell (Z = 2) and one in the asymmetric unit (Z′ = 1). The crystal structure is established through the C–H⋯O hydrogen bond dimer with a R22(10) graph-set motif [C8–H8B⋯O1, 2.495(1) Å, 165.1(1)°] and the C–H⋯O hydrogen bond propagated along the a-axis [C2–H2⋯O1, 2.284(2) Å, 157.5 (1)°] (Fig. 2, Table 2). This molecular assembly also prefers C–H⋯π interaction through the methylene group of the ester with the indolizine moiety [C13–H13B⋯Cg1, 2.771(2) Å, 153.5(1)°] (Fig. S37, Table S2†) and π⋯π interactions [Cg1⋯Cg2, 3.863(9) Å; Cg1⋯Cg2, 3.863(9) Å] (Fig. S38, Table S3†) stabilized the molecular assembly for effective crystal packing.
In summary, it is interesting to note that the crystal structures of 5a, 5b, 5d, 5l, and 5p prefer C–H⋯O hydrogen bond dimers, except 5n and 5h. It is also noteworthy that the phenyl-substituted derivatives (5a, 5b, and 5d) prefer a different type of dimer formation via C–H⋯O hydrogen bonds, which may be influenced by the R2 substituents. The indolizine derivatives, such as the p-nitrophenyl derivative (5h) and the p-toluene derivative (5n), prefer non-dimeric C–H⋯O interactions in their crystal packing. Furthermore, it is observed that the crystal structures of all the indolizine derivatives (5a, 5b, 5d, 5h, 5l, 5n, and 5p) favour C–H⋯π interactions between the methylene-ester moiety of the hydrogen atom and the indolizine ring. In contrast, the crystal structures of 5b and 5d exhibit an additional C–H⋯π interaction involving the methylene-ester moiety and the benzoyl group. Additionally, compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p, exhibit π⋯π (Cg⋯Cg) stacking interactions, which play a crucial role in the crystal packing (Fig. S38, Table S3†).
3.3 HS investigation and 2D fingerprint plots
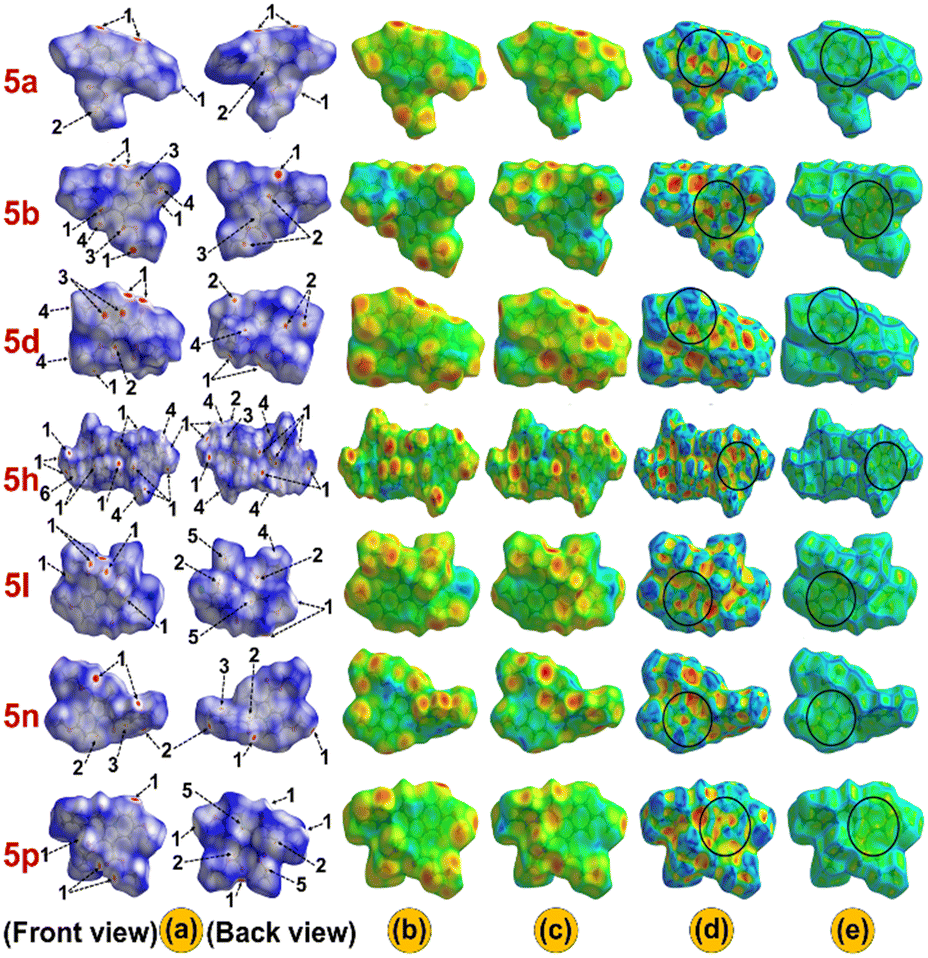
The HS of the single-crystal structures was analyzed with respect to dnorm, di, de, shape index, and curvedness to illustrate the characteristics of intermolecular interactions (Fig. 3). In the dnorm surfaces, red and blue areas denote shorter and longer contacts compared to the van der Waals radii, respectively, with white indicating contacts close to the van der Waals radii. Additionally, the red spots (labelled 1) on the dnorm surfaces indicate existing hydrogen bonding H⋯O interactions and contribute to the formation of dimers in specific crystal structures (Fig. S39†). Meanwhile, the areas labelled 2, especially in 5a, 5b, 5d, 5l, 5n and 5p, suggest the C⋯H interaction sites. Similarly, the light red spots (labelled 3) reveal the presence of C⋯C interactions, which may be associated with π⋯π interactions in the crystal packing. In contrast, the blue regions (labelled 4) indicate H⋯H interactions (Fig. 3(a)). The de and di surfaces represent the distances from the HS to the closest nuclei inside and outside the surface, respectively (Fig. 3(b) and (c)). Further, the shape index and curvedness surface analysis was carried out to investigate the π⋯π (Cg⋯Cg) interactions present in the system.61,62 The presence of red triangles in concave regions and blue triangles in convex regions as highlighted by the black circle on the shape index surface provides evidence of the π⋯π stacking and aromatic ring index, respectively (Fig. 3(d)). The HS, when mapped with curvedness, reveals flat regions outlined in blue, highlighting the contributions of the rings in π⋯π stacking interactions (Fig. 3(e)). Further, the 2D fingerprint plot depicted the percentage distribution of various interactions within the molecule, highlighting the presence of short contacts (Fig. S40(a)†). The 2D plot indicates that H⋯H interactions make up the highest contribution to the overall interactions i.e., 55.5%, 53.4%, 50.7%, 49.1%, 45.9%, 37.7%, and 35.4% in 5n, 5b, 5p, 5a, 5d, 5l, and 5h, respectively. Conversely, the contribution of H⋯O contact is relatively higher in 5h and 5d (33.0%, 27.6%) compared to 5a (18.3%), 5b (17.9%), 5l (24.5%), 5n (18.0%), and 5p (24.7%) on the HS (Fig. S40(b)†). Additionally, the contribution of C⋯C interactions to the total interactions is 2.2%, 5.0%, 1.4%, 2.7%, 3.3%, 5.1%, and 3.2% in 5a, 5b, 5d, 5h, 5l, 5n, and 5p, respectively, indicating the existence of π⋯π stacking within the compounds (Fig. S38†), as explained by the crystal structure analysis. The HS and 2D plots of compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p are in good agreement with the previously reported similar indolizine derivatives.9,35,63
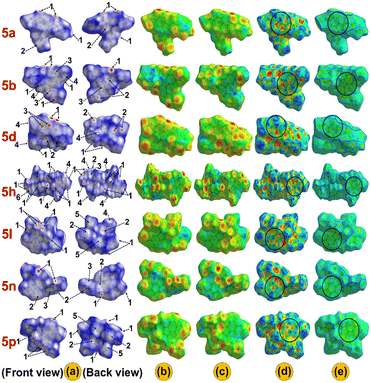 |
| | Fig. 3 Surfaces have been drawn over a) dnorm (front and back view), b) di, c) de, d) shape-index, and e) curvedness for the compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p. | |
3.4 Energy framework calculation
The energy framework calculation was carried out to construct the three-dimensional topology of interactions within a supramolecular structure. The scaling factors employed in generating electron densities for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p are as reported by Mackenzie et al.64,65 (Table S4†). The cut-off value for the threshold energy was set at 0 kJ mol−1, and the tube size was maintained at 100 in all energy frameworks. The interaction energies between molecular pairs are represented by cylinders connecting the molecules, with the cylinder radii proportional to the strength of these intermolecular interactions. The interaction energies between selected molecules within the cluster, formed around a single molecule at a radius of 3.8 Å with unit cell dimensions of 1 × 1 × 1, were analyzed. The visualizations of various energies like total interaction, dispersion, and electrostatic (coulombic) energies are depicted by blue, green, and red colored cylinders, respectively (Fig. S43†).
These energies with color-coded molecular pairs are illustrated in Fig. S41 and Tables S5–S11.† The total interaction energies for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p are −235.5 kJ mol−1, −288.1 kJ mol−1, −300.3 kJ mol−1, −356.3 kJ mol−1, −305.2 kJ mol−1, −191.5 kJ mol−1, and −319.7 kJ mol−1, respectively. Further, in all the compounds (5a, 5b, 5d, 5h, 5l, 5n, and 5p) the dispersion energy predominates over the electrostatic energy and has a major contribution to the total interaction energy, similar to the energy frameworks of reported indolizine derivatives.9,35,63 Notably, the addition of an extra ester moiety leads to an increase in the total interaction energy. In addition, electron-withdrawing groups like NO2 and Cl also raise the total interaction energy in these indolizine systems (Table S12†). Fig. S42† illustrates the dnorm mapped on the HS for the central molecule and energy frameworks.
3.5 Computational analysis
3.5.1. FMOs analysis and global reactivity descriptors.
The HOMO–LUMO energy gap is useful for determining the electronic arrangements of a molecule and plays a crucial role in analyzing its chemical reactivity.66,67 Further, the global chemical reactivity descriptors were calculated utilizing Koopmans' theorem.68 The positive phase is depicted in red in the three-dimensional orbital plots, while the negative phase is represented in green. In compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p, the HOMO electron cloud basically covers the indolizine ring, whereas the LUMO electron cloud is distributed over the indolizine and benzoyl ring (for 5a, 5b, 5d, 5l, 5n and 5p) except for 5h where the LUMO electron cloud covers majorly the benzoyl ring, which may be due to the presence of more electron withdrawing nitro group. This represents a shifting of the electron cloud from the indolizine moiety to the benzoyl ring during the HOMO–LOMO transition (Fig. 4).
 |
| | Fig. 4 Frontier molecular orbital (energy gap between bottom: HOMO, top: LUMO) of compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p. | |
Furthermore, the energy gap (ΔE = ELUMO − EHOMO) can also be used to define the stability and reactivity of synthesized compounds. Table S13† presents the HOMO and LUMO values for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p, along with the energy gaps and global chemical reactivity descriptors. Among the compounds, 5a has the largest energy gap, while 5h has the smallest. The high value for 5a indicates its high dynamic stability and being less prone to electron transition. The negative chemical potential (μ) values observed for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p suggest that they do not spontaneously disintegrate into their constituents.69 The compound 5h exhibits maximum chemical softness (S), indicating its high polarizability, which is further evidenced by its low chemical hardness (η) value.
3.5.2. MEP surface analysis.
The molecular electrostatic potential (MEP) surface is crucial for identifying and determining the reactive sites of the compounds for electrophilic or nucleophilic attacks. The MEP produces three-dimensional potential maps showing the isoelectron density surface of the molecule. The MEP maps for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p were generated, where red color denotes the most significant negative charge, highlighting a favourable site for an electrophilic attack. Conversely, blue color signifies the most significant positive charge, making it more susceptible to nucleophilic attack (Fig. 5). The reddish yellow regions (related to negative potential) over the carbonylic oxygen atoms in the above-mentioned compounds, with an additional region over oxygen atoms of the nitro group in 5h, indicate the possible sites for non-covalent interactions like weak hydrogen bonding.
 |
| | Fig. 5 The depiction of the molecular electrostatic potential for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p. | |
3.5.3. Non-covalent interactions analysis.
The reduced density gradients (RDGs) with low electron densities are basically associated with regions exhibiting weak non-covalent interactions (NCIs). Therefore, in order to assess and identify the regions implicated in the weak interactions in a molecular system, the NCI analysis utilizes the RDG values. The non-covalent interactions, especially HB interactions, can be effectively examined through NCI analysis. The plot of reduced density gradient (RDG) versus sign(λ2)ρ(r) elucidates the nature of such interactions. The bonded interactions have λ2 < 0, while non-bonded interactions have λ2 > 0. The 2D plots for compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p reveal that the intra-molecular hydrogen bonding interactions have sign(λ2)ρ(r) values ranging from −0.010 to −0.018 a.u. (Fig. 6). In contrast, the intermolecular hydrogen bonding interactions are comparatively weaker, which is evident from the sign(λ2)ρ(r) values around −0.006 to −0.013 a.u. (Fig. S44†). Further, the presence of the green isosurface between O and H atoms in the 3D plot suggests the HB formation. In summary, these C–H⋯O HB interactions have sign(λ2)ρ(r) values in the range of −0.006 a.u. to −0.018 a.u., which suggests their weak nature.
 |
| | Fig. 6 2D NCI plots (on the left) and 3D gradient isosurfaces (on the right) depict intra-molecular HB interactions within compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p. | |
3.5.4. QTAIM analysis.
HBs can be described qualitatively and quantitatively by quantum theory of atoms in molecules (QTAIM) analysis based on the topological investigation of electron density, proposed by Bader,70 which resulted in bond critical points (BCPs) and bond paths (BPs) for these interactions present in the crystal packing of the compounds 5a, 5b, 5d, 5h, 5l, 5n, and 5p (Fig. 7 and 8). The existence of HB interactions is clearly indicated by the (3; −1) BCPs along the H⋯O bond paths (BPs) in the systems. The electron density (ρ(r)) values at the BCPs of these HB interactions fall within the acceptable range of 0.002–0.035 a.u. Additionally, the positive values of the Laplacian of electron density (∇2ρ(r)) indicate the depletion of electronic charge along the interatomic path. Furthermore, the ratios of the absolute potential energy density to the kinetic energy density (|V(r)|/G(r)) are less than one, characteristic of closed-shell interactions essential for the existence of non-covalent interactions such as hydrogen bonding.71 The ∇2ρ(r) values for most of the HB interactions fall within the recommended range of 0.024–0.139 a.u. Moreover, the positive total energy (H(r)) values indicate the non-covalent nature of these H⋯O interactions (Table S14†). Furthermore, the correlation graph between the HB interaction energies (Eint), which has been calculated by implying the equation of Parthasarathi,72 and the bond distance (D), shows an inverse linear relationship with a high correlation coefficient (R2 = 0.952). A correlation coefficient of 0.912 indicates a linear relationship between the plot of Eint and ∇2ρ(r) (Fig. S45†). Again, the correlation coefficients of 0.927 and 0.951 for the inverse linear relationship between the graphs of ∇2ρ(r) and ρ(r) with D are illustrated in Fig. S46.† The shorter the C–H⋯O HB distances, the stronger the HB interactions, which is supported by the higher values of ρ(r) at BCPs along the HB BPs. Again, the topological parameters for intramolecular HBs, responsible for forming a pseudo six-membered ring along with other intermolecular HBs, are in good agreement with those of the reported indolizine derivatives.63
 |
| | Fig. 7 BCPs (small red dots) and BPs (orange line) for intra-molecular HB interactions in the molecular graphs of 5a, 5b, 5d, 5h, 5l, 5n, and 5p compounds. | |
 |
| | Fig. 8 BCPs (small red dots) and BPs (orange line) for inter-molecular HB interactions in the molecular graphs of 5a, 5b, 5d, 5h, 5l, 5n, and 5p compounds. | |
3.6 Anti-tubercular activity
The anti-tubercular activity of the target compounds (5a–p) was evaluated (in vitro) against three different MTB strains, namely H37Rv, rifampicin/isoniazid-resistant (MDR), and XDR (Table 3). Compound 5e, which features a nitro group attached to the 4-position of the benzoyl ring and a hydrogen atom at the 2-position of the indolizine ring, exhibited remarkable anti-tubercular activity. It displayed the most potent inhibitory effect against the drug susceptible H37Rv MTB strain, with a minimum inhibitory concentration (MIC) value of 4 μg mL−1. Four compounds (5a, 5f, 5h, and 5n) exhibited equal potency, displaying a minimum inhibitory concentration (MIC) value of 8 μg mL−1 against the H37Rv MTB strain. Compounds 5b, 5c, 5g, 5l, and 5p demonstrated significant anti-TB activity with an MIC value of 16 μg mL−1 against the same strain. Compound 5d displayed a moderate level of anti-TB activity, showing a MIC value of 32 μg mL−1 against the H37Rv MTB strain. On the other hand, no activity was observed for the remaining indolizines when tested against the H37Rv MTB strain.
Table 3
In vitro anti-tubercular activity of ethyl 7-methoxy-3-(4-substituted benzoyl)indolizine-1-carboxylate derivatives (5a–p) against H37Rv (susceptible), MDR and XDR strains of MTB
| Entry |
R1 |
R2 |
MIC (μg mL−1) |
| H37Rva (susceptible) |
MDR-MTBb |
XDR |
|
American Type Culture Collection (ATCC): 25177.
These isolates were found to be resistant to the first line of antibiotics, rifampicin (1 μg mL−1), and isoniazid (0.2 μg mL−1). Abbreviations: MDR-MTB, multidrug-resistant and XDR, extensively drug-resistant strains of Mycobacterium tuberculosis; MIC, minimum inhibitory concentration; NA, not active at the concentration range (0.2–128 μg mL−1). |
|
5a
|
H |
H |
8 |
8 |
NA |
|
5b
|
H |
CH3 |
16 |
16 |
NA |
|
5c
|
H |
C2H5 |
16 |
16 |
NA |
|
5d
|
H |
CO2CH2CH3 |
32 |
32 |
64 |
|
5e
|
NO2 |
H |
4 |
16 |
16 |
|
5f
|
NO2 |
CH3 |
8 |
16 |
16 |
|
5g
|
NO2 |
C2H5 |
16 |
16 |
NA |
|
5h
|
NO2 |
CO2CH2CH3 |
8 |
8 |
64 |
|
5i
|
Cl |
H |
NA |
NA |
NA |
|
5j
|
Cl |
CH3 |
NA |
NA |
NA |
|
5k
|
Cl |
C2H5 |
NA |
NA |
NA |
|
5l
|
Cl |
CO2CH2CH3 |
16 |
16 |
64 |
|
5m
|
CH3 |
H |
NA |
NA |
NA |
|
5n
|
CH3 |
CH3 |
8 |
8 |
128 |
|
5o
|
CH3 |
C2H5 |
NA |
NA |
NA |
|
5p
|
CH3 |
CO2CH2CH3 |
16 |
16 |
NA |
The current study revealed that the presence of a hydrogen, methyl, ethyl, or an ester substituent at the 2-position of the indolizine ring was favourable for attaining good antitubercular activity. Compounds featuring hydrogen and nitro groups at the para position of the benzoyl ring exhibited significant activity against H37Rv and MDR-MTB strains. However, methyl or chloro groups abolished activity for compounds 5i, 5j, 5k, 5m, and 5o against all strains. While most compounds were inactive against XDR-MTB, 5d, 5e, 5f, 5h, 5l, and 5n retained moderate activity, suggesting that they target different MTB molecular sites. For phenyl derivatives, compounds 5a (hydrogen), 5b (methyl), and 5c (ethyl) at the 2-position of the indolizine ring showed 4-fold and 2-fold greater activity compared to the compound 5d (ester). Similarly, for nitro derivatives, compounds 5e (hydrogen), 5f (methyl), and 5h (ester) exhibited 4-fold and 2-fold higher activity than 5g (ethyl) at the same position.
The structural alteration of the indolizine scaffold at 1- and 2-positions had a notable impact on the efficacy of the compound against various strains of MTB, including H37Rv, MDR, and XDR strains. The indolizine derivatives 5a–5h, 5l, 5n, and 5p were active against H37Rv and MDR-MTB but inactive against XDR-MTB, except for compounds 5d, 5e, 5f, 5h, 5l, and 5n (Table 3). The compounds 5d, 5h, and 5l with an ethyl ester group at the 2-position of the indolizine ring for phenyl, nitro, and chloro derivatives showed moderate activity against XDR-MTB, with 2-fold, 4-fold and 8-fold decrease in potency, respectively, as compared with the cellular activity against susceptible H37Rv MTB strain. However, compounds 5e and 5f of nitro derivatives showed similar activity against MDR and XDR-TB strains, but decreased in 4-fold and 2-fold potency compared with the cellular activity against the susceptible H37Rv MTB strain. The anti-tubercular activity profile of the tested compounds against MDR and XDR-MTB provided a clue for their mode of action. Indeed, the results demonstrated that the series of indolizine derivatives (5a–p) did not show activity against XDR-TB except for compounds 5d–5f, 5h, 5l, and 5n. In contrast, the series of compounds (5a–p) show good inhibition growth of H37Rv and MDR-MTB strains except for 5i–5k, 5m, and 5o. These findings and observations suggest that the series of indolizine derivatives are likely to inhibit different MTB molecular drug targets; such that the phenyl, chloro, and methyl derivatives of indolizine inhibit a common target that is different from that inhibited by the nitro derivatives. On the other hand, the nitro derivatives of the compounds that demonstrated activity against the XDR-TB strain could potentially engage with the identical molecular target as the phenyl, chloro, and methyl derivatives of indolizine. However, their interaction with the binding site of the target might differ.
3.7 ADME and toxicity study
All the potent compounds (5a–p) were subjected to ADME and toxicity predictions using SwissADME with pkCSM. Evaluation of a molecule's fate in the human body involves assessing its ADME (absorption, distribution, metabolism, and elimination) properties. Physicochemical and biopharmaceutical parameters were calculated to estimate the ADME parameters of the identified compounds. Analysis of the physicochemical features revealed that the molar refractivity ranged from 89.85 to 110.90, falling within the acceptable range (30–140). The topological polar surface area (TPSA) plays a critical role in balancing the lipophilicity and hydrophilicity of a drug candidate, ensuring that it meets optimal ADMET requirements for efficacy and safety. The TPSA ranged from 57.01 to 129.13 Å2, suggesting that the molecules tend to have better oral bioavailability, as they can more easily cross cell membranes via passive diffusion, and molecules with high TPSA are often more polar, reducing their ability to permeate lipophilic biological membranes. Compounds with TPSA < 90 Å2 (5e–5p) are more likely to cross the BBB, making it important in designing CNS-active drugs. The lipophilicity class, indicative of a molecule's solubility in lipophilic media, was assessed through iLOGP and SILICOS-IT. The iLOGP values fell within the acceptable range (−0.4 to +5.6), while SILICOS-IT values were highly favourable. Predicted intestinal absorption profiles indicated excellent absorption (>92%) for all identified leads. Water solubility, a crucial parameter affecting drug absorption and distribution, was evaluated through logS calculations. The calculated logS values (4.66–5.83) indicated moderate solubility, contributing to a good balance between permeability and solubility for oral drug administration. Predicted gastrointestinal (GI) absorption was high for all compounds. Permeability over human skin, an indicator of ADMET outcomes, ranged from −4.78 to −5.86, within acceptable limits. Metabolism predictions assessed interaction with cytochrome P450 enzymes (CYPs), revealing no significant inhibitory activity. Drug-likeness, an indicator of oral drug potential, was assessed using various filters. While some compounds exhibited moderate violations, others showed no violations and similar bioavailability scores (55%). Synthetic accessibility ranged from moderate to tough (0.363–0.82), and total renal clearance fell within normal values (0.739–0.984 ml min−1 kg−1). Renal OCT2 substrate activity was observed for 5a, 5b, and 5d but was negative for most compounds. Detailed analyses are provided in Table S15.†
Toxicity predictions were conducted for all analogues (5a–p) to assess their in silico toxicity profiles. The AMES test, utilized to determine the mutagenic potential of chemical compounds using bacteria, yielded favourable results for all compounds except 5d–5h and 5m which exhibited AMES toxicity. The maximum tolerated dose (human) ranged from −0.081 to −0.722log mg per kg per day, representing a moderate level of dosage according to established protocols. None of the compounds showed hERG I or II (human ether-a-go-go-related gene) inhibition, mitigating the risk of ventricular arrhythmia. Oral rat chronic toxicity (LD50) values ranged from 2.301 to 3.111 mol kg−1, while oral rat chronic toxicity (LOAEL) values ranged from 0.83 to 1.859log mg per kg bw per day, indicating a favourable safety profile. Although all analogues were predicted to be hepatotoxic, none showed skin sensitization. The comprehensive toxicity results for these analogues are summarized in Table S16.†
This study aims to further explore the structure–activity relationship (SAR) by examining the influence of different R1 and R2 substitutions on the indolizine scaffold, which may play a critical role in the anti-TB activity against H37Rv, MDR, and XDR strains of MTB (Scheme 1).
3.8 Structure–activity relationship (SAR) of the indolizine scaffold
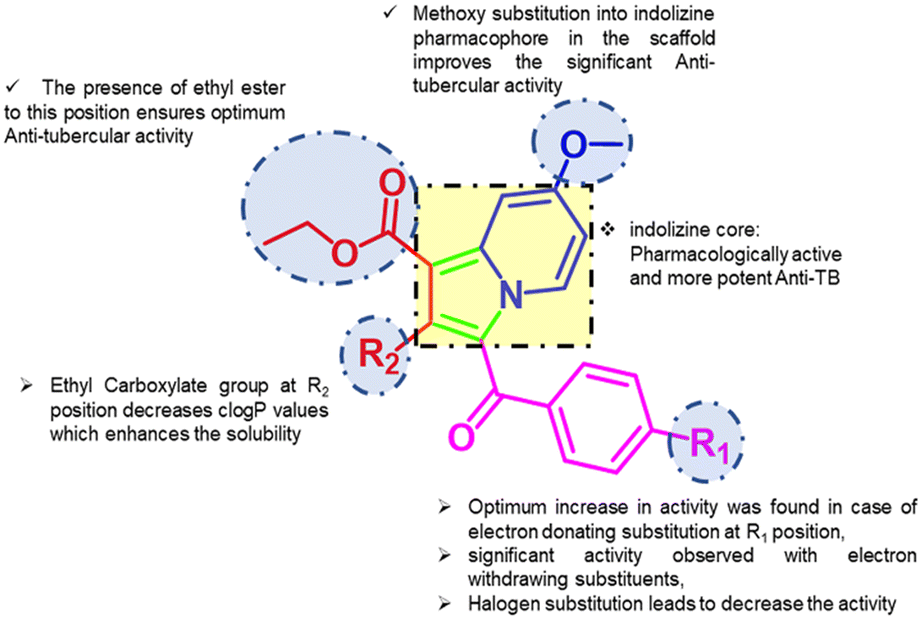
The significance of the indolizine core and its substituents in anti-tubercular potential has been evaluated through both earlier studies and the current research.23,24,27 This work focuses on the influence of various R1 and R2 substitutions on the indolizine scaffold (Fig. 9), which may play a critical role in anti-TB activity against H37Rv, as well as MDR and XDR strains of MTB. Findings from past literature and the present study suggest that the efficacy of indolizine derivatives in targeting both susceptible and drug-resistant MTB strains is highly dependent on the nature and position of these substituents, summarized as follows: the indolizine core is recognized for its potent pharmacological activity, particularly as an anti-tubercular agent. Incorporating a methoxy group into the indolizine scaffold significantly enhances its anti-TB potential. The presence of an ethyl ester at a specific position further ensures optimal activity against MTB. And current study suggested that an increase in anti-tubercular activity is observed with electron-withdrawing substitutions at the R1 position, while electron-donating groups also show considerable activity. However, halogen substitution at this position reduces the compound's efficacy (Table 3). Additionally, an ethyl carboxylate group at the R2 position reduces clogP values (Table 1), improving the solubility of the compounds, which is crucial for their bioavailability and effectiveness.
 |
| | Fig. 9 Summary of the structure–activity relationship (SAR) of the indolizine scaffold. | |
4. Conclusions
The anti-tubercular activity of indolizine derivatives (5a–p) was evaluated against three MTB strains: H37Rv, rifampicin/isoniazid-resistant (MDR), and XDR. Compound 5e, which has a nitro group on the benzoyl ring, exhibited the highest activity with a MIC of 4, 16, and 16 μg mL−1 against H37Rv, MDR, and XDR strains of MTB. Compounds 5a, 5f, 5h, and 5n also showed good activity (MIC: 8 μg mL−1). SAR analysis revealed that substitutions at the 2-position of the indolizine ring, such as hydrogen, methyl, ethyl, and ester groups, improved potency. Increased anti-tubercular activity was observed with electron-withdrawing substitutions at both the R1 and R2 positions. These findings suggest that the indolizine derivatives may inhibit different MTB molecular targets. Single crystal analysis revealed that the molecular assemblies of the indolizine derivatives are primarily governed by C–H⋯O hydrogen-bonded dimeric ring motifs, along with secondary interactions such as C–H⋯π and π⋯π interactions. The presence of hydrogen bonding is further confirmed by red spots and spikes in the 2D fingerprint plot of the Hirshfeld surface, indicating multiple interaction types. MEP analysis revealed sites for non-covalent interactions, including weak hydrogen bonds. Additionally, NCI and QTAIM analyses revealed the weak nature of intra- and intermolecular C–H⋯O hydrogen bonds in these derivatives in their solid state. In addition to these, a comprehensive ADMET analysis of all the compounds suggested a favourable toxicity profile and promising drug-like properties. These findings highlight the potential of these novel indolizine derivatives as promising candidates for further development in the fight against tuberculosis.
Data availability
CCDC 2380090–2380096 contain the supplementary crystallographic data for this paper. The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
Rahul D. Nagdeve: conceptualization, methodology, formal analysis, investigation, writing – original draft. Jyoti Swarup Thakur: software, validation, formal analysis, writing – review & editing. Sandeep Chandrashekharappa, Katharigatta N. Venugopala, Melendhran Pillay, Gourav Rakshit, and Pran Kishore Deb: methodology, resources, conceptualization. Pradip K. Mondal and Maurizio Polentarutti: single crystal X-ray data collection. Keshab M. Bairagi and Osama I. Alwassil: formal analysis, writing – review & editing. Susanta K. Nayak: supervision, funding acquisition, writing – review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
RDN thanks UGC-New Delhi, India, for providing financial assistance under UGC-JRF (JUNE18-136885) for the fellowship. RDN, JST, and SKN thank VNIT for providing research support. The authors are thankful for the single crystal data collection at the XRD1 beamline, Elettra Synchrotron, which was funded by a grant from the Italian Ministry of Foreign Affairs and International Cooperation and the Indian Department of Science & Technology. We also thank the Science and Engineering Research Board, India, (ECR/2016/-001820) for the polarized optical microscope with hot stage equipment facility. KNV thanks the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia (KFU242386), the Durban University of Technology, and the National Research Foundation (129330 and 129173), South Africa.
References
-
H. Mohajan, Tuberculosis is a Fatal Disease among Some Developing Countries of the World, 2014, pp. 18–31 Search PubMed.
- M. Zala, J. J. Vora and V. M. Khedkar, ACS Omega, 2023, 8, 20262–20271 CrossRef CAS PubMed.
-
A. Ohkado and S. Kato, in Pulmonary Tuberculosis and Its Prevention, Springer, 2022, pp. 3–31 Search PubMed.
- R. Panjabi, G. Comstock and J. Golub, Int. J. Tuberc. Lung Dis., 2007, 11, 828–837 Search PubMed.
- F. Stephanie, M. Saragih and U. S. F. Tambunan, Pharmaceutics, 2021, 13, 592 CrossRef CAS.
- A. A. Adeniji, K. E. Knoll and D. T. Loots, Appl. Microbiol. Biotechnol., 2020, 104, 5633–5662 Search PubMed.
- J. L. Khawbung, D. Nath and S. Chakraborty, Comp. Immunol., Microbiol. Infect. Dis., 2021, 74, 101574 Search PubMed.
- P. Mundhe, S. Kidwai, S. M. Saini, H. R. Singh, R. Singh and S. Chandrashekharappa, J. Mol. Struct., 2023, 1284, 135359 CrossRef CAS.
- K. N. Venugopala, S. Chandrashekharappa, P. K. Deb, C. Tratrat, M. Pillay, D. Chopra, N. A. Al-Shar'i, W. Hourani, L. A. Dahabiyeh and P. Borah, J. Enzyme Inhib. Med. Chem., 2021, 36, 1471–1486 CrossRef CAS PubMed.
- N. J. Ryan and J. H. Lo, Drugs, 2014, 74, 1041–1045 Search PubMed.
- V. A. Dartois and E. J. Rubin, Nat. Rev. Microbiol., 2022, 20, 685–701 CrossRef CAS PubMed.
- F. Andrade, D. Rafael, M. Videira, D. Ferreira, A. Sosnik and B. Sarmento, Adv. Drug Delivery Rev., 2013, 65, 1816–1827 Search PubMed.
- S. Parida, R. Axelsson-Robertson, M. Rao, N. Singh, I. Master, A. Lutckii, S. Keshavjee, J. Andersson, A. Zumla and M. Maeurer, J. Intern. Med., 2015, 277, 388–405 CrossRef CAS PubMed.
- P. Tiwari, G. S. Mangubhai, S. Kidwai, R. Singh and S. Chandrashekharappa, Chem. Biol. Drug Des., 2024, 103, e14512 CrossRef CAS.
- G. Stelitano, J. C. Sammartino and L. R. Chiarelli, Molecules, 2020, 25, 1239 CrossRef CAS.
- V. Dartois and C. E. Barry, Curr. Clin. Pharmacol., 2010, 5, 96–114 CrossRef CAS.
- D. U. Ganihigama, S. Sureram, S. Sangher, P. Hongmanee, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Eur. J. Med. Chem., 2015, 89, 1–12 CrossRef CAS.
- C. Sandeep, B. Padmashali and R. S. Kulkarni, Tetrahedron Lett., 2013, 54, 6411–6414 CrossRef CAS.
- C. Sandeep, B. Padmashali, R. S. Kulkarni, S. Mallikarjuna, M. Siddesh, H. Nagesh and K. Thriveni, Heterocycl. Lett., 2014, 4, 371–376 Search PubMed.
- G. Sumanth, S. M. Saini, K. Lakshmikanth, G. S. Mangubhai, K. Shivaprasad and S. Chandrashekharappa, J. Mol. Struct., 2023, 1286, 135561 CrossRef CAS.
- K. Lakshmikanth, S. M. Saini, S. T. Dorai and S. Chandrashekharappa, Tetrahedron, 2023, 133516 CrossRef CAS.
- K. N. Venugopala, S. Chandrashekharappa, S. Bhandary, D. Chopra, M. A. Khedr, B. E. Aldhubiab, M. Attimarad and B. Odhav, Curr. Org. Synth., 2018, 15, 388–395 CrossRef CAS.
- K. N. Venugopala, S. Chandrashekharappa, M. Pillay, H. H. Abdallah, F. M. Mahomoodally, S. Bhandary, D. Chopra, M. Attimarad, B. E. Aldhubiab and A. B. Nair, PLoS One, 2019, 14, e0217270 CrossRef CAS PubMed.
- M. A. Khedr, M. Pillay, S. Chandrashekharappa, D. Chopra, B. E. Aldhubiab, M. Attimarad, O. I. Alwassil, K. Mlisana, B. Odhav and K. N. Venugopala, J. Biomol. Struct. Dyn., 2018, 36, 2163–2178 CrossRef CAS.
- S. Chandrashekharappa, K. N. Venugopala, R. Venugopala and B. Padmashali, J. Appl. Pharm. Sci., 2019, 9, 124–128 CAS.
- K. N. Venugopala, C. Tratrat, S. Chandrashekharappa, M. Attimarad, N. Sreeharsha, A. B. Nair, S. Pottathil, R. Venugopala, O. H. A. Al-Attraqchi and M. A. Morsy, Indian J. Pharm. Educ. Res., 2019, 53, 545–552 CrossRef CAS.
- K. N. Venugopala, C. Tratrat, M. Pillay, F. M. Mahomoodally, S. Bhandary, D. Chopra, M. A. Morsy, M. Haroun, B. E. Aldhubiab and M. Attimarad, Antibiotics, 2019, 8, 247 CrossRef CAS.
- V. Uppar, S. Chandrashekharappa, A. I. Basarikatti, G. Banuprakash, M. K. Mohan, M. Chougala, K. K. Mudnakudu-Nagaraju, R. Ningegowda and B. Padmashali, J. Appl. Pharm. Sci., 2020, 10, 077–085 CAS.
- C. Sandeep, B. Padmashali, R. S. Kulkarni, K. N. Venugopala, R. Venugopala and B. Odhav, Asian J. Chem., 2016, 28, 1043–1048 CrossRef CAS.
- C. Sandeep, P. Basavaraj and S. Rashmi, J. Appl. Chem., 2013, 2, 1049–1052 CAS.
- V. Uppar, S. Chandrashekharappa, C. Shivamallu, S. P. Kollur, J. Ortega-Castro, J. Frau, N. Flores-Holguín, A. I. Basarikatti, M. Chougala and P. Sushma, Molecules, 2021, 26, 2722 Search PubMed.
- K. N. Venugopala, O. H. Al-Attraqchi, C. Tratrat, S. K. Nayak, M. A. Morsy, B. E. Aldhubiab, M. Attimarad, A. B. Nair, N. Sreeharsha and R. Venugopala, Biomolecules, 2019, 9, 661 CrossRef CAS PubMed.
- C. Sandeep, K. N. Venugopala, M. A. Khedr, B. Padmashali, R. S. Kulkarni, V. Rashmi and B. Odhav, Indian J. Pharm. Educ. Res., 2017, 51, 452–460 Search PubMed.
- S. Chandrashekharappa, K. N. Venugopala, C. Tratrat, F. M. Mahomoodally, B. E. Aldhubiab, M. Haroun, R. Venugopala, M. K. Mohan, R. S. Kulkarni and M. V. Attimarad, New J. Chem., 2018, 42, 4893–4901 RSC.
- K. N. Venugopala, S. Chandrashekharappa, C. Tratrat, P. K. Deb, R. D. Nagdeve, S. K. Nayak, M. A. Morsy, P. Borah, F. M. Mahomoodally and R. P. Mailavaram, Molecules, 2021, 26, 3550 Search PubMed.
- S. Chandrashekharappa, K. N. Venugopala, S. K. Nayak, R. M. Gleiser, D. A. García, H. M. Kumalo, R. S. Kulkarni, F. M. Mahomoodally, R. Venugopala and M. K. Mohan, J. Mol. Struct., 2018, 1156, 377–384 Search PubMed.
- C. Sandeep, K. N. Venugopala, R. M. Gleiser, A. Chetram, B. Padmashali, R. S. Kulkarni, R. Venugopala and B. Odhav, Chem. Biol. Drug Des., 2016, 88, 899–904 CrossRef CAS PubMed.
- O. I. Alwassil, S. Chandrashekharappa, S. K. Nayak and K. N. Venugopala, PLoS One, 2019, 14, e0223413 Search PubMed.
- A. Lausi, M. Polentarutti, S. Onesti, J. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi and D. Lamba, Eur. Phys. J. Plus, 2015, 130, 1–8 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS.
- O. Dolomanov, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 Search PubMed.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 Search PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. Turner, J. McKinnon, S. Wolff, D. Grimwood, P. Spackman, D. Jayatilaka and M. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 Search PubMed.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
-
R. A. Gaussian09, Gaussian Inc., Wallingford CT, 2009, vol. 121, pp. 150–166 Search PubMed.
- Y. Erdogdu, D. Manimaran, M. Güllüoǧlu, M. Amalanathan, I. Hubert Joe and S. Yurdakul, Opt. Spectrosc., 2013, 114, 525–536 CrossRef CAS.
-
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09 Revision A, 2009, vol. 1 Search PubMed.
- T. Lu and F. Chen, Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Martin, N. Morcillo, D. Lemus, E. Montoro, M. A. Telles, N. Simboli, M. Pontino, T. Porras, C. Leon, M. Velasco, L. Chacon, L. Barrera, V. Ritacco, F. Portaels and J. C. Palomino, Int. J. Tuberc. Lung Dis., 2005, 9, 901–906 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. Saeed, S. Ashraf, U. Flörke, Z. Y. D. Espinoza, M. F. Erben and H. Pérez, J. Mol. Struct., 2016, 1111, 76–83 CrossRef CAS.
- A. Saeed, Z. Ashraf, H. Nadeem, J. Simpson, H. Pérez and M. F. Erben, J. Mol. Struct., 2019, 1195, 796–806 CrossRef CAS.
- R. D. Nagdeve, J. S. Thakur, S. Chandrashekharappa, K. M. Bairagi, P. K. Deb, K. N. Venugopala, P. K. Mondal, M. Polentarutti, O. I. Alwassil and V. Mohanlall, J. Mol. Struct., 2024, 1308, 138080 CrossRef CAS.
- A. J. Edwards, C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Faraday Discuss., 2017, 203, 93–112 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- R. J. Xavier and E. Gobinath, Spectrochim. Acta, Part A, 2012, 86, 242–251 CrossRef CAS PubMed.
- H. Gökce and S. Bahçeli, Spectrochim. Acta, Part A, 2013, 114, 61–73 CrossRef.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- A. Bendjeddou, T. Abbaz, S. Maache, A. Gouasmia and D. Villemin, Int. J. Pharm. Sci. Rev. Res., 2016, 39, 136–143 Search PubMed.
-
R. Bader, Atoms in molecules “A quantum theory”, Clarendon, Oxford, UK, 1990 Search PubMed.
- U. Koch and P. L. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 Search PubMed.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2005, 109, 843–850 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jyoti Swarup
Thakur
a,
Jyoti Swarup
Thakur