
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polycondensations and cyclization of poly(L-lactide) ethyl esters in the solid state†
Steffen M.
Weidner
 a,
Andreas
Meyer
b,
Jana
Falkenhagen
a and
Hans R.
Kricheldorf
*c
a,
Andreas
Meyer
b,
Jana
Falkenhagen
a and
Hans R.
Kricheldorf
*c
aBAM, Federal Institute of Materials Research and Testing, Richard Willstätter Str. 11, D-12489 Berlin, Germany. E-mail: hrkricheldorf@chemie.uni-hamburg.de
bUniversität Hamburg, Institut für physikalische Chemie, Grindelallee 117, D-29147 Hamburg, Germany
cUniversität Hamburg, Institut für Technische und Makromolekulare Chemie, Bundesstr. 45, D-20146 Hamburg, Germany
First published on 27th November 2023
Abstract
The usefulness of seven different tin catalysts, bismuth subsalicylate and titan tetra(ethoxide) for the polycondensation of ethyl L-lactate (ELA) was examined at 150 °C for 6 d. Dibutyltin bis(phenoxides) proved to be particularly effective. Despite the low reactivity of ELA, weight average molecular masses (Mw) of up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 were found along with partial crystallization. Furthermore, polylactides (PLAs) of similar molecular masses were prepared via ELA-initiated ROPs of L-lactide by means of the four most effective polycondensation catalysts. The crystalline linear PLAs were annealed at 140 or 160 °C in the presence of these catalysts. The results of the transesterification reactions in the solid PLAs were studied by means of matrix-assisted laser desorption/ionization (MALDI TOF) mass spectrometry, gel permeation chromatography (GPC), differential scanning calourimetry (DSC) and small-angle X-ray scattering (SAXS). The results indicate that polycondensation and formation of cycles proceed in the solid state via formation of loops on the surface of the crystallites. In summary, five different transesterification reactions are required to explain all results.
500 were found along with partial crystallization. Furthermore, polylactides (PLAs) of similar molecular masses were prepared via ELA-initiated ROPs of L-lactide by means of the four most effective polycondensation catalysts. The crystalline linear PLAs were annealed at 140 or 160 °C in the presence of these catalysts. The results of the transesterification reactions in the solid PLAs were studied by means of matrix-assisted laser desorption/ionization (MALDI TOF) mass spectrometry, gel permeation chromatography (GPC), differential scanning calourimetry (DSC) and small-angle X-ray scattering (SAXS). The results indicate that polycondensation and formation of cycles proceed in the solid state via formation of loops on the surface of the crystallites. In summary, five different transesterification reactions are required to explain all results.
Introduction
Poly(L-lactide), PLA, is a biosourced and biodegradable polyester which has attracted increasing interest for various applications, so its technical production has meanwhile reached a level of approximately 700000 t pa.1–4 The technical production of PLA5 (and of polyglycolide6) is based on alcohol-initiated ring-opening polymerization (ROP) catalyzed by tin(II) 2-ethylhexanoate (SnOct2). The addition of alcohol allows for a rough control of the molecular masses via the LA/alcohol ratio and enhances the activity of the catalyst. For all these reasons, alcohol initiated ROP of LA and lactones has become the most widely studied method for the preparation of PLAs and polylactones. Far more than one hundred papers and patents dealing with the syntheses and mechanistic studies of homo- and copolyesters were published, and thus, only three review articles are cited.7–9
The polymerization mechanism and the influence of various experimental parameters on the course of this kind of ROP were the objectives of numerous studies.10–25 Based on the work of Kricheldorf et al.11–13 and Zhang et al.10 the coordination–insertion mechanism outlined in Scheme 1 has become the established ROP mechanism. The groups of Penczek14,15 and Jalabert16 contributed important details based on kinetic measurements and spectroscopic end group analyses. These authors found that the kinetic course obeys the definition of a living polymerization with low dispersities (Đ < 1.2) and full control of the molecular mass via the lactide/initiator ratio (LA/In). Furthermore, neat SnOct2 was found to be inactive, so alcohol is needed for activation. However, these studies14–17 were conducted in THF or toluene at temperatures of 80 °C or below, reaction conditions far from those for the technical production, which is performed in bulk at temperatures in the range of 130–180 °C.
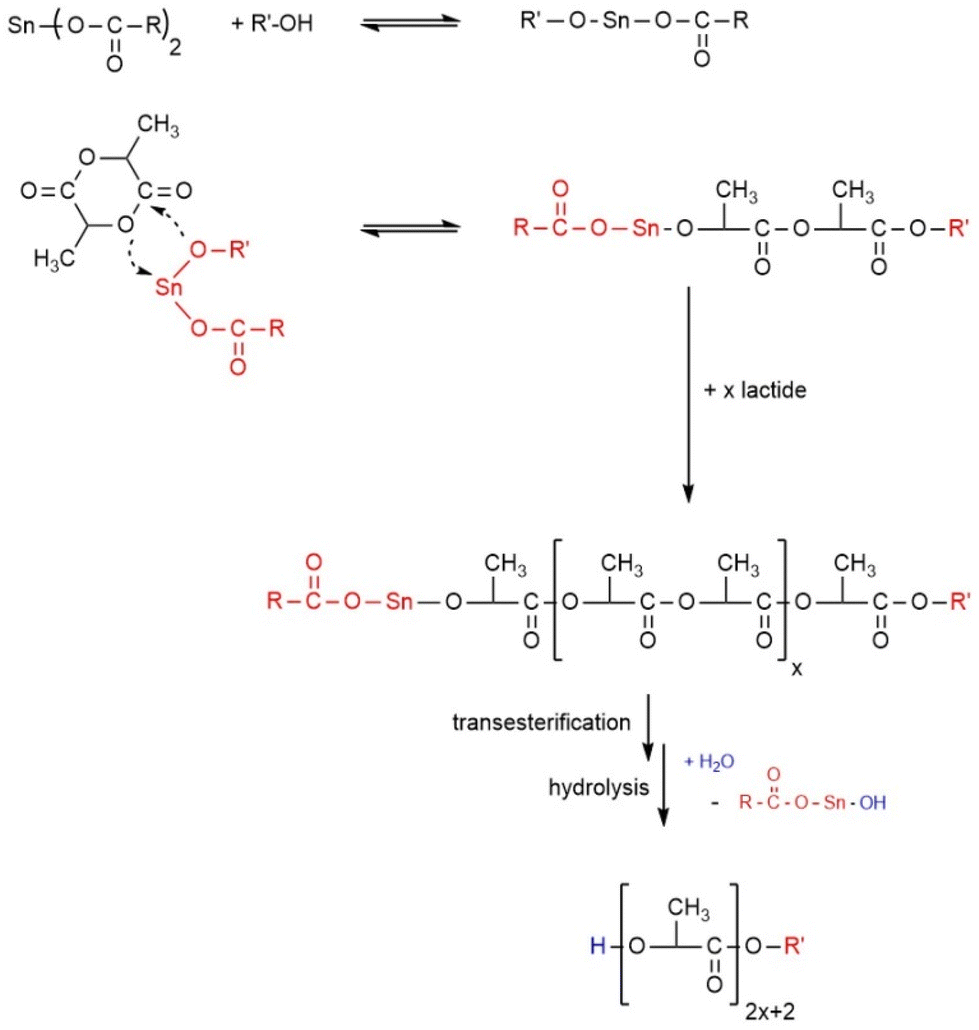 | ||
| Scheme 1 Mechanism of the alcohol-initiated and SnOct2-catalyzed ROP of L-lactide. | ||
Commercial PLAs possess dispersities Đ > 2 and typically contain a weight fraction of 1–3% of cyclic oligo- and polylactides, which indicates that the polymerization process performed under technical conditions is more complex than Scheme 1 or the studies of Kowalski14,15 or Jalabert16 suggest. A computer modelling of the technical polymerization process conducted in bulk at 130 °C was published by Yu et al. mainly with the purpose to model the molecular mass distribution (MWD).19,20 However, this mathematical approach did not include recent results of the authors.24,25
Studies have shown that neat SnOct2 is, in fact, a highly active catalyst when the polymerizations are conducted in bulk at 130 °C or above.26 The resulting PLAs have a cyclic topology in contrast to the alcohol-initiated chains, because of the ROPPOC mechanism (ROP + simultaneous polycondensation) catalyzed by neat SnOct2 (Scheme 2). This mechanism competes with the alcohol-initiated ROP, so decreasing the In/Cat ratio favours the formation of cyclic PLAs. Furthermore, three intermolecular transesterification mechanisms were detected which all become operating at temperatures below 130 °C. Moreover, it was found that not only temperature and time, but also the structure of the initiator has a significant influence on the nature and extent of transesterification reactions.15
 | ||
| Scheme 2 ROPPOC mechanism catalyzed by neat SnOct2. | ||
In all the previous academic publications and patents, the alkyl ester end groups resulting from initiation with alcohols were considered to be stable. Yet, quite recently the authors have found in another context that ethyl-L-lactate (ELA) can be oligomerized when heated with SnOct2 for more than one day, but the progress of the polycondensation was slow.27 This unexpected observation prompted the authors to study polycondensations of ethyl-L-lactate and crystalline oligolactides having ethyl ester end groups in more detail. Three aspects were in the focus of this work: first, a screening of various catalysts with respect to their activity; second, detection and identification of transesterification reactions that occur in the solid state across the surface of crystallites; third, detection of cyclic PLAs formed by condensation of crystallized linear PLAs.
Experimental
Materials
L-Lactide (LA), a product of Corbion Purac, was kindly supplied by Thyssen-Uhde AG (Berlin, Germany) and recrystallized from “Toluene 99.98%, extra dry” (ACROS Chemicals, Geel, Belgium). Its melting point was 98.5–98.8 °C, when measured by DSC with a heating rate of 5 K min−1. Ethyl-L-lactate (ELA) was purchased from Sigma Aldrich (Taufkirchen, Germany) and used as received. SnCl2, 4-toluene sulfonic acid (TSA) monohydrate, tin(II) 2-ethylhexanoate (purity >96%), titanium tetra-butoxide and bismuth subsalicylate (BiSub) were all purchased from Alfa Aesar and used as received. The catalysts dibutyltin bis(4-chloro-phenoxide) (BuSnPhCl), dibutyltin bis(4-cyanaophenoxide) and dibutyltin bis(pentafluorophenoxide) (BuSnPhF) were synthesized as described previously.28 Anhydrous dichloromethane and anisole were purchased from Thermo-Scientific Fisher (Schwerte, Germany).Polycondensations of ELA
The catalyst (0.15 mmol) was weighed into a 100 mL Erlenmeyer flask and ELA (150 mmol) was added together with a magnetic bar. The reaction vessel was closed with a “head” equipped with a gas outlet tube and a gas inlet tube which ended ca. 2 cm above the surface of ELA. Argon was slowly bubbled over the reaction mixture. After 6 d a sample was removed from the liquid or semi-crystalline reaction product and characterized in the virgin state.ELA-initiated ROP of L-lactide and annealing at 140 °C (LA/Cat = 1000/1)
Bu2Sn-bis(4-chlorophenoxide) (0.4 mmol) and ELA (1.4 mmol) were weighed into a 50 mL flame dried Erlenmeyer flask and L-lactide (42 mmol) and a magnetic bar were added under a blanket of argon. The closed reaction vessel was immersed in an oil bath thermostated at 140 °C. After 1 d the reaction vessel was destroyed, and the plaque of crystalline PLA was broken into 6 pieces. Two pieces were used for immediate characterization in Hamburg and Berlin, and four pieces were annealed under argon at 140 °C for 6 or 27 more days.Analogous experiments were performed with the other three catalysts.
Annealing at 160 °C (LA/Cat = 1000/1)
A crystalline PLA ethyl ester was prepared at 140 °C for 1 d as described above and broken into at least 6 pieces. Two pieces were used for characterization, two pieces were annealed at 160 °C for 6 d and two pieces were annealed at 160 °C for 27 d.Annealing at 160 °C with LA/Cat = 400/1
SnCl2 (0.1 mmol), ELA (140 mmol) and L-lactide (42 mmol) were weighed into a 50 mL Erlenmeyer flask under a blanket of argon and polymerized as described above. The crystalline PLA isolated after 1 d was broken into 6 pieces two of which were used for immediate characterization. Two pieces were annealed at 160 °C for 13 d and two pieces for 27 d. However, the samples isolated after 27 d had turned brown or black and a brownish syrup had been formed. Therefore, these samples were not characterized and are not listed in Table 4.Analogous experiments with similar results were performed with the other three catalysts.
All annealing experiments were conducted under argon.
ELA-initiated ROP and equilibration of lactide in anisole
Bu2Snbis(pentafluorophenoxide) (0.04 mmol), ELA (1.2 mmol) and L-lactide (42 mmol) were weighed into a flame dried 50 mL Erlenmeyer flask under a blanket of argon. A magnetic bar and anisole (14 mL) were added. The reaction vessel was immersed in an oil bath thermostated at 140 °C. After 28 d, the reaction mixture was diluted with dichloromethane (20 mL) and precipitated into ligroin (500 mL). The precipitated PLA was isolated by filtration and dried at 60 °C in vacuo. Yield: 92%, Mn:13200, Mw: 22000, with 0.1 mmol of the catalyst a yield of 91% and Mn = 5500 and Mw = 14500 were obtained.
Measurements
1H NMR measurements were performed with a Bruker Avance 400 III in 5 mm sample tubes, using CDCl3 containing TMS as the solvent and shift reference.MALDI TOF mass spectra were obtained with an Autoflex Max mass spectrometer (Bruker Daltonik, Bremen, Germany). All spectra were recorded in the positive ion linear mode. The MALDI stainless steel targets were prepared from chloroform solutions of poly(L-lactide) (3–5 mg mL−1) doped with potassium trifluoroacetate (2 mg mL−1 in THF). Typically, 20 μL of the sample solution, 2 μL of the potassium salt solution and 50 μL of the matrix solution (DCTB, trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile, 20 mg mL−1 in CHCl3) were pre-mixed in an Eppendorf vial. A droplet (1 μL) of this solution was deposited on the MALDI target and, after evaporation of the solvent, inserted in the mass spectrometer. 8000 single spectra were recorded and accumulated from 4 different places of each spot.
For Electrospray Ionization (ESI) mass spectrometry a Q-TOF Ultima ESI-TOF mass spectrometer (Micromass) running at 4 kV capillary voltage and 35 V cone voltage, at a source temperature of 120 °C and a desolvation temperature of 150 °C was used for all measurements. The mass spectrometer was operated in the positive ion mode. The samples (0.05–0.1 mg ml−1 in trichloromethane/methanol/70:30/v:v) were directly injected with a flow rate of 5 μL min−1. For data recording and evaluation, MassLynx Software (Waters) was used. For the representation of a spectrum in general, scans were accumulated for 0.5 min (scan time 1 min, up to 100000 spectra per s). Here only the low molar mass fraction up to m/z 1200 was considered.
GPC measurements were performed in chloroform on a LC 1200 (Agilent, USA) instrument kept at 40 °C. The flow rate was 1 mL min−1 and a refractive index detector was used. Samples were automatically injected (100 μL, 2–4 mg mL−1 in chloroform). For instrument control and data calculation, Win GPC software (Polymer Standards Service – PSS, Mainz, Germany) was applied. The calibration was performed using polystyrene standard sets (Polymer Standards Service – PSS, Mainz). The number average (Mn) and weight average (Mw) data listed in Tables 1–3 are uncorrected. Fractionation experiments were done manually by collecting the eluents at the end of the capillary using glass vials.
| Exp. No. | Catalyst | DP (NMR) | M n (NMR) | M n (GPC) | M w (GPC) | T m (°C) | ΔHm (J g−1) |
|---|---|---|---|---|---|---|---|
| 1 | SnCl2 | 15 | 1100 | 3200 | 7000 | — | — |
| 2 | SnCl2 + TSA | 27 | 1900 | 4100 | 7900 | — | — |
| 3 | SnOct2 | 20 | 1450 | 2600 | 5200 | — | — |
| 4 | SnBiph | 14 | 1000 | 1500 | 2900 | — | — |
| 5 | BuSnPhCl | 29 | 2900 | 5800 | 11300 |
161.5 | |
| 6 | BuSnPhCN | 30 | 2200 | 5200 | 9500 | 161.7 | |
| 7 | BuSnPhF | 35 | 2500 | 4600 | 10500 |
161.0 | |
| 8 | Ti(OEt)4 | 14 | 1000 | 2100 | 3600 | — | — |
| 9 | BiSub | 20 | 1450 | 3200 | 6700 | — | — |
| Exp. no. | Catalyst | Time (d) | M n | M w | T m (°C) | ΔHm (J g−1) | Cryst.a (%) | L (nm) | l c (nm) |
|---|---|---|---|---|---|---|---|---|---|
a Calculated with a 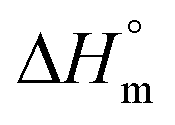 of 115 J g−1.
b The PLA prepared at140 °C for 1 d served as the starting material.
c Test of reproducibility. of 115 J g−1.
b The PLA prepared at140 °C for 1 d served as the starting material.
c Test of reproducibility.
|
|||||||||
| 1A | SnOct2 | 3 h | 8900 | 13000 |
— | — | — | — | |
| 1Bb | SnOct2 | 1 | 10200 |
13500 |
170.6 | 79.0 | 69 | 19.3 | 13.4 |
| 1C | SnOct2 | 7 | 12600 |
18500 |
176.2 | 91.6 | 80 | ||
| 1D | SnOct2 | 28 | 14500 |
23500 |
180.5 | 99.0 | 86 | 16.0 | 13.6 |
| 2A | SnCl2 | 3 h | 8200 | 10000 |
— | — | — | — | — |
| 2B | SnCl2 | 1 | 9000 | 11500 |
168.8 | 81.7 | 71 | 19.3 | 13.5 |
| 2C | SnCl2 | 7 | 10500 |
15000 |
174.4 | 81.0 | 70 | ||
| 2D | SnCl2 | 28 | 13000 |
22000 |
178.5 | 96.3 | 83 | 15.6 | 13.0 |
| 3A | BuSnPhCl | 3 h | 8100 | 9600 | — | — | — | — | |
| 3BX | BuSnPhCl | 1 | 10000 |
14000 |
171.1 | 86.1 | 75 | 17.6 | 13.2 |
| 3BYc | BuSnPhCl | 1 | 9700 | 13200 |
170.3 | 84.7 | 73 | — | — |
| 3C | BuSnPhCl | 7 | 12000 |
18000 |
175.3 | 85.4 | 74 | ||
| 3D | BuSnPhCl | 28 | 13800 |
22500 |
177.8 | 94.6 | 82 | 16.6 | 13.1 |
| 4A | BuSnPhF | 3 h | 8100 | 10400 |
— | — | — | — | — |
| 4BX | BuSnPhF | 1 | 9800 | 13500 |
170.5 | 81.8 | 71 | 17.8 | 12.6 |
| 4BYc | BuSnPhF | 1 | 9700 | 13000 |
171.0 | 80.7 | 70 | — | — |
| 4C | BuSnPhF | 7 | 11500 |
17000 |
174.4 | 87.8 | 77 | — | — |
| 4D | BuiSbPhF | 28 | 13300 |
21500 |
176.6 | 95.0 | 82 | 16.2 | 13.1 |
| Exp. no. | Catalyst | Temp. (°C) | Time (d) | M n | M w | T m (°C) | ΔHm (J g−1) | Cryst. (%) |
|---|---|---|---|---|---|---|---|---|
| 1A | SnOct2 | 140 | 1 | 9 800 | 13500 |
170.6 | 79.0 | 69 |
| 1B | SnOct2 | 160 | 6 | 10800 |
22700 |
177.7 | 67.5 | 59 |
| 1C | SnOct2 | 160 | 13 | 12500 |
28000 |
181.8 | 93.6 | 81 |
| 2A | SnCl2 | 140 | 1 | 8 200 | 10000 |
168.8 | 81.7 | 71 |
| 2B | SnCl2 | 160 | 6 | 12400 |
23000 |
177.3 | 83.0 | 72 |
| 2C | SnCl2 | 160 | 13 | 13000 |
25000 |
181.6 | 92.0 | 80 |
| 3A | BuSnPhCl | 140 | 1 | 10200 |
14000 |
172.1 | 86.1 | 74 |
| 3B | BuSnPhCl | 160 | 6 | 12900 |
27000 |
179.4 | 84.1 | 73 |
| 3C | BuSnPhCl | 160 | 13 | 20000 |
39000 |
184.4 | 94.7 | 83 |
| 4A | BuSnPhF | 140 | 1 | 9 800 | 13500 |
170.5 | 81.8 | 71 |
| 4B | BuSnPhF | 160 | 6 | 11700 |
22800 |
177.3 | 77.3 | 67 |
| 4C | BuSnPhF | 160 | 13 | 19000 |
34000 |
181.5 | 94.4 | 83 |
X-ray scattering experiments were performed using our in-house SAXS/WAXS apparatus equipped with an Incoatec™ X ray source IμS and Quazar Montel optics. The wavelength of the X ray beam was 0.154 nm and the focal spot size at the sample position was 0.6 mm2. The samples were measured in transmission geometry and were recorded with a Rayonix™ SX165 CCD-Detector. For the WAXS measurements, the sample–detector distance was 0.1 m, allowing us to detect an angular range of 2Θ = 5°–33°. The recording time for each WAXS pattern was 300 s. The SAXS measurements were performed at a sample–detector distance of 1.6 m and the accumulation was 20 minutes. DPDAK, customizable software for reduction and analysis of X-ray scattering datasets, was used for gathering 1D scattering curves.29 For the evaluation of the crystallinity of the samples the data were imported in Origin™ and analysed with the curve fitting module. After subtracting of the instrumental background, the integral intensity of the crystalline reflections was divided by the overall integral intensity to determine the crystallinity Xc. The SAXS curves were converted into Kratky plots. The long periods of the lamellar domains were determined by from q values of the reflection maxima.
Results and discussion
Polycondensation of ethyl-L-lactate: screening of catalysts
All polycondensations of L-lactate were conducted at 150 °C for two reasons. Firstly, previous studies have shown that at a temperature of 130 °C or below the progress of the polycondensation was too slow for the purpose of this work. In addition to a screening of various catalysts it was a goal of this study to reach number average molecular masses Mn in the range of 4000–5000 for comparison with the ROP of condensation experiments discussed below. Secondly, the volatility of the monomer (boiling point 165 °C) had to be taken into account, because argon was slowly bubbled through the reaction mixture to remove the liberated ethanol. Due to the long reaction time, this measure caused a loss of ELA around 30–40% at 150 °C, and thus, a higher temperature was not advisable.Two classes of catalysts were used, namely acidic catalysts such as SnCl2 and 4-toluene sulfonic acid (TSA), which show a proton-catalyzed transesterification mechanism (Scheme S1, ESI part†), and neutral transesterification catalysts, operating via a coordination–insertion mechanism (Scheme S2, ESI part†). The chemical structures and acronyms of the catalysts are compiled in Scheme 3. The reaction mixtures were characterized by GPC measurements, by MALDI TOF mass spectrometry and in some cases by ESI mass spectroscopy (No. 5–7, Table 1).
 | ||
| Scheme 3 Structure and acronyms of tin catalysts discussed in this work. | ||
The GPC measurements revealed that the best three catalysts (No. 5–7, Table 1) gave the intended Mn values in the range of 4000–6000. Cyclic esters were not detectable in the MALDI TOF mass spectra, two examples of which are presented in Fig. S1 (ESI part).†
However, a tiny fraction of cycles below m/z 800 was detected using ESI mass spectrometry. Hence, these experiments gave two results, which were decisive for the further studies. Firstly, the ethyl ester group proved to be reactive enough to enable syntheses of low molar mass PLAs, when reaction times of one week or longer are applied. Secondly, only dibutyltin bisphenoxides showed a satisfactory reactivity, whereas Ti(OBu)4, a widely used transesterification catalyst, gave a surprisingly poor result. For the subsequent studies four catalysts were selected, which according to increasing reactivity, obeyed the following order.
| SnOct2 < SnCl2 < BuSnPhF ∼ BuSnPhCl | (1) |
SnOct2 was included, because this tin salt is the most widely used catalyst for alcohol-initiated ROPs of LA7–9 (incl. the technical production of PLA) and because the authors have used this catalyst in several recent studies concerning transesterification reactions in solid PLA. In this connection, it is of interest that the aforegiven order of reactivities in polycondensation of ELA is not identical to the reactivities observed for ROPs of LA. For example, in ROPs, the reactivity of SnOct2 is higher than that of SnCl2,30 and the reactivity of dibutyltin bisphenoxides obeys the following order:
BuSnPhCl < BuSnPhCN < BuSnPhF.28
ROP of L-lactide at 140 °C
Since the experiments with ELA have demonstrated that the ethyl ester group is reactive enough for polycondensation reactions, when enough time and suitable catalysts come into play, the main part of this work had the purpose to explore the chemical reactions going on in PLAs having ethyl ester end groups along with CH-OH end groups. As the starting materials for these studies four ROPs of LA were initiated with ELA at a LA/In ratio of 30/1 and a temperature of 140 °C (1A, 2A, 3A, 4A, Table 2). These conditions were selected for the following reasons. Complete polymerizations (evidenced by 1H NMR spectra) yield PLAs having a degree of polymerization (DP) of 60 and under the given polymerization conditions a low dispersity was expected and indeed found (Đ ∼ 1.2–1.3 with the exception of 1A).The GPC measurements yielded number average molecular masses (Mn) in the range of 8100–8900 which at first glance overestimate the real Mn of 4300 Da for two reasons. First, it is known for decades that polystyrene calibrated GPC measurements overestimate the Mn of aliphatic polyesters by 50–80% due to a lower hydrodynamic volume, and in the case of PLA in chloroform this overestimation amounts to 50–55%. Hence a theoretical Mn of 4300 corresponds to a “GPC-Mn” of 6400–6600. Second, the GPC curves display a shoulder at higher molecular masses (Fig. 1), which results from beginning polycondensation as discussed below. This shoulder is particularly strong in the case of SnOct2, and the molecular mass data of the SnOct2-catlyzed PLA were in fact the highest values (1A, Table 2).
 | ||
| Fig. 1 GPC elution curves of PLAs prepared at 140 °C for 3 h with: (A) SnOct2 (1A, Table 2); (B) BuSnPhCl (3A, Table 2). | ||
In addition to the expected monomodal mass distribution with low dispersity, the MALDI TOF mass spectra revealed a large predominance of the odd-numbered chains. The fraction of even-numbered chains resulting from transesterification reactions depends, as expected, on the catalyst as demonstrated in Fig. 2. Alcohol-initiated ROPs of LA free of transesterification reaction will necessarily yield PLA chains consisting of dimeric repeat units. When ELA is used as the initiator, these PLAs will exclusively consist of odd-numbered chains due to incorporation of a single lactyl unit, and thus, the appearance of even-numbered chains clearly indicates the existence of transesterification reactions. Transesterification reactions in solution or in the melt resulting in broadening of the molecular mass distribution are quite normal, but the odd–even equilibration under investigation is quite unusual for three reasons. First, distribution of the new species obeys exactly the narrow distribution of the parent PLA chain and does not cause broadening of the MWD. Hence, it must result from an exchange of one or three lactyl units. Second, under the conditions of the present work, this even–odd equilibration involves even all crystallized chains and is not restricted to the initial melt or the amorphous phase. This observation has already been made in previous experiments performed with various alcohols at 130 °C. Third, the existence of this unusual even–odd equilibration was also observed by other research groups for alcohol-initiated and SnOct2-catalyzed ROPs of LA in solution, and it was found that it proceeds slowly even at 70 °C. On the other hand, the recent work demonstrates that 24 hours are needed for completion at 140 °C (Fig. 3).
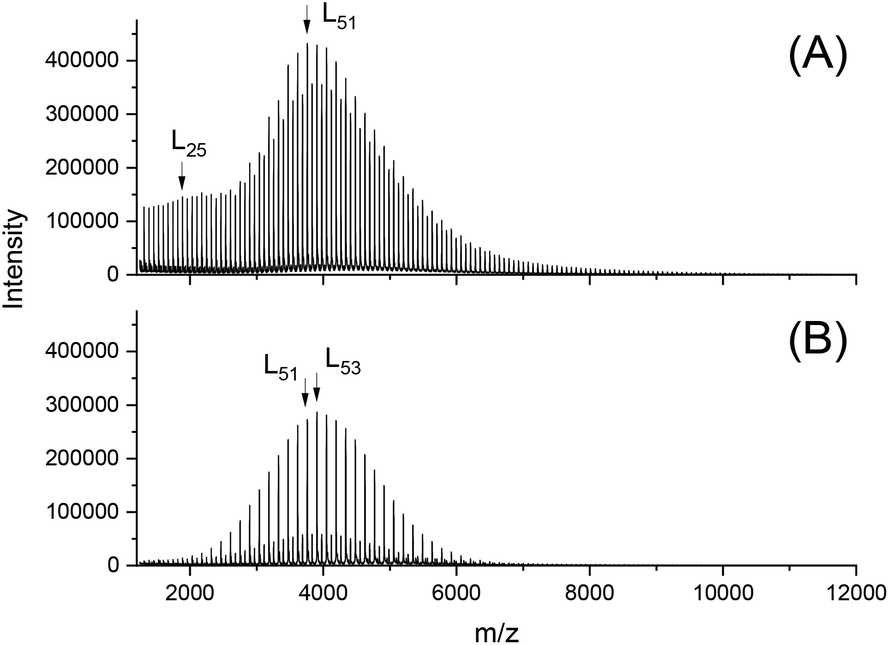 | ||
| Fig. 2 MALDI-TOF mass spectra of PLAs prepared at 140 °C for 3 h with: (A) SnOct2 (1A, Table 2); (B) BuSnPhCl (3A, Table 2). | ||
 | ||
| Fig. 3 MALDI TOF mass spectra of PLAs prepared at 140 °C for 1 d with: (A) SnOct2 (1B, Table 2); (B) BuSnPhCl (3B, Table 2). | ||
These observations suggest that the odd–even equilibration involves a low energy of activation in combination with a low frequency factor.
Quite interesting is the finding that when the extent of equilibration is taken as a measure of the catalytic activity the order of reactivity of the four catalysts obeys the following order:
| BuSnPhCl ∼ BuSnPhF < SnCl2 < SnOct2 | (2) |
This order is exactly opposite to the order of reactivities observed for the polycondensation of ELA (eqn (1)) and thus indicates another reaction mechanism. A detailed discussion of this even–odd equilibration mechanism has recently been published, and the afore-mentioned observations do not justify a formulation of a new mechanism along with a more detailed discussion.
Concerning the structure of the crystallites obtained by the ROP with a LA/In ratio of 30/1 the following published facts were taken into consideration. According to the X-ray studies of Wasanasuk and Tashiro31 ten lactyl units have a length of 2.9 nm in the helical conformation characteristic for the α-modification of PLA, which is the thermodynamically most stable crystal modification and usually adopted at temperatures above 120 °C.32 Therefore, the crystallized PLA chains having a DP of 60 have a length of around 19 nm.
The SAXS measurements of this work (listed in Table 2) indicate a crystal thickness (lc) of around 13 nm. Since both the DP of 60 and lc represent an average of a broader population of chains lengths and crystals, it may be assumed that the shorter PLA chains will be embedded in the crystal lattice in the form extended chains, whereas the longer chains will form one-fold. The antiparallel alignment which for thermodynamic reasons is characteristic of the stable α-modification32 has in turn led to the consequence that both flat surfaces of the crystallites are covered with an alternating array of CH(CH3)OH and COOEt end groups in both dimensions as schematically illustrated in Schemes 4 and 5.
 | ||
| Scheme 4 2D illustration of an ELA-initiated PLA crystallite made up of extended chains (A) and the results of potential condensation reactions (B). | ||
 | ||
| Scheme 5 2D illustration of an ELA-initiated PLA crystallite made up of once-folded chains (A) and results of potential condensation reactions (B). | ||
For the sake of clarity, Schemes 4 and 5 are a simplification in three directions. First the CH–OH end groups are in reality CH(CH3)–OH groups. Second, not all chain-ends sticking out from the surface of the crystallites have identical lengths. Third, extended chains and folded chains do presumably not form separate crystals but coexist in the same crystal. It was the purpose of this work to elucidate, if and to what extent the end groups and loops sticking out from the surface undergo condensation and transesterification reactions.
At this point, the terminology used in this paper should be defined to avoid misunderstanding. All tin-catalyzed reactions discussed in this work may be understood as transesterification reactions. (Poly)condensation and cyclization reactions may be classified as alcoholytic transesterifications (see Scheme S2†), whereas all other transesterifications may be classified as ester–ester interchange reactions (Scheme S3†). Regardless of the classification, it is typical for the tin chemistry of esters that all these reactions are versions of a coordination–insertion mechanism since neither radicals nor ions are involved as intermediates.
Annealing with (poly)condensation of PLA ethyl esters at 140 °C
The first series of annealing experiments was conducted at 140 °C with a LA/Cat ratio of 1000/1 (Table 2). The GPC curves of all four PLAs annealed for 1 d displayed an increase of the shoulder observed after 3 h and in the case of SnOct2 (1B, Table 2) the intensity of the former shoulder reached the same level as the first maximum (Fig. 4A). In addition to this second maximum a new shoulder was formed. All other samples (2B, 3B, 4B) showed the same trend but with significantly lower intensity as illustrated in Fig. 4B and Fig. S2.†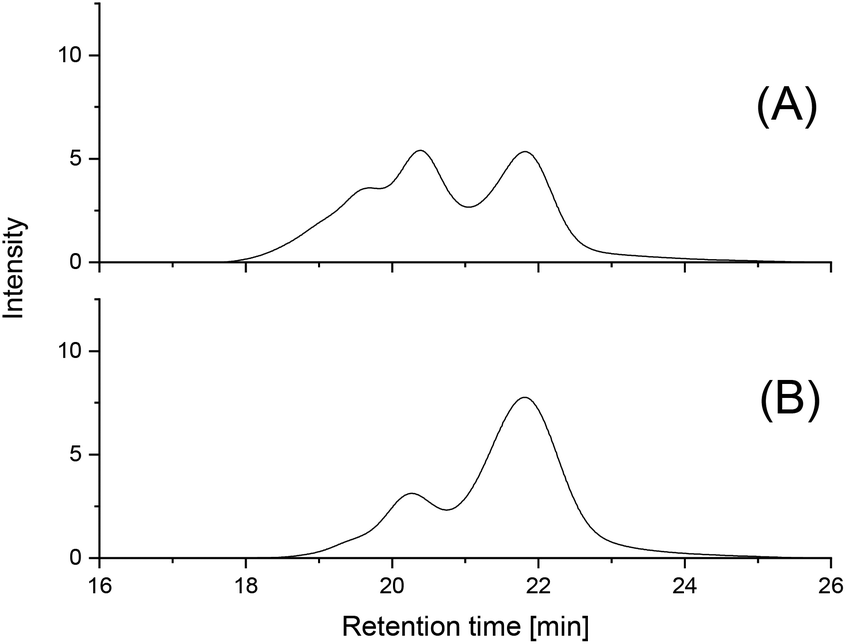 | ||
| Fig. 4 GPC elution curves of PLAs prepared at 140 °C for 1 d with: (A) SnOct2 (1B, Table 2); (B) BuSnPCl (3B, Table 2). | ||
To elucidate the origin of these shoulders the PLA prepared with BuSnPhF (4B, Table 2) was fractionated by GPC, and the fractions were characterized by MALDI TOF mass spectroscopy. As demonstrated in Fig. S3,† the main maximum was found between m/z 3600 and 4700 with a top around m/z 4050 (fractions 8–6) and a new weaker maximum around m/z 8000 in agreement with the mass spectrum of the non-fractionated original sample. The maximum around m/z 8000 Da indicates a doubling of the original PLA chains. This observation suggests that the polycondensation mainly proceeded by the formation of loops on the surface of the crystallites as outlined in Schemes 4 and 5. This interpretation was confirmed by the GPC elution curves obtained after 7 and 28 d and their fractionation. After 7 d, the GPC curves of all PLAs prepared with the four different catalysts were nearly identical and Fig. 5A is a representative example. Again, a further increase of the former shoulders yielding new maxima was found and a fourth shoulder appeared. This process continued upon annealing for 28 d. All four PLA samples isolated after 28 d at 140 °C showed identical GPC elution curves, an example of which is presented in Fig. 5B.
 | ||
| Fig. 5 GPC elution curves of PLAs prepared at 140 °C with BuSnPhCl: (A) after 7 d (3C, Table 2); (B) after 28 d (3D, Table 2). | ||
The MALDI TOF mass spectra recorded after annealing for 28 d gave the following results. First a slight broadening of the mass distribution towards low molecular masses was detectable and the strongest effect was observed for the SnOct2-catalyzed sample followed by SnCl2 (Fig. 6). When the mass range between m/z 7000 and 9000 was examined, a weak maximum was found, which exclusively consisted of linear chains, which were obviously formed by doubling of the original PLA chains (Fig. S4†). Fractionation of the 28 d samples 2D and 4 D (Table 2) revealed more information in this direction. The positions of the fractions are illustrated in Fig. S5,† and the mass spectra of the individual fractions obtained from the SnCl2 catalyzed sample (2D) are presented in Fig. 7. These mass spectra display several weak maxima with masses around m/z 8000 D, 12000, 16000, 20000, 24000 and even 28000. The analogous SEC curve and MALDI TOF mass spectra of a fractionated PLA prepared with BuSnPhF are displayed in Fig. S6 and S7.† Both series of maxima cannot be explained by random transesterification of all chains, but it can be explained by doubling tripling, quadrupling (etc.) of the original PLA chains via formation of loops on the surface of the crystallites as outlined in Schemes 4 and 5. Therefore, these GPC and mass spectrometric measurements prove that polycondensation via LA ethyl ester end groups is possible in the solid state.
 | ||
| Fig. 6 MALDI-TOF mass spectra of PLAs prepared at 140 °C for 28 d with: (A) SnCl2 (2D, Table 2); (B) BuSnOPF (4D, Table 2). | ||
 | ||
| Fig. 7 MALDI-TOF mass spectra of a PLA prepared at 140 °C for 28 d with SnCl2 (2D, Table 2) after fractionation (the corresponding SEC trace is shown in Fig. S5†). | ||
This interpretation is supported by a ROP experiment conducted under the conditions of the Table 2 experiments, but with addition of anisole. Anisole is a good solvent of PLA at high temperatures, and thus, random transesterification and equilibration of the resulting PLA chains could occur. The results are illustrated in Fig. S8 and S9.† The mass spectrum does not display a maximum around m/z 8000 but displays the exponential decay of the mass distribution typical of a reversible polycondensation. The GPC curve has the expected shape of a monomodal broad mass distribution.
Finally, the SAXS measurements deserve discussion. When cyclic PLAs were annealed at 160 or 170 °C in the presence of cyclic tin catalysts, an enormous increase of the crystal thickness (lc) was observed, in several cases up to a factor of 2.5 within 1 or 2 days. In contrast to these previous results, the SAXS data listed in Table 2 demonstrate that despite a time interval of 27 d the crystal thickness did not increase regardless of the catalyst (two representative examples of SAXS curves are displayed in Fig. S10†). On the other hand, increasing crystallinity and higher melting temperatures were found upon longer annealing. According to the Gibbs–Thomsen eqn (3)Tm not only depends on the crystal thickness but also on the surface free energy (σe) which is reduced upon smoothing of the surface.
 | (3) |
 ,
, 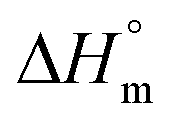 – melting temperature and melting enthalpy of an ideal crystal; r – density of the crystal, lc – thickness of the lamellar crystal, σe – surface free energy)
– melting temperature and melting enthalpy of an ideal crystal; r – density of the crystal, lc – thickness of the lamellar crystal, σe – surface free energy)
Scheme 6 illustrates two transesterification mechanisms that explain the removal of relatively long chain ends from the surface of the crystallites. It is certainly not far-fetched to assume that the formation of small loops is the most effective contribution to the smoothing of the surface. Smoothing of the surface has the additional consequence that denser and higher order of crystal packing in the spherulites becomes possible.
 | ||
| Scheme 6 Two transesterification mechanisms leading to the removal of longer chain ends. | ||
This consequence is evident from higher order reflections in the SAXS curves (Fig. S10B†). Even a third order reflection was observable in the SAXS curves of all four samples. To the best knowledge of the authors, such a high degree of order has not yet been reported in the literature of PLAs. However, it is also remarkable that all samples display a second and third order reflection already after 1d (Fig. S8A†). This high order after relatively short annealing is most likely due to the low dispersity of the molecular masses. It may have two consequences, namely a narrow distribution of crystal thickness, and second, long chain ends sticking out from the surface are rare and tie molecules connecting two crystallites are almost absent. In other words, the SAXS data are in good agreement with the properties and reactions of the PLA ethyl esters studied in this work.
Annealing and polycondensation of PLA ethyl esters at 160 °C
A second series of polycondensation experiments was studied at 160 °C (Table 3), whereupon the same starting materials were used as for the 140 °C series (Table 2). The GPC measurements revealed two trends, namely increasing molecular masses (roughly by a factor of two) and increasing dispersities (1.8–2.0). The shape of the elution curves changed in the same way as observed for 140 °C, but the shoulders and new maxima were not well separated. However, the most interesting results came from the MALDI TOF mass spectra, which displayed two trends. First, a substantial broadening of the molecular mass distribution towards low molar mass chains was found (Fig. 8). This trend corresponds to lower Mn values and a substantial broadening of the molecular mass distribution as measured by GPC (Table 3). The extent of the MWD broadening varied considerably with the nature of the catalyst. When the extent of the MWD broadening is taken as measure of the catalytic activity, SnOct2 proves to be the most active catalyst and the order of reactivity obeys eqn (4), which is exactly contrary to that of eqn (1).| BuSnPhCl < BuSnPhF < SnCl2 < SnOct2 | (4) |
 | ||
| Fig. 8 MALDI-TOF mass spectra of PLAs prepared at 160 °C for 13 d with: (A) SnOct2 (1C, Table 3), (B) SnCl2 (2C, Table 3), (C) BuSnPhCl (3C, Table 3), and (D) BuSnOPF (4C, Table 3). | ||
The low molar mass species most likely result from “disproportionation” (via transesterification) of longer chains which exist in the amorphous phase or form the outer walls of the crystallites (via lateral attack of the catalysts).
The second trend concerns the mass range of m/z 7000–9000, where a series of new peaks became detectable, namely the peaks of cycles (Fig. 9). The ratio of cycles relative to linear chains varied with the reactivity of the catalyst according to eqn (1). This finding may be understood as a piece of evidence for a condensation reaction as the origin of the cyclization process as outlined in Scheme 5. The masses of these cycles are nearly twice as high as those of the starting materials and suggest that they were formed via loop-closing according to Scheme 5(b).
 | ||
| Fig. 9 MALDI-TOF mass spectra (segments) of PLAs prepared at 160 °C for 13 d with: (A) SnOct2 (1C, Table 3), (B) SnCl2 (2C, Table 3), (C) BuSnPhCl (3C, Table 3), and (D) BuSnPhF (4C, Table 3). | ||
In summary, these results indicate that two kinds of transesterification reactions were operating under the same reaction conditions, namely the condensation reaction, where the activity of the catalysts obeys eqn (1) and ester–ester interchange reactions, where the activity of the catalysts obeys eqn (4)
After detection of cycles in the mass spectra, the authors designed a second series of annealing/polycondensation experiments at 160 °C, but now with a higher concentration of catalysts using a LA/Cat ratio of 400/1. For this series new starting materials were prepared at 140 °C with a higher catalyst concentration. A first result of this measure were higher molecular masses obtained after 1 d (A experiments, Table 4) relative to the molecular masses of the A experiments in Table 3. After 14 d a conspicuous decrease of Mn was found accompanied by a dramatic increase of the dispersity (up to values around 4). The mass spectra again indicate an enormous broadening of the mass distribution towards low molar mass chains (Fig. 10). Yet, the most interesting result was the appearance of the mass peaks of cycles in the range of m/z 7000–8000. Their abundance had somewhat increased relative to the experiments with a LA/Cat ratio of 1000/1 (Table 3), but in contrast to the previous experiments (Table 3) the mass peaks displayed now a clear “saw-tooth pattern” (STP) (Fig. 10). STPs were observed by the authors under a variety of conditions, when cyclic PLAs were annealed in the presence of cyclic tin catalysts, but they were never observed for crystallites made up of linear PLAs. As discussed recently, the STPs indicate the formation of a smooth surface under thermodynamic control for crystallites consisting of extended rings in the mass range of m/z 3000–15000. These extended-ring crystallites with a smooth surface represent the thermodynamically most perfect and stable form of PLA crystals in the mentioned mass range. Hence, the appearance of a STP in the mass spectra of PLAs annealed at 160 °C indicates the formation of crystallites primarily or exclusively consisting of extended rings.
| Exp. no. | Catalyst | Temp. (°C) | Time (d) | M n | M w | T m (°C) | ΔHm (J g−1) | Cryst. (%)a |
|---|---|---|---|---|---|---|---|---|
a Calculated with a 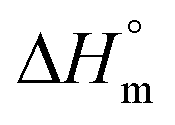 of 115 J g−1. of 115 J g−1.
|
||||||||
| 1A | SnOct2 | 140 | 1 | 12000 |
18500 |
181.7 | 76.3 | 66 |
| 1B | 160 | 13 | 8000 | 36000 |
— | — | — | |
| 2A | SnCl2 | 140 | 1 | 8700 | 11500 |
181.5 | 92.0 | 80 |
| 2B | 160 | 13 | 8000 | 36000 |
— | — | — | |
| 3A | BuSnPhCl | 140 | 1 | 11500 |
17500 |
184.4 | 92.7 | 80 |
| 3B | 160 | 13 | 6500 | 31000 |
— | — | — | |
| 4A | BuSnPhF | 140 | 1 | 12000 |
17000 |
181.6 | 93.8 | 82 |
| 4B | 160 | 13 | 18000 |
40 500 | — | — | — | |
 | ||
| Fig. 10 MALDI-TOF mass spectra of PLAs prepared at 160 °C for 14 d with a LA/Cat = 400/1 and: (A) SnCl2 (2B, Table 4), (B) SnOct2 (1B, Table 4), and (C) BuSnPhF (4B, Table 4). | ||
The formation of extended-ring crystallites on the basis of the afore-mentioned transesterification mechanisms is hard to understand and an additional reaction pathway should be proposed, the “wandering loops”, schematically formulated in Scheme S4.† When OH groups neighboring a loop do react via an alcoholytic transesterification and an ethyl ester group via an ester interchange mechanism a sequence of such transesterification reactions allows a migration of a loop across the surface of the crystallite. In this way extended-rings may accumulate in a certain area of a crystallite and finally transform the entire crystallite or part of it into an extended-ring crystal. The formation of extended-ring crystals allows, in turn, a modification of their surface towards a STP. The driving force behind this process is thermodynamic optimization of crystallites. This interpretation is supported by the detection that extended-ring crystallites are the most perfect and thermodynamically stable kind of PLA crystals in the mass range below 20000 Da.33 Furthermore, the authors have quite recently shown that linear and cyclic PLAs, when formed simultaneously, crystallize separately from the same reaction mixture regardless, if the polymerization is conducted in solution or in the melt.34,35
Finally, it should be mentioned that ESI mass spectrometry allowed for the detection of small amounts of cyclic PLAs with masses below m/z 1500 after annealing at 160 °C, whereas cyclic oligomers were barely detectable after annealing at 140 °C as exemplarily demonstrated in Fig. S11.† These cyclic oligomers are most likely the result of “backbiting” or end-to-end cyclization of linear oligomers in the amorphous phase, in analogy to the formation of cyclic oligomers in the polycondensation of ELA (Table 1). The observation that these cyclic oligomers appear at 160 °C, but not at 140 °C, agrees with the recent results of the authors which demonstrate that “back-biting” requires higher temperatures than intermolecular transesterification.27,30,36
When the cycle-to-linear ratio is taken as a measure of the catalytic activity, the order of catalysts obeys eqn (2) with SnOct2 as the most active species. This order is opposite to that of eqn (1) and confirms that the mechanism yielding the cyclic oligomers is different from the condensation mechanism yielding the cyclic PLAs with masses around m/z 8000. In summary, the linear PLAs annealed at 160 °C generate two different populations of cycles via different reaction pathways.
Conclusions
The results of the present work allow for the following conclusions. First, dibutyltin bisphenoxides were identified as particularly effective polycondensation catalysts for lactide alkyl esters. Second, under the influence of these and other tin-based transesterification catalysts, various transesterification reactions can proceed on the surface of crystallites, so that higher molecular masses and cyclic polylactides are formed.Third, crystallites exclusively consisting of linear PLAs can be transformed into crystallites almost exclusively consisting of extended cycles. Fourth, smoothing of the crystal surfaces by formation of loops enhances the crystallinity and favors a dense, rather perfect 3d-order of crystallites in the spherulites. The driving force behind all these processes has two sources, a gain in entropy due to liberalization of ethanol and short oligomers and a gain in enthalpy from increasing crystallinity and denser packing of crystallites.
Author contributions
HRK: conceptualization, supervision, investigation, and writing – original draft; SMW: investigation, methodology, data curation, visualization, and writing – review & editing, AM: investigation, data curation, and visualization, JF: investigation, data curation, and visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Bundesanstalt für Materialforschung und – prüfung (BAM) for technical support. The authors also thank A. Myxa (BAM) for the GPC measurements and S. Bleck (Univ. of Hamburg) for the DSC data.References
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. López-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, John Wiley & Sons, 2011 Search PubMed.
- M. L. Di Lorenzo and R. Androsch, in Advances in Polymer Science, 282, ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, Cham, 2018 Search PubMed.
- S. P. Dubey, H. A. Abhyankar, V. Marchante, J. L. Brighton and K. Blackburn, Int. Res. J. Pure Appl. Chem., 2016, 1–20 CAS.
- J. A. Byers, A. B. Biernesser, K. R. Delle Chiaie, A. Kaur and J. A. Kehl, in Synthesis, Structure and Properties of Poly(L-lactid acid), ed. M. L. Di Lorenzo and R. Androsch, Springer, Cham, 2017, vol. 279, pp. 67–118.
- E. Frazza and E. Schmitt, J. Biomed. Mater. Res., 1971, 5, 43–58 CrossRef CAS PubMed.
- S. Corneillie and M. Smet, Polym. Chem., 2015, 6, 850–867 RSC.
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2022, 13, 1618–1647 RSC.
- J. Li, J. Ding, T. J. Liu, L. Yan and X. Chen, in Industrial Applications of Poly(lactic acid), ed. M. L. DiLoreno and R. Androsch, 2018 Search PubMed.
- X. C. Zhang, D. A. Macdonald, M. F. A. Goosen and K. B. Mcauley, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2965–2970 CrossRef CAS.
- H. R. Kricheldorf, J. M. Jonté and M. Berl, Macromol. Chem. Phys., 1985, 12, 25–28 CrossRef CAS.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- S. Penczek, A. Duda, A. Kowalski, J. Libiszowski, K. Majerska and T. Biela, Macromol. Symp., 2000, 157, 61–70 CrossRef CAS.
- M. Jalabert, C. Fraschini and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1944–1955 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- S. Sosnowski and P. Lewinski, Polym. Chem., 2015, 6, 6292–6296 RSC.
- Y. C. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS.
- Y. C. Yu, E. J. Fischer, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2014, 53, 7333–7342 CrossRef CAS.
- D. Pholharn, Y. Srithep and J. Morris, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 213, 012022 Search PubMed.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, RSC Adv., 2021, 11, 14093–14102 RSC.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2022, 223, 2100464 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., 2022, 60, 3222–3231 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J, 2023, 185, 111822 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2020, 11, 2595–2604 RSC.
- H. R. Kricheldorf, S. M. Weidner and J. Falkenhagen, Polym. Chem., 2022, 13, 1177–1185 RSC.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli and P. Müller-Buschbaum, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2018, 219, 1800445 CrossRef.
- K. Wasanasuk and K. Tashiro, Polymer, 2011, 52, 6097–6109 CrossRef CAS.
- B. Lotz, Adv. Polym. Sci., 2018, 279, 273–302 CrossRef.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, Polymer, 2022, 263, 125516 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Polymer, 2023, 276, 125946 CrossRef CAS.
- S. M. Weidner, A. Meyer and H. Kricheldorf, Polymer, 2023, 285, 126355 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and J. Falkenhagen, Polym. Chem., 2021, 12, 5003–5016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01232h |
| This journal is © The Royal Society of Chemistry 2024 |