
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiatomic catalysts for Fenton and Fenton-like reactions: a promising platform for designing/regulating reaction pathways†
Fan
Mo
,
Qixing
Zhou
 *,
Chenghao
Li
,
Zongxin
Tao
,
Zelin
Hou
,
Tong
Zheng
,
Qi
Wang
,
Shaohu
Ouyang
and
Sihui
Zhan
*,
Chenghao
Li
,
Zongxin
Tao
,
Zelin
Hou
,
Tong
Zheng
,
Qi
Wang
,
Shaohu
Ouyang
and
Sihui
Zhan
Key Laboratory of Pollution Processes and Environmental Criteria (Ministry of Education), Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Carbon Neutrality Interdisciplinary Science Center, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: zhouqx@nankai.edu.cn
First published on 28th June 2023
Abstract
The optimization of the single-atom catalyst (SAC) performance has been the hot spot for years. It is widely acknowledged that the incorporation of adjacent single-atom sites (diatomic catalysts (DACs)) can enable synergistic effects, which can be used in cascade catalysis, dual-function catalysis, and performance regulation of intrinsic active sites. DACs have been widely applied in the CO2 reduction reaction (CO2RR), oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), etc.; however, their application is limited in Fenton or Fenton-like reactions. This perspective summarizes the most advanced achievements in this field, followed by the proposed opportunities in further research, including regulation of the magnetic moment, inter-atomic distance effect, strain engineering, atomic cluster (AC)/nanoparticle (NP) modification, etc. It is demonstrated that this perspective can contribute to the DAC application in Fenton or Fenton-like reactions with innovative design and mechanisms being put forward.
1 Introduction
1.1 Construction of multiple active sites – optimization of the reaction pathway
Single-atom catalysts (SACs) have exhibited unique characteristics (e.g., unusual configuration and high atomic utilization) in heterogeneous catalysis.1 However, it is extremely difficult to synchronously achieve the high-efficiency catalysis of two-step or multi-step reactions for SACs. Dinuclear-site catalysts with two active centers can independently interact with different intermediates and catalyze successive reaction steps, significantly improving the activities and selectivity of multiple-step catalytic reactions. Hu et al. (2021) fabricated a Fe-based catalyst including duet Fe3C and FeN NPs for the hetero-e-Fenton reaction. The Fe3C and FeN NPs can mediate the electroreduction of O2 to H2O2 and the H2O2 activation towards ˙OH, respectively (Fig. 1a). However, nanoparticle (NP) or atomic cluster (AC) catalysts usually suffer from inferior catalytic activity, due to the low atomic exposure and thus ineffective utilization of active sites.2 To solve this issue, a dual-atomic-site catalyst (CoFe DAC) was fabricated, which can synergistically catalyze ˙OH electro-generation. Specifically, Co SACs can promote O2 reduction to H2O2 intermediates, and Fe SACs were responsible for H2O2 activation to ˙OH3 (Fig. 2a). In addition, there are reactions focusing on simultaneous reactant enrichment and catalytic elimination near the active center, also highlighting the importance of constructing dual-function sites. For example, surface oxygen vacancies and Co sites can capture H2O2/peroxymonosulfate (PMS) (i.e., reduction reaction) and contaminants, respectively, decreasing the migration distance and thus increasing the degradation performance.4,5 Besides, dual-function sites can be used to drive simultaneous PMS oxidation and reduction reactions, for example, PMS can be oxidized at Co sites (e.g., generating 1O2) and reduced at Bi sites (i.e., generating SO4˙− and ˙OH) in Co-doped Bi2O2CO3.6 Therefore, the incorporation of the second atomic dispersed active center can be a promising method to improve the efficiency of Fenton or Fenton-like reactions.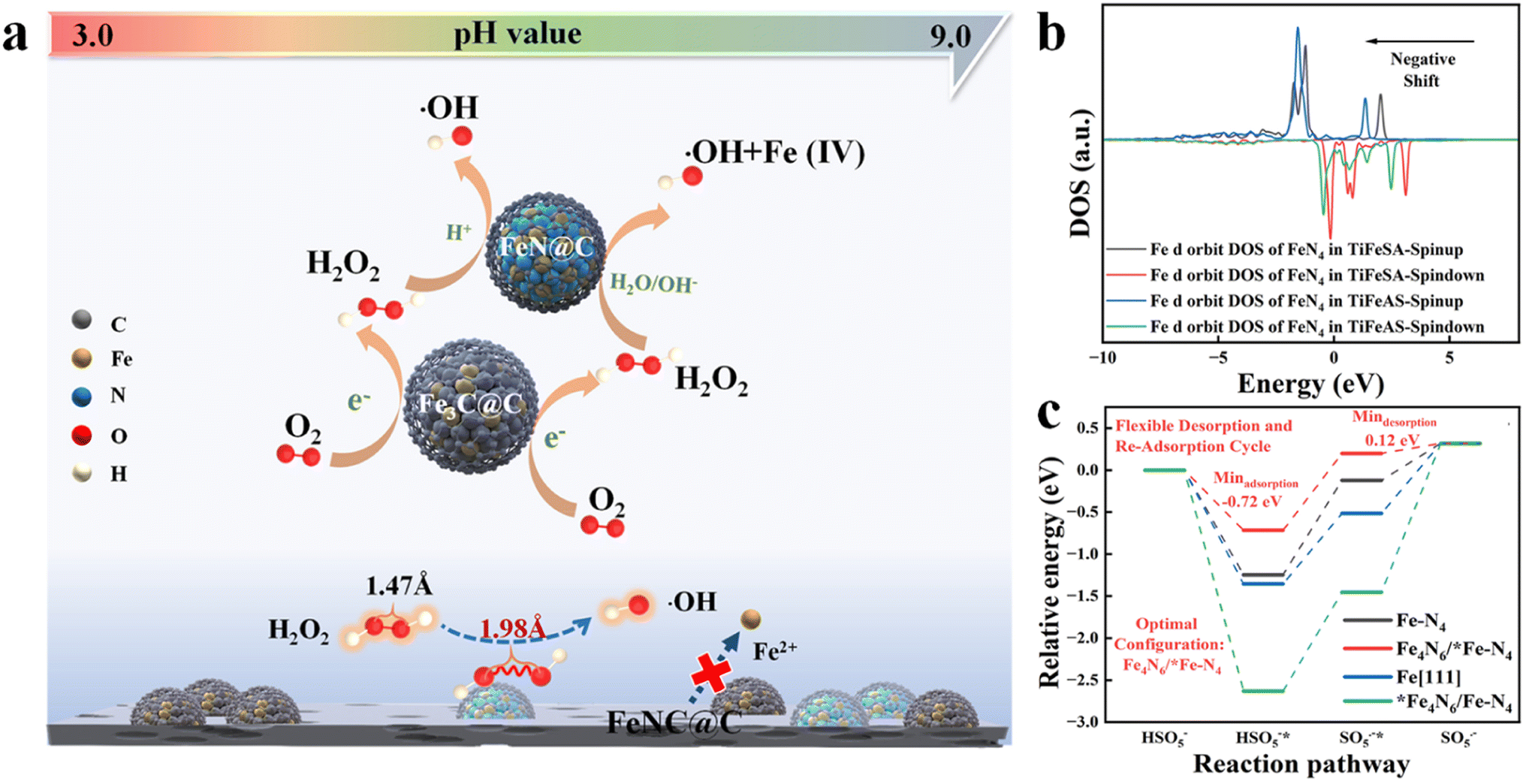 | ||
| Fig. 1 Probable role of Fe3C and FeN nanoparticles in the FeNC@C catalyst for enhanced heterogeneous electro-Fenton performance, reprinted (adapted) with permission from ref. 2 copyright 2021 ACS. (b) The Fe d orbit DOS of FeN4 in TiFeSA and TiFeAS. (c) Reaction energy diagrams for PMS oxidation on Fe–N4, Fe4N6/*FeN4, Fe[111], and *Fe4N6/FeN4, reprinted (adapted) with permission from ref. 1. Copyright 2023 PNAS. | ||
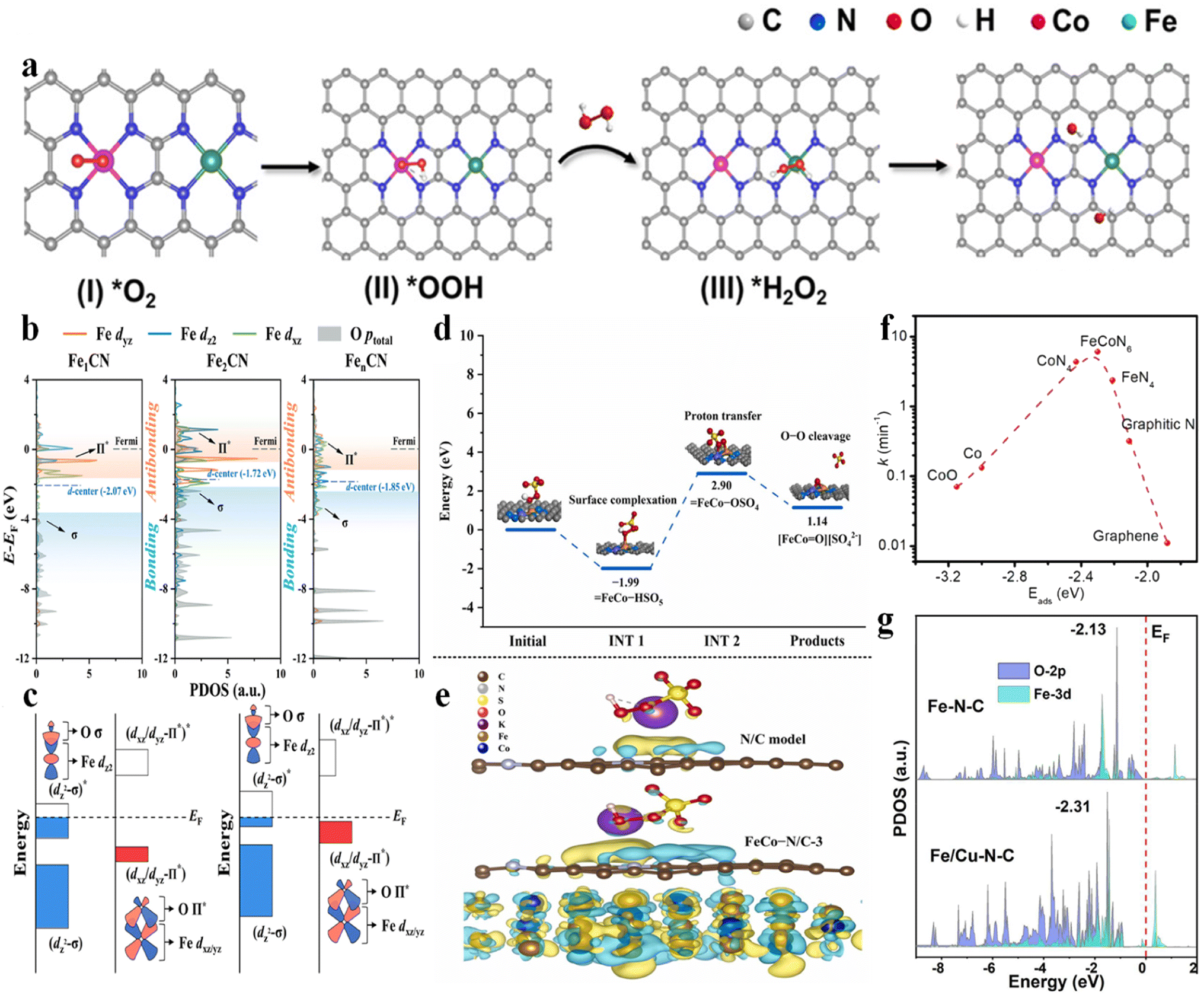 | ||
| Fig. 2 (a) (I–IV) Proposed mechanism of ˙OH electro-generation from the ORR on Co–N4/Fe–N4(CoFe), with permission from ref. 3 copyright 2023 ACS. (b) Projected density of states (PDOS) for Fe-3dyz/dz2/dxz orbitals and O-p orbitals of adsorbed PMS, with permission from ref. 9 copyright 2022 ACS. (c) Schematic illustration of the orbital interaction between Fe-3d (dz2 and dxz/yz) and O of adsorbed PMS (Π* and σ) of Fe sites in Fe1CN and Fe2CN, with permission from ref. 9 copyright 2022 ACS. (d) Potential energy profiles of the pathway for the generation of high-valent metal-oxo species, with permission from ref. 10 copyright 2022 Elsevier. (e) The calculated charge density difference diagrams of PMS adsorbed on the N/C model and FeCo–N/C-3. Isosurface contour is 0.0003e per bohr3. The light yellow and light blue correspond to the electron accumulation and electron depletion, respectively, with permission from ref. 10 copyright 2022 Elsevier. (f) Volcano plot of the reaction rate as a function of Eads of PMS at various active sites, with permission from ref. 7 copyright 2021 Elsevier. (g) PDOS values calculated for the O-2p and Fe-3d orbitals in Fe–N–C and Fe/Cu–N–C, with permission from ref. 13 copyright 2022 Elsevier. | ||
1.2 Incorporation of other atomic sites – strengthened reaction dynamics over the intrinsic active center
In addition, the incorporation of the second active center can effectively regulate the reaction activity when the interaction between the original catalytic site and intermediates is too weak or too strong. For example, Mo et al. (2023) reported that the incorporation of Fe ACs around the Fe single atoms (SAs) can significantly regulate the electronic structure of Fe SAs (i.e., regulated d-band center, Fig. 1b) and thus the interaction between Fe SAs and PMS (i.e., decreased adsorption energy, Fig. 1c), enabling rapid PMS adsorption and SO5˙−desorption,1 however, suffering from the drawbacks of low catalytic activity of ACs. Fe4N6 failed to function as active sites due to the high binding energy (Fig. 1c). Under these situations, the preparation cost may be reduced when substituting the ACs or NPs with SAs. For example, the coupling of Fe and Co sites (i.e., FeCoN6) optimized the adsorption strength between PMS and Fe sites (i.e., 2.30 eV), compared to the sole Fe (i.e., 2.14 eV) or Co SACs (i.e., 2.43 eV), enabling a moderate interaction intensity and thus efficient reactant activation and product desorption7 (Fig. 2f). Therefore, the incorporation of heteroatom sites may effectively regulate the electronic configuration of active centers. To sum up, DACs have rich reaction centers, and would show better catalytic activity and customized selectivity toward Fenton or Fenton-like reactions than SACs. However, the applications of DACs in Fenton or Fenton-like reactions are less focused. Therefore, this perspective aims at summarizing the most recent advances in DAC-mediated Fenton or Fenton-like reactions with the underlying mechanisms being revealed. Most importantly, promising research directions are proposed, expecting further development in this field.2 Most recent advances of DACs in Fenton or Fenton-like reactions
Several researchers have investigated the applications and mechanisms of DACs in Fenton or Fenton-like reactions, but their exploration is still in the initial stage.2.1 Heterogeneous electro-Fenton reactions
Qin et al. (2023) investigated the application of Co–Fe DACs in heterogeneous electro-Fenton reactions.3 Co–Fe DACs were easily fabricated through high-temperature annealing of Co, Fe-doped ZIF-8 precursors, followed by HNO3 leaching at 60 °C. The DFT calculation showed a lowest energy barrier for *H2O2 formation on Co–N4(CoFe) (0.31 eV) and highest adsorption energy of H2O2 on Fe–N4(CoFe) with a binding energy of −0.48 eV. Therefore, the synergistic effect can be established as follows: the atomically dispersed Co sites can enhance the O2 reduction to H2O2 intermediates. Subsequently, the single-atom Fe sites can catalyze the transformation of H2O2 to ˙OH (Fig. 2a). This synergism enabled a higher ˙OH production rate of 2.4 mM L−1 min−1 gcat−1 compared to Fe-NC (1.0 mM L−1 min−1 gcat−1) and Co-NC (0.8 mM L−1 min−1 gcat−1). Simultaneously, the energy consumption was only 19.0 kW h kg−1 COD−1 compared to other electrochemical technologies (29.7–68.0 kW h kg−1 COD−1).2.2 PMS/peroxydisulfate (PDS) activation system
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O through FeCo–HSO5 → FeCo–OSO4 (proton transfer) → FeCoO (O–O heterolytic cleavage) (Fig. 2d), with an energy barrier of 2.05 eV, which was significantly lower than those of high-valent Fe–O (3.28 eV) and high-valent Co–O (4.54 eV) species. Simultaneously, the FeCo alloy can provide electrons to the carbon lattice (Fig. 2e), reducing the Eads of PMS on the N/C and facilitating self-decomposition of PMS to generate 1O2. Therefore, the dual active sites enhanced the Fenton-like activity.10 A similar catalyst was reported by Yang et al. (2020), showing synergistic effects between the optimal FeCoN6 configuration and pyrrolic N as the adsorption site. A volcano-shaped relationship between the Eads of PMS and the rate constant was established, showing optimum adsorption energy at 2.31 eV, which was close to the Eads of PMS on the FeCoN6 site (Fig. 2f), highlighting the pivotal role of the Fe(N3)–Co(N3) coordination.7 Zhu et al. (2023) fabricated Fe–Co DACs using NH2-UIO-66 as a porous carbon support, showing favorable synergism as well. Specifically, the higher adsorption energy of PMS at FeCoN6 (−0.958 eV (–SO3 moiety near Fe atom) or −0.918 (–SO3 moiety near Co atom) eV) was verified compared to FeN4 (−0.568 eV) and CoN4 (−0.471 eV), enabling high PMS concentration at the catalyst surface. In addition, the Fe–Co DACs possess higher DOS near the Fermi level compared to the Fe or Co SAC counterparts, inducing superior reactivity for PMS activation (i.e., favorable electron exchange from the lone electron pair of O to the metal 3d vacant orbital and vice versa).11 Besides, a spin-state reconstruction mechanism on Fe–Co DACs was used to elucidate the performance enhancement. The isolated Fe–Co DACs significantly increased the spin state of the Co center, while lowering the spin state of the Fe center, compared to the Co SAC and Fe SAC counterparts. These variations favorably resulted in the moderate adsorption energy of PMS on Fe–Co DACs (−1.71 eV on Co and −1.91 eV on Fe, moderate) compared to Fe SACs (−2.08 eV, too high) and Co SACs (−1.64 eV, too low).12
O through FeCo–HSO5 → FeCo–OSO4 (proton transfer) → FeCoO (O–O heterolytic cleavage) (Fig. 2d), with an energy barrier of 2.05 eV, which was significantly lower than those of high-valent Fe–O (3.28 eV) and high-valent Co–O (4.54 eV) species. Simultaneously, the FeCo alloy can provide electrons to the carbon lattice (Fig. 2e), reducing the Eads of PMS on the N/C and facilitating self-decomposition of PMS to generate 1O2. Therefore, the dual active sites enhanced the Fenton-like activity.10 A similar catalyst was reported by Yang et al. (2020), showing synergistic effects between the optimal FeCoN6 configuration and pyrrolic N as the adsorption site. A volcano-shaped relationship between the Eads of PMS and the rate constant was established, showing optimum adsorption energy at 2.31 eV, which was close to the Eads of PMS on the FeCoN6 site (Fig. 2f), highlighting the pivotal role of the Fe(N3)–Co(N3) coordination.7 Zhu et al. (2023) fabricated Fe–Co DACs using NH2-UIO-66 as a porous carbon support, showing favorable synergism as well. Specifically, the higher adsorption energy of PMS at FeCoN6 (−0.958 eV (–SO3 moiety near Fe atom) or −0.918 (–SO3 moiety near Co atom) eV) was verified compared to FeN4 (−0.568 eV) and CoN4 (−0.471 eV), enabling high PMS concentration at the catalyst surface. In addition, the Fe–Co DACs possess higher DOS near the Fermi level compared to the Fe or Co SAC counterparts, inducing superior reactivity for PMS activation (i.e., favorable electron exchange from the lone electron pair of O to the metal 3d vacant orbital and vice versa).11 Besides, a spin-state reconstruction mechanism on Fe–Co DACs was used to elucidate the performance enhancement. The isolated Fe–Co DACs significantly increased the spin state of the Co center, while lowering the spin state of the Fe center, compared to the Co SAC and Fe SAC counterparts. These variations favorably resulted in the moderate adsorption energy of PMS on Fe–Co DACs (−1.71 eV on Co and −1.91 eV on Fe, moderate) compared to Fe SACs (−2.08 eV, too high) and Co SACs (−1.64 eV, too low).12
The Fe–Cu DACs were fabricated through hydrothermal synthesis (Fe(acac)3/Cu(acac)3@ZIF-8) and pyrolysis (Fe/Cu–N–C),13 showing an enhanced chloramphenicol removal rate from 0.073 (Fe–N–C) to 0.093 min−1 (Fe/Cu–N–C). The synergistic effects can be illustrated as follows: on the one hand, the electron would transfer from Cu to Fe, enabling the generation of low-valence Fe for peroxydisulfate (PDS) activation. On the other hand, the d-band center of Fe-3d downshifted from −2.13 eV (Fe–N–C) to −2.31 eV (Fe/Cu–N–C) when adding Cu–N–C, thus promoting the adsorption (−2.97 eV vs. −2.35 eV on Fe–N–C) and activation of PDS (Fig. 2g). An et al. (2023) reported the application of Fe–Cu DACs in PMS activation as well. Theoretical simulations illustrated that the electron transfer from Cu to Fe optimized the Fe 3d orbital distribution, showing the decreased d-band center position from −0.456 eV to −0.693 eV, promoting the adsorption and activation of PMS. In addition, the SMX was easily adsorbed on pyrrolic N nearby the Fe–Cu diatomic sites, decreasing the migration distance of active species (i.e., SO4˙−, ˙OH, O2˙−, high-valent metal-oxo species, and metal-peroxo intermediates in this system).14
Studies dealing with the application of DACs in Fenton or Fenton-like reactions are still in its infancy (i.e., only 9 published papers), requiring further validation and exploitation. Due to the characteristics of dual active centers of DACs, these catalysts can efficiently mediate multiple-step reactions (e.g., H2O2 generation and activation),3 achieve dual-function catalysis (e.g., simultaneous PMS oxidation and reduction),6 or strengthen intrinsic activity (e.g., optimizing the realistic active center by incorporating co-catalytic sites). The innovative viewpoints for DAC optimization are listed in the following part.
3 Future outlook
Several theories and methods aiming at improving SAC activity have been put forward, e.g., regulation of the magnetic moment, interatomic distance effect, strain engineering, heteroatomic engineering, coordination regulation, ligand design, etc., which may lay the theoretical foundation for fabricating innovative DACs applied in Fenton or Fenton-like reactions.153.1 Regulation of the magnetic moment
Magnetic moment variation can lead to a significant alteration in reaction activity. The introduction of Pd ACs around Fe SAs achieved the spin transition of low spin (LS) to intermediate spin (MS) for Fe SAs. To be specific, the electron would transfer from Fe SAs to Pd ACs due to the higher electronegativity of Pd compared to Fe2+, leading to the redistribution of Fe 3d-orbital electrons and an increasing spin state of the Fe dz2 orbital. In terms of the dz2 orbital perpendicular to the Fe–N–C plane, the originally empty orbital of LS Fe(II) would change into a partially occupied one in the MS configuration, thereby effectively tuning the orbital overlapping with oxygen-containing intermediates. Interestingly, the O2 molecule was adsorbed on Fe SAs in an end-on fashion (Pauling model) with the O–O bond elongated by 0.08 Å. Whereas, the side-on adsorption configuration (Griffiths model) was preferred on Fe SAs modified with Pd ACs with the O–O bond being elongated by 0.136 Å, which lowered the cleavage barrier of the O–O bond. In addition, the removal of OH* was accelerated upon the incorporation of Pd ACs (Fig. 3a).16 The cleavage barrier of the O–O bond is also important for the Fenton or Fenton-like reactions as the H2O2, PMS, or PDS activation required the peroxide bond dissociation as well. Specifically, the adsorption between the O-containing group and active center (e.g., O–H bond dissociation, O–O bond formation, and O–O bond dissociation) is an important process for the OER (e.g., O–O bond formation (ESI† eqn (3) and (8)) and O–H bond dissociation (ESI eqn (2), (4), (6), and (9)†)) and ORR (e.g., O–O bond dissociation (ESI eqn (3) and (8)†)). Similarly, these processes are key reactions for PMS activation (e.g., O–O bond dissociation in PMS reduction and O–H bond dissociation in PMS oxidation), PDS activation (i.e., O–O bond dissociation), and H2O2 activation (e.g., O–O bond dissociation). Therefore, energy-related reactions, especially the OER and ORR, can be used as important references in designing DACs. Thus, optimizing the magnetic moment can potentially regulate the Fenton or Fenton-like reaction activity through influencing the interaction between active sites and O-containing intermediates.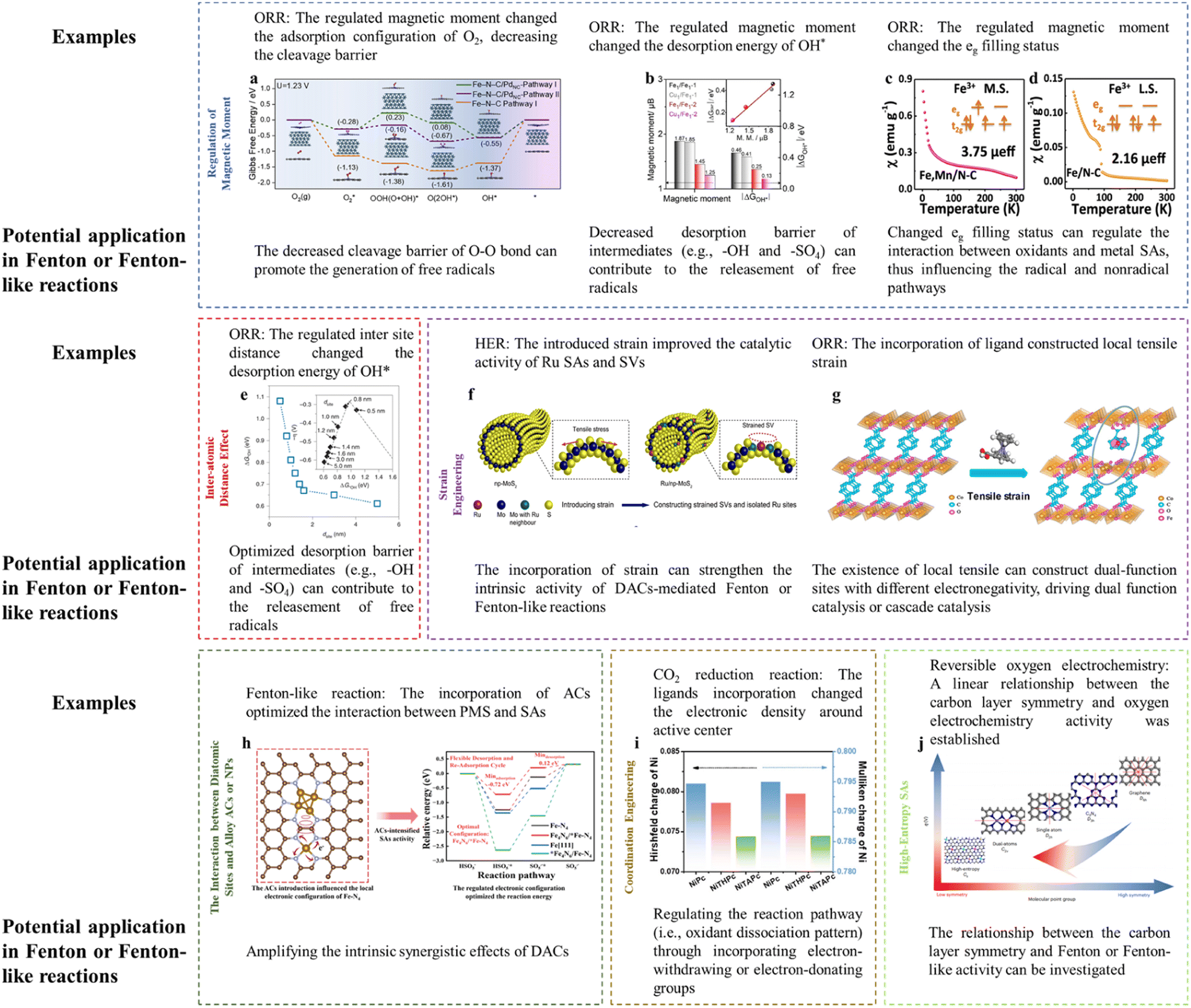 | ||
| Fig. 3 (a) Gibbs free energy diagram of the ORR after the consideration of the solvent effect. Reprinted (adapted) with permission from ref. 16. Copyright 2022 Cell Press. (b) Comparison of the magnetic moment and ΔGOH*. Reprinted (adapted) with permission from ref. 17. Copyright 2022 Wiley. (c and d) Magnetic susceptibility of (c) Fe–Mn/NC, (d) Fe/NC (M. S. represents medium-spin, L. S. represents low-spin), with permission from ref. 18 copyright 2021 Springer Nature. (e) The dsite-dependent ΔGOH* obtained by DFT calculations. (Inset) Volcano plot of calculated overpotentials for the ORR against ΔGOH*. Reprinted (adapted) with permission from ref. 20. Copyright 2021 Springer Nature. (f) Illustration of the construction of Ru/np-MoS2. Reprinted (adapted) with permission from ref. 21. Copyright 2021 Springer Nature. (g) Crystal structures of CoBDC and CoBDC FcCA models obtained from DFT simulations. The microreactor is composed of directly coordinated carboxyl oxygen atoms of FcCA (Co1) and unsaturated coordinative Co2, together with the FcCA linker labeled by the circle. Reprinted (adapted) with permission from ref. 23. Copyright 2021 ACS. (h) The regulatory role of Fe ACs in the Fe SA-mediated PMS oxidation reaction. Reprinted (adapted) with permission from ref. 1. Copyright 2023 PNAS. (i) Hirshfeld and Mulliken charges of the Ni atom in NiPc, NiTHPc, and NiTAPc. Reprinted (adapted) with permission from ref. 30. Copyright 2021 Wiley. (j) The relationship between symmetry and electrocatalytic performance, with permission from ref. 32 copyright 2023 Springer Nature. Notably, high-resolution figures can be viewed in ESI (Fig. 3a–j corresponding to Fig. S4–S11†). | ||
In addition, the magnetic moment is closely associated with the intermediate desorption. For example, the desorption energy of –OH (ΔGOH*) was proved to be roughly linearly correlated with the Fe magnetic moment (Fig. 3b). The incorporation of Cu atoms adjacent to Fe centers reduced the Fe magnetic moment, accompanied by an improved ORR activity (i.e., decreased energy barrier for OH* desorption).17 Specifically, the Fe SACs with Cu incorporation showed increased dz2 electron filling near the Fermi energy level (marked by the red circle, appearing as a decreased magnetic moment) (Fig. S3a and b†), indicating more active electrons for reduction reaction. Thus, the desorption energy of the OH* (i.e., potential decomposition product of H2O2 or PMS) can also be optimized through regulation of the magnetic moment, enabling rapid reaction dynamics.
The adsorption configuration of PMS, H2O2, PDS, etc. on SAs was proved to be metal–O interaction. For example, Mi et al. (2021) proposed that the Co SAs would interact with PMS through the interaction of O on the –SO4 side, therefore, highlighting the pivotal role of metal–O interaction in Fenton or Fenton-like reactions. Also, radical and nonradical pathways are closely associated with the binding energy between metal SAs and O sites in oxidants. Promisingly, this interaction can be regulated through the magnetic moment as well. Furthermore, the adjacent atomically dispersed Mn–N moieties can activate the FeIII sites through modulation of the spin-state and electronic configuration, showing that the FeIII with one eg electron (t2g4eg1) can readily penetrate into the antibonding π-orbital of O.18 In detail, the zero-field cooling (ZFC) temperature-dependent magnetic susceptibility was measured. The calculated effective magnetic moment of Fe–Mn/NC and Fe/NC was 3.75 μeff and 2.16 μeff, respectively (Fig. 3c and d). Therefore, the number of unpaired d electrons (n) of FeIII ions was determined using the following equation:
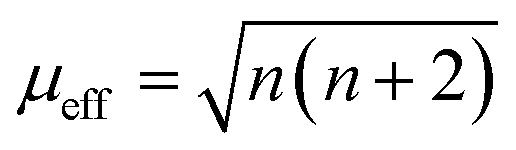 | (1) |
The n value of Fe/N–C was 1.3, suggesting the low-spin state of FeIII ions without eg filling, enabling a strong interaction between the FeIII/O2 interaction due to the fact that no electron occupied the σ* antibonding orbital of FeN4. Whereas, an increased n value3 was determined for Fe–Mn/NC, indicating single eg filling. These results indicated that the unusual low-spin state of neighboring MnIII moieties permitted FeIII in FeN4 to achieve the ideal eg filling, endowing the catalysts with optimal binding energy with O2 and thus favorable ORR activity. Hence, this strategy can be potentially used to regulate the metal–O interaction, thus regulating the radical and non-radical pathways.
Therefore, the incorporation of second metal sites (i.e., constructing DACs) can optimize the magnetic moment (i.e., spin state) of the intrinsic metal active site through the 3d electron interaction, especially the orbital filling of dz2. In further studies, the exact correlation between the magnetic moment of the active center and Fenton or Fenton-like reactions should be established, especially focusing on the adsorption configuration, energy barrier for the peroxide bond dissociation, and –OH desorption. Particularly, the trade-off between the energy barrier for O–O dissociation and intermediate desorption (e.g., –SO4 or –OH) should be deeply investigated.
3.2 Inter-atomic distance effect
DACs with direct M–M interaction (e.g., M1–M2–N6)7 and indirect M–M interaction (e.g., M–N4/M–N4)3 were both prepared for Fenton or Fenton-like reactions. Simultaneously, the advanced SAC synthesis field is devoted to improve the mass loading of metal sites (i.e., exceeding 20% through a two-step annealing method).19 In addition to the metal loading changes, the inter-site distance would change accordingly, which is not deeply investigated to date. This scientific problem would be extremely interesting in the diatomic configuration.As discussed in the “Regulation of the Magnetic Moment”, O–O bond cleavage and OH* desorption are important in both the ORR and Fenton or Fenton-like reactions. Therefore, the study focusing on the ORR can be used for reference. For example, Jin et al. (2021) reported the effect of Fe inter-atomic distance in regulating the ORR activity.20 The results showed that the inter-site distance would induce valence changes and distorted D4h symmetry (changed pre-edge peaks and pre-edge absorption density, as confirmed by XANES analysis), spin state changes (D1 sites, assigned to low-spin Fe(II)–N4 or high-spin Fe(III)–N4 accounted for 56 and 63% when the Fe inter-site distance was 1.9 and 0.5 nm, respectively, as confirmed by Mössbauer spectroscopy), spin moment (marked decrease in the on-site magnetic moment when the Fe inter-site distance was lower than ca. 1.6 nm, as verified by the density-functional theory (DFT)), and changed eg electron filling (a higher degree of filling for the eg electron would weaken the interaction intensity of OH*, as verified by the DFT). The experimental results showed improved activity when decreasing the distance from 1.2 to 0.7 nm (optimum), below which the intrinsic activity would be slightly diminished, which can be ascribed to the optimized ΔGOH* (Fig. 3e).
Specifically, the XANES analysis identified that the decreased inter-atomic distance led to the decreased valence state and more distorted D4h symmetry. It was demonstrated that the electronic activity would increase with decreased basal-plane symmetry. Therefore, the more distorted D4h symmetry regulated by the inter-atomic distance may enhance the ORR performance, originating from the lowered aromaticity and more localized electronic structure. The inter-atomic distance-induced spin state changes verified by Mössbauer spectroscopy can potentially influence the dz2 orbital filling, thus influencing the ORR efficiency. Similar underlying mechanisms can be established for the magnetic moment, which is important for the overlap between the metal dz2 orbital and pz orbital of oxygen-related adsorbates. The downshift of the energy of the dz2 orbital when two Fe atoms get closer allows a higher orbital filling for the eg electron, thereby resulting in a lower on-site spin moment. Consequently, the increased filling degree of the partially occupied d orbitals would weaken the bonding strength between the metal site and adsorbed hydroxyl intermediates (e.g., OH*). Therefore, the correlation between the inter-atomic distance and GOH* can be established through the changed basal symmetry, spin state, magnetic moment, etc. Similarly, the inter-atomic distance in the DAC configuration can possibly influence the Fenton or Fenton-like reaction activity, originating from the varied valence state, basal symmetry, dz2 orbital filling (i.e., magnetic moment and spin state), etc. To solve this issue in the Fenton or Fenton-like reaction field, the following aspects may be focused or considered:
(i) The inter-site distance can be easily regulated through changing the metal precursor concentration, i.e., the site density-mediated active center distance, which should be examined by inductively coupled plasma mass spectrometry (ICP-MS). Particularly, acid etching should be performed to remove metal ACs or NPs. For example, Jin et al. (2021) placed the catalysts in 0.5 M H2SO4 solution for 8 h to remove the aggregated metal ACs and NPs.20
(ii) To precisely assess the atomic dispersion of DACs, ion-beam thinning should be conducted to reduce the thickness of entities (where all metal atoms were able to be counted), followed by high-angle annular dark-field image (HAADF-STEM) analysis and X-ray absorption spectroscopy (XAS).
(iii) Most importantly, the inter-site distance can be estimated by statistical distribution (i.e., HAADF-STEM images taken in a thin area) and in situ surface interrogation scanning electrochemical microscopy (SI-SECM).
(iv) Subsequently, the correlational analysis between site density and reaction activity can be established, mainly focusing on the valence using XAS analysis, spin state using Mössbauer spectroscopy analysis, and spin moment and intermediate binding energy using DFT.
3.3 Strain engineering
The synergistic effects of DACs are widely verified, however, it is still desirable to amplify their synergism and thus promote the practical application. Promisingly, the incorporation of strain (i.e., the construction of a high-curvature surface) into DACs can optimize the electronic configuration.21 To the best of our knowledge, limited studies have paid attention to this strategy, in order to strengthen the Fenton or Fenton-like reaction efficiency, thus requiring the investigation and validation of strain engineering in this field.In this part, several examples dealing with the hydrogen evolution reaction (HER) and ORR were referenced to provide insights for catalyst-mediated Fenton or Fenton-like reactions. In Fenton or Fenton-like reactions, the strain construction can amplify the known synergistic effects of DACs. When introducing the curvature, the mass transport of H2O2 from Co SAs to Fe SAs and their respective catalytic efficiency (i.e., Co SA-mediated H2O2 generation and Fe SA-mediated H2O2 activation) may be facilitated in Co–Fe DAC-catalyzed electro-Fenton reaction.3 For example, the incorporation of tensile stress on Ru SAs and S vacancies (SVs) strengthened their intrinsic activity. The reaction density around SVs and H2O dissociation/H2 evolution on Ru SAs was promoted, respectively (Fig. 3f). Similar results were reported for the high-curvature surface-loaded FeN4 (i.e., introducing compressive stress), showing the down-shifted d-band center and thus optimized bonding strength towards oxygenated intermediates.22 This strategy can be useful in regulating the DAC-mediated Fenton or Fenton-like reactions, as the optimized d-band position is a critical factor in influencing the activation efficiency of oxidants (e.g., H2O2, PMS, or PDS).1
Promising substrates that can be applied to exert pressure are listed as follows:
(i) Nanoporous substrates that can introduce curvature, for example, the nanoporous Au with the Au ligament, followed by the coverage or loading of DAC materials.
(ii) Helical carbon structure with abundant high-curvature surface, which can be obtained by carbonization of helical polypyrrole that was templated from self-assembled chiral surfactants, followed by metal loading.
(iii) MOFs with the incorporation of other ligands, for example, when substituting partial terephthalic acid (BDC) in Co-BDC with ferrocene carboxylic acid (FcCA), the crystal lattice CoBDC FcCA with tensile strain was obtained, generating Co coordinated with the carboxyl oxygen atom of FcCA (Co1) and unsaturated coordinative Co2 (Fig. 3g). Under this situation, the intermediate spin state (t2g6eg1) Co can be constructed, showing the optimal OH* adsorption energy and OOH* formation energy.23
3.4 Interaction between diatomic sites and alloy ACs or NPs
Recently, several reports have highlighted the role of in situ formation of ACs or NPs, and proposed the viewpoints as follows:(i) The role of ACs/NPs in SAC-mediated catalytic reaction was ignored.24 Specifically, the transformation of Cu SAs to ACs/NPs was observed during electrochemical reduction of nitrate to ammonia. Interestingly, the aggregated Cu NPs would reversibly disintegrate into SAs and then be restored to the Cu–N4 structure upon being exposed to an ambient atmosphere.24
(ii) The contribution of ensembled multiple atom sites in chemical reactions should be highly regarded.25 Specifically, the contribution of various surface Pd species, ranging from SAs to ACs and NPs, in dehydrogenation of dodecahydro-N-ethylcarbazole (DNEC, for H2 transportation and utilization) was determined.25 Interestingly, the results showed that the Pd SAs were inactive to this reaction. The fully exposed Pd ACs with an average Pd–Pd coordination number of 4.4 can actively activate the reactant and promote product desorption, due to the fact that this reaction required multiple metal sites to initiate the DNEC adsorption. Whereas, the strong interaction between the reactant and crystalline surface (i.e., Pd(111)) enabled a sluggish desorption process.
Theoretically, the incorporation of ACs or NPs can amplify the synergistic effects of DACs, increasing the charge asymmetry, thus influencing the reaction pathways. For example, the AC-induced charge redistribution of Fe–N–C was verified, showing a down-shifted d-band center and decreased Bader charge, thus strengthening the interaction between Fe–N–C and PMS. In detail, the incorporation of ACs optimized the HSO5− oxidation and SO5˙− desorption steps, accelerating the reaction progress1 (Fig. 3h). Therefore, the reinforcement of the catalytic efficiency of DACs by incorporating ACs or NPs can be expected. Definite catalyst types are listed as follows, especially in dual-metal forms.
(i) Metal 1 and metal 2 ensembled DACs, accompanied by the M1/M2 alloy NPs or ACs. For example, the coupled FeN3–CoN3 moiety and FeCo alloy moiety loaded on N-doped C were reported, working separately to generate high-valence metal (i.e., FeCo–O) and 1O2.10
(ii) The M1/M2 alloy NP or AC supported M1 SAs or M2 SAs. For example, the CoPt alloy-strengthened Pt SAs. The Pt SA aided by the CoPt alloy showed high d-band occupation for promoting the HER reaction kinetics.26
(iii) The M1 NP or AC supported M1 SAs coupled with M2 NP or AC supported M2 ACs. This strategy is extremely less explored, which presents wide prospects. For example, the constructed Fe4@FeN4 and Ni4@NiN4 showed decreased adsorption energy of OH* for Fe SAs modified with Fe ACs and increased adsorption energy of OH* for Ni SAs modified with Ni ACs, promoting the ORR and OER activities, respectively.27
3.5 Coordination engineering
Several in-plane structure engineering strategies were widely studied and previously reported, such as the regulation of coordination atoms, coordination number, etc. Analogous to the above discussion, these strategies can also effectively regulate the electronic configuration, for example, the d-band center, charge status (i.e., Bader charge, valence number, etc.), etc. Undoubtedly, the bonding energy between intermediates and active centers can be optimized, showing optimal adsorption energy and a decreased energy barrier. These types of fabrication methods can be summarized as follows, which can be used in DAC performance optimization.(i) The doping of heteroatoms with different electronegativities. For example, the Fenton-like activity (i.e., PMS activation activity) can be enhanced or reduced by integrating electron-deficient boron (B) or electron-rich phosphorus (P) into the carbon substrate, respectively. Specifically, the long–range interaction with B atoms decreased the electronic density of single-atom Cu sites and down-shifted the d-band center, leading to the optimized adsorption energy for PMS activation.28
(ii) The optimization of the coordination number. For example, the coordination number of single-atom Ni can be easily regulated through changing the annealing temperature for the ZIF precursor, generating Zn–N3 and Zn–N4 at 900 and 800 °C, respectively.29 Theoretically, the different coordination numbers would significantly regulate the DAC performance, through influencing the intermediate formation.
(iii) The incorporation of ligands. Two types of reaction pathways can be expected for the PMS activation reaction (e.g., PMS oxidation and PMS reduction), depending on the electronic density of the active center. Typically, PMS would be oxidized at electron-deficient sites and reduced at electron-rich sites.6 For example, PMS was reduced at Bi sites and oxidized at Co sites in Co-doped in Bi2O2CO3, generating SO4˙−/˙OH and 1O2, respectively.6 Promisingly, the electron status of the active center can be easily regulated by incorporating an organic ligand. Polymer modification (poly-4-vinylpridine (P4VP)) on Co sites in cobalt phthalocyanine (CoPc) through Co-pyridine coordination was observed, enabling the transformation of Co(II) to more nucleophilic Co(I). In addition, the incorporation of electron-donating groups (e.g., hydroxyl or amino) can induce electronic localization at the Ni site, increasing its electronic density (Fig. 3i). In contrast, electron-withdrawing groups would lead to the electron delocalization of the Ni site.30 Therefore, this strategy can undoubtedly be used to design the PMS reaction pathway.
3.6 High-entropy SAs
DACs should be highly regarded due to the intrinsic drawbacks of SACs, i.e., single-function active centers. This strategy can be extended to multiple-atom systems, due to the fact that there typically existed limited active centers and other heteroatoms would participate in the catalytic system as cocatalysts. For example, triple-atom sites (Co–O–Bi) were fabricated through Co doping in Bi2O2CO3. The O sites would function as electron mediators, whereas, the Co and Bi would initiate reduction function (PMS → SO4˙− and ˙OH) and oxidation function (PMS → 1O2), respectively.6 In addition, the incorporation of Co optimized the binding between Fe sites and PMS. In this situation, the Fe sites and Co sites would function as catalytic sites and co-catalytic sites, respectively.7 However, the excessive complex system (i.e., multiple-atom system) may be avoided as the optimal interaction strength can be obtained especially when only introducing two metals.31 Otherwise, the mechanism interpretation and high cost would be the significant problems.The recent study proposed an innovative and interesting concept (i.e., high-entropy single atom). Researchers embedded five single atoms (i.e., Fe, Mn, Co, Ni, and Cu) with two sources of N, effectively minimizing the local symmetry to destabilize the π-electron network of graphitic carbons, thus endowing the catalysts with ideal binding energies (e.g., neither too strong nor too weak) (Fig. 3j).32 Specifically, graphitic carbons with sp2 hybridization showed unique aromaticity with delocalized π electrons, derived from the high symmetry (D6h) of single graphene layers. However, π-conjugated electron network-protected stable graphitic carbons are extremely stable and inert towards chemical reactions. Several strategies such as heteroatom doping, defects, edge, and morphology modification have been developed to break this symmetry, so as to increase reactivity. The electrocatalytic activity enhancement has been verified when decreasing the basal-plane symmetry from D6h to D2h and C2v. Therefore, incorporating several metal atoms can ideally decrease the symmetry to a higher level. Hence, an interesting theory or principle can be established, we should focus on the local structure when fabricating DACs, however, may pay attention to the holistic characteristics when incorporating several metal heteroatoms (i.e., carrier engineering). Intriguingly, the symmetry of carbon layers and Fenton or Fenton-like reactions may be focused, aiming at establishing a correlation.
3.7 Trade-off between activity and stability: the pivotal role of in situ characterization technology
It is widely acknowledged that SAs would be the active sites during numerous reactions.33–35 However, the transformation of Cu SAs to ACs/NPs was observed during electrochemical reduction of nitrate to ammonia. Interestingly, the aggregated Cu NPs would reversibly disintegrate into SAs and then be restored to the Cu–N4 structure upon being exposed to an ambient atmosphere,24 thus highlighting the pivotal role of in situ formation of ACs/NPs in catalytic reactions. In another study dealing with single-atom Pt on CeO2 for CO oxidation, interesting results were reported as well. The dynamic evolution of two types of single-atom Pt on CeO2, i.e., adsorbed Pt1 in Pt/CeO2 and square planar Pt1 in PtATCeO2, fabricated at 500 °C and 800 °C (atom-trapping method), respectively, was investigated. Adsorbed Pt1 in Pt/CeO2 was mobile with the in situ formation of Pt ACs during CO oxidation, however, showing high reactivity with near-zero reaction order in CO. In contrast, PtATCeO2 with strongly anchored Pt1 sites led to relatively low reactivity with a positive reaction order in CO. It's worth noting that both Pt/CeO2 and PtATCeO2 in CO transformed Pt1 to Pt NPs, but retaining their initial Pt1 state after re-oxidative treatments.36 Therefore, the changed atomic sites during the reaction can be considered as realistic active centers to some extent. Atomic catalysts with high stability may face inferior reactivity. These issues should be further focused with advanced in situ characterization technology, such as in situ XAS, in situ X-ray photoelectron spectroscopy (XPS), and in situ electron microscopy.4 Conclusion
DACs are promising candidates in Fenton or Fenton-like reactions. On the one hand, the diatomic configuration is conducive to the achievement of cascade reaction, bifunctional reaction, etc. On the other hand, the incorporation of the second active center can strengthen the intrinsic active sites through influencing the electronic configuration. In consideration of the limited focus on DAC-mediated Fenton or Fenton-like reactions (i.e., only 9 published papers), several innovative developmental directions were put forward. Specifically, the in-depth mechanisms in designing DACs included regulation of the magnetic moment (i.e., spin state), inter-atomic distance, and strain and incorporation of NPs/ACs and heteroatoms/ligands with specific electronic characteristics, through influencing the valence electron state, basal plane symmetry, orbital filling (e.g., dz2 electron), etc., eventually regulating the reaction pathways (e.g., cascade reaction, bifunctional reaction, oxidation reaction, reduction reaction, etc.) due to the optimized interaction (e.g., adsorption configuration) between metal sites and O-containing intermediates. It is suggested that DACs can effectively design and regulate the Fenton or Fenton-like reactions, especially for multi-step reactions, thus representing a new avenue in Fenton-based water treatment technologies.Author contributions
All authors contributed to the writing – review & editing of the manuscript. Particularly, Qixing Zhou was responsible for the funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China as a Shandong joint fund project (grant No. U1906222), the Ministry of Science and Technology of People’s Republic of China as a key technology research and development program project (grant no. 2019YFC1804104), and the Tianjin Science and Technology Bureau as a key science and technology supporting project (grant no. S19ZC60133).References
- F. Mo, et al., The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2300281120 CrossRef CAS PubMed.
- J. Hu, et al., Duet Fe3C and FeNx sites for H2O2 generation and activation toward enhanced electro-Fenton performance in wastewater treatment, Environ. Sci. Technol., 2021, 55, 1260–1269 CrossRef CAS PubMed.
- X. Qin, et al., Highly efficient hydroxyl radicals production boosted by the atomically dispersed Fe and Co sites for heterogeneous electro-Fenton oxidation, Environ. Sci. Technol., 2023, 57, 2907–2917 CrossRef CAS PubMed.
- S. Zhan, et al., Efficient Fenton-like process for pollutant removal in electron-rich/poor reaction sites induced by surface oxygen vacancy over cobalt–zinc oxides, Environ. Sci. Technol., 2020, 54, 8333–8343 CrossRef CAS PubMed.
- H. Zhang, C. Li, L. Lyu and C. Hu, Surface oxygen vacancy inducing peroxymonosulfate activation through electron donation of pollutants over cobalt-zinc ferrite for water purification, Appl. Catal., B, 2020, 270, 118874 CrossRef CAS.
- Q. Zhou, et al., Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2300085120 CrossRef CAS PubMed.
- J. Yang, et al., A highly efficient Fenton-like catalyst based on isolated diatomic Fe-Co anchored on N-doped porous carbon, Chem. Eng. J., 2021, 404, 126376 CrossRef CAS.
- M. Li, et al., Diatomic Fe–Fe catalyst enhances the ability to degrade organic contaminants by nonradical peroxymonosulfate activation system, Nano Res., 2023, 16, 4678–4684 CrossRef CAS.
- J. Li, et al., Modulating the electronic coordination configuration and d-band center in homo-diatomic Fe2N6 catalysts for enhanced peroxymonosulfate activation, ACS Appl. Mater. Interfaces, 2022, 14, 37865–37877 CrossRef CAS PubMed.
- Z. Zhao, et al., Construction of dual active sites on diatomic metal (FeCo–N/C-x) catalysts for enhanced Fenton-like catalysis, Appl. Catal., B, 2022, 309, 121256 CrossRef CAS.
- C. Zhu, et al., Heterogeneous Fe-Co dual-atom catalyst outdistances the homogeneous counterpart for peroxymonosulfate-assisted water decontamination: New surface collision oxidation path and diatomic synergy, Water Res., 2023, 241, 120164 CrossRef CAS PubMed.
- Z. Zhao, et al., Improved electronic structure from spin-state reconstruction of a heteronuclear Fe-Co diatomic pair to boost the Fenton-like reaction, Environ. Sci. Technol., 2023, 57, 4556–4567 CrossRef CAS PubMed.
- H. Wu, et al., Synergistic effects for boosted persulfate activation in a designed Fe–Cu dual-atom site catalyst, Chem. Eng. J., 2022, 428, 132611 CrossRef CAS.
- S. An, L. Wang, G. Li, B. Zhu and Q. Jin, Well-defined Fe/Cu diatomic catalysts for boosted peroxymonosulfate activation to degrade organic contaminants, Sep. Purif. Technol., 2023, 316, 123827 CrossRef CAS.
- H. Zhang, X. Jin, J. M. Lee and X. Wang, Tailoring of active sites from single to dual atom sites for highly efficient electrocatalysis, ACS Nano, 2022, 16, 17572–17592 CrossRef CAS PubMed.
- X. Wei, et al., Tuning the spin state of Fe single atoms by Pd nanoclusters enables robust oxygen reduction with dissociative pathway, Chem, 2023, 9, 181–197 CAS.
- T. He, et al., Theory-guided regulation of FeN4 spin state by neighboring Cu atoms for enhanced oxygen reduction electrocatalysis in flexible metal–air batteries, Angew. Chem., Int. Ed. Engl., 2022, 134, e202201007 Search PubMed.
- G. Yang, et al., Regulating Fe-spin state by atomically dispersed Mn–N in Fe–N–C catalysts with high oxygen reduction activity, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- X. Hai, et al., Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- Z. Jin, et al., Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- K. Jiang, et al., Rational strain engineering of single-atom ruthenium on nanoporous MoS(2) for highly efficient hydrogen evolution, Nat. Commun., 2021, 12, 1687 CrossRef CAS PubMed.
- J. Yang, et al., Compressive strain modulation of single iron sites on helical carbon support boosts electrocatalytic oxygen reduction, Angew. Chem., Int. Ed. Engl., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- F. He, et al., Metal-organic frameworks with assembled bifunctional microreactor for charge modulation and strain generation toward enhanced oxygen electrocatalysis, ACS Nano, 2022, 16, 9523–9534 CrossRef CAS PubMed.
- J. Yang, et al., Potential-driven restructuring of Cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- C. Dong, et al., Fully exposed palladium cluster catalysts enable hydrogen production from nitrogen heterocycles, Nat. Catal., 2022, 5, 485–493 CrossRef CAS.
- W. Yang, et al., Tuning the cobalt–platinum alloy regulating single-atom platinum for highly efficient hydrogen evolution reaction, Adv. Funct. Mater., 2022, 32, 2205920 CrossRef CAS.
- Z. Wang, et al., Cooperation between Dual Metal Atoms and Nanoclusters Enhances Activity and Stability for Oxygen Reduction and Evolution, ACS Nano, 2023, 17, 8622–8633 CrossRef CAS PubMed.
- X. Zhou, et al., Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119492119 CrossRef CAS PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Rational fabrication of low-coordinate single-atom Ni electrocatalysts by MOFs for highly selective CO2 reduction, Angew. Chem., 2021, 133, 7685–7689 CrossRef.
- K. Chen, et al., Ligand engineering in nickel phthalocyanine to boost the electrocatalytic reduction of CO2, Adv. Funct. Mater., 2021, 32, 2111322 CrossRef.
- J. D. Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Design of Co-Cu diatomic site catalysts for high-efficiency synergistic CO(2) electroreduction at industrial-level current density, Angew. Chem., Int. Ed. Engl., 2022, 61, e202212329 CAS.
- X. Lei, et al., High-entropy single-atom activated carbon catalysts for sustainable oxygen electrocatalysis, Nat. Sustain., 2023 DOI:10.1038/s41893-023-01101-z.
- P. Yang, Y. Long, W. Huang and D. Liu, Single-atom copper embedded in two-dimensional MXene toward peroxymonosulfate activation to generate singlet oxygen with nearly 100% selectivity for enhanced Fenton-like reactions, Appl. Catal., B, 2023, 324, 122245 CrossRef CAS.
- H. Feng, et al., Porphyrin-based Ti-MOFs conferred with single-atom Pt for enhanced photocatalytic hydrogen evolution and NO removal, Chem. Eng. J., 2022, 428, 132045 CrossRef CAS.
- Y. Zhang, et al., Single-atom Cu anchored catalysts for photocatalytic renewable H2 production with a quantum efficiency of 56, Nat. Commun., 2022, 13, 58 CrossRef CAS PubMed.
- Z. Zhang, et al., Memory-dictated dynamics of single-atom Pt on CeO(2) for CO oxidation, Nat. Commun., 2023, 14, 2664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02872k |
| This journal is © The Royal Society of Chemistry 2023 |