
Milk exosomes with enhanced mucus penetrability for oral delivery of siRNA†
Matthew R.
Warren
a,
Chenzhen
Zhang
a,
Armin
Vedadghavami
a,
Krister
Bokvist
b,
Pradeep K.
Dhal
 c and
Ambika G.
Bajpayee
*ad
c and
Ambika G.
Bajpayee
*ad
aDepartments of Bioengineering, Northeastern University, Boston, MA 02115, USA. E-mail: warren.ma@husky.neu.edu; zhang.chenz@husky.neu.edu; vedadghavami.a@husky.neu.edu
bSanofi SA Global R&D, Frankfurt am Main, Germany. E-mail: krbo13@hotmail.com
cSanofi SA Global R&D, Waltham, MA 02451, USA. E-mail: Pradeep.dhal@sanofi.com
dMechanical Engineering, Northeastern University, Boston, MA 02115, USA. E-mail: a.bajpayee@northeastern.edu; Tel: +1 617-373-7018
First published on 7th December 2020
Abstract
Bovine milk-derived exosomes have recently emerged as a promising nano-vehicle for the encapsulation and delivery of macromolecular biotherapeutics. Here we engineer high purity bovine milk exosomes (mExo) with modular surface tunability for oral delivery of small interfering RNA (siRNA). We utilize a low-cost enrichment method combining casein chelation with differential ultracentrifugation followed by size exclusion chromatography, yielding mExo of high concentration and purity. Using in vitro models, we demonstrate that negatively charged hydrophobic mExos can penetrate multiple biological barriers to oral drug delivery. A hydrophilic polyethylene glycol (PEG) coating was introduced on the mExo surface via passive, stable hydrophobic insertion of a conjugated lipid tail, which significantly reduced mExo degradation in acidic gastric environment and enhanced their permeability through mucin by over 3× compared to unmodified mExo. Both mExo and PEG-mExo exhibited high uptake by intestinal epithelial cells and mediated functional intracellular delivery of siRNA, thereby suppressing the expression of the target green fluorescence protein (GFP) gene by up to 70%. We also show that cationic chemical transfection is significantly more efficient in loading siRNA into mExo than electroporation. The simplicity of isolating high purity mExo in high concentrations and equipping them with tunable surface properties, demonstrated here, paves way for the development of mExo as an effective, scalable platform technology for oral drug delivery of siRNA.
1. Introduction
Oral delivery of drugs is the most preferred mode of therapeutic intervention. Particularly for the treatment of chronic diseases, significant patient compliance with orally-administered therapeutics arises from the flexibility of dosing, ability for self-administration, simplicity of formulations and storage capability.1–3 However, the oral bioavailability of macromolecular drugs such as proteins, peptides, nucleic acids etc. is significantly limited due to several biological barriers, which include the propensity for enzymatic and acidic degradation in the gastrointestinal (GI) tract and considerably limited permeation across the intestinal mucosal and epithelial cell layers.4–6 Although permeation enhancers like caprates have shown some promise with a GLP-1 peptide drug approved for clinical use, the oral bioavailability of the drug from this formulation is only about 1%.7 While particles such as liposomes can encapsulate and protect therapeutics against enzymatic degradation, the negatively charged, hydrophobic mucosal membrane lining the intestinal epithelium presents significant steric hindrance to the diffusive transport of such particulate systems.8 Thus, an efficient, versatile encapsulation system for orally delivered therapeutics remains elusive.Exosomes are cell-derived, membranous vesicles present in nearly all bodily fluids. With sizes ranging between 35–120 nm in diameter, exosomes are composed of a phospholipid bilayer derived from the membrane of the cell of origin.9 Initially thought to be a means of waste disposal for the cell, exosomes have recently gained attention due to their natural role of shuttling molecular cargos (e.g. DNA, small RNAs, proteins, and lipids) between distant cells in the body. Due to their capacity for protection and precise delivery of bioactive macromolecules, exosomes have been identified as a promising drug delivery platform, including for oral delivery.10,11 For instance, exosomes have been evaluated for the delivery of small-interfering RNA (siRNA).12,13 Delivery of siRNA presents a significant challenge due to several factors; for instance, these large (∼13 kDa), negatively charged macromolecules face a steep thermodynamic barrier in crossing cell membranes. Moreover, systemically-delivered siRNAs are prone to enzymatic degradation by circulating RNAse.14 While traditional synthetic vehicles for siRNA delivery such as liposomes and polymeric nanoparticles face limited tissue targeting ability and toxicity concerns,15 exosomes hold significant promise as a non-immunogenic, non-inflammatory, and biocompatible delivery system due to their inherent compatibility with recipient cells.
There remain considerable challenges to translating exosome-based therapeutics into clinical use. For instance, there is no standard source of exosomes, as they are found in nearly all bodily fluids.9 Many studies use exosomes isolated from cell culture media;16–18 however, culture media typically suffers from a low yield of exosomes. Moreover, there are immunological safety concerns with repurposing vesicles derived from immortalized cell lines for use in humans.19 Exosomes derived from bovine milk have recently gained interest as drug carriers, in particular for oral delivery, since bovine milk represents a low cost, scalable source from which a high yield of exosomes can be extracted.20 Due to frequent consumption by the majority of the population, bovine milk is considered safe,2 and bovine milk-derived exosomes exhibit low systemic immunogenicity and inflammatory properties in vivo.20,21 Moreover, milk exosomes are naturally suited for intestinal uptake.22,23 In fact, it has been proposed that milk exosomes evolved as a means for the transfer of biologically active molecules from mother to child via the oral route.2 Recent studies have shown that milk-derived exosomes and their microRNAs are stable under degrading gastric conditions (low pH) and exhibit significant bioavailability following oral administration.2,24,25
In this study, we have engineered highly pure bovine milk exosomes (mExo) with modular surface tunability as vehicles for the oral delivery of siRNA. We employ a low-cost enrichment process that combines casein chelation with differential ultracentrifugation followed by size exclusion chromatography to yield mExo of high purity and concentration. A polyethylene glycol (PEG) coating was introduced on the mExo surface via passive, stable hydrophobic insertion of a conjugated lipid tail in order to provide improved stability in the stomach's acidic environment and facilitate transport through multiple physiological barriers to oral delivery, including the intestinal mucus and cells. PEGylation reduced mExo degradation in the gastric environment and significantly enhanced mExo permeability through intestinal mucin without affecting their uptake by the intestinal epithelial cells. Subsequently, we have directly compared the loading efficiency of commonly used methods (including cationic chemical transfection reagents and electroporation) for the encapsulation of siRNA into the lumen of milk exosomes; these methods were assessed for any downstream adverse effects on the integrity of mExo membrane. Finally, we have demonstrated that siRNA-loaded, surface-modified mExo can functionally deliver siRNA and silence a target gene in vitro, making them promising naturally-derived carriers for oral delivery of siRNA.
2. Methods
2.1 Materials
Fat-free bovine milk (Hood) was purchased from a local supermarket. EDTA was purchased from Quality Biological (Gaithersburg, MD). Polycarbonate, round-bottom centrifuge tubes, micro-bicinchoninic acid (BCA) assay kit, dibenzocyclooctyne-Cy5 (DBCO-Cy5) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene glycol)-2000] (DSPE-PEG(2000)-azide) were bought from Thermo Fisher (Waltham, MA). qEV original 35 nm size exclusion chromatography columns were bought form Izon Science (Christchurch, New Zealand). Western blotting reagents were purchased as follows: Laemmli sample buffer from Alfa Aesar (Haverhill, MA), animal-free blocking buffer from Cell Signaling Technology (Danvers, MA), horseradish peroxidase (HRP) substrate from Millipore (Burlington, MA). The ExoGlow Protein fluorescent labeling kit and the ExoFect transfection kit were bought from System Biosciences (Palo Alto, California). Cell culture media components were purchased as follows: high-glucose Dulbecco's modified Eagle's medium (DMEM), non-essential amino acids, fetal bovine serum and GlutaMAX from Gibco (Carlsbad, CA), trypsin-EDTA from Thermo Fisher. Lipofectamine-2000 (Lipofect) reagent and Ambion Silencer Cy3-conjugated siRNA were purchased from Thermo Fisher. Bovine serum albumin, fluorescein isothiocyanate isomer I (FITC), 100 kDa molecular weight cut-off (MWCO) Amicon® Ultra Centrifugal Filters, purified type II porcine mucin, phosphate buffered saline (PBS) and other salts and reagents were purchased from Sigma-Aldrich (St Louis, MO). GFP-targeting siRNA duplex was purchased from Horizon Discovery.2.2 Milk exosome harvesting
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 35000g, and 70000g for 1 h each in 36 mL round-bottom ultracentrifuge tubes on an ultracentrifuge (Sorvall WX80, Thermo Fisher), recovering the supernatant and discarding the pellet between steps in order to remove protein aggregates and larger contaminating vesicles. The supernatant was recovered, filtered through 0.22 μm syringe filters, and subjected to ultracentrifugation at 100000g for 2 h to pellet milk extracellular vesicles (mEVs). The mEV pellet was resuspended in 600 μL of PBS, which was immediately run through a commercial size exclusion chromatography (SEC) column (Izon qEV, 35 nm pore size) to remove any contaminating vesicles as well as proteins and protein aggregates, with 500 μL fractions being collected. Following individual fraction characterization, mExo fractions (8–9) were pooled, and aliquots were frozen at −80 °C for downstream applications. An overview of the isolation and purification process is shown in Fig. 1.
000g, 35000g, and 70000g for 1 h each in 36 mL round-bottom ultracentrifuge tubes on an ultracentrifuge (Sorvall WX80, Thermo Fisher), recovering the supernatant and discarding the pellet between steps in order to remove protein aggregates and larger contaminating vesicles. The supernatant was recovered, filtered through 0.22 μm syringe filters, and subjected to ultracentrifugation at 100000g for 2 h to pellet milk extracellular vesicles (mEVs). The mEV pellet was resuspended in 600 μL of PBS, which was immediately run through a commercial size exclusion chromatography (SEC) column (Izon qEV, 35 nm pore size) to remove any contaminating vesicles as well as proteins and protein aggregates, with 500 μL fractions being collected. Following individual fraction characterization, mExo fractions (8–9) were pooled, and aliquots were frozen at −80 °C for downstream applications. An overview of the isolation and purification process is shown in Fig. 1.
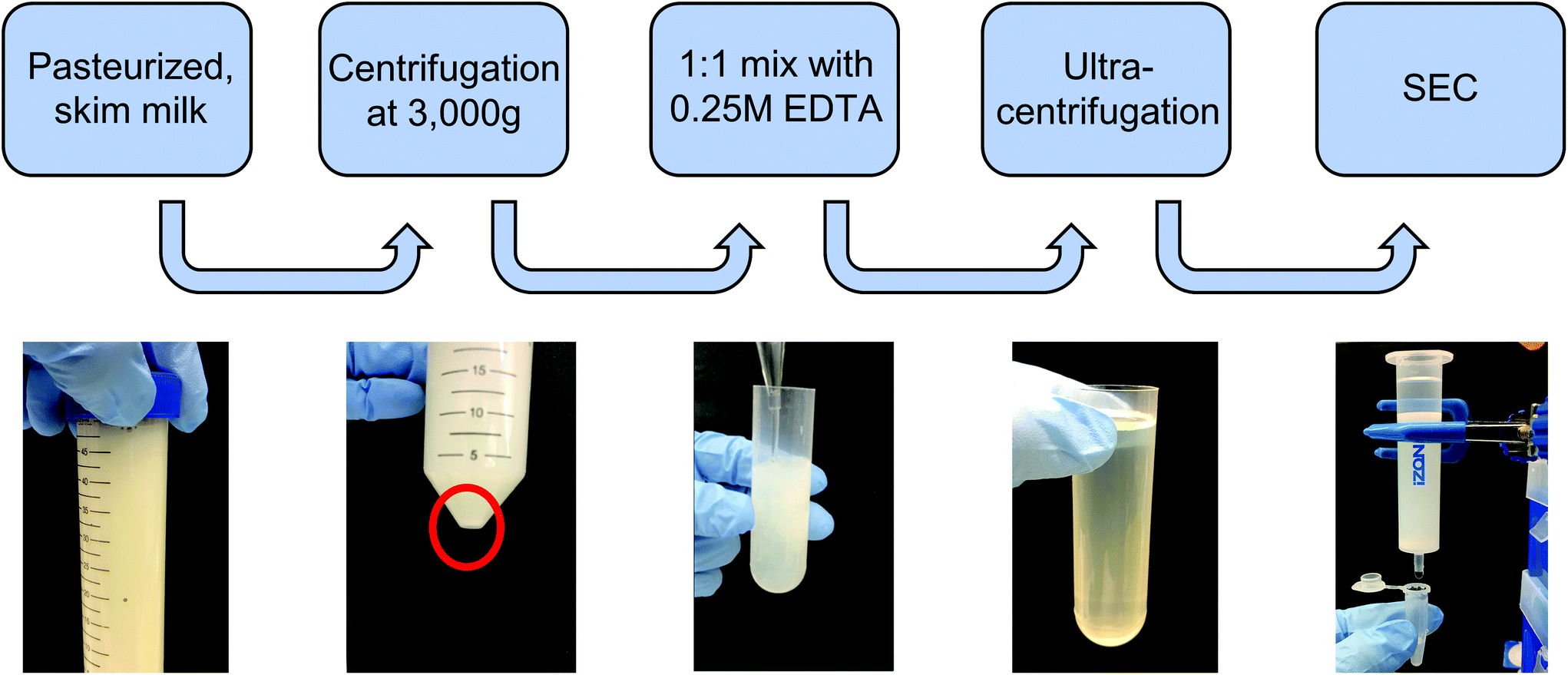 | ||
| Fig. 1 Isolation and purification process of exosomes from bovine milk. Pasteurized milk samples were first diluted with PBS and centrifuged at 3000g to pellet cells and debris. The supernatant was then treated with an equal volume of 0.25 M EDTA to chelate casein micelles and casein-coated exosomes. This was followed by differential ultracentrifugation to remove large protein aggregates and contaminating microvesicles. The sample was subsequently centrifuged at 100000g for 2 h to pellet extracellular vesicles (mEV). Finally, the resuspended mEV pellet was purified by size-exclusion chromatography (SEC) using a commercially available column. | ||
2.3 Exosome characterization
:200 ratio in 20 nm-filtered PBS with 0.8% Tween-20 surfactant and 200 nm diameter polystyrene calibration beads (1010 particles per mL final concentration). For testing, 5 μL of sample was loaded into Spectradyne TS-300 or TS-400 cartridges, and at least 15 acquisitions (n ≥ 20000 particles) were measured per sample. Individual sample size distribution data was processed using the Spectradyne software for calibration and filtering of false-positive particle detection events. The filtering criteria were as follows: events with <60 μs transit time and <210 nm measured diameter were accepted, and the automated background subtraction feature was implemented. Filtered data was then calibrated for diameter and concentration based on the distribution of 200 nm control beads, and final concentration values were determined by integration of the size distribution curve. Fraction purity was calculated as the ratio of particle count to μg of total protein (from BCA) in corresponding samples.
:1 ratio of Laemmli sample buffer. Reduced proteins were electrophoresed using the BioRad system for 45 min at 150 V in NuPAGE 4–12% precast Bis-Tris gels. After separation, proteins were transferred to a nitrocellulose membrane (pre-treated with methanol) in a BioRad blotting module at 75 V for 2.5 h. After confirmation of successful transfer with Ponceau-S staining dye, membranes were blocked using animal-free blocking buffer for 1 h, and then treated overnight with primary antibodies (1:1000 dilution of mouse anti-TSG101, sc-7964; 1:1000 mouse anti-Flot1, sc-74566; 1:600 mouse anti-Alix, sc-53540; Santa Cruz). After washing thrice in PBS-Tween 20 washing buffer, membranes were incubated for 1 h in secondary antibody (1:1000 dilution of mouse IgGk binding protein-HRP, sc-516102, Santa Cruz). Finally, membranes were washed, incubated for 5 min in HRP substrate, and chemiluminescence was imaged using a BioRad ChemiDoc CCD imaging system.
2.4 Surface modification of mExo with polyethylene glycol (PEG)
:4 final molar ratio. This solution was then incubated for 6 h at room temperature with light shaking to facilitate the click reaction. After the click reaction, the solution containing DPA-Cy5 and excess DBCO-Cy5 was added directly to exosome samples for insertion.
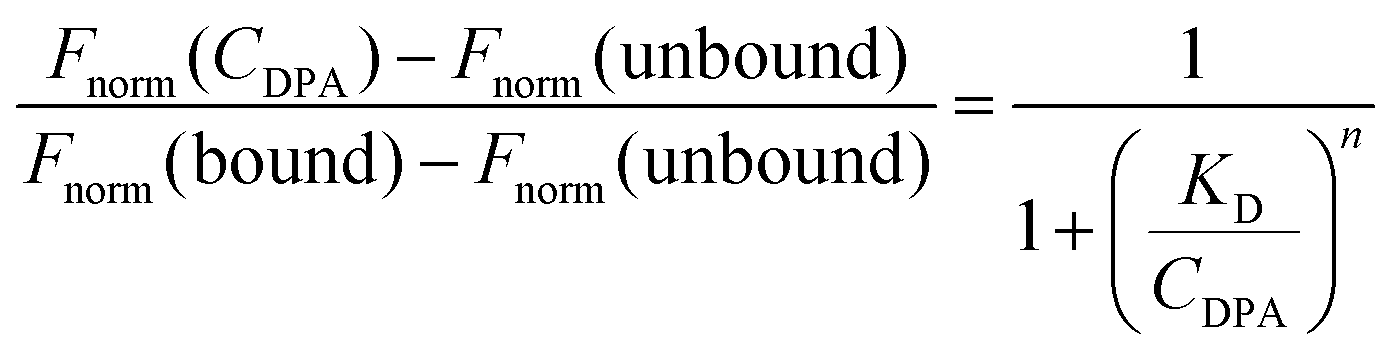
When quantifying surface loading efficiency, DPA-Cy5 was used in place of DPA, using the same loading process. To quantify DPA surface loading, we carried out the process described above using various molar ratios of DPA to mExo particles – 5000:1, 10000:1, and 20000:1 – all reacted in 1.5 mL mixtures, followed by purification by UFC. Cy5 fluorescence was measured in each final PEG-mExo sample using a plate reader and was compared to a standard curve of DBCO-Cy5 to quantify the concentration of DPA inserted into the mExo surface. The number of DPA molecules per mExo particle was then estimated using the following formula:

It was assumed that all Cy5 signal retained in the UCF filters was from DPA inserted into the mExo membrane. However, to account for possible background signal from DPA-Cy5 micelles retained in the filter as well as excess DBCO-Cy5 associated with the mExo surface, control mixtures were prepared with no DPA (only DBCO-Cy5 and mExo) and no mExo (only DPA-Cy5), followed by purification and quantification of retained Cy5 signal as described above. Cy5 signal from both of these control samples was subtracted from measurements of samples with graded DPA:mExo molar ratios.
where Fnorm(unbound) is the normalized fluorescence of only unbound mExo, Fnorm(bound) is normalized fluorescence of the mExo-PEG complex at saturated bound state and Fnorm(CDPA) is normalized fluorescence measured at titration concentration of DPA.
 000:1 molar ratio was incubated in either PBS or a biosimilar mucin gel for various times at 30 °C with light shaking. Samples were subsequently subjected to two 10 min UFC spins on 100 kDa filters at 4000g to elute DPA-Cy5 molecules that had partitioned out of the mExo membrane. Cy5 signal was measured in the eluted sample for each time point and compared to a DBCO-Cy5 standard curve. In parallel, free DPA-Cy5 was incubated in PBS and mucin gel for 1 h at the same concentration as PEG-mExo and separated by the same process to define a point of comparison for 100% DPA-Cy5 dissociation from the mExo membrane. This accounts for the possibility that diffusion of DPA-Cy5 molecules through UFC spin filters may be hindered by the presence of mucin proteins. Thus, dissociation of DPA-Cy5 from PEG-mExo at each time point was quantified as the percentage of eluted DPA-Cy5 compared to 100% dissociation controls for both PBS and mucin. Biosimilar mucin was prepared by reconstituting purified type-II porcine intestinal mucin in 20 mM HEPES/20 mM NaCl via light stirring overnight at 4 °C, as previously described.28
000:1 molar ratio was incubated in either PBS or a biosimilar mucin gel for various times at 30 °C with light shaking. Samples were subsequently subjected to two 10 min UFC spins on 100 kDa filters at 4000g to elute DPA-Cy5 molecules that had partitioned out of the mExo membrane. Cy5 signal was measured in the eluted sample for each time point and compared to a DBCO-Cy5 standard curve. In parallel, free DPA-Cy5 was incubated in PBS and mucin gel for 1 h at the same concentration as PEG-mExo and separated by the same process to define a point of comparison for 100% DPA-Cy5 dissociation from the mExo membrane. This accounts for the possibility that diffusion of DPA-Cy5 molecules through UFC spin filters may be hindered by the presence of mucin proteins. Thus, dissociation of DPA-Cy5 from PEG-mExo at each time point was quantified as the percentage of eluted DPA-Cy5 compared to 100% dissociation controls for both PBS and mucin. Biosimilar mucin was prepared by reconstituting purified type-II porcine intestinal mucin in 20 mM HEPES/20 mM NaCl via light stirring overnight at 4 °C, as previously described.28
2.5 In vitro evaluation of PEG-mExo for oral delivery
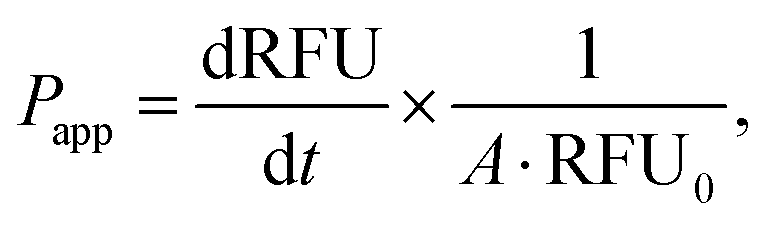
where RFU is the acceptor chamber fluorescence reading, RFU0 is the starting solution fluorescence, and A is the surface area of the transwell membrane.
000 cells per well of a 96-well plate and allowed to adhere for 36 h. 12.5–100 μg of fluorescently-labelled mExo were added to 200 μL of media and incubated with cells for 2.5 h. After uptake, cells were washed three times with cold PBS, detached using phenol-red free 0.25% trypsin-EDTA, and fluorescence was read at 490/520 nm excitation/emission. Uptake was quantified by comparing cell fluorescence to a standard curve of a known weight of labelled mExo suspended in PBS, using cells incubated in mExo-free media as a blank. Uptake data was fit to the Michaelis–Menten kinetic equation, modelling mExo uptake by endocytosis as a saturable process. Data was fit to the following equation:  , where Ru is the rate of mExo transport, Vmax is the maximum Ru at which endocytosis saturates, and Km is the Michaelis–Menten constant, defined as the mExo concentration at which Ru = 0.5 × Vmax.
, where Ru is the rate of mExo transport, Vmax is the maximum Ru at which endocytosis saturates, and Km is the Michaelis–Menten constant, defined as the mExo concentration at which Ru = 0.5 × Vmax.
Uptake of mExo and PEG-mExo into Caco-2 cells was also measured using fluorescence imaging. Cells were seeded at a density of 20000 cells per well of a 96-well plate and allowed to adhere for 36 h. 130 μg of fluorescently-labelled mExo and dual-labelled PEG-mExo (prepared as described in Methods 2.4.4) were added to 200 μL of media and incubated with cells for 2.5 h. After uptake, cells were washed three times with cold PBS and fixed for 10 min in 4% paraformaldehyde. Cell nuclei were then stained by incubating in DAPI for 5 min. Fixed, stained cells were then washed three times with cold PBS and imaged using a Nikon Eclipse Ts2R fluorescence microscope with constant exposure time and 10× magnification. Images were processed using ImageJ software. To account for potential sources of background signal in images with single- and dual-labels, cells were incubated with free DBCO-Cy5, DPA-Cy5, and ExoGlow dye at both high and low concentrations, aligning with concentrations of these moieties in mExo samples pre- and post-purification. These cells were imaged as described above to evaluate the extent to which these potential sources of background signal contribute to single- and dual-labelled uptake images.
2.6 In vitro gene silencing by mExo-siRNA
000g for 5 min, and pellets were resuspended in 100 μL PBS for downstream loading analysis, or 50 μL complete DMEM for cell culture studies.
000 cells per well in 48-well plates in antibiotic-free media. mExo-siRNA formulations were prepared as described above, using 80 pmol of siGFP and 5 μL of Lipofect in the initial mixture, and diluted to 12 μg per well mExo protein for addition to cells. Simultaneously, cells were treated with lower concentrations of the mExo-siRNA formulation to investigate a dose-dependent silencing response. To account for silencing by Lipofect-siRNA complexes that co-precipitate with mExo during Exo-Quick TC purification, the loading/purification process was carried out in the absence of mExo, and the resulting samples (termed “Residual Lipofect”) were also evaluated for silencing capability. In parallel, Lipofectamine-siRNA complexes were added directly to cells as a positive control (termed “Lipofect-siRNA”). In a separate experiment, PEG-mExo-siRNA was prepared by mixing purified mExo-siRNA with 20000:1 molar excess DPA via dropwise addition to a 1.5 mL reaction mixture with continuous stirring, followed by incubation at 37 °C for 1 h with light shaking. 12 μg per well of mExo-siRNA and PEG-mExo-siRNA was added directly to cells.
For all silencing experiments, mExo internalization and gene knockout occurred over 72 h. After this time, cells were imaged using the FITC filter of a Nikon Eclipse Ts2R fluorescence microscope, with constant exposure time. To quantify silencing of GFP, cell FITC fluorescence was measured using a Beckman Coulter Cytoflex flow cytometer. Briefly, following siRNA treatment, cells were detached from the well plate with 0.25% trypsin-EDTA and washed twice by centrifugation and resuspension in PBS. siRNA conditions were tested in duplicate, and 10000 events were acquired per sample and constricted to a custom FC/SC gate as designated by control cells.
3. Results
3.1 Isolation, purification and characterization
Exosomes were harvested from bovine milk using differential ultracentrifugation followed by purification using SEC (Fig. 1). The total protein content of each SEC fraction (500 μL) was determined by BCA assay (Fig. 2A). The elution profile shows a peak in total protein content in fractions 7–11, with fractions 8 and 9 containing maximum concentrations. This fraction range corresponds to the elution of exosomes (mExo) and other milk extracellular vesicles (mEV), which is in agreement with the manufacturer's instructions. This protein elution profile was consistently observed across multiple batches of mExo isolation, suggesting that SEC using the qEV column is a reliable and reproducible method for isolating exosomes from bovine milk. The trend towards increased protein content in later fractions (12–16) suggests the presence of contaminating proteins, aggregates, and smaller vesicles in these fractions. As a result, only mEV fractions (7–11) were used for further studies. | ||
| Fig. 2 Physical characteristics of individual mEV fractions following size exclusion chromatography. (A) Total protein concentration of individual SEC fractions 1–16, measured by BCA assay. (B) Particle concentration in exosome fractions (7–11), measured by Spectradyne nCS1. (C) Purity of milk exosome (mExo) fractions, represented as particle-to-total protein ratio. (D) Representative size distribution of pre-SEC mEV sample and pooled fractions 8 and 9, measured by Spectradyne nCS1. (E) Transmission electron microscopy images of pooled mExo sample showing spherical morphology. Scale bars on left and middle images represent 100 nm and 500 nm, respectively. Data are presented as mean ± 95% confidence intervals (calculated by t-distribution). | ||
The particle concentrations for mEV and mExo fractions were determined by using the high-resolution resistive pulse sensing technique (Fig. 2B). The majority of particles were observed in fractions 8 and 9. In comparison to the particle concentration in the pre-SEC samples, nearly all mExo particles were recovered in fractions 8 and 9. Although some particles (<0.25 × 1012 particles per mL) were observed in later fractions (10–11), these fractions contain less than 10% of the total particles isolated from a single SEC run. Moreover, these particles contain a considerably high protein concentration, indicating a particle population of significantly lower purity (as expressed by the particle-to-protein ratio). Interestingly, purity of particles decreased with increased elution volume (Fig. 2C), with exosome fractions exhibiting 2.2-fold higher purity than pre-SEC samples. Taken together, these results suggest that an exosome population of significantly higher purity is present in fractions 8 and 9 compared to pre-SEC samples, while recovering nearly all particles present in the initial sample.
The size distribution of pooled mExo samples (fractions 8–9) was measured by resistive pulse sensing and confirmed by negative-stain transmission electron microscopy (TEM) (Fig. 2D and E). A majority of measured particles displayed a diameter between 65–90 nm, with a median diameter of approximately 70 nm (10th percentile of 62.2 nm, 90th percentile of 90.4 nm). This is supported by TEM, which displays particles in this size range and reveals the spherical morphology of the isolated exosomes. From 18 mL of bovine milk, 650 μg of purified mExo was recovered in 1 mL final volume. The results on the physical properties of the purified mExo samples are summarized in Table 1.
| Yield | ||
| Particle concentration (1012 particles per mL) | 1.92 ± 0.38 | |
| Total protein (μg mL−1) | 641 ± 40 | |
| Purity | ||
| Particle per protein ratio (109 particles per μg protein) | 3.0 ± 0.6 | |
| Size | ||
| Median diameter (nm) | 70.5 ± 3.3 | |
| 10th percentile | 62.2 ± 4.8 | |
| 90th percentile | 90.4 ± 2.4 | |
3.2 Proteome and protein biomarkers
Western blotting was performed on individual mExo fractions (8–11) to qualitatively confirm the presence of exosomes based on common protein biomarkers (Fig. 3). The samples were probed for the presence of two proteins related to the endosomal sorting complex required for transport (ESCRT) – TSG-101 and Alix – and two exosome-associated membrane proteins – Flotillin-1 and CD63.36 For comparison, pre-SEC mEV samples were also analyzed for these markers. All four diagnostic markers were present in mEV samples, strongly supporting the presence of exosomes in the crude samples before SEC. Following purification, fractions 8 and 9 contained all biomarkers. Conversely, fractions 10 and 11 did not show the presence of TSG-101 or Alix. This suggests that mExo are present in lesser amounts in fractions 10 and 11, further supporting the lower purity of those fractions (Fig. 3A). Therefore, due to the ubiquitous presence of exosome protein biomarkers, as well as higher purity and particle content in fractions 8 and 9, these fractions were pooled to generate final milk exosome samples for further investigations.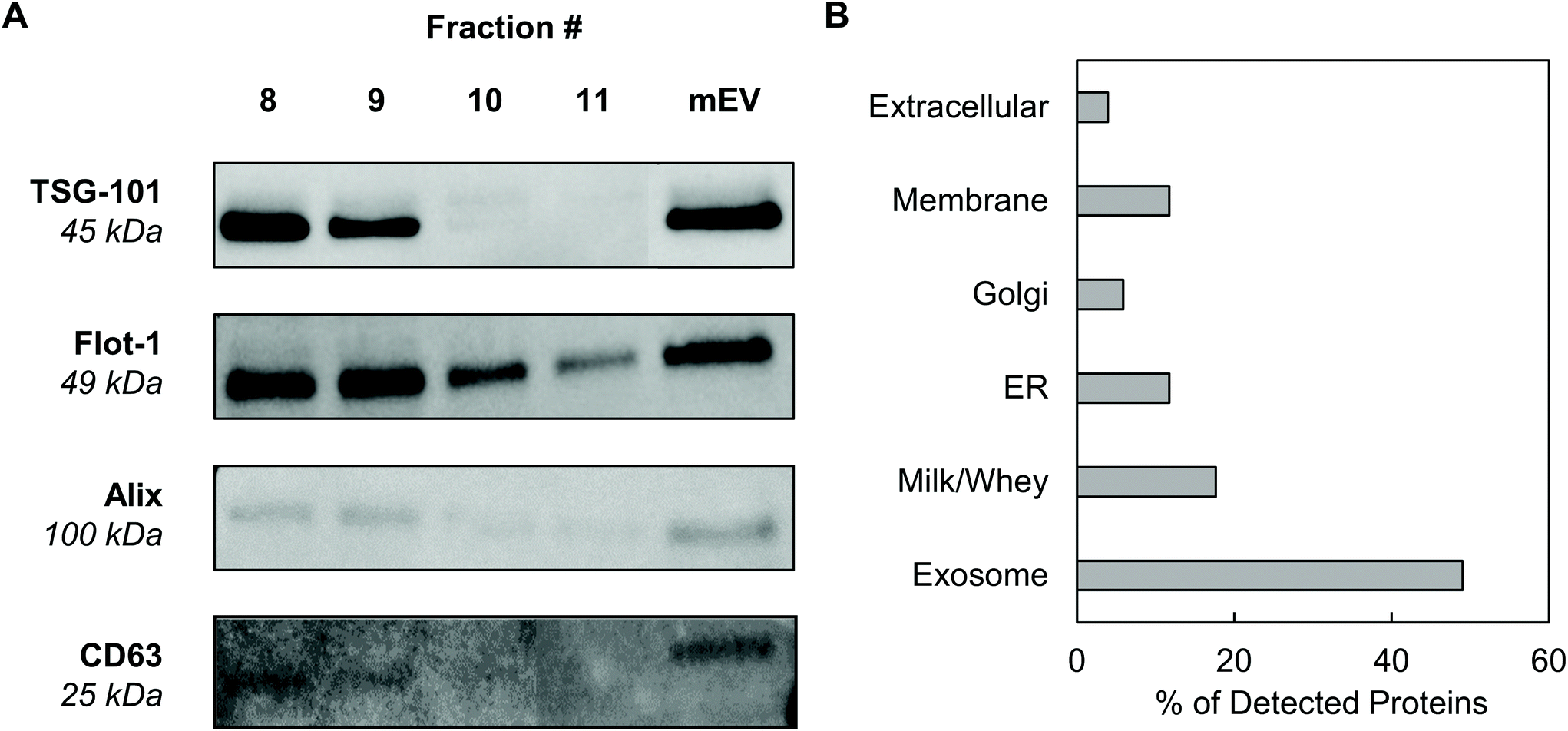 | ||
| Fig. 3 Screening for the presence of protein exosome biomarkers in mExo samples. (A) Western blotting for the presence of a variety of exosome markers, including membrane proteins CD63 and Flotillin-1 and ESCRT proteins (TSG-101 and Alix). Individual fractions after SEC are compared to the resuspended mEV pellet after ultracentrifugation and before SEC. (B) Breakdown of the categories of proteins present in exosome proteomics analysis by mass spectrometry. | ||
Mass spectrometry was performed to identify the proteome of final pooled mExo samples (see Table 2). Raw spectral data from mass spectrometry was processed to generate a list of observed proteins (ESI Table S1†). In total, the presence of 214 proteins was observed. After careful analysis, the list was narrowed down to 51 high-confidence proteins (for which two or more unique peptide sequences were detected). Proteins unrelated to EVs, such as cell debris markers, were excluded. A full list of the mExo proteome is presented in the ESI.† Out of the 51 shortlisted proteins in the sample, 25 were considered to be related to EVs (i.e. previously reported exosome biomarkers, proteins related to extracellular vesicle biogenesis, and membrane proteins), 9 proteins were deemed milk proteins, and the remaining 17 proteins were cell-related (Fig. 3B). Proteins were categorized and screened for following the recommendation of the International Society of Extracellular Vesicles.36
| Classification | Location | Family | Proteins |
|---|---|---|---|
| Exosome marker | Membrane receptor | Tetraspanins | CD9, CD63, CD81 |
| Membrane/lipid bound | Rab proteins | Rab 11b, 15, 18, 7a, 1b, 5c | |
| Annexins | Annexin A1, A2, A5, A7 | ||
| Cytosolic | Heat shock proteins | Hsc70, Hsp70 | |
| Eukaryotic translation elongation factor (EEF) | EEF1α2, EEF2 | ||
| Lacto proteins | Lactoadherin (MFG-38), lactotransferrin, lactoperoxidase | ||
| Fatty acid synthase | FASN | ||
| Protein zeta/delta | YWHAE | ||
| Milk-Related Proteins | Surrounding solution, cytosol, membrane and lipid bound | Caseins | α-Casein, B-casein, κ-casein |
| Albumins | Serum albumin, lactalbumin | ||
| Lipoproteins | Apolipoproteins A-I, A-IV, E | ||
| Others | Immunoglobulin, B-lactoglobulin | ||
| Negative controls (not present in sample) | Cytosolic, membrane | Endoplasmic reticulum | Calnexin, endoplasmin (HSP90B1) |
| Transmembrane | Integrin-B1, CD40, CD62P | ||
| Golgi | GM130 | ||
| Mitochondria | Cytochrome C | ||
Mass spectrometry confirmed the presence of commonly reported exosome biomarkers related to the formation and secretion of exosomes from donor cells, including tetraspanins (CD9, CD81), heat shock proteins (Hsp70), Rab proteins and annexins. Additionally, a variety of milk-related proteins were found in the final exosome samples, including casein, albumins, lipoproteins and lactoglobulin, which are potentially remnants from the ultracentrifugation process. Importantly, proteomic data was screened for the presence of proteins reported as microvesicle- and cell debris-related contaminants, such as ER-related calnexin and endoplasmin (HSP90B1); membrane-associated integrin-b1, CD40 and CD62P; Golgi marker GM130; and mitochondrial cytochrome C (see Table 2, row 3).36 None of these proteins were found in proteomic data, suggesting high purity of final samples and absence of major contaminants such as cellular debris or non-exosome vesicles.
3.3 PEGylation of mExo membrane by hydrophobic insertion
Stable insertion of PEG chains into the mExo lipid bilayer was achieved by using a conjugated, bivalent lipid tail (DSPE-PEG-Azide or DPA; Fig. 4).37 Azide-derivatized PEG was chosen as a modular moiety for mExo surface engineering, as the terminal azide group enables simple conjugation of a variety of peptides or proteins by using azide–alkyne click chemistry, enabling modulation of mExo surface properties.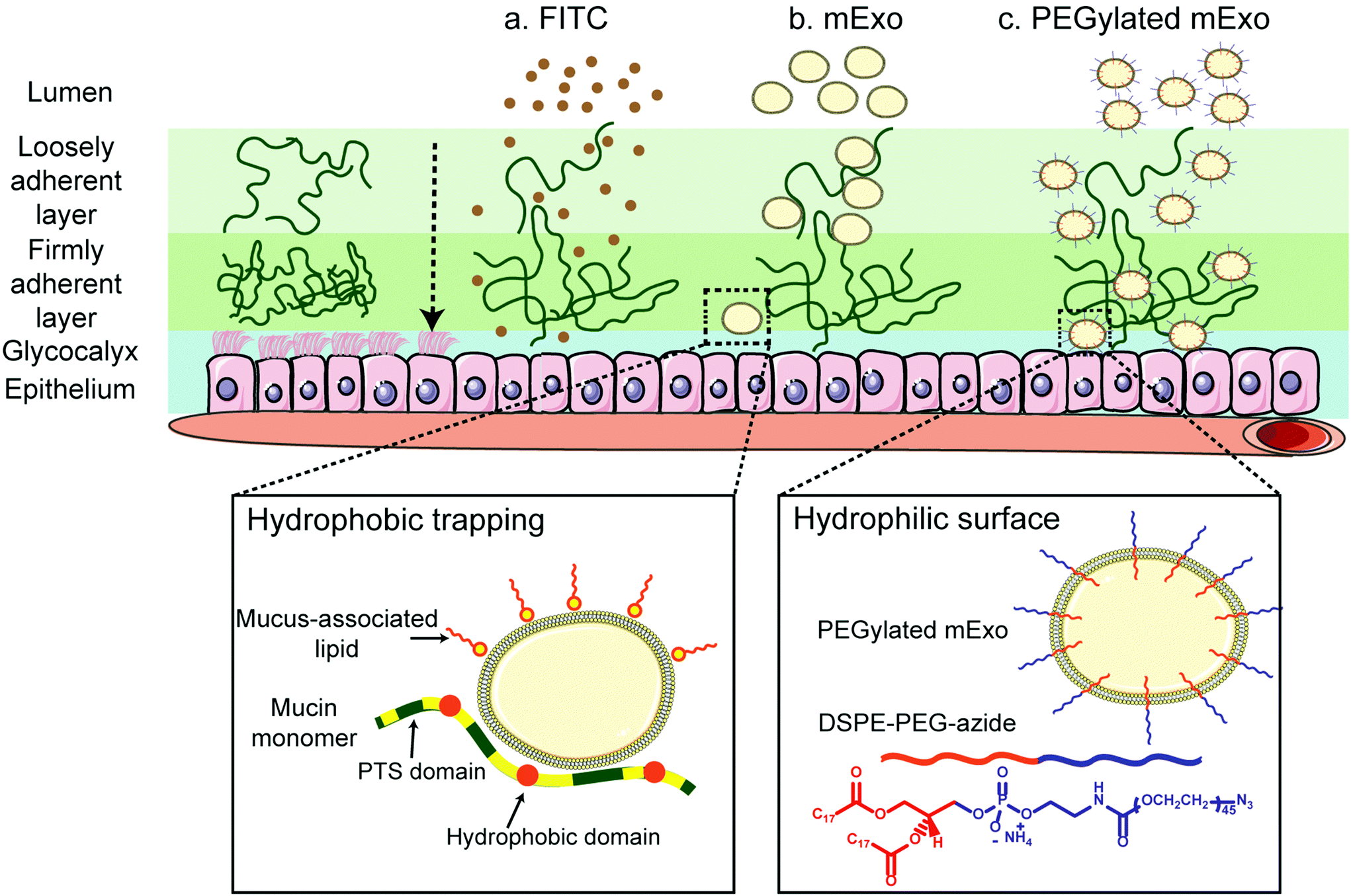 | ||
| Fig. 4 Enhanced transport of mExo through intestinal mucus by surface PEGylation. Intestinal mucus presents a significant barrier to oral drug delivery. Transport of unmodified mExo through the mucin is slowed down due to hydrophobic interactions between mExo lipid bilayer and hydrophobic domains of mucin. mExo was PEGylated by inserting the hydrophobic end of DSPE-PEG-azide (DPA) into its bilayer to enhance its effect on transport properties across intestinal mucus. | ||
mExo mixed with DPA-Cy5 at a molar ratio of 20000 DPA per mExo particle resulted in approximately 394 PEG chains incorporated onto each mExo particle (Fig. 5A, ESI Fig. S1†). To confirm hydrophobic insertion of DPA into the mExo lipid bilayer, the affinity of the binding event between mExo and DPA was measured using MST and the resultant binding curve was fit to the Hill model. The dissociation constant (KD) of the binding was estimated to be 347 μM, which is consistent with previous reports of hydrophobic incorporation with a lipid bilayer,38 confirming the insertion of DPA onto mExo. Additionally, the Hill coefficient (n) was determined as n = 1.55, which suggests the binding of DPA with mExo is cooperative (n > 1). Consequently, binding of one DPA molecule on mExo surface may facilitate binding of other DPA molecules on mExo (Fig. 5B). Successful PEGylation of mExo was further affirmed though confocal imaging of dual labeled PEG-mExo. The overlap of the channels showed that spatial distribution of DPA-Cy5 was focused in the vicinity of green mExo, confirming insertion of DPA-Cy5 on mExo surface (Fig. 5C). PEGylation did not affect the size of mExo but shielded the negative zeta potential values by 53% (72.8 ± 1.4 nm diameter and −5.4 ± 0.1 mV zeta potential for PEG-mExo compared to 70.5 ± 3.3 nm diameter and −8.4 ± 0.9 mV zeta potential for unmodified mExo). The DSPE hydrophobic anchor remained inserted in the mExo membrane over 1 h in PBS, and dissociated by only 10% in presence of hydrophobic mucin proteins (Fig. 5D), indicating suitable stability of the anchor over expected time scales of transport in mucus. PEGylation also protected mExo significantly from degradation in a low pH environment, mimicking human stomach conditions (Fig. 5E). 28% and 47% exosome protein loss at pH 4.5 and 2.2 was observed respectively for PEG-mExo, which was about 2 times lower than that measured for unmodified mExo.
 | ||
| Fig. 5 PEGylation of mExo and characterization. (A) The number of DPA-Cy5 inserted on mExo surface for different mixing ratios (* vs. 5k mixing ratio; p < 0.05). (B) Binding affinity of DPA with the mExo bilayer using microscale thermophoresis. (C) Confocal microscopy images of dual-labeled PEG mExo. mExo is marked green; DPA is labeled red. Spatial overlap of green and red signal confirms insertion of DPA-Cy5 into the mExo membrane. (D) Stability of the DPA hydrophobic anchor on mExo surface over 1 h in the presence and absence of mucin. (E) Tolerability of unmodified and PEGylated mExo in conditions mimicking infant (pH = 4.5) and adult (pH = 2.2) stomach acidity. (* vs. corresponding condition at pH 7.4, # vs. mExo; p < 0.05). | ||
3.4 In vitro mucus penetration and uptake in intestinal epithelium
By using two separate transport models, we evaluated the ability of mExo and PEG-mExo to cross two different biological barriers to oral delivery of macromolecular drugs. These include (i) translocation across an in vitro model of the intestinal mucus layer and (ii) uptake by an intestinal epithelial cell layer. mExo were labelled using the ExoGlow internal protein labelling kit rather than a conventional lipophilic membrane-anchoring dye (i.e. DiO or PKH67) to eliminate any potential interference arising from exosome–cell surface protein interactions, as well as to track mExo internal contents rather than membrane lipids. Unmodified mExo showed excellent mucus penetration behavior: the measured apparent permeability coefficient (Papp) of the unmodified mExo was found to be 13.2-fold higher than FITC dye (a representative model for small molecule drugs). Importantly, PEGylated mExo exhibited significantly enhanced mucus penetrability, increasing measured Papp by 3.2-fold (Fig. 6A and B).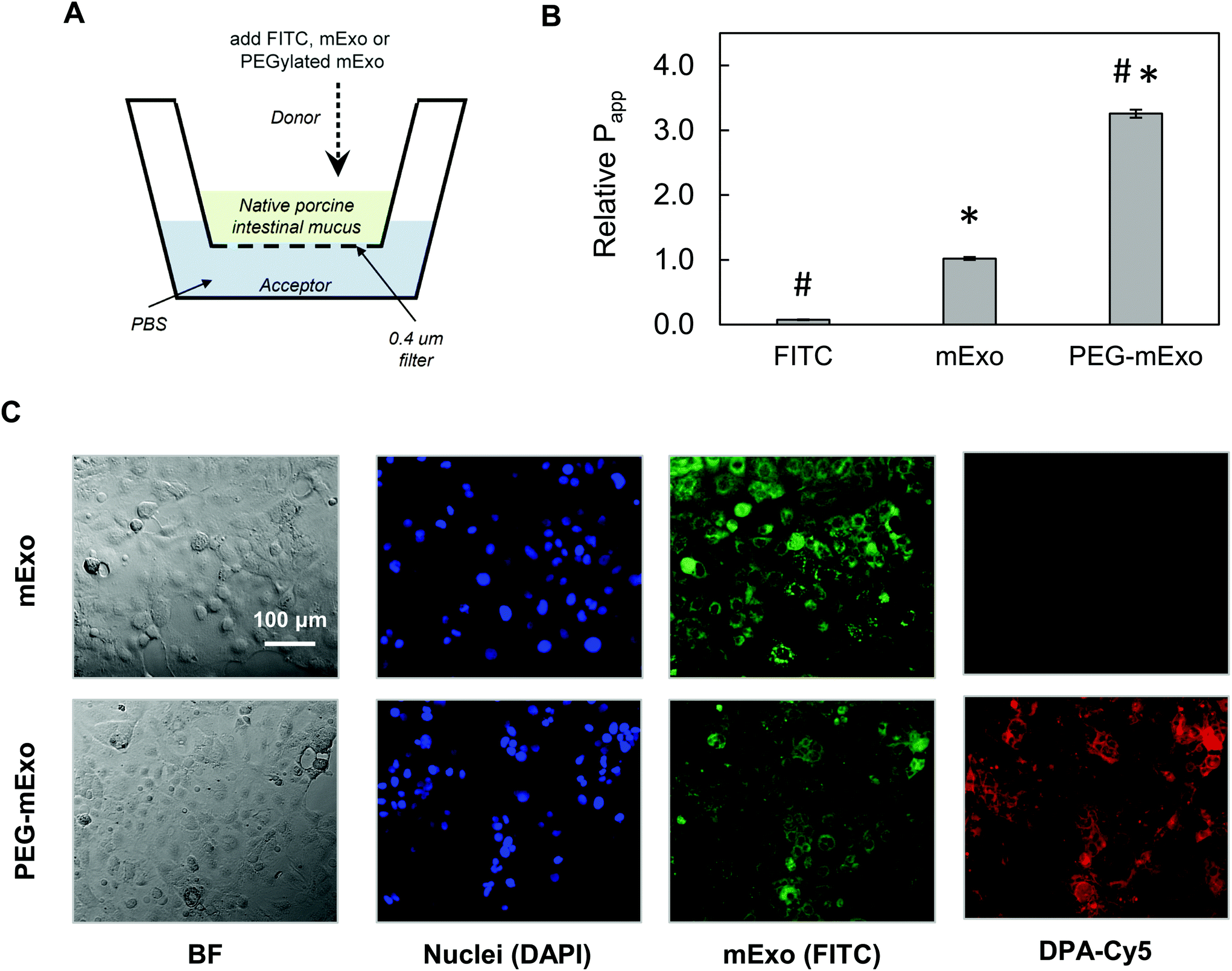 | ||
| Fig. 6 Transport of mExo and PEG-mExo through porcine intestinal mucus and uptake in human intestinal epithelial (Caco-2) cells. (A) A transwell setup was used for evaluating FITC, mExo and PEG-mExo permeability in porcine intestinal mucin. (B) Relative permeability coefficients (Papp) of unmodified mExo and PEG-mExo in mucus. (* vs. FITC, # vs. mExo; p < 0.05). (C) Uptake of mExo and PEG-mExo in Caco-2 cells after incubation for 2.5 h. mExo is labeled green, DPA in PEG-mExo is labeled red and cell nuclei are in blue. Control images accounting for background signal from free fluorophores are shown in ESI Fig. S3.† | ||
The in vitro intestinal uptake of unmodified mExo and PEG-mExo was investigated using Caco-2 cells. By modelling mExo internalization rate as a saturable process, we found that the rate of mExo uptake begins to saturate at treatment concentrations higher than 130 μg based on fitting to Michaelis–Menten model (ESI Fig. S2†). This concentration was used in subsequent imaging studies. Considerable uptake was observed after 2.5 h for both mExo and PEG-mExo, as evident from green signal that spatially aligns with DAPI staining of cell nuclei (Fig. 6C). High amounts of DPA-Cy5 red signal was also visible in PEG-mExo condition, confirming that PEGylation does not interfere with cell surface–exosome interactions and endocytosis internalization mechanisms.
3.5 siRNA loading and gene silencing
To directly compare siRNA internalization efficiency, mExo were loaded with siRNA using multiple methods, including electroporation and two different chemical transfection reagents: Lipofectamine and ExoFect. Initially, we assessed the widely-used electroporation method using the Invitrogen Neon pipette-based transfection system.12,39 However, despite screening a range of parameters (voltage, pulse number), we found that loading was ineffective with this method (ESI Fig. S4†). Such low loading by electroporation has previously been attributed to siRNA aggregation, obscuring past reports of high loading efficiencies by this method.40 Therefore, chemical exosome transfection reagents were chosen for further studies.Lipofect exhibited >2× higher loading efficiency than Exofect (Fig. 7A). To account for residual, non-fused siRNA-vehicle complexes co-precipitating with exosomes during purification by Exo-Quick TC, we carried out the loading process described in Methods 2.6.1 and 2.6.2 in the absence of mExo. While Lipofect-siRNA complexes showed minimal presence following Exo-Quick TC precipitation, ExoFect-siRNA complexes presented a fluorescent signal about 50% as strong as the ExoFect + mExo reading (Fig. 7A). This suggests that the high reading of Cy3-siRNA in purified ExoFect-loaded mExo samples is obscured by unloaded siRNA complexes that co-precipitate with the exosome particles, supporting Lipofect to be a more effective transfection agent. Therefore, Lipofect-mediated loading was further optimized to produce particles for downstream silencing studies. It was found that a 4% v/v Lipofect:mExo ratio resulted in about 58% siRNA loading efficiency, comparable to that achieved with twice the Lipofect volume (8% v/v) (Fig. 7B), and thus 4% v/v Lipofect:mExo ratio was used to formulate siRNA-loaded mExo for gene silencing experiments. Finally, despite the positive charge of non-fused siRNA-vehicle complexes (i.e. in the absence of mExo), mExo loaded by both methods revealed similar surface charge and particle size distributions as unmodified mExo (Fig. 7C and D). This suggests that the use of these chemical transfection agents for loading did not produce any large particle aggregates or alter mExo surface charge by fusion with the cationic transfection reagents.
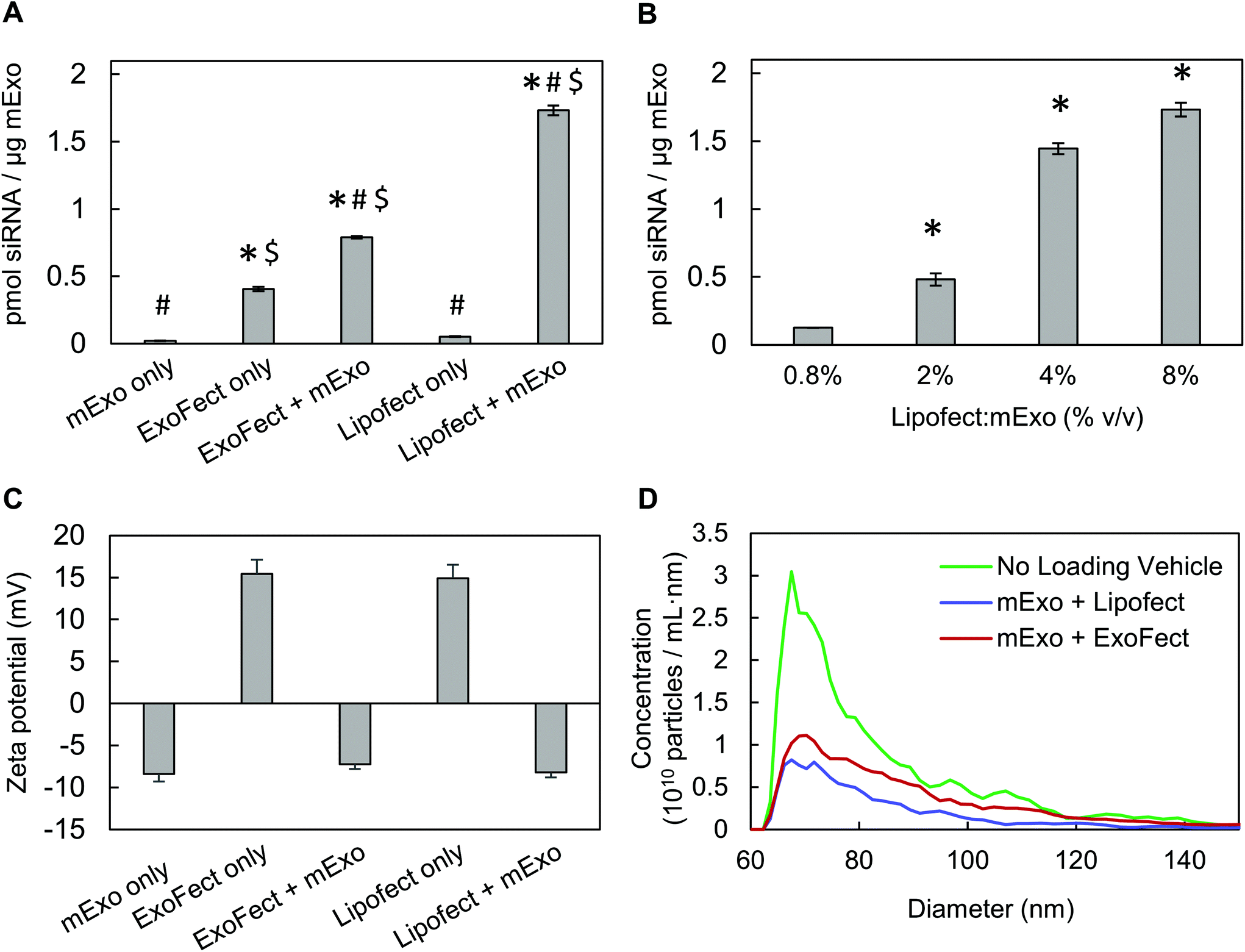 | ||
| Fig. 7 Characterization of mExo-siRNA loaded via ExoFect and Lipofectamine (A) comparison of relative efficiencies of intra-luminal siRNA loading into mExo using two chemical transfection reagents, ExoFect and Lipofectamine-2000 (* vs. Lipofect only, # vs. ExoFect only, $ vs. mExo only; p < 0.05). (B) Optimization of mExo siRNA loading using Lipofect (* vs. 0.8% Lipofect:mExo (% v/v); p < 0.05). (C) Change in particle surface charge following siRNA loading by ExoFect and Lipofectamine. (D) Change in particle size following loading by both methods. Data are presented as mean ± 95% bootstrap confidence intervals. | ||
The in vitro gene silencing properties of unmodified and PEGylated Exosome-Lipofectamine-siRNA formulations (hereby mExo-siRNA) were evaluated using GFP-expressing HEK293 cells. Treatment with high concentrations of mExo-siRNA resulted in a significant decrease (71%) in GFP fluorescence compared to untreated cells (Fig. 8A). Further, flow cytometry measurements revealed a dose-dependent decrease in GFP fluorescence. Importantly, at identical concentrations (12 μg per well), PEG-mExo showed only slightly reduced silencing (60%) compared to unmodified mExo (71%, Fig. 8B), suggesting that surface PEGylation does not considerably hinder cytosolic release of siRNA cargo. To confirm that the observed levels of GFP silencing were attributable to mExo-encapsulated siRNA rather than residual siRNA-Lipofect complexes in the sample, cells were treated with similar concentrations of residual Lipofect-siRNA, which was prepared by carrying out the siRNA loading process in the absence of mExo. There was a negligible decrease in cell fluorescence using such formulations. Similarly, no gene silencing was observed by treating cells with naked siRNA, either with or without adding mExo (ESI Fig. S5A†). These results suggest that the observed silencing with the mExo-siRNA formulation can indeed be attributed to exosome-mediated siRNA delivery. Finally, under all conditions, more than 85% of all cell events fell within the initially set custom gate, suggesting little adverse effects of mExo-siRNA or Lipofect on cell morphology at the concentrations studied (ESI Fig. S5B–C†).
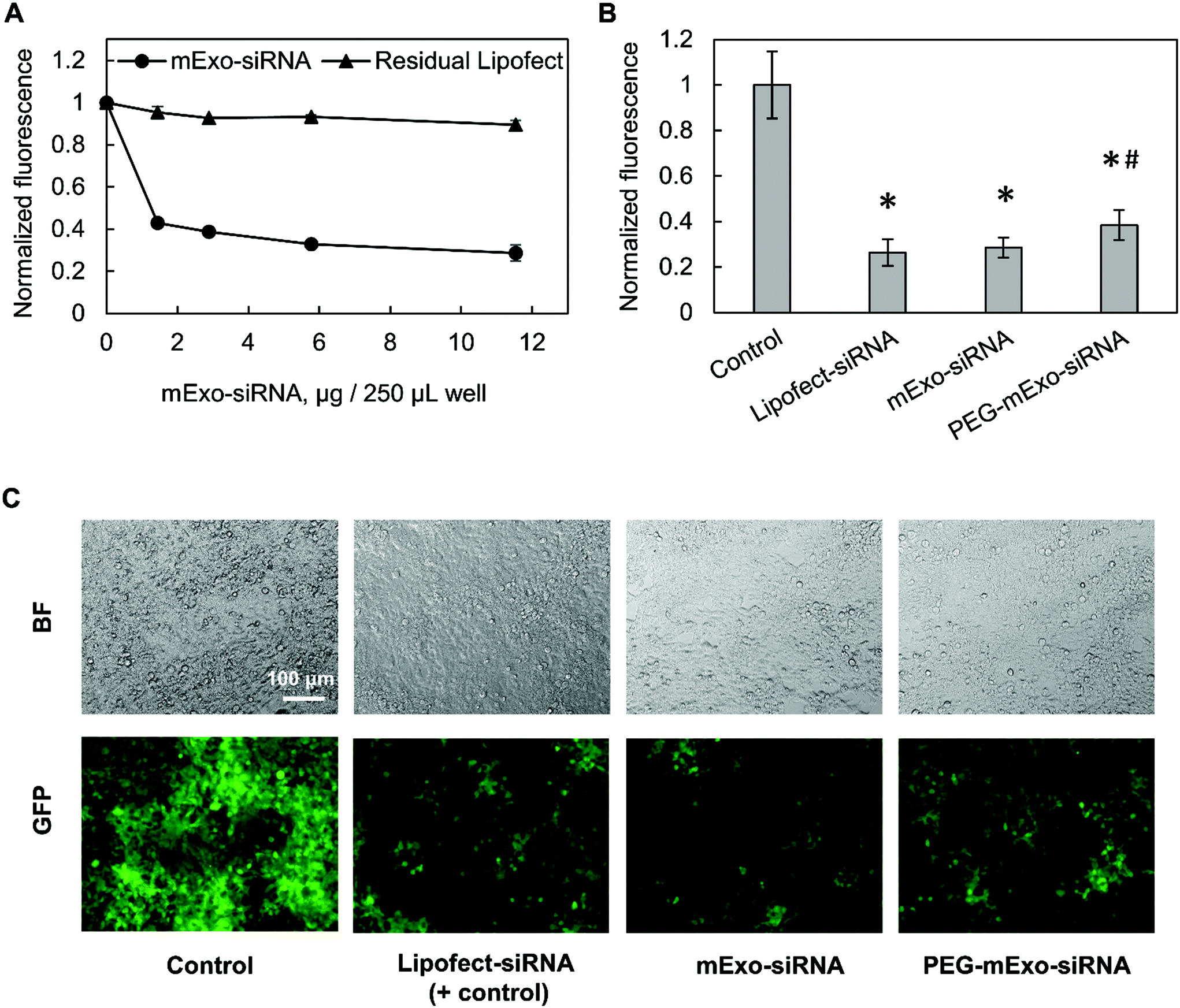 | ||
| Fig. 8 mExo-mediated siRNA delivery and silencing of GFP in HEK293 cells. (A) Dose-dependent response of exosome-mediated GFP silencing, compared to residual Lipofect-siRNA complexes left over from the purification process. (B) Comparison of silencing between unmodified and PEGylated mExo (* vs. Control, # vs. mExo-siRNA; p < 0.05). (C) Fluorescence microscopy imaging showing silencing of GFP in confluent HEK293 cells. White scale bar represents 100 μm. | ||
4. Discussion
Here we have engineered high purity milk exosomes (mExo) with modular surface tunability for improved oral delivery of macromolecular drugs such as siRNA. Using appropriate in vitro assays including intestinal mucus and the Caco-2 epithelial cell layer, we have demonstrated that negatively charged mExo can penetrate multiple biological barriers to oral delivery. PEGylation by hydrophobic anchoring allowed us to cloak mExo with a hydrophilic surface, which led to enhanced mucus permeability by more than threefold while not hindering cellular uptake. Furthermore, we have shown that mExo encapsulation facilitated intracellular siRNA delivery and downstream functional gene knockdown in HEK293 cells – when targeting the GFP reporter gene, significant (up to 70%) gene silencing was observed in a dose-dependent manner. HEK293 cells were chosen as a model cell type for selective uptake of molecules from the gastrointestinal tract after penetration through the intestinal mucosal layer and epithelium. There have been several prior reports describing the use of engineered exosomes to encapsulate siRNA for delivery to a variety of cell types.10,41 For example, Alvarez-Erviti et al. have used cell-derived exosomes to encapsulate siRNA to deliver cargo across the blood brain barrier.12 Similarly, Aqil et al. have used folic acid functionalized exosomes to encapsulate siRNA for targeted delivery to tumor cells.13 However, in comparison to cell- and plasma-derived vesicles,42 milk exosomes have garnered less attention for siRNA delivery, in particular via the oral route.In previous reports, differential ultracentrifugation and density-gradient centrifugation have been considered adequate for isolating milk exosomes.20–22,43 In our studies, we have employed size-exclusion chromatography as an additional purification step following ultracentrifugation27 (Fig. 1). This process produced exosomes with a 2.2-fold increase in purity, and nearly 100% recovery of mExo particles was achieved from the column (Fig. 2, Table 1). An important part of this isolation process is the use of EDTA prior to ultracentrifugation, because EDTA chelates casein micelles that naturally form around Ca2+ cores in milk.26,27 Immediately following ultracentrifugation, there is likely a considerable amount of free casein trapped in the mEV pellet, which is subsequently removed from exosomes via SEC, eluting in later fractions (12+).
We have demonstrated a high yield of milk exosomes isolated by the process used herein (1.92 ± 0.38 × 1012 particles per mL). Furthermore, these particles exhibit spherical morphology, with a particle size of about 70 nm diameter (Fig. 2). The measured particle-to-protein ratio of 3.0 ± 0.6 × 109 particles per μg protein is consistent with a previous theoretical estimate, which has been calculated based on the protein content of cell membranes; according to this estimate, pure exosome samples should contain approximately 2 × 109 particles per μg protein.44 While the Spectradyne nCS-1 is highly sensitive in capturing polydispersity in particle count and size distribution for particles like exosomes, it is restricted by a lower sensitivity limit of 65 nm for TS-400 cartridges (with a larger nano-constriction) and 50 nm for TS-300 cartridges. In this study, we have used TS-300 cartridges to measure the size distribution down to 50 nm. However, due to frequent clogging of the smaller constriction by particles, TS-400 cartridges were used for higher-throughput particle count measurements. For accuracy, we have reported particles that are reliably measured in the 65–120 nm size range. However, since exosomes can be as small as 35 nm diameter,9 there may be more particles of sizes smaller than 65 nm present in analyzed samples, which might have been excluded from the final measurements during data filtering. Spectradyne nCS-1 measurements offer a high-accuracy compliment to traditional optical measurements (i.e. dynamic light scattering and nanoparticle tracking analysis) for the characterization of mExo particles.
Analysis of the protein content of mExo by both western blotting and mass spectrometry indicated the presence of several known exosome marker proteins (e.g. ESCRT proteins, tetraspanins, etc.). In addition, an abundance of milk proteins (e.g. casein, apolipoproteins) carried over from the purification process were also present. It is possible that the presence of milk proteins could interfere with downstream processes for modifications of mExo, such as surface anchoring of functional moieties with a lipid tail,16,18 click chemistry,45 or siRNA encapsulation. Therefore, incorporation of SEC purification was important to reduce these protein impurities (Fig. 2). This extra purification step facilitated isolation of mExo particles that could be subjected to efficient PEG anchoring (Fig. 5) and siRNA loading (Fig. 7).
Intestinal mucus is a hydrated, viscoelastic and dynamic gel layer composed of high molecular weight glycoproteins, which contain negatively charged domains and hydrophobic, cysteine-rich cross-linking regions.46 This mucosal layer presents a barrier to oral drug delivery,8 and encapsulation systems must be capable of rapid trans-mucosal transport to achieve clinical translatability. Given their size and negative surface charge, exosomes would intuitively be expected to be inefficient at penetrating through mucus layers due to charge–charge repulsions and steric hindrance. However, the results reported herein suggest that unmodified milk exosomes exhibit increased trans-mucosal penetration compared to a representative small molecule (FITC). Hydrophobic interactions with mucus proteoglycans can hinder diffusion, rendering small hydrophobic molecules less able to penetrate mucus than larger, more hydrophilic species.4 The strategy of exosome particle encapsulation has been used previously to overcome this obstacle and enable transport of paclitaxel, a highly hydrophobic small molecule that would likely exhibit minimal permeability across the intestinal mucosal layer.43 In our studies, we set out to enhance mucus penetrability of the exosome carrier itself. Strategies for increasing nanoparticle mucus transport rate typically involve imparting a hydrophilic surface coating (e.g. PEGylation) to minimize particle interaction with hydrophobic domains of mucin and mucus-associated lipids.8,34,47 Here, by simple mixing, the lipophilic segment of DPA was automatically inserted into the lipid bilayer of mExo, making it hydrophilic and shielding its net negative charge. The resulting modified mExo showed 3.2-fold higher mucus permeability compared to native mExo (Fig. 6); moreover, PEGylated mExo showed increased tolerability in acidic conditions mimetic of the stomach (Fig. 5). Thus, surface PEGylation holds potential to significantly improve oral bioavailability of mExo-encapsulated drugs.
Various approaches have been taken to introduce functional molecules to the surface of exosomes and EVs, including direct conjugation to native surface proteins48,49 and click chemistry to exosomes engineered with azide-containing lipids45 or proteins.50 In contrast, lipophilic insertion of surface peptides via a hydrophobic anchor such as DSPE is emerging as a simple, fast and modular technique for exosome surface engineering.17,18,37 A post-insertion approach offers advantages over alternative strategies in that it supersedes the need for genetic engineering of producer cells and does not interfere with exosome surface proteins that are critical for recipient cell endocytosis and immune system evasion.11,51 We verified hydrophobic insertion of DPA into the mExo surface by using microscale thermophoresis and measured a KD of 347 μM for binding between DPA and mExo, which is in agreement with the previously measured hydrophobic binding affinity of liposomes decorated with glycosphingolipids towards cellular lipid bilayers (∼500 μM).38 Additionally, the spatial overlap of DPA-Cy5 and mExo in dual-label confocal images further confirmed insertion of DPA on the mExo surface. The incorporated terminal azide groups impart modular surface tunability via a chemical handle for straightforward conjugation of different molecules such as proteins, peptides, dyes and other agents to the mExo surface via azide–alkyne click chemistry. In this study, we have used dibenzocyclooctyne (DBCO)-Cy5 as the functional alkyne to link a fluorescent dye to the mExo surface for particle tracking. In principle, a variety of biologically active reagents including proteins and peptides can be covalently linked to the mExo surface using this approach. For instance, mucus penetrability could be further enhanced by functionalizing mExo with zwitterionic peptides,28,31,46 mimicking surface properties of mucus-penetrating viruses.8 Such viruses utilize weak, reversible charge–charge interactions to transiently bind to weakly negatively charged mucin constituents,46 which enable rapid penetration through the full thickness of the mucin.28,52–54
A key design challenge in repurposing exosomes as an siRNA delivery vehicle is the efficient incorporation of high molecular weight RNA oligonucleotides into the exosome core without disrupting the lipid membrane. This is a limitation of exosomes compared to other vectors like liposomes and nanoparticles, since siRNA can be loaded into synthetic delivery vehicles during their preparation process. In this study, several previously used methods were tested for siRNA loading into mExo, with varying degrees of success. Electroporation using a next-generation transfection system (the Invitrogen Neon system, as opposed to traditional cuvette-based machines) was not effective in loading milk exosomes with siRNA, despite previous reports of success.12,39,42 It is possible that milk exosomes are impenetrable by electroporation, or the high voltage electric fields may have caused mExo to rupture. Nevertheless, electroporation has been reported with results of low loading efficiency (∼5%),13 and the focus of further experimentation was therefore directed toward chemical transfection methods for siRNA encapsulation. Lipofectamine was found to be the most effective transfecting agent, resulting in an estimated loading efficiency of 58%. No change in mExo zeta potential was observed, which suggests that most siRNA was incorporated into the core of the mExo particles and siRNA adherence to the exosome surface was minimal. As a proof of principle using GFP as the target gene, we demonstrated that mExo-encapsulated siRNA molecules are capable of efficient gene silencing in HEK293 cells, with and without surface PEGylation. Our findings are in agreement with previously-reported successes using milk exosomes or extracellular vesicles in silencing oncogenes such as K-Ras13 in mice tumor xenograft models and B-catenin in hepatocellular carcinoma cells in vitro.55 Thus, the results reported herein further support the utility of mExo as suitable delivery vectors for siRNA-mediated knock down of therapeutically relevant genes.
Previous work has identified milk exosome cell uptake as an energy-dependent process via clathrin- and caveolae-dependent mechanisms.56 Notably, prior studies have reported markedly decreased uptake of milk exosomes into intestinal cells following protease treatment and in the presence of endocytosis inhibitors and carbohydrate competitors, which highlights the importance of interactions between mExo surface proteins and cell membrane receptors/glycoproteins to facilitate endocytosis.22 Our results show that PEGylation did not affect cellular uptake of mExo (Fig. 6 and ESI Fig. S6†) but yielded a slightly reduced silencing effect compared to unmodified mExo (Fig. 8). To exhibit a knockdown effect, mExo must exploit a mechanism to avoid lysosomal degradative pathways and release intraluminal siRNA contents into the cytosol.51 Furthermore, with surface-engineered exosome-siRNA platforms, it is critical to preserve endosomal escape functionality via surface protein interactions, especially considering that siRNA delivered by lipid-based vehicles have shown less than 2% endosomal escape efficiency.57 Although the intrinsic ability for unmodified exosomes to partially avoid lysosomal degradation has been directly observed previously58,59 and is apparent by the silencing capability by mExo-siRNA, it is unknown whether the endosomal escape of mExo-encapsulated siRNA may be attenuated by dense surface PEGylation. Whether the disparity in silencing with PEGylation observed herein is attributable to reduced cell uptake or less efficient endosomal escape of PEG-mExo remains to be elucidated. Nevertheless, PEG-mExo exhibited silencing capability within 15% of unmodified mExo, supporting this modality as an efficient vehicle for oral siRNA delivery.
5. Conclusion
We have demonstrated high-purity bovine milk exosomes with tunable surface properties as a new class of naturally derived drug delivery systems, which can be used to successfully encapsulate siRNA for oral delivery to silence intracellular gene targets. We show that mExo exhibit the ability to efficiently diffuse across mucosal barriers and can be taken up by the intestinal epithelium, two key barriers to oral drug delivery. Furthermore, we have shown that PEGylation of the mExo surface is achievable by stable, passive hydrophobic insertion, and that a PEG coating improves mucin penetration capability and particle stability in acidic conditions representative of the stomach. Incorporation of different functional reagents on the mExo surface by click chemistry offers an efficient and modular method to create functionalized mExo particles with tuned surface properties. This combined with a simplified isolation procedure offers a modular and scalable platform to generate highly purified exosome vectors for the delivery of macromolecular drugs across biological barriers. Future studies should focus on evaluating transmucosal transport of mExo surface-modified with electrically charged and mucus-penetrating peptides via this click chemistry platform.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Sanofi Innovation Award and by the National Institute of Health Trailblazer R21 grant EB028385. We would like to thank Dr Jiahe Li for his guidance and expertise, Dr Rebecca Carrier for providing porcine mucin, and the lab of Dr Mansoor Amiji for allowing access to their ultracentrifugation equipment and for guidance in experimental protocols.References
- E. B. Odom, K. B. Patel and D. C. Odom, Inpatient Care versus Subacute Care for Long Term Intravenous Antibiotics: Cost from the Patient Perspective, Journal of Academic Hospital Medicine, 2016, vol. 8, issue 3 Search PubMed.
- J. L. Betker, B. M. Angle, M. W. Graner and T. J. Anchordoquy, J. Pharm. Sci., 2019, 108, 1496–1505 CrossRef CAS.
- J. Shepard, W. Ward, A. Milstone, T. Carlson, J. Frederick, E. Hadhazy and T. Perl, JAMA Surg., 2013, 148, 907–914 CrossRef.
- S. K. Lai, Y. Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171 CrossRef CAS.
- A. Hayward, T. Bensel, H. Mazdiyasni, J. Rogner, A. R. Kirtane, Y. L. Lee, T. Hua, A. Bajpayee, J. Collins, S. McDonnell, C. Cleveland, A. Lopes, A. Wahane, R. Langer and G. Traverso, Sci. Rep., 2018, 8, 11816 CrossRef.
- A. R. Kirtane, T. Hua, A. Hayward, A. Bajpayee, A. Wahane, A. Lopes, T. Bensel, L. Ma, F. Z. Stanczyk, S. Brooks, D. Gwynne, J. Wainer, J. Collins, S. M. Tamang, R. Langer and G. Traverso, Sci. Transl. Med., 2019, 11(521), eaay2602 CrossRef.
- C. Twarog, S. Fattah, J. Heade, S. Maher, E. Fattal and D. J. Brayden, Pharmaceutics, 2019, 11, 78 CrossRef CAS.
- T. L. Carlson, J. Y. Lock and R. L. Carrier, Annu. Rev. Biomed. Eng., 2018, 20, 197–220 CrossRef CAS.
- M. Colombo, G. Raposo and C. Thery, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS.
- X. Luan, K. Sansanaphongpricha, I. Myers, H. Chen, H. Yuan and D. Sun, Acta Pharmacol. Sin., 2017, 38, 754–763 CrossRef CAS.
- J. L. Hood, Nanomedicine, 2016, 11, 1745–1756 CrossRef CAS.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS.
- F. Aqil, R. Munagala, J. Jeyabalan, A. K. Agrawal, A. H. Kyakulaga, S. A. Wilcher and R. C. Gupta, Cancer Lett., 2019, 449, 186–195 CrossRef CAS.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS.
- H. Bahadar, F. Maqbool, K. Niaz and M. Abdollahi, Iran. Biomed. J., 2016, 20, 1–11 Search PubMed.
- S. A. A. Kooijmans, J. Gitz-Francois, R. M. Schiffelers and P. Vader, Nanoscale, 2018, 10, 2413–2426 RSC.
- M. S. Kim, M. J. Haney, Y. Zhao, D. Yuan, I. Deygen, N. L. Klyachko, A. V. Kabanov and E. V. Batrakova, Nanomedicine, 2018, 14, 195–204 CrossRef CAS.
- T. J. Antes, R. C. Middleton, K. M. Luther, T. Ijichi, K. A. Peck, W. J. Liu, J. Valle, A. K. Echavez and E. Marban, J. Nanobiotechnol., 2018, 16, 61 CrossRef.
- W. Whitford and P. Guterstam, Future Med. Chem., 2019, 11, 1225–1236 CrossRef CAS.
- R. Munagala, F. Aqil, J. Jeyabalan and R. C. Gupta, Cancer Lett., 2016, 371, 48–61 CrossRef CAS.
- M. Somiya, Y. Yoshioka and T. Ochiya, J. Extracell. Vesicles, 2018, 7, 1440132 CrossRef.
- T. Wolf, S. R. Baier and J. Zempleni, J. Nutr., 2015, 145, 2201–2206 CrossRef CAS.
- G. Carobolante, J. Mantaj, E. Ferrari and D. Vllasaliu, Pharmaceutics, 2020, 12, 226 CrossRef CAS.
- S. Shandilya, P. Rani, S. K. Onteru and D. Singh, J. Agric. Food Chem., 2017, 65, 9506–9513 CrossRef CAS.
- Y. Liao, X. Du, J. Li and B. Lonnerdal, Mol. Nutr. Food Res., 2017, 61(11), 1700082 CrossRef.
- T. Morcol, Q. He and S. J. D. Bell, Biotechnol. Prog., 2001, 17, 577–582 CrossRef CAS.
- K. Vaswani, Y. Q. Koh, F. B. Almughlliq, H. N. Peiris and M. D. Mitchell, Reprod. Biol., 2017, 17, 341–348 CrossRef.
- L. D. Li, T. Crouzier, A. Sarkar, L. Dunphy, J. Han and K. Ribbeck, Biophys. J., 2013, 105, 1357–1365 CrossRef CAS.
- D. E. Beasley, A. M. Koltz, J. E. Lambert, N. Fierer and R. R. Dunn, PLoS One, 2015, 10, e0134116 CrossRef.
- T. L. Carlson, H. Yildiz, Z. Dar, J. Y. Lock and R. L. Carrier, PLoS One, 2018, 13, e0209151 CrossRef.
- W. Shan, X. Zhu, W. Tao, Y. Cui, M. Liu, L. Wu, L. Li, Y. Zheng and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 25444–25453 CrossRef CAS.
- H. Friedl, S. Dunnhaupt, F. Hintzen, C. Waldner, S. Parikh, J. P. Pearson, M. D. Wilcox and A. Bernkop-Schnurch, J. Pharm. Sci., 2013, 102, 4406–4413 CrossRef CAS.
- A. C. Groo, P. Saulnier, J. C. Gimel, J. Gravier, C. Ailhas, J. P. Benoit and F. Lagarce, Int. J. Nanomed., 2013, 8, 4291–4302 Search PubMed.
- J. Leal, T. Dong, A. Taylor, E. Siegrist, F. Gao, H. D. C. Smyth and D. Ghosh, Int. J. Pharm., 2018, 553, 57–64 CrossRef CAS.
- J. Orloff and J. Bloom, 18–05 Introduction to Probability and Statistics, Spring 2014. Massachusetts Institute of Technology: MIT OpenCouseWare, https://ocw.mit.edu/. License: Creative Commons BY-NC-SA.
- J. Lotvall, A. F. Hill, F. Hochberg, E. I. Buzas, D. Di Vizio, C. Gardiner, Y. S. Gho, I. V. Kurochkin, S. Mathivanan, P. Quesenberry, S. Sahoo, H. Tahara, M. H. Wauben, K. W. Witwer and C. Thery, J. Extracell. Vesicles, 2014, 3, 26913 CrossRef.
- S. A. A. Kooijmans, L. A. L. Fliervoet, R. van der Meel, M. Fens, H. F. G. Heijnen, P. M. P. van Bergen En Henegouwen, P. Vader and R. M. Schiffelers, J. Controlled Release, 2016, 224, 77–85 CrossRef CAS.
- A. Kunze, M. Bally, F. Hook and G. Larson, Sci. Rep., 2013, 3, 1452 CrossRef.
- T. N. Lamichhane, R. S. Raiker and S. M. Jay, Mol. Pharm., 2015, 12, 3650–3657 CrossRef CAS.
- S. A. A. Kooijmans, S. Stremersch, K. Braeckmans, S. C. de Smedt, A. Hendrix, M. J. A. Wood, R. M. Schiffelers, K. Raemdonck and P. Vader, J. Controlled Release, 2013, 172, 229–238 CrossRef CAS.
- M. Lu, H. Xing, Z. Xun, T. Yang, P. Ding, C. Cai, D. Wang and X. Zhao, Asian J. Pharm. Sci., 2018, 13, 1–11 CrossRef.
- J. Wahlgren, T. D. L. Karlson, M. Brisslert, F. Vaziri Sani, E. Telemo, P. Sunnerhagen and H. Valadi, Nucleic Acids Res., 2012, 40, e130 CrossRef CAS.
- A. K. Agrawal, F. Aqil, J. Jeyabalan, W. A. Spencer, J. Beck, B. W. Gachuki, S. S. Alhakeem, K. Oben, R. Munagala, S. Bondada and R. C. Gupta, Nanomedicine, 2017, 13, 1627–1636 CrossRef CAS.
- E. D. Sverdlov, Bioessays, 2012, 34, 873–875 CrossRef CAS.
- M. Wang, S. Altinoglu, Y. S. Takeda and Q. Xu, PLoS One, 2015, 10, e0141860 CrossRef.
- A. Vedadghavami, C. Zhang and A. G. Bajpayee, Nano Today, 2020, 34, 100898 CrossRef CAS.
- S. K. Lai, D. E. O’Hanlon, S. Harrold, S. T. Man, Y. Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(5), 1482–1487 CrossRef CAS.
- T. Tian, H.-X. Zhang, C.-P. He, S. Fan, Y.-L. Zhu, C. Qi, N.-P. Huang, Z.-D. Xiao, Z.-H. Lu, B. A. Tannous and J. Gao, Biomaterials, 2018, 150, 137–149 CrossRef CAS.
- S. Shi, T. Li, X. Wen, S. Y. Wu, C. Xiong, J. Zhao, V. R. Lincha, D. S. Chow, Y. Liu, A. K. Sood and C. Li, Bioconjugate Chem., 2019, 30, 2675–2683 CrossRef CAS.
- P. Zhang, B. Dong, E. Zeng, F. Wang, Y. Jiang, D. Li and D. Liu, Anal. Chem., 2018, 90, 11273–11279 CrossRef CAS.
- D. E. Murphy, O. G. de Jong, M. Brouwer, M. J. Wood, G. Lavieu, R. M. Schiffelers and P. Vader, Exp. Mol. Med., 2019, 51, 1–12 CAS.
- A. Vedadghavami, E. K. Wagner, S. Mehta, T. He, C. Zhang and A. G. Bajpayee, Acta Biomater., 2019, 93, 258–269 CrossRef CAS.
- A. G. Bajpayee and A. J. Grodzinsky, Nat. Rev. Rheumatol., 2017, 13, 183–193 CrossRef CAS.
- C. C. Young, A. Vedadghavami and A. G. Bajpayee, Bioelectricity, 2020, 2, 68–81 CrossRef.
- A. Matsuda and T. Patel, Methods Mol. Biol., 2018, 1740, 187–197 CrossRef CAS.
- F. Aqil, R. Munagala, J. Jeyabalan, A. K. Agrawal and R. Gupta, AAPS J., 2017, 19, 1691–1702 CrossRef CAS.
- M. Maugeri, M. Nawaz, A. Papadimitriou, A. Angerfors, A. Camponeschi, M. Na, M. Holtta, P. Skantze, S. Johansson, M. Sundqvist, J. Lindquist, T. Kjellman, I. L. Martensson, T. Jin, P. Sunnerhagen, S. Ostman, L. Lindfors and H. Valadi, Nat. Commun., 2019, 10, 4333 CrossRef CAS.
- T. Tian, Y. Wang, H. Wang, Z. Zhu and Z. Xiao, J. Cell. Biochem., 2010, 111, 488–496 CrossRef CAS.
- W. Heusermann, J. Hean, D. Trojer, E. Steib, S. von Bueren, A. Graff-Meyer, C. Genoud, K. Martin, N. Pizzato, J. Voshol, D. V. Morrissey, S. E. Andaloussi, M. J. Wood and N. C. Meisner-Kober, J. Cell Biol., 2016, 213, 173–184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01497d |
| This journal is © The Royal Society of Chemistry 2021 |