
Recent progress in ligand-centered homogeneous electrocatalysts for hydrogen evolution reaction
Geng-Geng
Luo
 *a,
Hai-Lin
Zhang
a,
Yun-Wen
Tao
b,
Qiao-Yu
Wu
a,
Dan
Tian
cd and
Qichun
Zhang
*c
*a,
Hai-Lin
Zhang
a,
Yun-Wen
Tao
b,
Qiao-Yu
Wu
a,
Dan
Tian
cd and
Qichun
Zhang
*c
aKey Laboratory of Environmental Friendly Function Materials Ministry of Education, College of Materials Science and Engineering, Huaqiao University, Xiamen 361021, P.R. China. E-mail: ggluo@hqu.edu.cn
bDepartment of Chemistry, New York University, New York, NY 10003, USA
cSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: qczhang@ntu.edu.sg
dKey Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech. University, 30 South Puzhu Road, P.R. China
First published on 17th December 2018
Abstract
Electrocatalytic hydrogen evolution reaction (HER) is a critical process in a sustainable energy conversion system. Many metal complexes as small-molecule catalysts for electrochemical H2 evolution have been reported over recent decades, which have made a great contribution to a detailed understanding of their catalytic mechanisms. In most reported mechanistic pathways, metal hydride species are always considered as key intermediates during H2 formation, with the metal centre at the active site docking both electrons and protons. These metal-centred HER routes need to employ metal ions with open coordination sites, which can accommodate multiple redox states to involve two-electron hydride transfer for H2 production. By comparison, an alternative approach, specifically focusing on redox-active ligand-centred electrocatalytic HER, has garnered increasing attention over the last six years. This review highlights recent progress in metal and metal-free ligand-centred homogeneous electrocatalytic HER since 2012 and discusses it according to two categories: organic ligand-centred and inorganic ligand-centred HER. In addition, some existing issues and suggestions for future directions in this catalytic area have been provided.
 Geng-Geng Luo | Geng-Geng Luo received his Ph.D. in Chemistry from Xiamen University in China in 2009. He studied for one year at the University of Rochester (USA) with Prof. Richard Eisenberg in the area of artificial light-harvesting and catalytic systems. Later on, he visited Prof. Zhang's group as a visiting scholar and conducted his research there for one year at Nanyang Technological University (Singapore). Currently, he is an associate professor at Huaqiao University and his research interests focus on coordination chemistry, supramolecular assembly and renewable energy catalysis. |
 Hai-Lin Zhang | Hai-Lin Zhang obtained her Bachelor of Engineering degree in Applied Chemistry from Huaqiao University in 2017. She is currently pursuing her Master of Science degree under the guidance of associate Prof. Geng-Geng Luo. Her current research interests are focused on molecular catalysis and photochemistry. |
 Yun-Wen Tao | Yun-Wen Tao obtained his B.S. in Biotechnology at the University of Electronic Science and Technology of China in 2014 and completed his Ph.D. on theoretical and computational chemistry in 2018 at Southern Methodist University, USA, under the supervision of Prof. Dieter Cremer and Prof. Elfi Kraka. He worked as a post-doc with Prof. Tamar Schlick at New York University on RNA design from June to December in 2018. His research interest covers method development and application of computational chemistry for deeper insight of chemical bonding and similarity. |
 Qiao-Yu Wu | Qiao-Yu Wu is a senior undergraduate student at Huaqiao University and is currently pursuing his Bachelor of Engineering degree in Applied Chemistry. Under the guidance of Dr Geng-Geng Luo, his main research interests are in the incorporation of water and CO2 reduction photo/electro-catalysts into molecular containers for solar fuel generation. |
 Dan Tian | Dan Tian received her Ph.D. in the field of Chemistry from Nankai University (China) in 2015. She then studied for two years at Nanjing Tech University in China with Prof. Ling Huang and Prof. Wei Huang in the area of lanthanide-doped upconversion MOFs and photophysical properties of solid-state boron-dipyrromethene dyes. She is currently a postdoctoral researcher working in collaboration with Prof. Qichun Zhang at Nanyang Technological University (Singapore). Her current interests are focused on the coordination chemistry and aggregation induced emission of small molecules. |
 Qichun Zhang | Qichun Zhang obtained his B.S. at Nanjing University in China in 1992, M.S. in physical organic chemistry (organic solid lab) at the Institute of Chemistry, Chinese Academy of Sciences in 1998, M.S. in organic chemistry at the University of California, Los Angeles (USA), and completed his Ph.D. in inorganic chemistry at University of California Riverside in 2007. Then, he joined Prof. Kanatzidis's group at Northwestern University as a Postdoctoral Fellow (2007–2008). In 2009, he joined the School of Materials Science and Engineering at Nanyang Technological University (NTU, Singapore) as an assistant professor. In 2014, he was promoted to associate professor with tenure, and later that year, he became an adjunct associate professor in the Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University. He received a TCT fellowship in 2013 and a lectureship from National Taiwan University in 2014. Currently, he is an associate editor of J. Solid State Chemistry, the International Advisory Board member of Chemistry – An Asian Journal, an Advisory board member of Journal of Materials Chemistry C, an Advisory board member of Materials Chemistry Frontiers, and an Advisory board member of Inorganic Chemistry Frontiers. Also, he has been a Guest Editor of Inorganic Chemistry Frontiers (2016–2017), Guest Editor of Journal of Materials Chemistry C (2017–2018), and Guest Editor of Inorganic Chemistry Frontiers (2017–2018). Currently, he is a fellow of the Royal Society of Chemistry and a highly-cited researcher for 2018. He has published more than 300 papers and 4 patents (H-index: 62). |
1. Introduction
The energy crisis and anthropogenic climate change related to the use of fossil fuels have signalled an urgent need to find carbon-neutral, clean, and renewable energy alternatives.1 Hydrogen (H2) has been considered as an ideal and environment-friendly molecular fuel due to its high energy density and its sole combustion product being water.2 Although there are many ways to produce H2, electrochemical hydrogen evolution reaction (HER) is known as a key enabling technology for large-scale H2 production. In this regard, HER is deceptively a very simple reaction (2H+ + 2e− → H2), however, an efficient and durable catalyst is required to increase the reaction kinetics by lowering the high activation energy of H2 generation, i.e., reducing the electrochemical overpotential.3Besides natural biocatalysts in the form of hydrogenases (H2ases), current efficient catalytic systems for electrochemical H2 production include solid/heterogeneous and molecular/homogeneous ones. Heterogeneous catalytic systems are more difficult to study because the properties of catalysts highly rely on local variations in their surface morphology. By contrast, homogeneous molecule-type catalysts have become a widely-pursued research topic because their catalytic behaviours can be tuned at the molecular level to provide detailed mechanistic insights, which are difficult to obtain from the heterogeneous ones.
During the past twenty years, intense efforts have been put into the development of earth-abundant-metal based small molecule electrocatalysts for H2 evolution, and several review articles with a particular emphasis on the structure–property relationship relevance to catalysis have been published.4–6 Among all research on catalysed H2 production, the mechanistic investigation on proton reduction catalysis is very important because it can provide us with some guidelines to design better molecular catalysts in the future.
In most cases, H2 production catalysed by metal complexes is widely believed to happen through a common metal hydride intermediate with metal ([Mn+]) ions at the reactive centre, formed by either consecutive or coupled proton and electron transfer, followed by two possible mechanisms for the formation of the subsequent H–H bond (Scheme 1). The first mechanism involves a homolytic or bimetallic route, where a metal–hydride species ([Mn+–H]) can react with another metal hydride to release one H2via reductive elimination. Alternatively, a heterolytic or monometallic pathway can also occur, where the metal hydride [Mn+–H] is further reduced and protonated to evolve one H2. In the latter pathway, two protons and two electrons are delivered to one single metal centre and a putative metal hydride complex ([M(n−1)+–H]) is formed.4
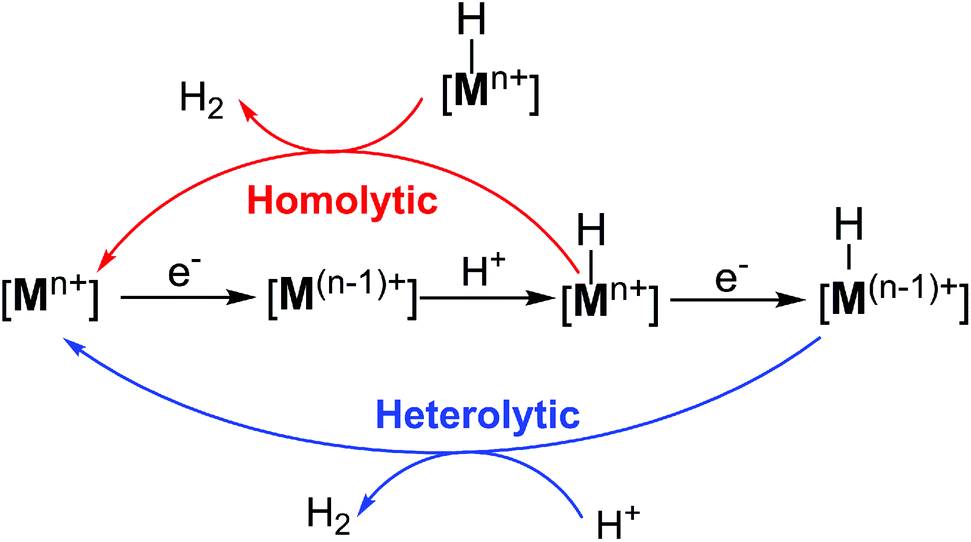 | ||
| Scheme 1 Proposed homolytic and heterolytic mechanisms for H2 evolution via the formation of a metal–hydride intermediate. Note that M is designated as an active metal centre. | ||
While deeply understanding a rich variety of metal–hydride-based catalytic mechanisms has led to significant advances in the design of efficient H2-evolving molecular catalysts, these traditional metal-centred HER routes require the use of metal ions that should have available open coordination sites and are capable of accommodating multiple redox states for the two-electron hydride transfer for H2 production.4 Thus, it is highly desirable to explore new alternative pathways in which metal hydrides are not necessary to enter the catalytic cycle.
In this respect, redox-active ligands beyond the reactivity of metals have received continuous attention. Initial interest in ligand hydride systems was partially inspired by biological reduction processes, where nicotinamide adenine dinucleotide phosphate or flavin adenine dinucleotide transfers ligand hydrides to an unsaturated substrate bound in a natural enzyme active site.7 Recent scientific efforts have been put into investigating redox-active ligand hydride systems and their utilization in catalytic reduction processes of C–H activation, water oxidation, alcohol oxidation, H2 oxidation, and CO2 activation, and in helping to mediate multielectron reactions.8–11 However, to date, there are relatively few reports on the redox-active ligand-centred homogeneous catalysis of electrochemical H2 production.
This review summarizes the research progress in the field of metal and metal-free ligand-centred electrocatalytic generation of H2 within the past six years. We will describe two classes of ligand-centred H2-evolving catalysis: organic ligand-centred and inorganic ligand-centred HER. Their electrocatalytic properties as well as structural and mechanistic features will be summarized and discussed in detail. Although the development of ligand-centred electrocatalytic H2 evolution is not yet in the mainstream of current research in HER, we believe that this alternative approach might lead to more interesting discoveries and creative ideas in the design of better H2-evolving molecular catalysts in the coming years.
2. Metal and metal-free organic ligand-centred electrocatalytic HER
2.1 Metal complexes with organic ligand-centred electrocatalytic HER
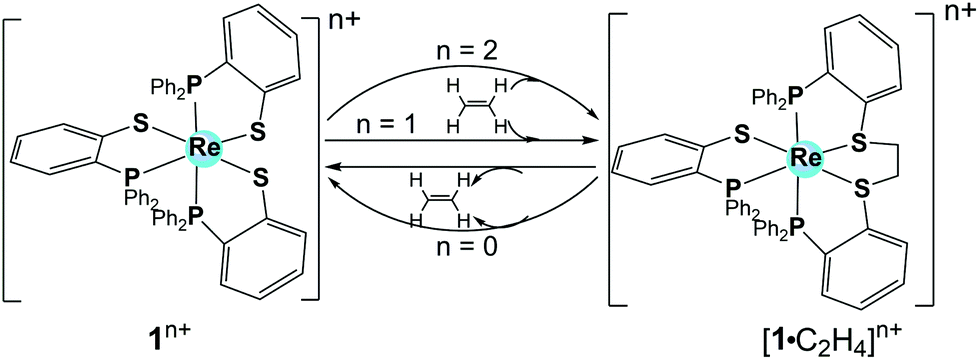 | ||
| Fig. 1 Representation of 1 with different oxidation states and the corresponding ethylene (C2H4) binding adducts. | ||
In view of the similarities of the frontier molecular orbitals between ethylene and molecular hydrogen, 1 was later studied by Grapperhaus et al. for electrocatalytic H2 activation and formation.14 In the presence of base (triethylamine), 1 oxidizes H2 in CH2Cl2 with a turnover frequency (TOF) of 4 ± 1 s−1 and an overpotential of 0.67 V. When using weak acid (CH3COOH) or strong acid (H2SO4) as the proton source, 1 reduces the protons to H2 in CH2Cl2 with acid-independent rate constant of 32 ± 3 s−1 and an overpotential of 0.38 V (Table 1). The rate constant (32 ± 3 s−1) and overpotential are relatively low and remind us that catalysis at low overpotentials usually leads to low TOFs. A bulk electrolysis experiment of CH3COOH (0.05 M) solution in CH2Cl2 at an applied potential of −1.8 V vs. Fc/Fc+ in the presence of 1 afforded a turnover number (TON) of H2 evolution of 54 in a 6 h experiment with 73% faradaic efficiency. The rate of catalysis with a TON of 6 h−1 is sustained over the 6 h electrolysis experiment, indicating the robustness of this electrocatalyst.
| Catalyst | Redox-active organic ligand | Catalytic potential (Ep)b | Proton source | Solvent | OPc |
k
obs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
Ref. |
|---|---|---|---|---|---|---|---|
| a Conditions: GC working electrode. b Catalytic potential (Ep) of 1, 4 and 6–9vs. Fc/Fc+; Ep of 2, 3 and 5vs. SCE. c For 1, 6–8, OP = overpotential = |Ep − EθHA|; for 2–3, OP was determined from a bulk electrolysis experiment; for 5, OP = overpotential = |Ecat/2vs. NHE – 0.059pH|. d The observed rate constant (kobs) = TOF for 1, 6–8 was calculated in the acid-independent region according to the equation: ic/ip = {(2/0.4463) × [(RTkobs)/(Fv)]1/2}, where ic is the steady state catalytic peak current in the presence of an acid and ip is the peak current measured in the absence of an acid. For 2–3, kobs = TOF was determined from a bulk electrolysis experiment; for 9, kobs was obtained from foot-of-the-wave analysis. e Not determined. | |||||||
| 1 | Diphenylphosphino-benzenethiolate | −1.7 V | CH3COOH | CH2Cl2 | 0.38 V | 32 s−1 | 14 |
| 2 | Bis(imino)pyridine | −1.2 V | LutH+ | THF | 0.78 V | 19.4 s−1 | 17 |
| 2 | Bis(imino)pyridine | −1.2 V | DMAPH+ | THF | 0.54 V | 2.1 s−1 | 17 |
| 3 | Bis(pyrazolyl)pyridine | −1.26 V | [HNEt3]BF4 | THF | 4.85 s−1 | 18 | |
| 4 | Porphyrin | −1.8 V | Benzoic acid | THF | 19 | ||
| 4 | Porphyrin | −1.37 V | Tosic acid | THF | 19 | ||
| 5 | Pyrazinedithiolate | −0.8 to −1.0 V | Acidic water | H2O | 0.330–0.400 V | 23 | |
| 6 | Bis(thiosemicarbazone) | −1.7 V | CH3COOH | CH3OH | 0.756 V | 1170 s−1 | 25 |
| 6 | Bis(thiosemicarbazone) | −2.3 V | CH3COOH | CH3CN | 1.074 V | 11700 s−1 |
25 |
| 7 | Bis(thiosemicarbazone) | −1.7 V | CH3COOH | CH3CN | 0.80 V | 10000 s−1 |
26 |
| 7 | Bis(thiosemicarbazone) | −1.9 V | CH3COOH | DMF | 0.76 V | 5100 s−1 | 26 |
| 8 | Bis(thiosemicarbazone) | −2.1 V | CH3COOH | CH3CN | 1.43 V | 1320 s−1 | 25 |
| 9 | Tetra(pentafluorophenyl) porphyrin | −1.3 V | Tosic acid | THF | 1.02 V | 0.528–0.891 s−1 | 30 |
To evaluate the catalytic reaction kinetics of 1, the rate law and H/D kinetic isotope effect (KIE) were measured. The rate law of the reaction displays a first-order dependence on the catalyst concentration and a second-order dependence on acid concentration, resulting in an overall third-order rate constant of 184 M−2 s−1. Reactions with deuterated acids display a large KIE of 9 ± 1, irrespective of deuterated acid (CD3CO2D and CF3SO3D). The similarities of the TON and KIE values in the strong and weak acids further support a common rate-determining step. The as-determined large KIE value suggests that the rate-determining step may be related to the release of H2 with significant catalyst–hydrogen bond breaking occurring at the transition state. Metal hydrides are always formed or proposed in HER when catalysed by H2ase enzymes and synthetic electrocatalysts. An inverse KIE with the values ranging from 0.54 to 0.57 was previously reported by Gray et al.,15 which was attributed to metal hydride formation via proton-coupled electron transfer in a rate-determining step. Compared with the inverse KIE values observed for metal hydrides, the relatively-high KIE value for 1 indicated a clearly different HER mechanism for H2 evolution. Two hypothetical catalytic cycles have been proposed by Grapperhaus et al. for this electrocatalytic H2 production by 1, namely the [ECCE] and [CECE] mechanism (with E standing for an electron transfer process and C relating to a chemical step, in this case protonation) (Scheme 2). Both mechanisms involve a key ligand–hydride as the hydrogen-evolving complex, which does not require metal hydride species and is suggested based on the similarities to the redox-regulated, ligand-centred binding of ethylene to 1.14,16
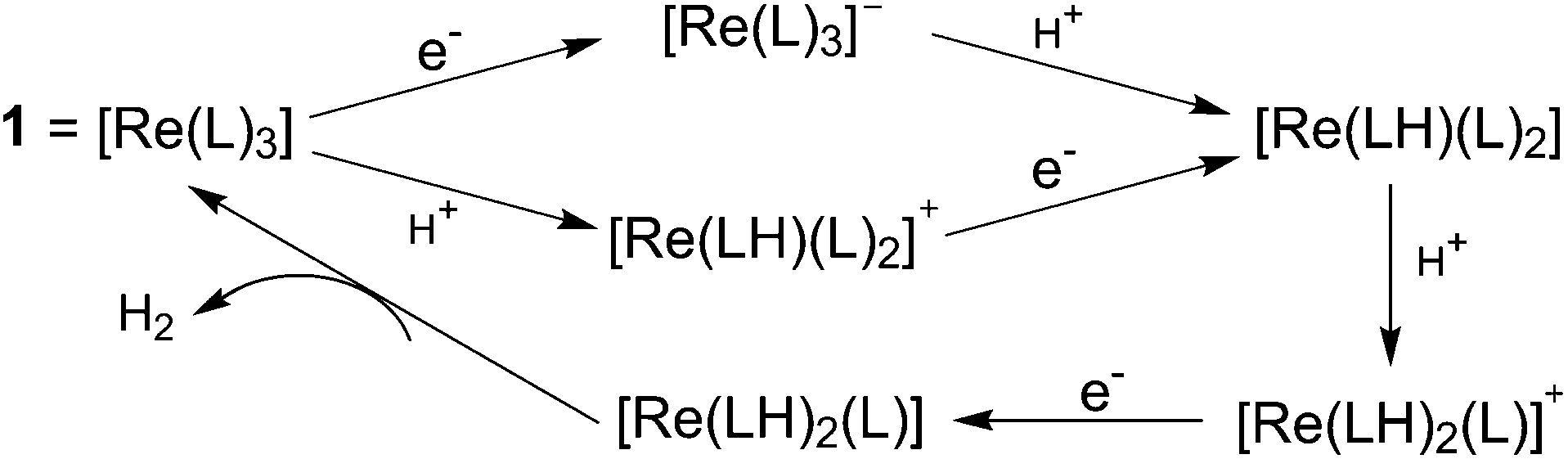 | ||
| Scheme 2 Proposed [ECCE] and [CECE] mechanisms for proton reduction catalysed by 1 = [ReL3] (where L = dppbt (2-diphenylphosphino-benzenethiolate)) with a common ligand–hydride as a key species. | ||
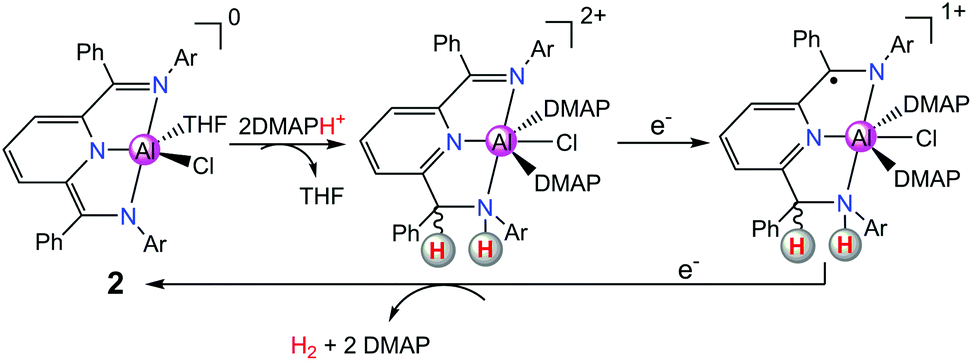 | ||
| Scheme 3 A proposed [CCEE] mechanism for proton reduction catalysed by 2 with a doubly protonated intermediate. | ||
In order to further design new Al complexes with redox-active ligands for HER, in another recent report, Berben's group used another Al(III) complex [(iPrPz2P)AlCl2(THF)][AlCl4] (3 in Scheme 4, where iPrPz2P = bis(pyrazolyl)pyridine) with a redox non-innocent ligand iPrPz2P as a molecular catalyst for the electrochemical production of H2.18 Compound 3 exhibits a catalytic peak at −1.26 V vs. SCE in THF with salicylic acid or [HNEt3]+ as the source of proton. Compound 3 was proposed to produce H2 catalytically via the [EECC] reaction mechanism through ligand-centred HER (Scheme 4). The pathway involving two-electron reduction and single-ligand protonation of 3 affords a key intermediate [(iPrHPz2P−)AlCl2], where each of the electron- and proton-transfer events is ligand-centred. Further protonation of the ligand–hydride [(iPrHPz2P−)AlCl2] would formally close the catalytic cycle for H2 production.
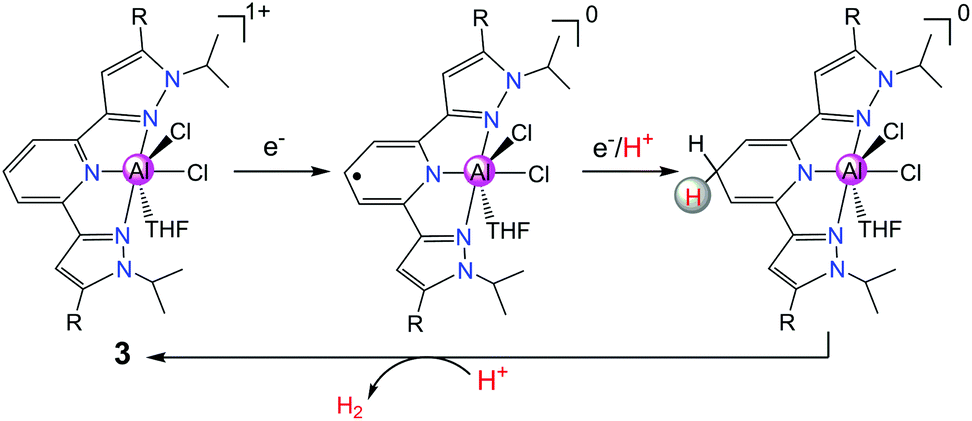 | ||
| Scheme 4 A proposed [EECC] mechanism for proton reduction catalysed by 3 with a ligand–hydride as a key intermediate. | ||
Although the above-proposed HER mechanisms by Al(III) complexes of iPrPz2P and PhI2P are based on ligand-centred proton and electron transfer, the former facilitates reduction before ligand protonation, while the latter is protonated twice before the reduction happens to liberate H2. These results indicated that the control of ligand donor atom pKa values can tune the HER mechanism of ligand-based hydride generation and transfer.
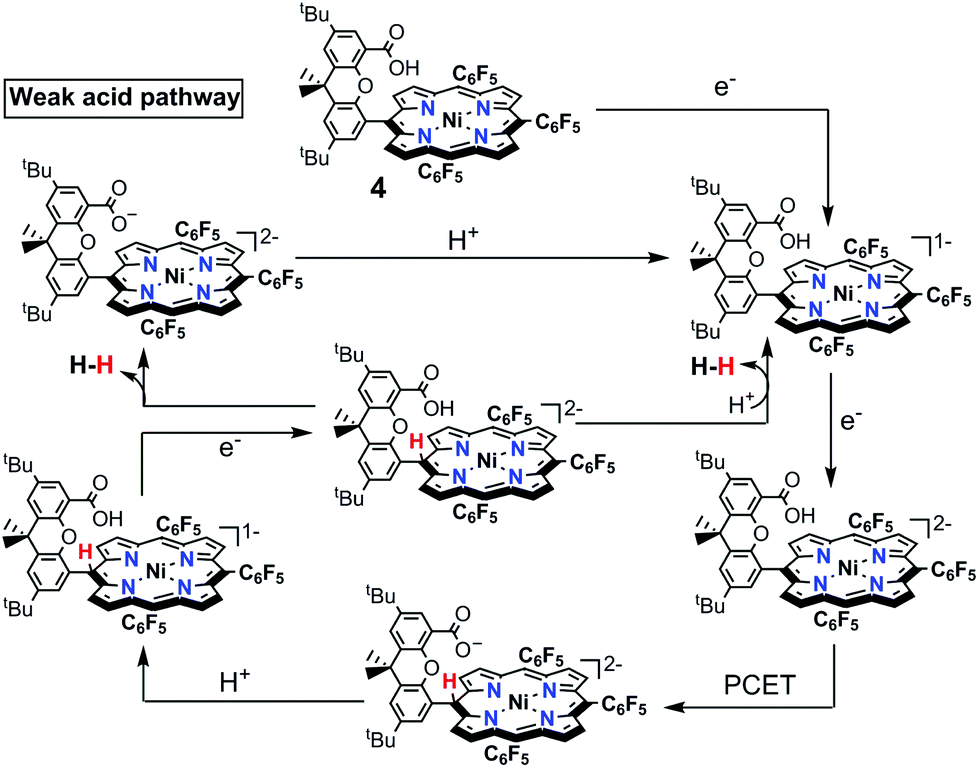 | ||
| Scheme 5 Proposed mechanisms catalysed by 4 with weak benzoic acid. | ||
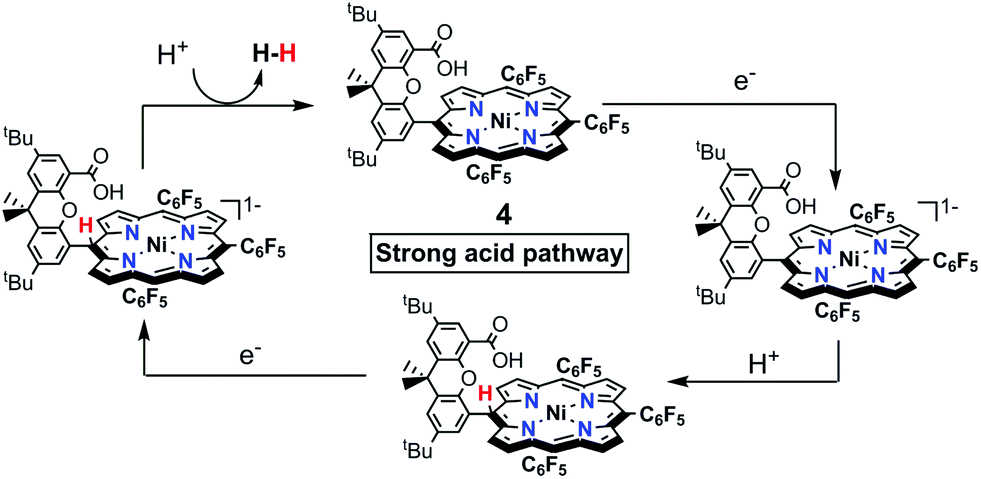 | ||
| Scheme 6 A proposed mechanism catalysed by 4 with strong Tosic acid. | ||
Results from protein crystallography22 revealed that the active centre of [NiFe]-H2ase contains a NiS4 core (Fig. 2a), which has inspired the synthesis of many [NiFe]-H2ase mimic complexes. Among them, Yamauchi and Sakai et al. recently reported that a Ni(II) pyrazinedithiolate complex [Ni(dcpdt)2]2− (5 in Fig. 2b, dcpdt = 5,6-dicyanopyrazine-2,3-dithiolate) having a NiS4 core similar to the active site of [NiFe]-H2ase can serve as an efficient molecular electrocatalyst for water reduction to generate H2.23 This bio-inspired molecular catalyst 5 was shown to effectively reduce protons in pH 4–6 buffered water with relatively low overpotentials (0.33–0.4 V) for HER. Moreover, in a 24 h controlled potential electrolysis at −0.95 V vs. SCE in an aqueous acetate buffer solution at pH 5, 5 was reported to catalyse H2 evolution with a faradaic yield of ca. 100% and a TON of >2 × 104 mol H2 per mol cat on a GCDE, indicating its excellent durability for long-term use in electrochemical HER. A combined electrochemical and theoretical study suggested a possible catalytic mechanism involving a diprotonated one-electron-reduced species (i.e., [NiII(dcpdt)(dcpdtH2)]− or [NiII(dcpdtH)2]−), which was a ligand-based reduction accompanied by protonation of the pyrazine donors formed at pH < 6.4, proposed as critical intermediates for electrocatalytic HER. The ligand-based proton-coupled electron-transfer pathway leads to electrocatalytic HER without applying the highly negative potential required to generate low-valent nickel intermediates.
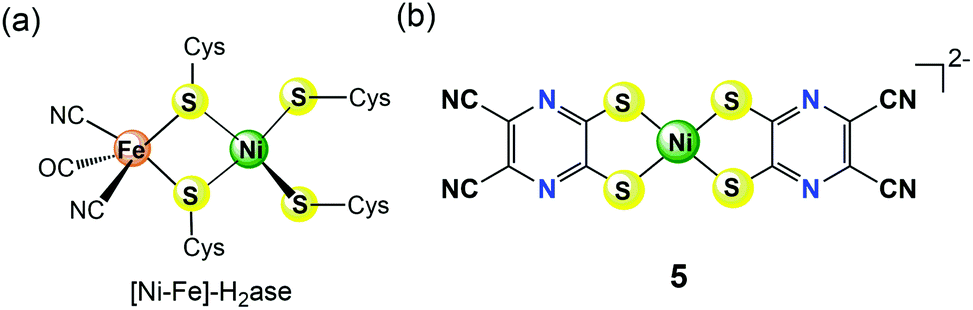 | ||
| Fig. 2 Chemical structures of the active site of [NiFe]-H2ase (a) and nickel(II)-dithiolate complex 5 containing redox-active pyrazinedithiolate ligands (b) discussed in the text. | ||
700 s−1 in CH3CN at an overpotential of 0.756 V and 1.074 V by using CH3COOH as the proton source. A bulk electrolysis experiment showed a TON of H2 evolution of 37 in a 2.5 h experiment with a faradaic yield of 85% at a controlled potential of −1.7 V vs. Fc/Fc+ in a CH3OH solution of 6 and CH3CO2H (12 mM) on a GCDE. Further mechanistic insights were obtained by the reduction of deuterated acid (CD3CO2D) substrates. Reactions with CD3CO2D display a small KIE of 1.2, which is distinct from the inverse KIE found by Nocera et al. and the large KIE observed by Grapperhaus et al.,14,15 indicating a clearly different mechanism for H2 evolution. The simulated kinetic and thermodynamic parameters revealed that the catalysis proceeds via ligand-centred processes involving the homocoupling of two neutral Zn(HL˙) radicals and the hetero-coupling of a neutral Zn(HL˙) radical with the cationic radical [Zn(H2L˙)]+. Further DFT calculations elucidated protonation at the hydrazino nitrogen, which is favoured relative to other potential basic sites within 6. The formation of H2 is realized via ligand hydride–proton coupling while avoiding traditional metal–hydride intermediates (Scheme 7).
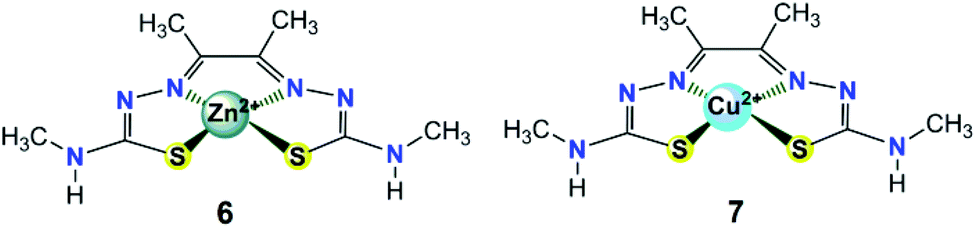 | ||
| Fig. 3 Chemical structures of zinc(II) and copper(II) complexes 6–7 with redox non-innocent bis(thiosemicarbazone) ligands discussed in the text. | ||
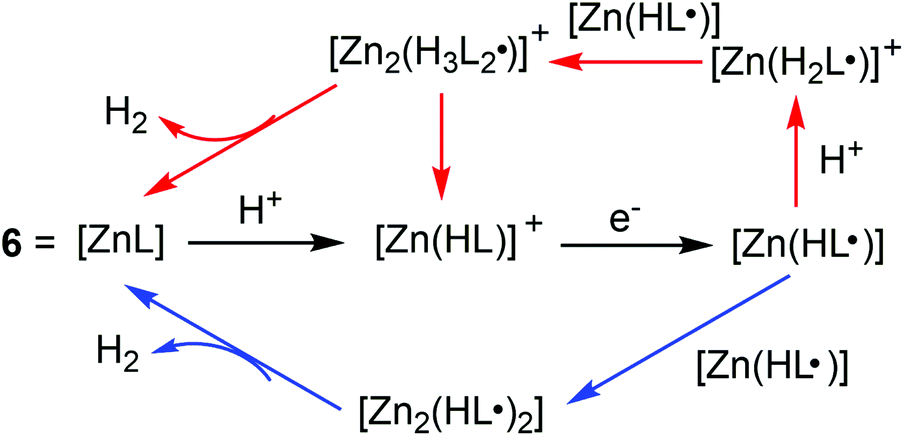 | ||
| Scheme 7 Possible catalytic pathways for HER by 6 = [ZnL] (where L = diacetyl-bis(N-4-methyl-3-thiosemicarbazide)) through hetero-coupling of a neutral Zn(HL˙) and cationic [Zn(H2L˙)] + and homocoupling of neutral Zn(HL˙) radicals (blue arrows). | ||
In addition to [ZnL], another Cu(II) complex containing the same diacetyl-bis(N-4-methyl-3-thiosemicarbazide) ligand (7, in Fig. 3) was also subsequently synthesized and assessed by Grapperhaus et al. as a H2-evolving electrocatalyst.26 Complex 7 exhibits a maximum TOF of 10000 s−1 in CH3CN and 5100 s−1 in DMF at an overpotential of 0.80 and 0.76 V, respectively. Controlled potential electrolysis confirmed 7 as a good electrocatalyst to produce H2 with a minimum faradaic efficiency of 81% and TON as high as 73 while showing no sign of degradation over 23 h. The HER kinetics was probed using deuterated acid (CD3CO2D), demonstrating a KIE of 7.54, which is different from that of the ZnII catalyst (KIE = 1.2).25 A possible HER mechanism catalysed by 7 was examined by DFT computational studies. In the proposed mechanism, an initial chemical event involves the protonation of the hydrazino nitrogen on the ligand. This is followed by an electrochemical step assigned as a copper-centred reduction resulting in a d10 Cu(I) species [Cu(HL)] that is isoelectronic with the Zn(II) complex of bis(thiosemicarbazone) [Zn(HL)]+.25 A second protonolysis of [Cu(HL)] occurs at the opposing hydrazino nitrogen of the ligand to yield [Cu(H2L)]+. Further one-electron reduction of [Cu(H2L)]+ leads to the formation of the H2-evolving complex [Cu(H2L)], through rapid intramolecular rearrangement. The overall [CECE] catalytic cycle for this process is given in Scheme 8.
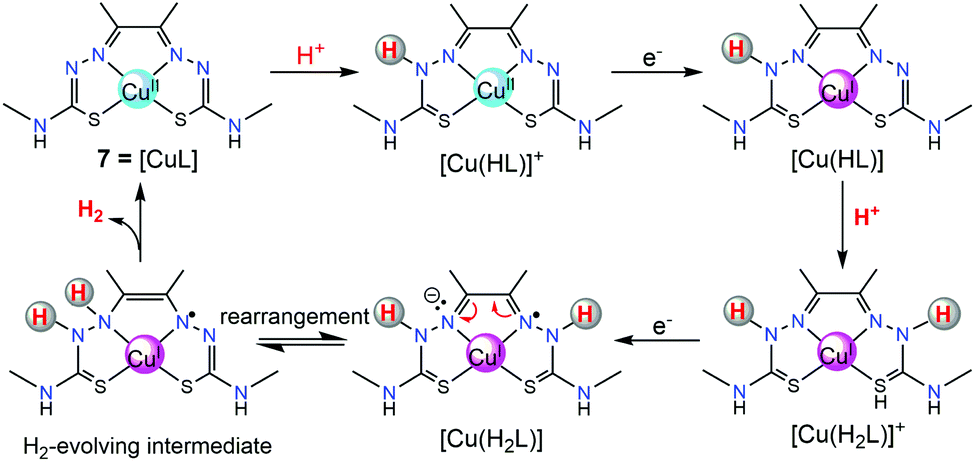 | ||
| Scheme 8 Plausible [CECE] mechanism for proton reduction by 7. | ||
It has to be mentioned that the identity of metal ions may affect the type of HER mechanism. The impact of the metal is manifested in the initial protonation and reduction sites. For example, when the metal source was changed to Ni(II) or Co(II) ion, the corresponding metal complexes were also found to be efficient molecular electrocatalysts for H2 production. However, in these cases, the HER mechanisms were proposed to be metal-centred, i.e. in the formation of metal hydrides, there is no ligand hydride as a key step in the proposed HER mechanism.27,28
2.2 Metal-free organic molecules as molecular electrocatalysts for HER
In recent years, most research efforts on significant homogeneous catalysis have been concentrated on the development of catalysts using non-precious metals such as iron, cobalt, nickel, or molybdenum to produce H2.4–6 A HER catalytic system utilizing metal-free organic molecules as electrocatalysts is relatively uncommon. However, organic compounds as molecular catalysts may provide lower manufacturing costs, superior synthetic flexibility, and greater chemical stability. In fact, some simple organic molecules have been shown to be competent mediators and catalysts for electrochemical CO2 reduction.29 Thus, it is expected that organic compounds can also be employed as molecular catalysts for electrochemical H2 production.In 2016, Grapperhaus et al. investigated the free ligand diacetyl-bis(N-4-methyl-3-thiosemicarbazone) (8, in Fig. 4) towards its applicability for electrocatalytic HER.25 This organic compound shows a catalytic peak at −2.10 V (vs. Fc/Fc+) in anhydrous CH3CN with CH3COOH as the proton source. Under acid-saturated conditions, 8 displayed the highest TOF of 1320 s−1 with a high overpotential of 1.430 V. The high TOF of catalyst 8 comes at the price of a high overpotential (more than 1.4 V). In addition, the TOF value (1320 s−1) of 8 is apparently lower compared to that of the corresponding Cu(II)-complex (10000 s−1) and Zn(II)-complex (11700 s−1).25,26 A potential explanation for this behaviour was partially attributed to the Lewis acidities of Cu(II) and Zn(II), which can balance the charge of the anionic ligand, promote the protonation, and lower the reduction potential. Notably, this is the first example of a homogeneous and metal-free organic molecule acting as an efficient electrocatalyst for HER, which may provide a new direction towards the development of abundant organic compounds as H2-evolving catalysts for the replacement of metals.
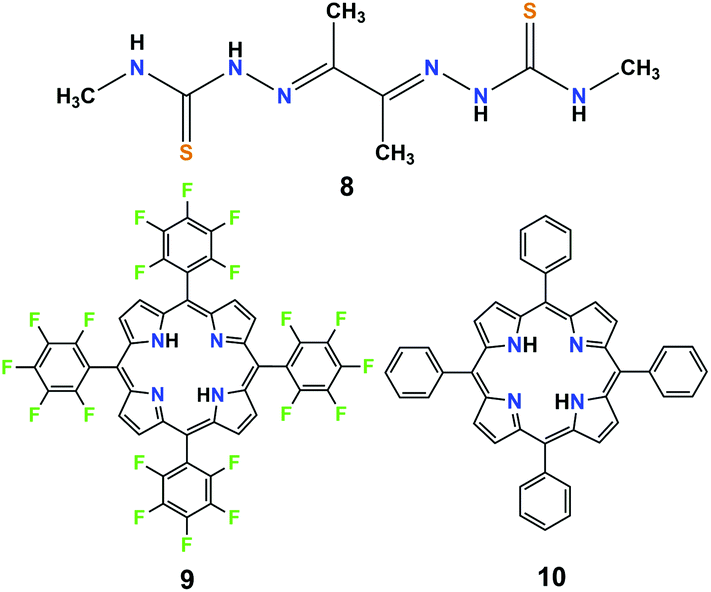 | ||
| Fig. 4 Chemical structures of metal-free organic molecule-based H2-evolving electrocatalysts (8–10) discussed in the text. | ||
It is well-known that metal-free porphyrin compounds can exhibit rich multi-electron redox chemistry. Two basic nitrogen atoms of imine in the porphyrin core can potentially produce a diprotonated porphyrin species, indicating that free-base porphyrins can be electrochemically active. In 2018, Villagrán et al. reported a metal-free porphyrin catalyst, which is a similar and comparable electrocatalytic HER complex.30 The free-base meso-tetra(pentafluorophenyl) porphyrin (9, in Fig. 4) was employed to electrocatalyze H2 evolution using p-toluenesulfonic acid (HOTs) as the proton source in THF. The cyclic voltammogram (CV) on a GCDE of 9 in THF showed two reversible reduction waves with midpoint potentials of E1/2 = −1.14 V and E1/2 = −1.54 V vs. Fc/Fc+, assigned to the first and second reductions to produce the porphyrin radical anion [9]˙− and the dianion species [9]2−, respectively. Upon successive addition of strong acid HOTs (pKa (HOTs) = 8.7 in CH3CN)31 as the proton source, the first reduction wave of 9 remains unchanged, while a new electrocatalytic current for the proton reduction appeared at the onset of the catalytic wave (−1.31 V), from which the overpotential for HER with 9 was determined to be 1.02 V. Controlled potential electrolysis of 9 at −1.31 V (vs. Fc/Fc+) on a GCDE was conducted over the course of 40 min, and more than 3.2 equiv. of charge was passed, which yielded H2 at 90% faradaic efficiency. Foot-of-the-wave analysis was employed to measure the HER kinetics. The as-observed rate constant kobs values of 0.528, 0.742 and 0.891 s−1 at an acid concentration [H+] of 0.4, 0.9 and 1.2 mM were calculated from the slope of the linear region near the foot of the wave. Plotting kobsversus [H+] gives a linear relationship, suggesting a first order dependence on [H+]. A KIE of 1.16 was observed when using deuterated HOTs. Interestingly, as a control, a second metal-free meso-tetraphenylporphyrin (10, in Fig. 4) was also evaluated as a HER electrocatalyst under the same experimental conditions. However, the achieved electrocatalytic property based on this porphyrin 10 was very low. The analyses of the shape of the CVs, spectroelectrochemical experiments, together with DFT computational studies, are consistent with the most favourable mechanistic process for HER in an [ECEC] sequence (Scheme 9). Although complex 9 is the first metal-free porphyrin molecule serving as an efficient H2-evolving electrocatalyst, the disadvantages are that the HER occurs with a large overpotential (>1.0 V) and must be carried out using strong acid as the proton source.32
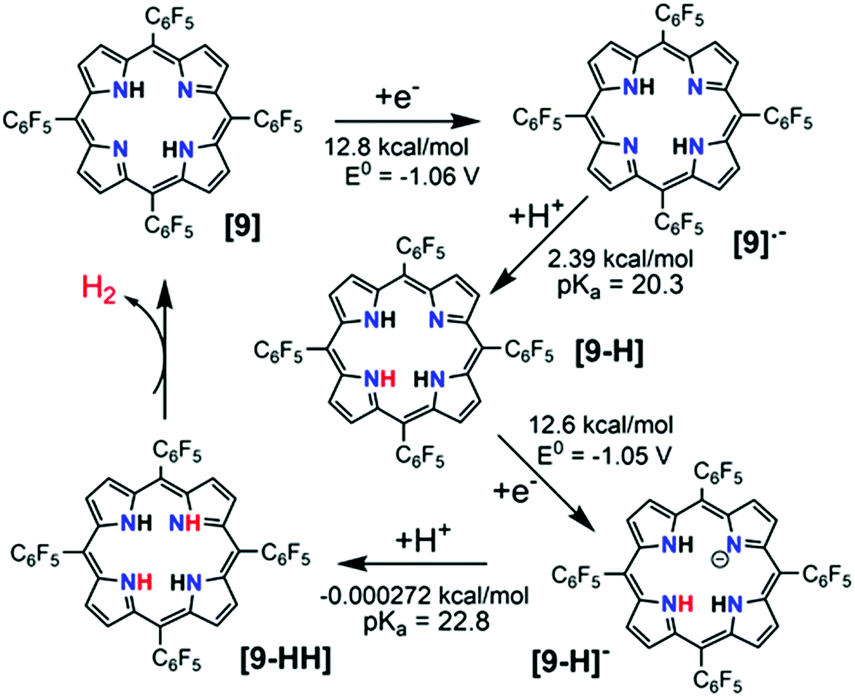 | ||
| Scheme 9 Proposed catalytic cycle by 9 for H2 generation, following an [ECEC] mechanism. | ||
3. Molybdenum-disulfide complexes for inorganic ligand-centred electrocatalytic HER
3.1 A molybdenum(IV)-disulfide complex for disulfide inorganic ligand-centred electrocatalytic HER in aqueous solution
In recent years, as one of the two-dimensional (2D) inorganic materials, molybdenum sulfide (MoS2) has been identified as a promising material for non-precious heterogeneous electrocatalysis in the HER under acidic conditions.33 However, the desired performance of MoS2 is often limited by the density of active sites for the HER, which are thought to be localized to rare edges, where disulfide linkages or triangular MoS2 units are exposed and can absorb H species with a small free energy while the bulk form is inert.34 The use of biomimetic molecular electrocatalysts mimicking these rare edges is an attractive approach because these catalysts may retain elecrocatalytic activity at a small size and maximize the amount of active sites. Additionally, some major advantages of well-defined biomimetic catalysts include: (1) traditional molecular characterization techniques, such as MS, NMR spectroscopy, and single-crystal X-ray diffraction, can be employed to probe the local structures of catalysts; (2) they allow for a fine tuning of their catalytic properties through ligand modifications; and (3) these bio-inspired catalysts may provide detailed mechanistic insights into the active sites, which is difficult to achieve in nanoparticles.Long et al. previously identified a Mo(IV)-oxo pentapyridine complex [(PY5Me2)MoO]2+ (where PY5Me2 = [2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine]) and its ability to electrocatalytically generate gaseous H2 either from neutral pH water or from sea water.35 A MoII/MoIV mechanism for HER catalyzed by the MoIV-oxo complex was proposed, in which a [(PY5Me2)Mo(H2O)]2+ species is responsible for the water splitting to release OH− and H2. Along these lines, the same group subsequently reported a well-defined MoIV-disulfide(S22−) pentapyridine complex [(PY5Me2)MoS2]2+ (11, in Fig. 5a and b), which is a highly-active yet stable molecular electrocatalyst for HER in acidic aqueous solution.36 In an acetate buffer (pH = 3) on a Hg pool electrode, the CV of complex 11 shows an irreversible reductive process at −0.58 V vs. NHE corresponding to an overpotential of 0.4 V, which is consistent with catalytic proton reduction. A 23 h controlled potential electrolysis experiment of 11 at an overpotential of 0.78 V in aqueous acetate buffer (pH = 3) in a double-compartment cell generated H2 at 100% faradaic efficiency. The effectiveness of this system is impressive: the lower-bound value for the TON calculated by using the bulk solution of electrocatalyst molecules afforded ∼3.5 × 103 moles of H2 per mole of catalyst, and the calculated upper-limit TON value reached 1.9×107 moles of H2 per mole of electrocatalyst, assuming a constant monolayer on the electrode. The molecular electrocatalyst 11 remains stable under these experimental conditions and its catalytic efficiency is comparable to those of the biocatalysts H2ases.
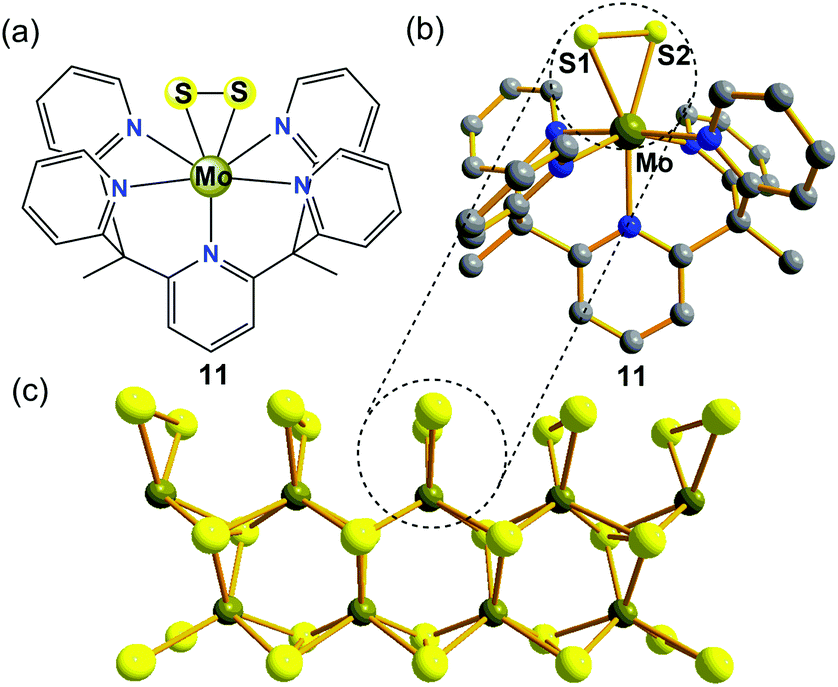 | ||
| Fig. 5 (a) Chemical structure of a molybdenum(IV)-disulfide pentapyridine complex 11 with an inorganic ligand platform (S22−) discussed in the text. (b) The single-crystal X-ray structure of 11. (c) A model of the 2D inorganic material MoS2 highlighting the exposed triangular MoS2 units on the edge sites. | ||
It is important to note that complex 11 represents the first example of a side-on bound S22− on Mo(IV) that mimics the structure of the proposed triangular MoS2 active surface sites (Fig. 5c). It was expected that the findings from MoIV-S22−11 are encouraging because they have important implications for the further design of structural and functional biomimetic molecular catalysts of other layered transition-metal dichalcogenides (MoSe, WS2, WSe, etc.), in the same way that bioinorganic chemists distil the structure and the reactivity of H2ase enzymes by resembling the active core groups in metalloenzymes.
To provide further insight into the HER mechanism catalysed by 11, Dang et al. performed detailed DFT calculations to explore possible reaction intermediates and various proton reduction pathways.37 For the classical HER mechanisms catalysed by well-defined molecular catalysts in acidic water, metal hydride intermediates are often invoked as key species in the context of H2-evolving catalysts. However, the results from the theoretical calculations revealed that H2 evolved through the traditional protonation of the metal centre to generate a molybdenum hydride is thermodynamically unfavourable. The preferred pathways are attributed to the formation of active sulfur hydrides via two- or three-electron reduction from the molybdenum(IV)-disulfide mimic 11 (Scheme 10). Moreover, theoretical calculations also suggested that two-electron and three-electron reduced sulfur hydride intermediates may react with a hydrated proton to generate H2 with the help of water acting as a bridge for H2 evolution from the intermolecular H+/H− coupling between sulfur hydride complexes and hydrated protons.37
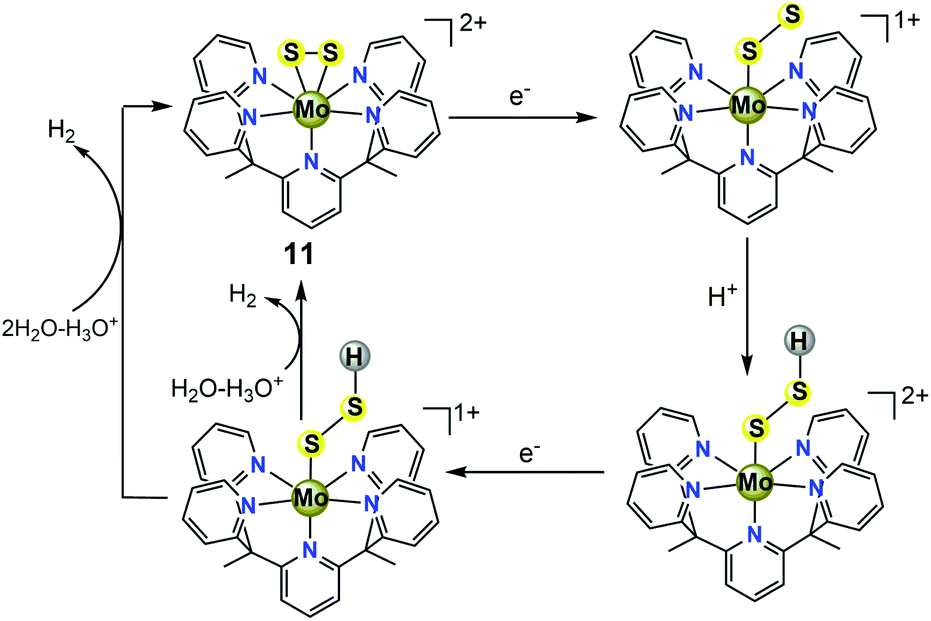 | ||
| Scheme 10 Two-electron reduction mechanism for 11-catalyzed HER. | ||
3.2 Molybdenum(VI)-disulfide complexes with disulfide inorganic ligand-centred electrocatalytic HER in organic media
Inspired by the original MoS2 biomimetic work of Long and Chang,36 Wu et al. later successfully prepared and well-characterized a molybdenum(VI)-disulfide complex (12, in Fig. 6a) containing two redox-active S22− inorganic ligands, which has been employed as a solution electrocatalyst for HER using CF3CO2H as the proton source.38 The O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mo(S2)2 moiety in 12 (see Fig. 6b for the crystal structure) mimics the terminal edges in MoS2 that are active sites for HER. Different from the MoIV ion in 11, the oxidation state of the metal centre in 12 is +6. In the absence of acid, the CV of 12 in DMF showed an irreversible voltammetric response at −1.59 V vs. Fc/Fc+, assigned to two one-electron reduction processes, as suggested by constant-potential coulometry. Calculated charge density plots indicated that the reduction process is primarily S–S ligand-centred. The addition of the strong acid CF3CO2H (pKa(CF3CO2H) = 6.0 in DMF)39 to the solution of 12 in DMF resulted in an increase of the cathodic current at the near potential of the S–S reduction. In the acid-saturated regime, 12 exhibited a TOF of 95 s−1 with an overpotential of 0.617 V. In a controlled potential electrolysis, complex 12 catalysed H2 production at 83% faradaic efficiency on a GCDE. Further studies on two other high-valance Mo(VI)-disulfide derivatives (13 and 14, in Fig. 6a) revealed that simply varying substitutions at the para-position of 2,2′-bipyridine ligand affects the basicity of the S–S ligand, thereby changing the catalytic activities of H2 production including the TOFs and overpotentials.38 The TOF values of H2 evolution increased from 227 to 246 s−1 when the electron-donating group at the para-position of the 2,2′-bipyridine ligand was changed from MeO to t-butyl, with overpotentials varying from 0.806 to 0.86 V using CF3CO2H as the proton source in DMF solution. The electrocatalytic results are summarized in Table 2. The basicity of the S22− ligands was affected by varying substitutions on the 2,2′-bipyridine, indicating that during the electrocatalytic reaction, the S–S inorganic ligand may be the initial site of protonation.
Mo(S2)2 moiety in 12 (see Fig. 6b for the crystal structure) mimics the terminal edges in MoS2 that are active sites for HER. Different from the MoIV ion in 11, the oxidation state of the metal centre in 12 is +6. In the absence of acid, the CV of 12 in DMF showed an irreversible voltammetric response at −1.59 V vs. Fc/Fc+, assigned to two one-electron reduction processes, as suggested by constant-potential coulometry. Calculated charge density plots indicated that the reduction process is primarily S–S ligand-centred. The addition of the strong acid CF3CO2H (pKa(CF3CO2H) = 6.0 in DMF)39 to the solution of 12 in DMF resulted in an increase of the cathodic current at the near potential of the S–S reduction. In the acid-saturated regime, 12 exhibited a TOF of 95 s−1 with an overpotential of 0.617 V. In a controlled potential electrolysis, complex 12 catalysed H2 production at 83% faradaic efficiency on a GCDE. Further studies on two other high-valance Mo(VI)-disulfide derivatives (13 and 14, in Fig. 6a) revealed that simply varying substitutions at the para-position of 2,2′-bipyridine ligand affects the basicity of the S–S ligand, thereby changing the catalytic activities of H2 production including the TOFs and overpotentials.38 The TOF values of H2 evolution increased from 227 to 246 s−1 when the electron-donating group at the para-position of the 2,2′-bipyridine ligand was changed from MeO to t-butyl, with overpotentials varying from 0.806 to 0.86 V using CF3CO2H as the proton source in DMF solution. The electrocatalytic results are summarized in Table 2. The basicity of the S22− ligands was affected by varying substitutions on the 2,2′-bipyridine, indicating that during the electrocatalytic reaction, the S–S inorganic ligand may be the initial site of protonation.
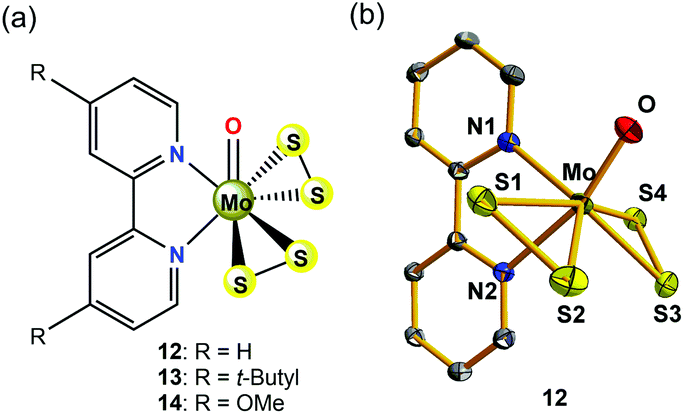 | ||
| Fig. 6 (a) Chemical structures of molybdenum(VI)-sulfur complexes 12–14 with redox non-innocent inorganic sulfide scaffolds discussed in the text. (b) Representation of the single-crystal X-ray structure of 12 (all H atoms omitted for clarity). | ||
| Catalyst | Redox-active inorganic ligand | Catalytic potential (Ep)b | Proton source | Solvent | OPc |
k
obsd |
Ref. |
|---|---|---|---|---|---|---|---|
| a Conditions: Hg pool working electrode for 11, and GC working electrode for 12–14. b Catalytic potential (Ep) of 11vs. NHE and Ep of 12–14vs. Fc/Fc+. c For 11, OP = overpotential = |Eonvs. NHE – 0.059pH|; for 12–14, OP = overpotential = |Ep − EθHA|. d k obs = TOF for 12–14 was calculated in the acid-independent region according to the equation: ic/ip = {(2/0.4463) × [(RTkobs)/(Fv)]1/2}. ic is the peak current in the presence of an acid and ip is the peak current in the absence of an acid. For 11, kobs = TOF was determined from a bulk electrolysis experiment. | |||||||
| 11 | Disulfide | −0.58 V | pH 3, acetate buffer | H2O | 0.40 V | 280 s−1 | 36 |
| 12 | Disulfide | −1.63 V | CF3COOH | DMF | 0.62 V | 95 s−1 | 38 |
| 13 | Disulfide | −1.71 V | CF3COOH | DMF | 0.86 V | 246 s−1 | 38 |
| 14 | Disulfide | −1.77 V | CF3COOH | DMF | 0.81 V | 227 s−1 | 38 |
From the consideration of electrochemical measurements coupled with DFT calculations,38,40 the electrocatalytic HER mechanism of the molecular MoS2-mimicking complex 12 shown in Scheme 11 could be described as follows: (1) the reaction mechanism initially involves two one-electron reductions of the inorganic ligand S22− in 12 to generate a subsulfide (S23−) species. The S23− species is hypothesized to contain a special 2c/3e S–S σ half bond. Of note, the term ‘subsulfide’ and the corresponding S–S 2c/3e σ half bond were firstly coined by Berry.41 (2) The next step is the protonation of the S23− ligand. The theoretical pathway showed that the second protonolysis occurs preferentially at the MoO moiety acting as a proton relay. (3) Protonated S23− acts as the hydride and the protonated MoO acts as the proton source. The combination of the hydride and proton affords H2 and the regeneration of 12. The overall catalytic cycle closes with an [EECC] mechanism (Scheme 11).
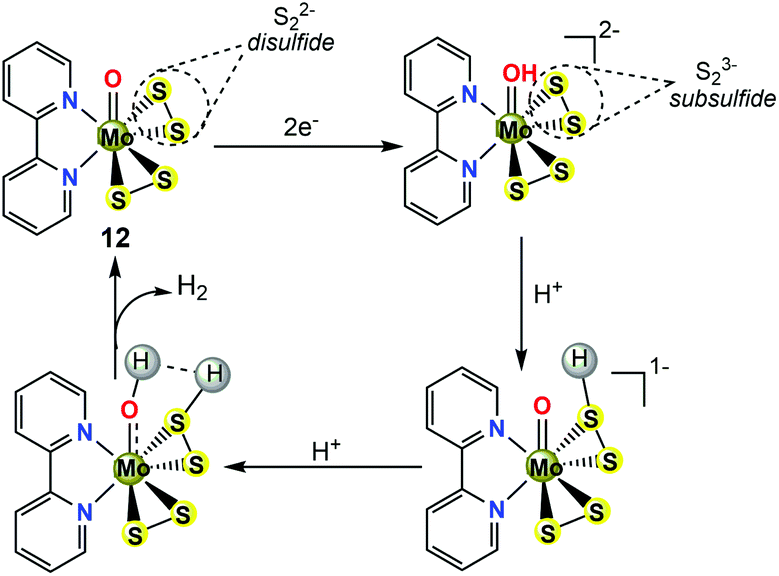 | ||
| Scheme 11 A proposed catalytic cycle for H2 generation by 12, following an [ECEC] mechanism. | ||
4. Conclusions and outlook
In this review, we have summarized the fundamental aspects and recent advances of the metal and metal-free ligand-centred homogeneous electrocatalytic HER. Although reasonable progress has been made in this field of electrocatalysis, there still exist several issues to be addressed. For example, low catalytic turnover rates and large overpotentials required by most H2-evolving molecular electrocatalysts (see Tables 1 and 2) are independent of the strength of the proton sources used. Typical examples are the metal-free organic molecular catalysts for electrocatalytic H2 formation, which show relatively large overpotentials (exceeding 1.0 V). Another crucial problem is that most ligand-centred HER processes can only be conducted in organic media with the requirement of extra moderate and strong acids as proton sources. The use of organic solvents and extra acids for the electrochemical reduction of protons will impede these catalyst systems in future large scale applications.While current research in catalytic H2-evolving mechanisms is focused on the development of metal-centred HER, we believe that redox-active ligand-centred HER should not be overlooked. A few important directions in this field to be followed for the future design of HER catalysts are provided as follows: (i) homogeneous catalytic production of H2 has long been dominated by transition-metal hydrides. In very recent advances, partly inspired by naturally biological systems, chemists have reported two purely organic molecules, which are potentially useful as electrocatalysts for H2 production.25,30 It is clear that there is a huge amount of unexplored potential for metal-free organic compounds in H2 production catalysis. These metal-free hydride catalyst systems may be viable alternatives to transition-metal hydride catalysts because the catalytic activity and overpotential of organic molecules can be further tuned and improved by varying the electronic and/or structural parameters of organic compounds. Thus, the development of H2-evolving catalysis without metals to allow the reaction to operate at a low overpotential might be an active topic in the coming years. (ii) As in the examples outlined above, metal and metal-free ligand-centred electrocatalytic HER is currently the subject of intensive investigations. By contrast, systems involving photochemical ligand-centred HER have received much less attention,42 and therefore it is an important issue to be investigated in the context of ligand-centred HER. (iii) Biomimetic molybdenum disulphide complexes have been performed as electrocatalysts for the HER. Therefore, another suggestion is to continue to design and synthesize structural and functional biomimetic molecular catalysts of other layered transition-metal dichalcogenides (MoSe, WS2, WSe, etc.), in much the same way that one mimics the rare edges of MoS2. (iv) Current mechanistic investigations of these ligand-centered proton reduction catalysis systems heavily rely on computational quantum chemical methods. However, the precise determination of mechanistic routes requires the isolation and characterization of some ligand hydride species. Therefore, apart from single-crystal X-ray structure determination of stable intermediates isolated from catalytic systems, a combination of spectroscopy and catalysis measurements should be performed for in situ identification of some unstable reactive ligand–hydride intermediates to gain more knowledge on the catalytic mechanism of these systems. It is our view that a better understanding of the mechanisms of H2 formation and deep insight into the basic structure–function relationship of the ligand catalytic centre will aid the effective design of highly efficient catalysts based on ligand-centred HER.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the National Natural Science Foundation of China (Grant No. 21641011, 21601086), the Natural Science Foundation of Fujian Province (Grant No. 2015J01053, 2015H0024) and the Natural Science Foundation of Jiangsu Province (BK20160994) for financial support of this work. Q. Z. acknowledges financial support from AcRF Tier 1 (RG 111/17, RG 2/17, RG 114/16, RG 8/16) and Tier 2 (MOE 2017-T2-1-021), Singapore. Q. Z. is also grateful for the support from the Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm201833), Jilin University, China. G. L. also thanks the Provincial Innovative Foundation Project for undergraduates (201810385072).References
- (a) M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef; (b) A. J. Brad and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef; (c) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- (a) R. Coontz and B. Hnson, Science, 2004, 305, 957 CrossRef CAS; (b) W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- A. Phuruangrat, D. J. Ham, S. Thongtem and J. S. Lee, Electrochem. Commun., 2009, 11, 1740–1485 CrossRef CAS.
- (a) R. E. Adams, T. A. Grusenmeyer, A. L. Griffith and R. H. Schmehl, Coord. Chem. Rev., 2018, 362, 44–53 CrossRef CAS; (b) S. J. C. Robinson and D. M. Heinekey, Chem. Commun., 2017, 53, 669–676 RSC; (c) D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed; (d) J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC; (e) D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed; (f) R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC; (g) V. S. Thoi, Y.-J. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC; (h) M. Wang, L. Chen and L.-C. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC.
- (a) Z.-H. Pan, Y.-W. Tao, Q.-F. He, Q.-Y. Wu, L.-P. Cheng, Z.-H. Wei, J.-H. Wu, J.-Q. Lin, D. Sun, Q. Zhang, D. Tian and G.-G. Luo, Chem. – Eur. J., 2018, 24, 8275–8280 CrossRef CAS PubMed; (b) A. Xie, J. Zhu and G.-G. Luo, Int. J. Hydrogen Energy, 2018, 43, 2772–2780 CrossRef CAS; (c) G.-G. Luo, Y.-H. Wang, J.-H. Wang, J.-H. Wu and R.-B. Wu, Chem. Commun., 2017, 53, 7007–7010 RSC; (d) G.-G. Luo, K. Fang, J.-H. Wu and J. Mo, Chem. Commun., 2015, 51, 12361–12364 RSC.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373–8426 CrossRef CAS PubMed.
- (a) J. Berg, J. Tymoczko and L. Stryer, Biochemistry, W. H. Freman and Co., 6th edn, 2007, pp. 565–566 Search PubMed; (b) C. A. Raines, Photosynth. Res., 2003, 75, 1–10 CrossRef CAS PubMed.
- (a) D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC; (b) W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356–3358 CrossRef CAS PubMed; (c) A. L. Smith, K. I. Hardcastle and J. D. Scoper, J. Am. Chem. Soc., 2010, 132, 14358–14360 CrossRef CAS PubMed; (d) P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed; (e) C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS; (f) M. Nielsen, A. Kammer, D. Cozzula, H. Junge, S. Gladiali and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9593–9597 CrossRef CAS PubMed; (g) M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788–789 CrossRef CAS PubMed; (h) H. Kotani, T. Sugiyama, T. Ishizuka, Y. Shiota, K. Yoshizawa and T. Kojima, J. Am. Chem. Soc., 2015, 137, 11222–11225 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- (a) A. McSkimming and S. B. Colbran, Chem. Soc. Rev., 2013, 42, 5439–5488 RSC; (b) S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- J. R. Dilworth, A. J. Hutson, S. Morton, M. Harman, M. B. Hursthouse, J. Zubieta, C. M. Archer and J. D. Kelly, Polyhedron, 1992, 11, 2151–2155 CrossRef CAS.
- C. A. Grapperhaus, K. Ouch and M. S. Mashuta, J. Am. Chem. Soc., 2009, 131, 64–65 CrossRef CAS PubMed.
- A. Z. Haddad, D. Kumar, K. O. Sampson, A. M. Matzner, M. S. Mashuta and C. A. Grapperhaus, J. Am. Chem. Soc., 2015, 137, 9238–9241 CrossRef CAS PubMed.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS PubMed.
- W. Zhang, A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 2177–2187 CrossRef CAS PubMed.
- E. J. Thompson and L. A. Berben, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS PubMed.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS PubMed.
- (a) V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed; (b) X.-L. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- (a) C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed; (b) B. H. Solis, A. G. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, ACS Catal., 2014, 4, 4516–4526 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- K. Koshiba, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2017, 56, 4247–4251 CrossRef CAS.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40, 3005–3018 RSC.
- A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, J. Am. Chem. Soc., 2016, 138, 7844–7847 CrossRef CAS PubMed.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Brapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS.
- (a) T. Straistari, J. Fize, S. Shova, M. Réglier, V. Artero and M. Orio, ChemCatChem, 2017, 9, 2262–2268 CrossRef CAS; (b) R. Jain, A. A. Mamun, R. M. Buchanan, P. M. Kozlowski and C. A. Grapperhaus, Inorg. Chem., 2018, 57, 13486–13493 CrossRef CAS PubMed.
- (a) T. Straistari, R. Hardré, J. Fize, S. Shova, M. Giorgi, M. Réglier, V. Artero and M. Orio, Chem. – Eur. J., 2018, 24, 8779–8786 CrossRef CAS PubMed; (b) Y. Zhao, Y.-H. Wang, Q.-Y. Wu, J.-Q. Lin, S.-H. Wu, W.-H. Hou, R.-B. Wu and G.-G. Luo, Chin. J. Catal., 2018, 39, 517–526 CrossRef CAS.
- Y. Oh and X.-L. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- Y.-Y. Wu, N. Rodríguez-López and D. Villagrán, Chem. Sci., 2018, 9, 4689–4695 RSC.
- P. A. Jacques, V. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- The denotations of strong acids and moderate acids are as follows: pKa < 10 and 10 ≤ pKa ≥ 15. See ref. 4h.
- (a) C.-L. Tan, X.-H. Cao, X.-J. Wu, Q.-Y. He, J. Yang, X. Zhang, J.-Z. Chen, W. Zhao, S.-K. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed; (b) J. H. Han, M. Kwak, Y. Kim and J. Cheon, Chem. Rev., 2018, 118, 6151–6188 CrossRef CAS PubMed.
- (a) Y. Yan, B.-Y. Xia, Z.-C. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef CAS; (b) G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed; (c) G. Ertl, Angew. Chem., Int. Ed. Engl., 1990, 29, 1219 CrossRef.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- T.-L. Yang, S.-F. Ni, P. Qin and L. Dang, Chem. Commun., 2018, 54, 1113–1116 RSC.
- B. R. Garrett, S. M. Polen, K. A. Click, M. He, Z. Huang, C. M. Hadad and Y. Wu, Inorg. Chem., 2016, 55, 3960–3966 CrossRef CAS PubMed.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 432–444 CrossRef PubMed.
- B. R. Garrett, S. M. Polen, M. Pimplikar, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2017, 139, 4342–4345 CrossRef CAS PubMed.
- J. F. Berry, Acc. Chem. Res., 2016, 49, 27–34 CrossRef CAS PubMed.
- T. Matsumoto, H.-C. Chang, M. Wakizaka, S. Ueno, A. Kobayashi, A. Nakayama, T. Taketsugu and M. Kato, J. Am. Chem. Soc., 2013, 135, 8646–8654 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2019 |