DOI:
10.1039/C8CE01311J
(Paper)
CrystEngComm, 2018,
20, 6394-6405
Novel pharmaceutical salts of albendazole†
Received
5th August 2018
, Accepted 10th September 2018
First published on 11th September 2018
Abstract
Albendazole (ABZ) is a class II safe and effective antihelmintic drug in the benzimidazole group according to the BCS (Biopharmaceutics Classification System) with low solubility (9 mg L−1) and high permeability (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P 2.54). Novel salts and salt hydrates of ABZ are reported with benzene and p-toluene sulfonic acid (BSA, PTSA), as well as carboxylic acids such as oxalic acid (OA), maleic acid (MLE), L-tartaric acid (LTA), 2,6-dihydroxybenzoic acid (2,6-DHBA), and 2,4,6-trihydroxybenzoic acid (2,4,6-THBA). The products ABZ–BSA, ABZ–BSA–H, ABZ–PTSA, ABZ–PTSA–H, ABZ–OA–H and ABZ–2,6-DHBA were confirmed by single crystal X-ray diffraction. In the hydrate structures (designated as –H), the water molecule acts as a bridge in the hydrogen bonding network. The salt formation of ABZ–MLE, ABZ–LTA, and ABZ–2,4,6-THBA was confirmed by 15N ss-NMR based on the chemical shift change of ca. 50 ppm. The sulfonate salt hydrates exhibit 2D isostructurality, and position disorder in the thiopropyl group in the drug crystal structure was not observed in the salts. Crystal lattice energies were calculated for the MLE, LTA, and 2,4,6-THBA complexes of ABZ to confirm the molecular salt formation. The cocrystals of ABZ with the hydroxybenzene carboxylic acids are novel salts in the benzimidazole drugs class.
P 2.54). Novel salts and salt hydrates of ABZ are reported with benzene and p-toluene sulfonic acid (BSA, PTSA), as well as carboxylic acids such as oxalic acid (OA), maleic acid (MLE), L-tartaric acid (LTA), 2,6-dihydroxybenzoic acid (2,6-DHBA), and 2,4,6-trihydroxybenzoic acid (2,4,6-THBA). The products ABZ–BSA, ABZ–BSA–H, ABZ–PTSA, ABZ–PTSA–H, ABZ–OA–H and ABZ–2,6-DHBA were confirmed by single crystal X-ray diffraction. In the hydrate structures (designated as –H), the water molecule acts as a bridge in the hydrogen bonding network. The salt formation of ABZ–MLE, ABZ–LTA, and ABZ–2,4,6-THBA was confirmed by 15N ss-NMR based on the chemical shift change of ca. 50 ppm. The sulfonate salt hydrates exhibit 2D isostructurality, and position disorder in the thiopropyl group in the drug crystal structure was not observed in the salts. Crystal lattice energies were calculated for the MLE, LTA, and 2,4,6-THBA complexes of ABZ to confirm the molecular salt formation. The cocrystals of ABZ with the hydroxybenzene carboxylic acids are novel salts in the benzimidazole drugs class.
1. Introduction
Albendazole (ABZ, methyl 5-[propylthio]benzimidazole-2-carbamate) belongs to the benzimidazole family of drugs first marketed by http://en.wikipedia.org/w/index.php?title=Amedra_Pharmaceuticals&action=edit&redlink=1 under the brand name ALBENZA.1 ABZ is a safe and effective antihelmintic drug (400 mg dose) but has significant antitumor activity in hepatocellular cancer.2 Benzimidazoles exhibit low erratic availability as a result of its poor aqueous solubility.3 According to the Biopharmaceutics Classification System (BCS),4 ABZ is a class II drug of low solubility (9 mg L−1) and high permeability (logP 2.54), representing a dose number of 160 (which means that a single tablet of albendazole will fully dissolve when taken with 160 glasses of 250 mL water).5 Solid dispersions have been reported to improve the solubility of ABZ, such as polyvinyl pyrrolidone6 and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) complexes.7,8 There are no cocrystals or salts of ABZ reported in the published literature even though it is an essential drug. Two polymorphs, form I and II, of albendazole are reported as tautomers (CSD Refcode SUTWIO, BOGFUZ). There is positional disorder in the thiopropyl chain and tautomerism in these crystal structures. However, a crystal structure without disorder in the S-C3H7 chain is not reported.9,10 The positional disorder of the alkyl chain is solved in the crystal structures of the ABZ salts reported in this paper.
2. Experimental
Crystallization of salts
ABZ–BSA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) salt.
100.00 mg (0.377 mmol) of ABZ and 59.56 mg (0.377 mmol) BSA were ground in a mortar with a pestle for 30 min after adding 5 drops of nitromethane. The binary crystalline powder was dissolved in CH3NO2 solvent under hot conditions, filtered to remove the undissolved particles and then allowed to crystallize at room temperature. Plate morphology crystals were harvested under ambient conditions after 7 days. Anhydrous ABZ–BSA (1:1) salt was further prepared in bulk by nitromethane solvent fast evaporation using a rotary evaporator. Majority of the slow evaporation crystallization experiments in CH3NO2 resulted in a mixture of salt and salt hydrate, and the crystals are of different morphologies.
:1) salt.
100.00 mg (0.377 mmol) of ABZ and 59.56 mg (0.377 mmol) BSA were ground in a mortar with a pestle for 30 min after adding 5 drops of nitromethane. The binary crystalline powder was dissolved in CH3NO2 solvent under hot conditions, filtered to remove the undissolved particles and then allowed to crystallize at room temperature. Plate morphology crystals were harvested under ambient conditions after 7 days. Anhydrous ABZ–BSA (1:1) salt was further prepared in bulk by nitromethane solvent fast evaporation using a rotary evaporator. Majority of the slow evaporation crystallization experiments in CH3NO2 resulted in a mixture of salt and salt hydrate, and the crystals are of different morphologies.
ABZ–BSA–H (1:1:1) salt hydrate.
100.00 mg (0.377 mmol) of ABZ and 59.56 mg (0.377 mmol) of BSA were ground in a mortar with a pestle for 30 min after adding 5 drops of water, and then kept for crystallization in a solvent mixture of MeOH and water (1:1 v/v) at room temperature. Plate shaped crystals were harvested under ambient conditions after 7 days.
ABZ–PTSA (1:1) salt.
100.00 mg (0.377 mmol) of ABZ and 64.70 mg (0.377 mmol) of PTSA were ground in a mortar with a pestle for 30 min after adding 5 drops of MeOH, and then kept for crystallization in a solvent mixture of 5 mL of MeOH and nitromethane (1:1 v/v) at room temperature. Plate shaped crystals were harvested under ambient conditions after 3–4 days. The anhydrous material ABZ–PTSA (1:1) salt was further prepared in bulk by nitromethane solvent fast evaporation in a rotary evaporator.
ABZ–PTSAH (1:1:1) salt.
100.00 mg (0.377 mmol) of ABZ and 64.70 mg (0.377 mmol) of PTSA were ground in a mortar with a pestle for 30 min after adding 5 drops of water, and then kept for crystallization in a solvent mixture of 5 mL of MeOH and water (1:1 v/v) at room temperature. Plate shaped crystals were harvested under ambient conditions after 7 days.
ABZ–2,6-DHBA (1:1) salt.
ABZ, 100.00 mg (0.377 mmol), and 2,6-DHBA, 57.90 mg (0.377 mmol), were ground in a mortar with a pestle with a few drops of MeOH added for 30 min and crystallized in MeOH and nitromethane solvents. IR and PXRD results of the new solid material were different from those of the initial components.
ABZ–2,4,6-THBA (1:1) salt.
ABZ, 100.00 mg (0.377 mmol), and 2,4,6 THBA, 62.94 mg (0.377 mmol), were ground in a mortar with a pestle with a few drops of MeOH added for 30 min. Attempts to grow single crystals afforded a precipitate. The IR and PXRD results of the new solid material were different from those of the starting components.
ABZ–MLE (1:1) salt.
ABZ, 100.00 mg (0.377 mmol), and MLE, 43.77 mg (0.377 mmol), were ground in a mortar with a –pestle with a few drops of MeOH added for 30 min. The IR and PXRD results of the new solid material were different from those of the initial components. Attempts to grow single crystals were not successful.
ABZ–LTA (1:1) salt.
ABZ, 100.00 mg (0.377 mmol), and D-LTA, 56.55 mg (0.377 mmol), were ground in a mortar with a pestle with a few drops of MeOH added for 30 min. The IR and PXRD results of the new solid material were different from those of the initial components. Attempts to grow single crystals resulted in a precipitate.
Single crystal X-ray diffraction
Single crystal X-ray diffraction was carried out at 298 or 100 K on a Bruker SMART APEX-1 CCD area detector system equipped with a graphite monochromator and a Mo-Kα fine focus sealed tube (λ = 0.71073 Å) operated at 1500 W power (40 kV, 30 mA). A total of 2400 frames were collected at a scan width of 0.3° in the ω mode and an exposure time of 12 s per frame. The frames were integrated with the Bruker SAINT-Plus software using a narrow-frame integration algorithm, and the data was corrected for absorption effects using the multi-scan method (SADABS); further structures were solved and refined using the Bruker SHELX-TL (ref. 11) software. A check of the final crystallographic information file (.cif) was performed using PLATON.12 A few of the single crystals were mounted on an Oxford Diffraction Gemini X-ray diffractometer (Mo-Kα source, λ = 0.71013 Å) at 298 K. Data reduction was performed using CrysAlisPro 171.33.55 software. The X-Seed program13 and Mercury 3.1 (ref. 14) was used to prepare the final figures and packing diagrams. The crystallographic parameters of both crystal structures are summarized in Table 1. The hydrogen atoms on the heteroatoms (O–H, N–H) were located in the Fourier difference electron density maps for each crystal structure. The hydrogen bond distances (Table S1†) were neutron-normalized to fix the D–H distance to its accurate neutron value in the X-ray crystal structure. Cif files were deposited at the Cambridge Crystallographic Data Centre, CCDC No. 1834924–1834929.
Table 1 Crystallographic parameters of the ABZ new solid phases in this study
|
|
ABZ–BSA (2:2) |
ABZ–BSAH (1:1:1) |
ABZ–PTSA (1:1) |
ABZ–PTSAH (1:1:1) |
ABZ–OAH (1:1:1) |
ABZ–2,6-DHBA (2:2) |
| CCDC no. |
1834929
|
1834924
|
1834925
|
1834926
|
1834927
|
1834928
|
| Empirical formula |
C12H16N3O2S, C6H5O3S |
C12H16N3O2S, C6H5O3S, H2O |
C12H16N3O2S, C7H7O3S |
C12H16N3O2S, C7H7O3S, H2O |
C12H12N3O2S, C2HO4, H2O |
C12H8N3O2S, C7H6O4 |
| Formula weight |
423.50 |
441.50 |
437.52 |
455.54 |
369.35 |
818.74 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n |
P |
P21/n |
P21/n |
|
T (K) |
298 |
298 |
298 |
100 |
100 |
100 |
|
a (Å) |
8.5042(7) |
8.9020(6) |
15.2992(17) |
8.900(5) |
5.2659(7) |
17.176(5) |
|
b (Å) |
9.4678(7) |
9.2715(8) |
9.7420(9) |
9.573(5) |
28.337(4) |
9.293(3) |
|
c (Å) |
26.084(2) |
14.2635(12) |
15.8825(14) |
14.453(8) |
11.202(15) |
25.789(8) |
|
α (°) |
96 |
84 |
90.00 |
83 |
90 |
90.00 |
|
β (°) |
93 |
89 |
115 |
72 |
93 |
105 |
|
γ (°) |
102 |
63 |
90.00 |
63 |
90 |
90.00 |
|
V (Å3) |
2031.1(3) |
1053.47(15) |
2135.3(4) |
1047.3(10) |
1667.8(4) |
3958(2) |
|
D
calcd (g cm−3) |
1.385 |
1.392 |
1.361 |
1.445 |
1.471 |
1.374 |
|
μ (mm−1) |
0.296 |
0.292 |
0.284 |
0.296 |
0.237 |
0.204 |
|
θ range |
2.66 to 24.71 |
2.73 to 24.71 |
2.83 to 26.31 |
2.68 to 28.31 |
2.32 to 26.05 |
1.28 to 24.71 |
|
Z/Z'
|
2 |
1 |
1 |
1 |
1 |
2 |
| Range h |
−9 to +9 |
−10 to +10 |
−16 to +17 |
−11 to +11 |
−6 to 6 |
−20 to +20 |
| Range k |
−11 to +11 |
−10 to +9 |
−11 to +6 |
−11 to +10 |
−33 to 33 |
−10 to +10 |
| Range l |
−30 to +30 |
−16 to +15 |
−18 to +15 |
−18 to +18 |
−13 to +13 |
−30 to +30 |
| Reflections collected |
13242 |
6424 |
8071 |
10722 |
15227 |
35767 |
| Total reflections |
3638 |
4246 |
6906 |
3587 |
2841 |
6738 |
|
R
1 [I > 2σ(I)] |
0.0740 |
0.0694 |
0.0844 |
0.0639 |
0.0640 |
0.0778 |
| wR2 (all) |
0.2090 |
0.1945 |
0.2722 |
0.1748 |
0.1599 |
0.2216 |
| Goodness of fit |
1.090 |
1.046 |
0.991 |
1.017 |
1.326 |
1.100 |
| Diffractometer |
Oxford CCD |
Smart Bruker Apex-I |
Oxford CCD |
Smart Bruker Apex-I |
Smart Bruker Apex-I |
Smart Bruker Apex-I |
Powder X-ray diffraction
Bulk samples were analyzed by powder X-ray diffraction using a Bruker AXS D8 diffractometer (Bruker-AXS, Karlsruhe, Germany). Experimental conditions: Cu-Kα radiation (λ = 1.5418 Å), 40 kV, 30 mA, scanning interval 5–50° 2θ at a scan rate of 1° min−1, time per step 0.5 s.
Solid-state NMR spectroscopy
Solid-state 15N NMR spectra were recorded on a Bruker Ultra shield 400 spectrometer (Bruker BioSpin, Karlsruhe, Germany). A cross-polarization, magic angle spinning (CP-MAS) pulse sequence was used for spectral acquisition. Each sample was spun at a frequency of 5.0 ± 0.01 kHz, and the magic angle setting was calibrated by the KBr method. Approximately 100 mg of the fine crystalline sample was tightly packed into a zirconia rotor with the help of a Teflon stick up to the Kel-F cap mark. Each data set was subjected to a 5.0 Hz line broadening factor and subsequently Fourier transformed and phase corrected to produce a frequency domain spectrum. 15N CP-MAS spectra were recorded at 40 MHz and referenced to glycine N, and then the chemical shifts were recalculated to nitromethane (δglycine = −347.6 ppm).
Thermal analysis
DSC experiments were performed using a Mettler Toledo DSC 822e module. Samples were placed in vented aluminum sample pans for DSC. A typical sample size is 4–6 mg for DSC. The temperature range was 30–300 °C at 5 K min−1 for DSC. The samples were purged with a stream of dry N2 flowing at 80 mL min−1.
Vibrational spectroscopy
A Thermo-Nicolet 6700 FT-IR spectrometer (Waltham, MA, USA) was used to record the IR spectra. The IR spectra were recorded for the samples dispersed in KBr pellets.
3. Results and discussion
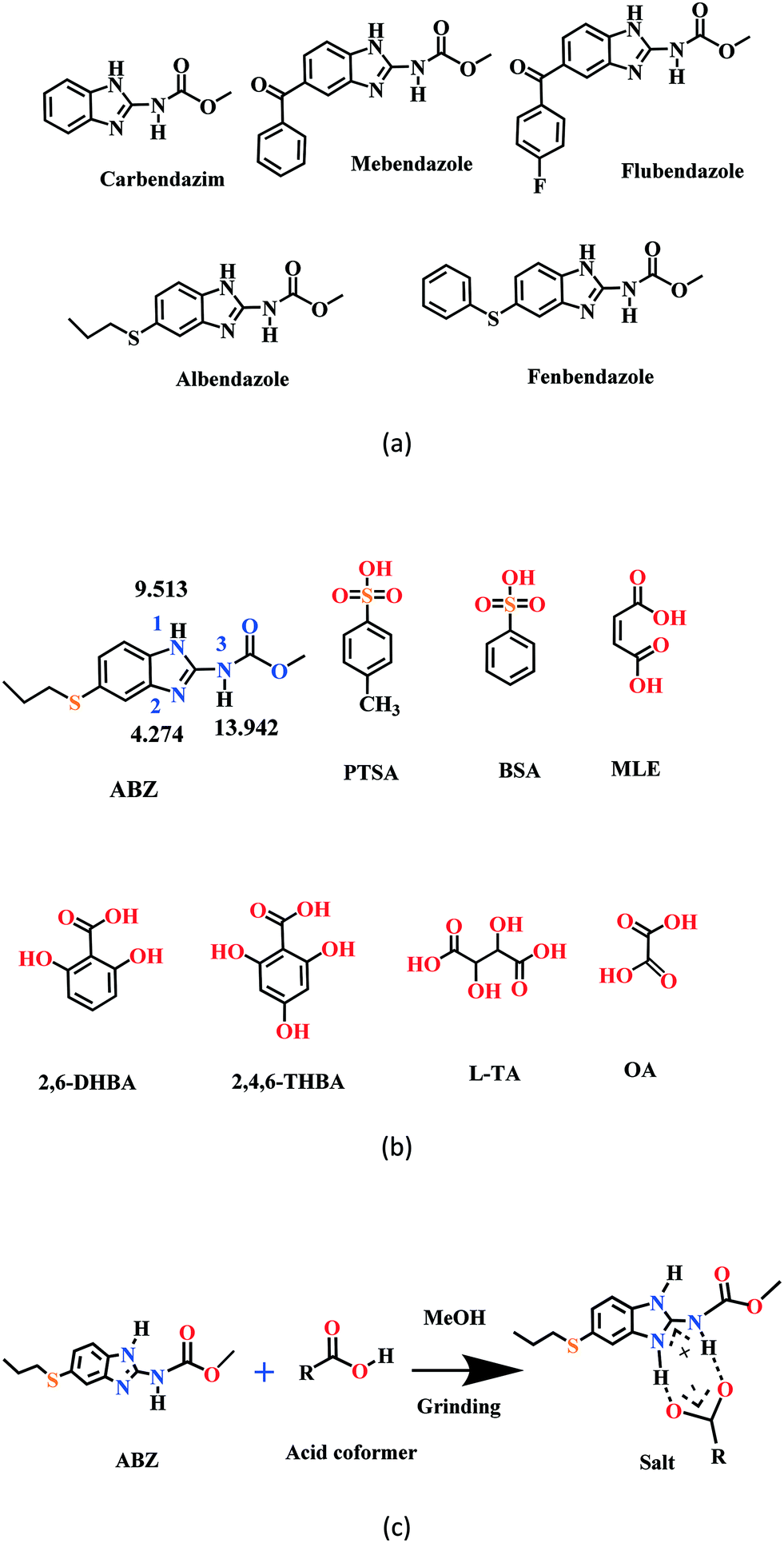
New solid phases of ABZ with different sulfonic acids such as benzenesulfonic acid (BSA) and p-toluenesulfonic acid (PTSA) and carboxylic acids such as oxalic acid (OA), 2,6-dihydroxybenzoic acid (2,6-DHBA), 2,4,6-trihydroxybenzoic acid (2,4,6-THBA), maleic acid (MLE) and L-tartaric acid (LTA) were prepared and characterized by IR, PXRD and DSC. The products with BSA, PTSA, OA, and 2,6-DHBA were determined to be salts, confirmed by single crystal X-ray diffraction. Good diffraction quality crystals could not be harvested for ABZ–2,4,6-THBA, ABZ–MLE, and ABZ–LTA, therefore, these phases were confirmed as salts by the chemical shift in their solid-state 15N NMR spectra. A few benzimidazole drugs are shown in Scheme 1a, and the structures of the coformers used in this study are shown in Scheme 1b. The supramolecular synthon in albendazole salts with proton transfer from the carboxylic acid group to the basic benzimidazole nitrogen of the R22(8) motif15–17 is shown in Scheme 1c. The sulfonate salts crystalize as anhydrates and hydrates (designated as –H in name abbreviations) during the solution crystallization experiments (Experimental section). Attempts to prepare an anhydrate salt of BSA was successful the first time, subsequent experiments gave the hydrate. For the carboxylic acid coformers, OA gave a salt hydrate exclusively and DHBA an anhydrate salt. Salts of mebendazole with carboxylic acids and sulfonic acids have been reported.18–20 Similarly, a flubendazole salt with maleic acid was published recently.21 Even though there are related drugs listed in Scheme 1a from literature reports, the crystal structures of albendazole salts are new observations in present study.
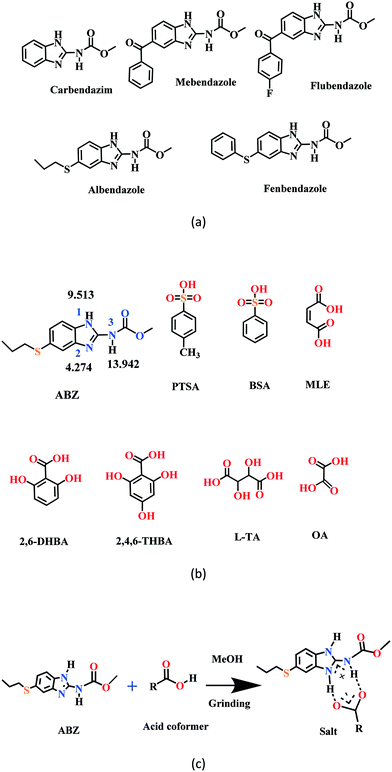 |
| | Scheme 1 (a) A few benzimidazole drugs. (b) Chemical structures of ABZ (along with their N pKa values, will be discussed in later sections) and the coformers reported in this study. (c) Ionic synthon of ABZ with carboxylic acid in an R22(8) motif. | |
Crystal structure analysis
The crystal structures of the two reported polymorphs of albendazole are discussed first.9 The ABZ molecules form a N–H⋯N dimer synthon in an R22(6) motif and extend via N–H⋯O hydrogen bonding to the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in both crystal structures. The pseudo-symmetry in the molecule due to the imidazole ring results in the disorder of the S–propyl side chain, and the two structures differ in the nature of their side chain disorder (Fig. 1). The two polymorphs are a result of benzimidazole tautomerism, reported for omeprazole polymorphs about a decade ago.22,23 The hydrogen bonds are stronger in the salts and cocrystals of ABZ (Table S1 for the crystallographic data and Table S2 for the hydrogen bonding details, see the ESI†).
O in both crystal structures. The pseudo-symmetry in the molecule due to the imidazole ring results in the disorder of the S–propyl side chain, and the two structures differ in the nature of their side chain disorder (Fig. 1). The two polymorphs are a result of benzimidazole tautomerism, reported for omeprazole polymorphs about a decade ago.22,23 The hydrogen bonds are stronger in the salts and cocrystals of ABZ (Table S1 for the crystallographic data and Table S2 for the hydrogen bonding details, see the ESI†).
 |
| | Fig. 1 ABZ crystal structures reported in the literature. ABZ exhibits sulphur attached alkyl disorder and the imidazole rings show tautomerism labelled as (a) form II (ref. 9) and (b) form I.10 CSD ref codes SUTWIO, BOGFUZ. | |
ABZ sulfonate salts
ABZ–BSA (1:1) salt.
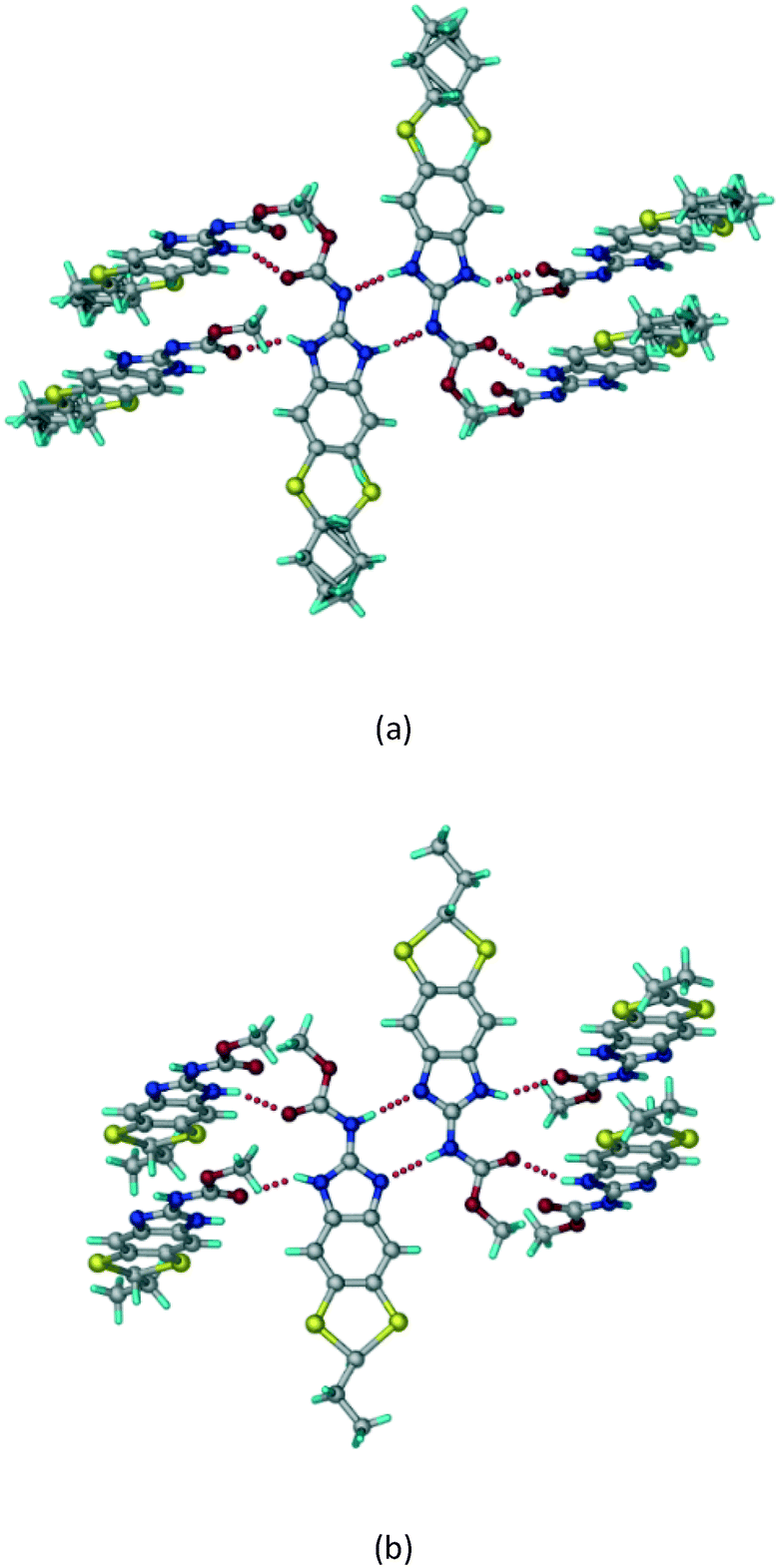
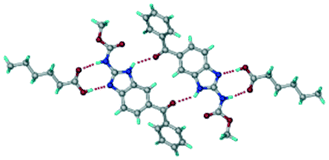
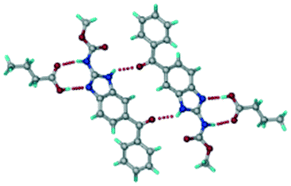
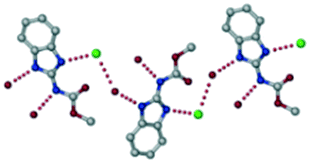
A salt of ABZ and benzenesulfonic acid (triclinic P space group) was crystallized with the proton transfer between BSA and the imidazole nitrogen resulting in an R22(8) ring motif.15–17 During the routine solution crystallization experiments in nitromethane, EtOAc, and CH3CN solvents, the anhydrate and hydrate salts were observed concomitantly. The polymorphs were distinguished by their crystal morphology as plates and rods, which were hand-picked for crystal data collection. The asymmetric unit contains two molecules of ABZ and BSA, and such salt aggregates are connected via C–H⋯O (C21–H21A⋯O1 3.139 Å; C9–H9B⋯O3, 3.164 Å) interactions of the ester dimer R22(10) ring (Fig. 2a). The complete molecular packing is shown in Fig. 2b.
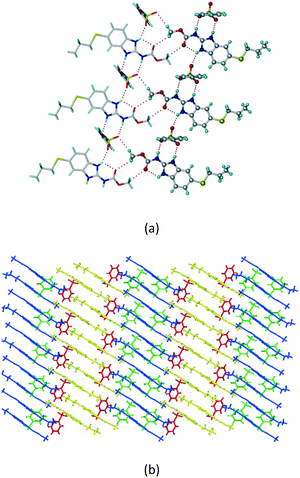 |
| | Fig. 2 (a) Ionic synthon of sulfonate–imidazole and ester dimers along the b-axis. (b) Molecular packing in the ABZ–BSA structure. Symmetry-independent molecules are shown by different shades. | |
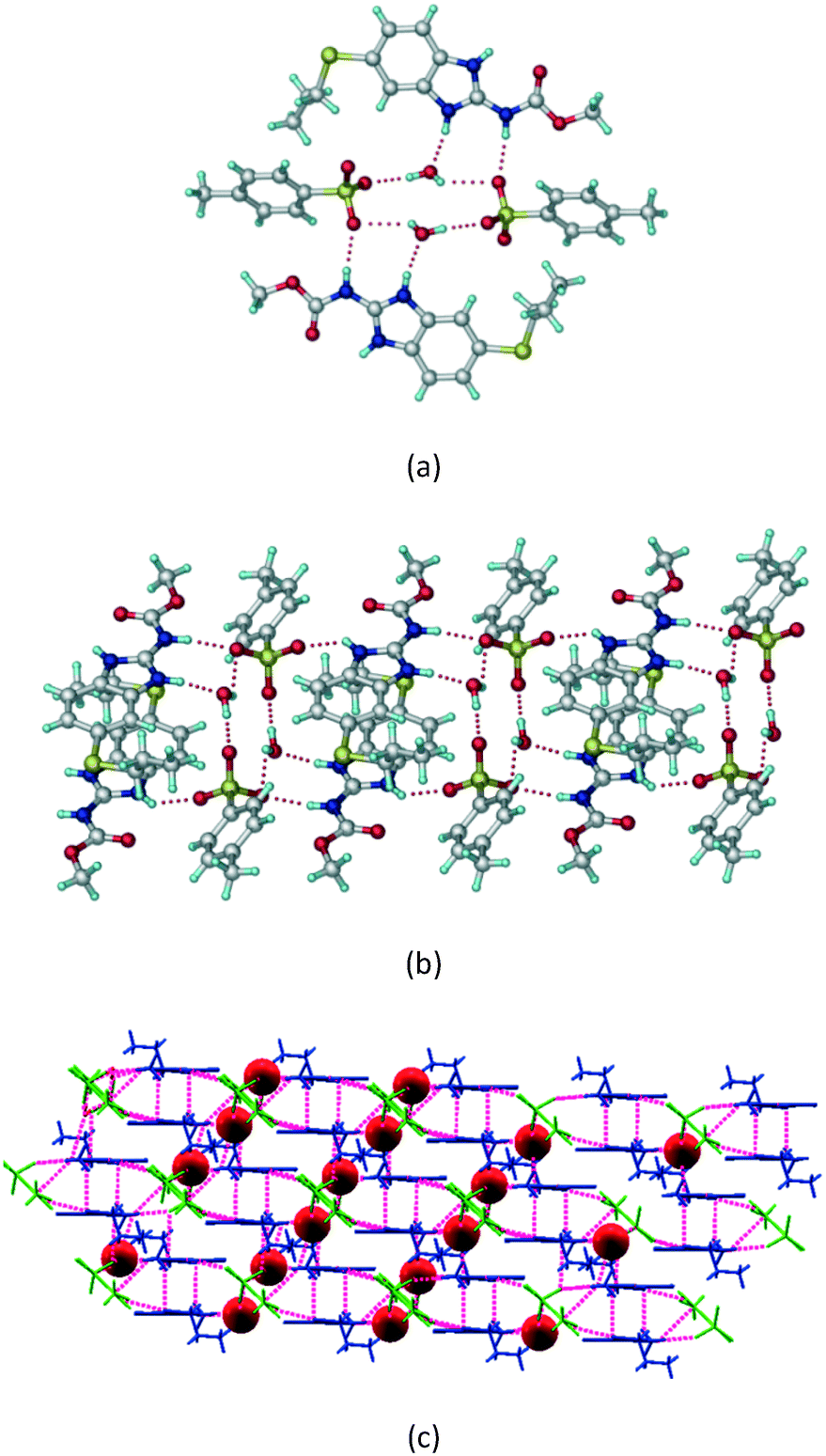
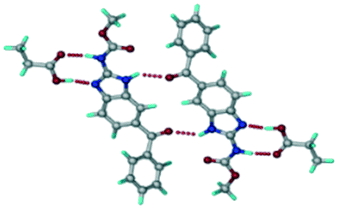
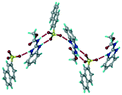
ABZ–BSA–H (1:1:1) salt hydrate.
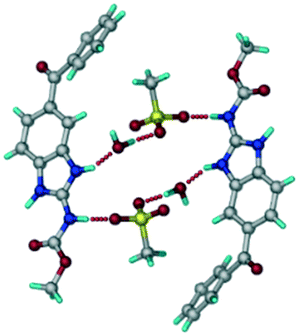
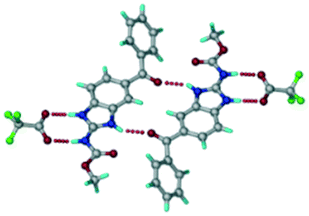
The ABZ–BSA salt crystallization in alcoholic solvents afforded a hydrate, whereas the crystallization in a CH3NO2–EtOAc (1:1) solvent mixture resulted in crystals of both the salt and its hydrate. ABZ–BSA–H (1:1:1) crystallized in the triclinic space group P. Water molecules act as a bridge between the ABZ and BSA molecules via N–H⋯O− (N3–H3A⋯O3, 2.798(4) Å, 173°), N–H⋯O (N1–H1A⋯O6, 2.687(4) Å, 169°) and O–H⋯O− (O6–H6A⋯O4, 2.686(6) Å, 162°; O6–H6B⋯O3, 2.834(4) Å 134°) bonds affording a network of R23(8)R44(12)R23(8) ring motifs (Fig. 3a). BSA and water ring motifs are sandwiched between the ABZ molecules and connected via N+–H⋯O− hydrogen bonds (Fig. 3b). The schematic packing with water shown as red spheres is shown in Fig. 3c.
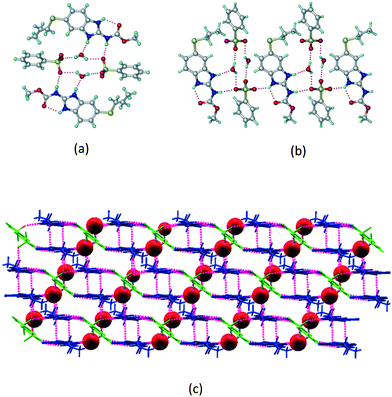 |
| | Fig. 3 (a) The bridging water molecules act as proton donors for the sulfonate and acceptors for N+–H resulting in the ring network R23(8)R44(12)R23(8). (b) The sulfonate–water molecules are bonded via N–H⋯O− hydrogen bonds to ABZ. (c) Complete packing of cations, anions and water molecules. | |
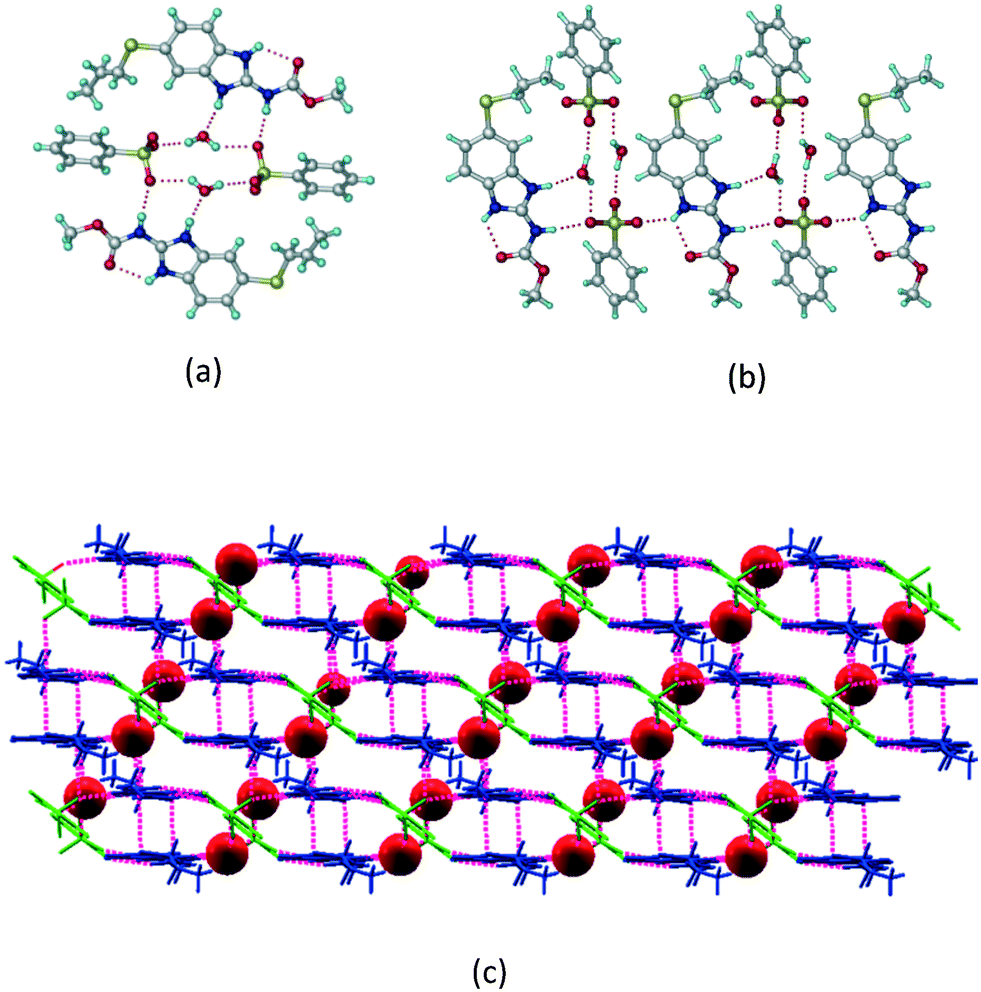
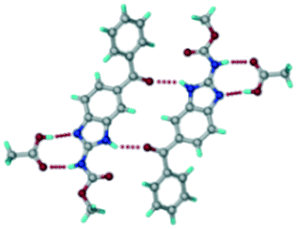
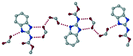
ABZ–PTSA (1:1) salt.
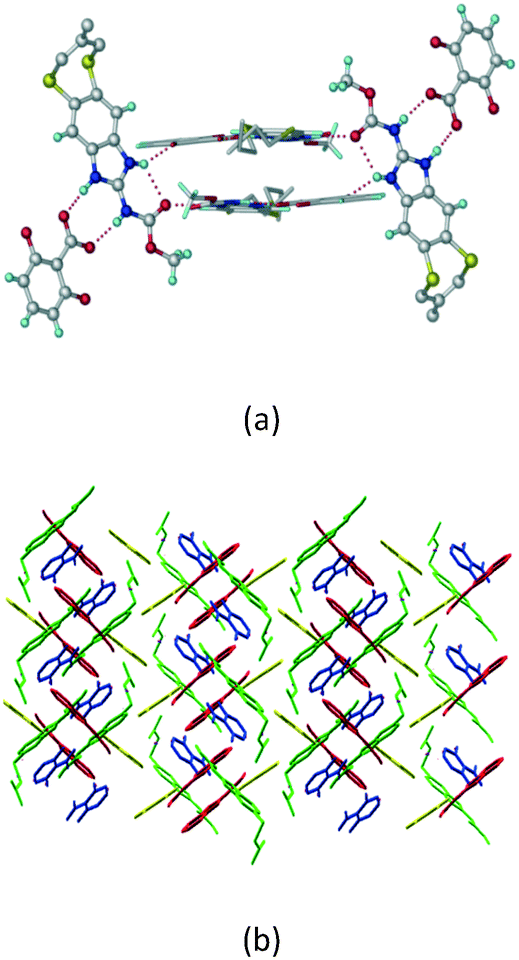
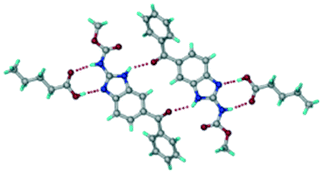
Crystals of the ABZ–PTSA (1:1) salt were obtained in nitromethane/EtOAc solvent (1:1 v/v) in the space group P21/n. The proton transfer from PTSA to the imidazole nitrogen atom of ABZ results in an R22(8) ring motif via imidazole N–H⋯O− (N1–H1A⋯O4, 2.763(5) Å, 169°) and amide N–H⋯O− (N3–H3A⋯O3, 2.703(5) Å, 164°) bonding with the sulfonate oxygen atoms of PTSA (Fig. 4a). The dimers of these units extend along the b-axis as chains. The imidazolium N+–H donor and O− acceptor of PTSA form an R22(8) motif with bifurcated (N2–H2A⋯O5, 2.820(5) Å, 146°) hydrogen bonds (Fig. 4b). Additionally, these binary adducts make 1D chains which extend through C–H⋯O− (C10–H10B⋯O3, 3.309(9) Å, 142°) and C–H⋯π (C9–H9A⋯π of PTSA, 2.932 Å; C18–H18⋯π of ABZ, 3.262 Å) interactions (Fig. 4c).
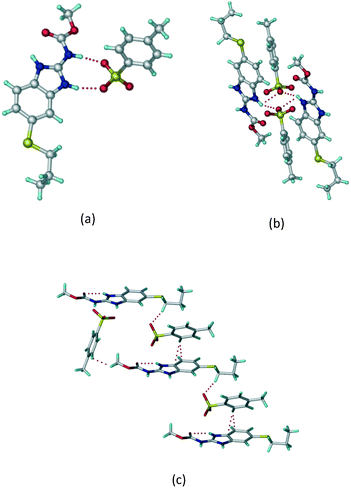 |
| | Fig. 4 (a) Proton transfer from p-toluenesulfonic acid to the ABZ secondary imidazole N atom results in an R22(8) ring motif in the ABZ–PTSA (1:1) salt. (b) The view of the R22(8) ring motif present in ABZ–PTSA. (c) Bimolecular units of ABZ–PTSA extended through N+–H⋯O− interactions along the b-axis. | |
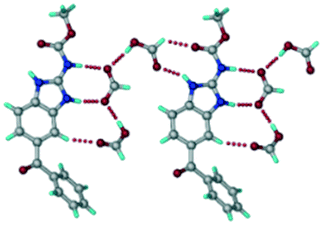
ABZ–PTSA–H (1:1:1) salt hydrate.
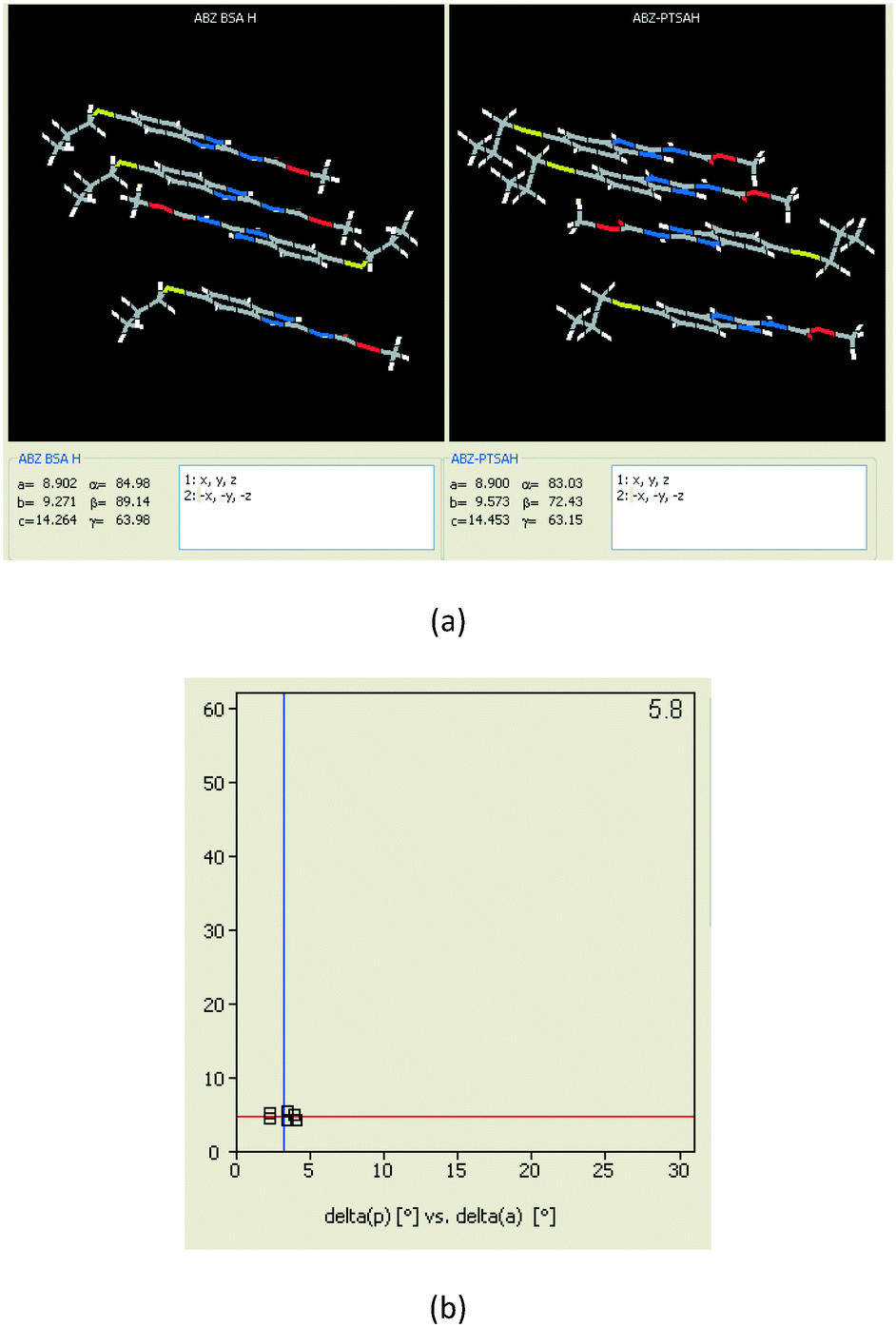
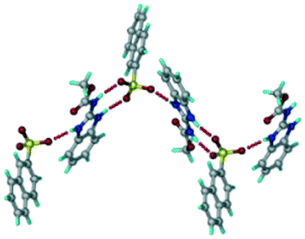
In addition to ABZ–PTSA, the salt hydrate ABZ–PTSA–H (1:1:1) crystallized (P space group) after a few crystallization cycles. The proton is transferred from PTSA to the imidazole nitrogen of ABZ (Fig. 5a). The hydrogen bonding in this structure is similar to that of the ABZ–BSA–H salt and also showed 2D isostructurality (Fig. 8). The calculated dissimilarity index of 5.8 confirms the 2D iso-structurality24,25 between them. The water molecule acts as a bridge between the ABZ and PTSA molecules via N–H⋯O− (N3–H3A⋯O4, 2.865(4) Å, 175°); N–H⋯O (N1–H1A⋯O6, 2.665(4) Å, 174°) and O–H⋯O− (O6–H6A⋯O3, 2.678(4) Å, 141°; O6–H6B⋯O4, 2.747(3) Å 145°) hydrogen bonds in the R23(8)R44(12)R23(8) motif. The oxygen of PTSA is connected through N+–H⋯O− (N2–H2A⋯O5 2.790(4) Å, 142°) H-bonds (Fig. 5b). Such motifs extend along the b-axis in a double helical structure and helical sheets (Fig. 5c).
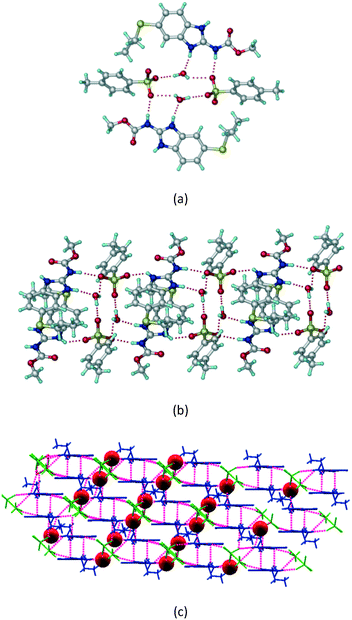 |
| | Fig. 5 (a) Bridging water molecules between ABZ and PTSA result in a basic ring unit. (b) The basic motifs are extended via N+–H⋯O− hydrogen bonds along the a-axis. (c) 2D packing of the ABZ–PTSAH basic ring motifs resulted in a stacked wave nature as that of ABZ–BSAH. | |
ABZ carboxylate salts
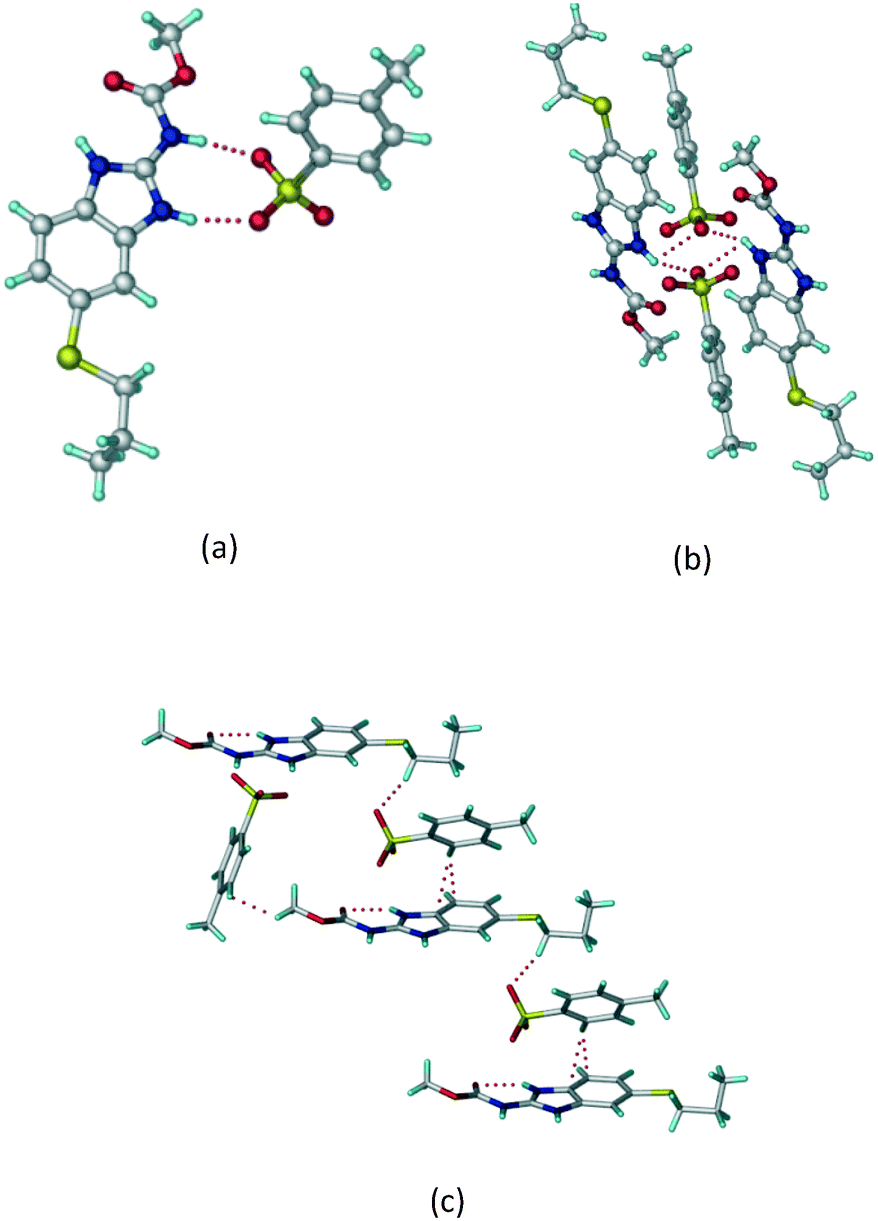
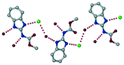
ABZ–OA–H (1:1:1) salt hydrate.
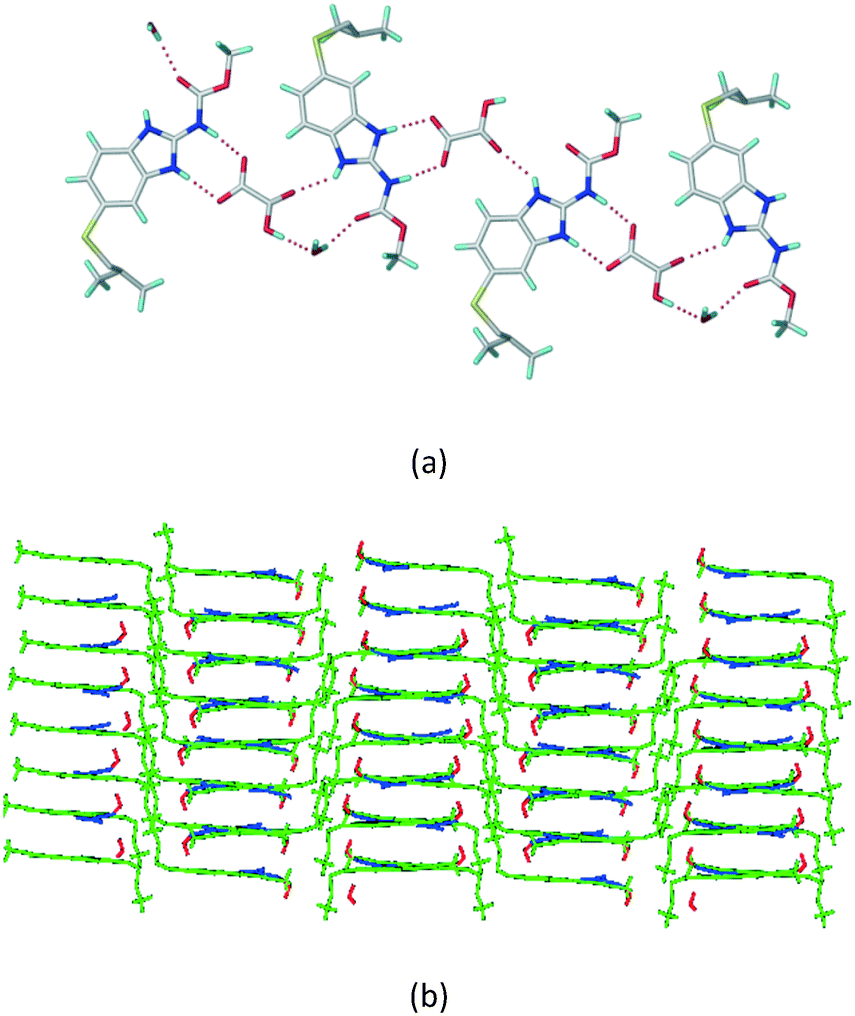
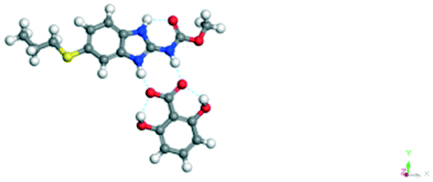
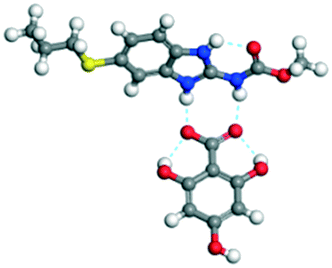
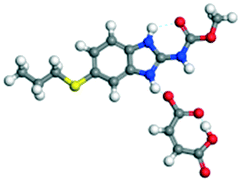
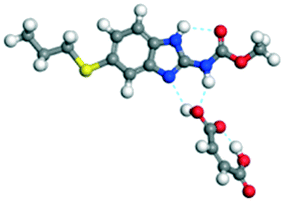
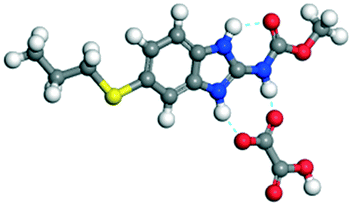
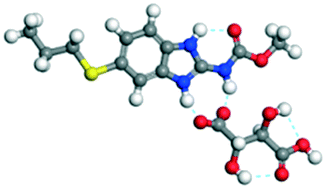
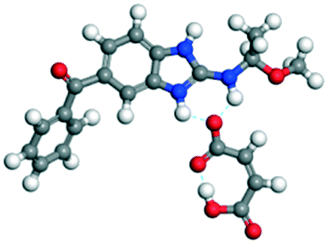
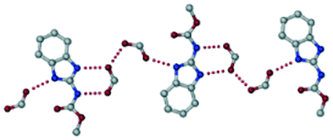
The dimers of ABZ are interrupted by oxalic acid coformers (P21/n space group). One of the OA protons is transferred to ABZ resulting in ABZ–OA–H, and the second COOH of OA is neutral. Heterodimers of the ABZ and OA are assembled via N–H⋯O− and N–H⋯O (N1–H1A⋯O4, 2.671(4) Å, 171°; N3–H3A⋯O3, 2.689(4) Å, 174°) hydrogen bonds in the R22(8) motif (Fig. 6a). The neutral COOH of the OA bonds with the second N–H of ABZ and water molecules through N+–H⋯O− and O–H⋯O− (N2–H2A⋯O5, 2.786(4) Å, 139°; O7–H7B⋯O1, 2.859(4) Å, 154°) hydrogen bonds (Fig. 6b).
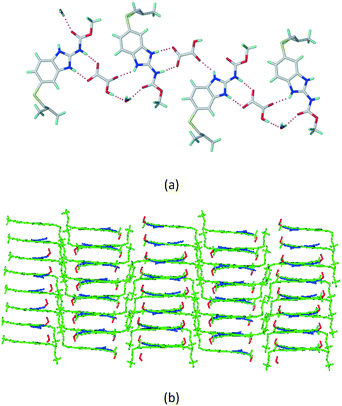 |
| | Fig. 6 (a) The carboxylate ion of OA forms a heterodimer with a protonated ABZ 1,3 di N–H moiety resulting in R22(8) motifs. (b) 2D packing of ABZ–OA–H results in a successive ladder type of packing. | |
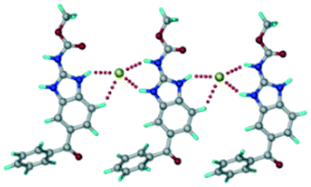
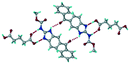
ABZ–2,6-DHBA (1:1) salt.
Cations of ABZ assemble with the carboxylate of 2,6-DHBA by N–H⋯O− (N3–H3A⋯O−4, 2.649(5) Å, 173°; N1–H1A⋯O−6, 2.703(4) Å, 175°; N5–H5A⋯O−11, 2.696(5) Å, 179°; N6–H6A⋯O12 2.680(5) Å, 169°) hydrogen bonds forming an R22(8) motif involving two molecules each in the asymmetric unit (space group P21/n) (Fig. 7a, shaded differently). The molecular planes are inclined at 77.9°, and such units extend via N+–H⋯O hydrogen bonds. 1D chains extend via weak C–H⋯π interactions to construct 2D sheet structures. The sulfur atom and n-butyl chains in the two molecules are split and modelled using the PART command. The hydrogen atoms attached to the alkyl chain are disordered.
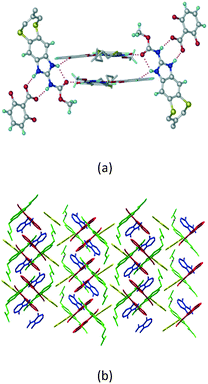 |
| | Fig. 7 (a) The basic unit present in the ABZ–2,6-DHBA salt in a 1:1 ratio with two molecules in the asymmetric unit. (b) 2D packing of the salt shows adjacent helical nature packing (H atoms are removed for clarity). | |
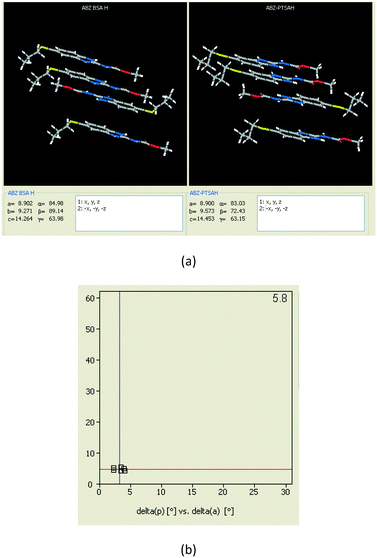 |
| | Fig. 8 (a) The XPac analysis of ABZ–BSA–H and ABZ–PTSA–H shows that four molecules are identically arranged in the supramolecular construct. (b) The interplanar angular deviation (δp, x-axis) vs. angular deviation (δa, y-axis) plot (in °) indicates a dissimilarity index of 5.8, which confirmed the 2D isostructurality of the two salt hydrates. | |
IR spectroscopy
The infrared spectroscopy26,27 results of ABZ showed characteristic peaks of an ester carbonyl (1712.3 cm−1) and a N–H stretch (3328.0 cm−1). The new solid phases ABZ–MA, ABZ–LTA, ABZ–2,6 DHBA, ABZ–2,4,6 THBA, ABZ–BSA, ABZ–PTSA and ABZ–PTSA–H exhibited a shift in their IR bands when compared to the starting materials. The red shift in the CO and N–H frequency peaks indicates stronger hydrogen bonding (spectra are shown in Fig. S1, ESI† and values are listed in Table 2). Chen et al.18 reported an MEB–MLE salt peak at 1745 cm−1 which is comparable with that of the ABZ–MLE adduct at 1746 cm−1.
Table 2 FT-IR stretching frequencies (νs, cm−1) of the ABZ salts
| Crystalline forms |
CO (ester carbonyl of ABZ) cm−1 |
N–H (Br) cm−1 |
O–H (Br) for coformer cm−1 |
Carboxylate/sulfonate (asym) cm−1 |
| ABZ |
1712.3 |
3328.0 |
— |
— |
| ABZ–BSA–H (1:1:1) |
1751.2 |
3259.7 |
3374.7 |
1250.5, 1127.5 |
| ABZ–PTSA (1:1) |
1762.1 |
3453.9 |
3397.8 |
1245.7, 1200.7 |
| ABZ–PTSA–H (1:1:1) |
1754.5 |
3458.1 |
3397.8 |
1232.5, 1181.8 |
| ABZ–OA–H (1:1:1) |
1751.3 |
3432.7 |
3439.8 |
1248.0 |
| ABZ–2,6-DHBA (1:1) |
1745.5 |
3467.2 |
3475.5 |
1253.3 |
| ABZ–2,4,6-THBA (1:1) |
1730.8 |
3545.1 |
3406.8 |
1144.0 |
| ABZ–MLE (1:1) |
1746.0 |
— |
— |
1261.3 |
| ABZ–LTA (1:1) |
1751.2 |
3319.3, 3264.5 |
3402.0 |
1244.0 |
Powder X-ray diffraction
Powder X-ray diffraction28–30 is the most frequently used technique to establish the formation of new crystalline materials. Quick comparison of the fingerprints of the product phase (cocrystal/salt) and the starting material peaks (drug, coformer) is done by visual inspection of the diffraction line patterns. The powder pattern of ABZ, the ground material with coformers, and the coformer plots indicate new sets of peaks. The bulk phase powder patterns of ABZ–BSA–H, ABZ–PTSA, ABZ–PTSA–H, ABZ–OA–H and ABZ–2,6-DHBA matches those of the calculated X-ray lines for the salts from the crystal structures (Fig. S2a–e, ESI†). A PXRD comparison of the ground material diffraction pattern of ABZ–2,4,6-THBA, ABZ–MLE and ABZ–LTA showed unique peaks at 2θ values which confirmed the new crystalline phases (Fig. S2f–h, ESI†).
Thermal analysis
A different melting point of the cocrystal/salt in the DSC thermogram is indicative of a new phase. The ABZ commercial sample used in the experiments melts at 198–202 °C; the DSC endotherms of the ABZ salts are shown in Fig. S3, ESI.† The melting points of the products along with their coformers are listed in Table 3. ABZ–MLE and ABZ–PTSA showed melting points in between those of their two components, whereas ABZ–LTA, ABZ–2,6-DHBA, and ABZ–2,4,6-THBA melted below their components' melting temperature.
Table 3 Melting points of the ABZ salts from the DSC thermograms
| Crystalline form |
m.p. (°C) of ABZ/coformer |
m.p. (°C) of salt |
| ABZ |
198–202 |
— |
| ABZ–BSA (1:1) |
103–106 |
159–170 |
| ABZ–BSA–H (1:1:1) |
103–106 |
83–92, 167–173 |
| ABZ–PTSA (1:1) |
103–106 |
159–170 |
| ABZ–PTSA–H (1:1:1) |
103–106 |
83–92, 167–173 |
| ABZ–OA–H (1:1:1) |
|
57–60, 141–146 |
| ABZ–2,6-DHBA (1:1) |
200–205 |
178–182 |
| ABZ–2,4,6-THBA (1:1) |
210–213 |
144–148 |
| ABZ–MLE (1:1) |
135–137 |
145–147 |
| ABZ–LTA (1:1) |
206–209 |
170–173 |
Solid state NMR (ss-NMR)
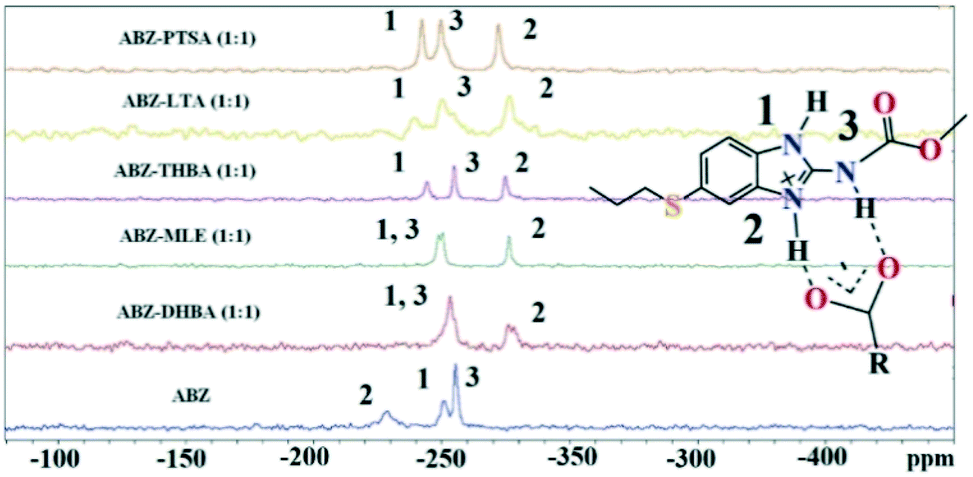
Solid state NMR of the polymorphs and cocrystals31–34 provides information about the short range order and hydrogen bonding. The positions of the hydrogen atoms in the crystalline molecular compounds exhibiting partial charge transfer are difficult to locate in the absence of X-ray diffraction. It is more complicated if the crystal structures of the organic solids are not available. The difficulty of growing good quality single crystals leads to the situation that 15N ss-NMR becomes an alternative choice to better understand their structure, even though neutron diffraction can produce more precise data, it is not routinely accessible using a laboratory diffractometer. However, 15N ss-NMR is challenging because the 15N isotope has a low natural abundance (0.368%). The difference in the chemical shifts (Δppm) of 15N is correlated with those of the salts to assess the hydrogen bond strength. Li et al. showed that 15N ss-NMR could be used for monitoring a cancer drug in dicarboxylic cocrystals and salts. Those reported results ranged from no transfer to partial transfer to complete transfer of the proton on the aromatic N of ABZ and ss-NMR was used to compare the single crystal structures.34 In addition, they showed that the N–H and N⋯O distances are a function of the difference in their 15N ss-NMR chemical shifts, Δppm of −20 to −60 for no proton transfer and −60 to −120 for proton transfer. The neutral N–H of imidazole was assigned as atom 1, protonated N–H as 2 and the carbamate N as atom 3 (Fig. 9, Table 4). 15N ss-NMR of the ABZ–PTSA salt (for which the single crystal data are available to confirm the salt state) and ABZ–LTA, ABZ–MLE, and ABZ–2,4,6-THBA (single crystal data could not be recorded) allowed the salt structure of ABZ to be assigned based on the chemical shift of the 15N (protonated) peak, which was at −274.14 ppm for ABZ–2,6-DHBA, −274.97 ppm for ABZ–2,4,6-THBA, −276.38 ppm for ABZ–MLE, −277.99 ppm for ABZ–LTA, and −275.75 ppm for ABZ–PTSA (for full chemical shift values see Table 4, −228.96 ppm for ABZ). N2 is involved in the protonation, hence an upfield shift is observed from −228 to ⋍275 ppm, whereas the other N atoms N1 and N3 were not involved in the protonation, but the hydrogen packing interactions changed when compared with the starting ABZ which resulted in a small change in ppm in the spectrum.
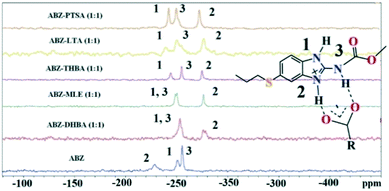 |
| | Fig. 9
15N ss-NMR of the ABZ salts. | |
Table 4
15N ss-NMR chemical shifts (ref. δglycine–347.6 ppm) of the ABZ salts. The protonated N2 site column is formatted in bold
| Salt |
δ N1 |
δ N2
|
δ N3 |
| ABZ |
−255.49 |
−228.96
|
−250.88 |
| ABZ–2,6-DHBA (1:1) |
−249.26 |
−274.14
|
−272.02 |
| ABZ–2,4,6-THBA (1:1) |
−244.37 |
−274.97
|
−254.94 |
| ABZ–MLE (1:1) |
−248.88 |
−276.38
|
−250.42 |
| ABZ–LTA (1:1) |
−242.01 |
−277.99
|
−250.06 |
| ABZ–PTSA (1:1) |
−245.79 |
−275.75
|
−253.23 |
ΔpKa rule
The ΔpKa rule35–37 predicts the product as cocrystal or salt based on the pKa of acid and base reacted during cocrystallization. If ΔpKa = pKa (conjugate acid of the base) − pKa (acid) is less than 0, the result is a cocrystal, a ΔpKa > 3 will result in a salt, and the range 0 < ΔpKa < 3 is an intermediate or grey zone of different proton states. The formation of salts in this study is consistent with a ΔpKa > 3. The pKa values were calculated using a Marvin pKa calculator considering a water medium38 (Table 5). The pKa range for the intermediate zone is continuously updated as additional data is made available, e.g. from the Cambridge Structural Database.39
Table 5 pKa values of ABZ and the coformersa
| Name |
pKa |
Difference |
Result |
|
pKa values were calculated considering a water medium using the Marvin pKa calculator, N2 is the active N for salt formation. https://www.chemaxon.com/marvin/sketch/index.php.
|
| ABZ |
N2 = 4.27, 9.51, −1.80 |
|
|
| BSA |
−2.36 |
6.63 |
Salt |
| PTSA |
−2.13 |
6.4 |
Salt |
| OA |
1.36, 4.11 |
2.91, 0.16 |
Salt |
| 2,6-DHBA |
1.51 |
2.76 |
Salt |
| 2,4,6-THBA |
1.82 |
2.41 |
Salt |
| MLE |
3.04, 5.91 |
1.13, −1.64 |
Salt |
| LTA |
3.86, 5.95 |
0.14, −1.68 |
Salt |
Complexation calculations
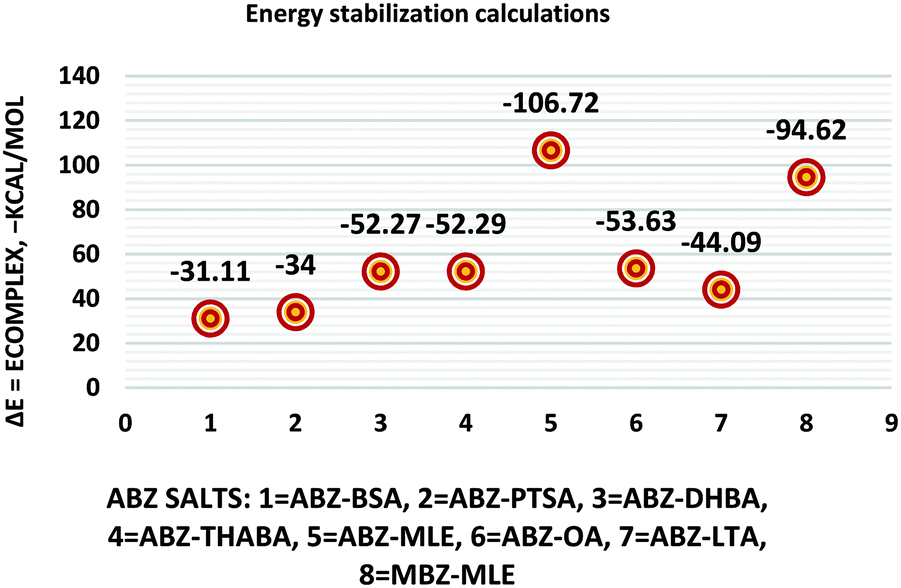
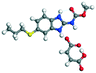
The molecular structure geometry was optimized by using the COMPASS II force field in Materials Studio. The association energy was calculated as the difference in energy of the optimized complex and the combined energy of the optimized individual molecules where Ecomplex is the energy of the optimized molecular complex, EABZ is the energy of the optimized ABZ molecule, and Ecoformer is the energy of the optimized coformer (eqn (1)). The sulfonate salts reported in present study showed that, they are stabilized by −30 to −35 kcal mol−1 whereas carboxylates are stabilized by −50 kcal mol−1 (except LTA, −44 kcal mol−1) during complex formation. However, MLE salt has the highest complexation energy of −106 kcal mol−1. This means that complexes of the carboxylate salts are more stable than those of the sulfonates. The stabilization energy of the ABZ–MLE cocrystal (no proton transfer) at −8.69 kcal mol−1 is much less compared to those of the mebendazole–maleic acid, and ABZ–MLE salts (−94.62, −106.72 kcal mol−1, with proton transfer). Based on the ss-NMR results and the calculated complexation energies, we surmise that ABZ–MLE is a salt, Table 6, Fig. 10.| | | ΔE = Ecomplex − (EABZ + Ecoformer) | (1) |
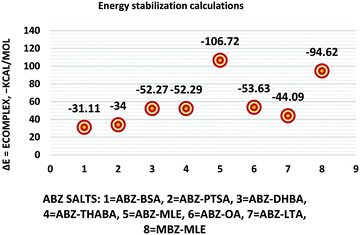 |
| | Fig. 10 Energy stabilization plot of the ABZ salts. | |
Table 6 Energy stabilization calculations for ABZ, the coformers and salts with their structures
| S. No. |
|
Calculated energy kcal mol−1 |
ΔE = Ecomplex − (EABZ + Ecoformer) kcal mol−1 |
Adduct structure |
| 1 |
ABZ |
−116.57 |
|
— |
| 2 |
BSA |
−7.12 |
−154.81 − (−123.70) = −31.11 |
|
| 3 |
PTSA |
−14.93 |
−165.50 − (−131.50) = −34.00 |
|
| 4 |
2,6-DHBA |
−55.56 |
−224.41 − (−172.14) = −52.27 |
|
| 5 |
2,4,6-THBA |
−78.58 |
−247.45 − (−195.16) = −52.29 |
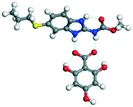
|
| 6 |
MLE |
−62.05 |
Salt −285.35 − (−178.63) = −106.72 |
|
| Co-crystal −187.32 − (−178.63) = −8.69 |
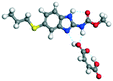
|
| 7 |
OA |
24.82 |
−145.38 − (−91.75) = −53.63 |
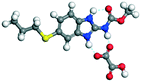
|
| 8 |
LTA |
20.72 |
−139.93 − (−95.84) = −44.09 |
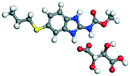
|
| 9 |
MBZ–MLE |
MBZ −70.82 |
−227.49 − (−132.87) = −94.62 |
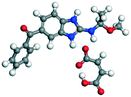
|
| VEVPUJ ref. 18 |
MLE −62.05 |
Salts and cocrystals of bendazole drugs reported in the literature
Mebendazole (MBZ) is an anthelmintic drug in the bendazole family. ABZ and MBZ differ in the substitution on the benzene portion, and MBZ–MLE salt crystal structures have been reported.18 MBZ forms a salt-cocrystal continuum adduct with formic acid, MBZH+–FA−–FA, and a salt with trifluoroacetic acid, whereas with other homologous acids such as acetic, propionic, butanoic, pentanoic, and hexanoic acids, it formed cocrystals.20 Bendazole salts with HCl, HBr, sulfonic acids such as methane, benzene, toluene, and naphthalene, and aliphatic carboxylic acids such as formic, maleic and oxalic have been reported.40–44 (Table 7) We present new salts of L-tartaric acid and hydroxy benzoic acids with ABZ in addition to those from previous studies.
Table 7 Literature reports of bendazole salts and cocrystals with their name, CSD Refcode, reference and packing
| S. No. |
Compound name, CSD Refcode |
Crystal structure |
Adduct |
Reference |
| 1 |
Mebendazole–hydrobromide, BMCBIB |
|
Salt |
40
|
| 2 |
Mebendazole mesylate monohydrate, MIJDIU |
|
Salt |
41
|
| 3 |
Mebendazole hydrochloride, NIZCIJ |
|
Salt |
42
|
| 4 |
Mebendazole hexanoic acid, PIDJOD |
|
Co-crystal |
20
|
| 5 |
Mebendazole pentanoic acid, PIDJUJ |
|
Co-crystal |
20
|
| 6 |
Mebendazole butanoic acid, PIDKAQ |
|
Co-crystal |
20
|
| 7 |
Mebendazole propionic acid, SAGQEW |
|
Co-crystal |
20
|
| 8 |
Mebendazole acetic acid, PIDKEU |
|
Co-crystal |
20
|
| 9 |
Mebendazole formate formic acid, PIDKIY |
|
Salt co-crystal adduct |
20
|
| 10 |
Mebendazole trifluoroacetate, PIDKOE |
|
Salt |
20
|
| 11 |
2-(Methoxy carbonylamino) benzimidazolium naphthalene-1-sulfonate, QESBOG |
|
Salt |
43
|
| 12 |
Methyl N-(1,3-benzimidazolium-2-yl)carbamate formate formic acid, RAVLAC |
|
Salt co-crystal adduct |
44
|
| 13 |
Methyl N-(1,3-benzimidazolium-2-yl)carbamate chloride dihydrate, RAVLEG |
|
Salt |
44
|
| 14 |
Mebendazole glutaric acid, VEVPOD |
|
Cocrystal |
18
|
| 15 |
Mebendazole maleic acid, VEVPUJ |
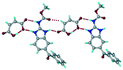
|
Salt |
18
|
| 16 |
Mebendazole methoxy(oxo)acetate, VEVQAQ |
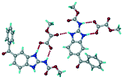
|
Salt |
18
|
4. Conclusions
New salts of the albendazole drug with BSA, PTSA, OA, 2,6-DHBA MLE, LTA, and 2,4,6-THBA are reported. The salts of BSA, PTSA, OA, and 2,6-DHBA were confirmed by single crystal diffraction whereas others were characterized through powder X-ray diffraction, IR and ss-NMR and DSC. In addition, BSA, PTSA and OA crystallize as hydrates, and anhydrates during crystallization were observed. The BSA and PTSA salt hydrates are 2D isostructural, confirmed by Xpac analysis. The ABZ butyl chain disorder observed in the parent structure is resolved in the salt crystal structures except in ABZ–2,6-DHBA. Proton transfer to afford a salt was confirmed by 15N ss-NMR by tracking the chemical shift of the imidazole N2 peak in the MLE, LTA and 2,4,6-THBA salts. The salt formation of ABZ–MLE was further confirmed by comparing the complexation energy of ABZ–MLE with that of MBZ–MLE. The ΔpKa rule of 3 was followed in the present study. These results add to the growing literature on the salts and cocrystals of anthelminthic bendazole drugs.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Financial and infrastructure support from the University Grants Commission, New Delhi (through the UPE and CAS programs), the Department of Science and Technology, New Delhi (through the PURSE and FIST programs), the JC Bose Fellowship (SR/S2/JCB-06/2009), the CSIR project on Pharmaceutical cocrystals (02(0223)/15/EMR-II), and the SERB scheme on Multi-component cocrystals (EMR/2015/002075) are gratefully acknowledged. GB thanks the UGC for a research fellowship.
References
- H. Mohammad, E. Pourgholami, A. Javed, W. E. Lisa, L. E. Ying and L. David, Cancer Chemother. Pharmacol., 2005, 55, 425–432 CrossRef PubMed.
- J. A. Bogan and S. E. Marriner, Vet. Res. Commun., 1983, 7, 271 CrossRef.
- K. Anand and S. Wakode, Int. J. Chem. Stud., 2017, 5, 350–362 CAS.
-
https://tsrlinc.com/resources/services/ (dated 24/07/2018).
- N. Jagadeeshbabu and A. Nanga, Cryst. Growth Des., 2011, 11, 2662–2679 CrossRef.
- S. Torrado, J. J. Torrado and R. Cadórniga, Int. J. Pharm., 1996, 140, 247–250 CrossRef CAS.
- V. L. Bassani and D. Kriege, J. Inclusion Phenom. Mol. Recognit. Chem., 1996, 25, 149–152 CrossRef.
- C. Moriwaki, G. L. Costa, C. N. Ferracini, F. F. Moraes, G. M. Zanin, E. A. G. Pineda and G. Matioli, Braz. J. Chem. Eng., 2008, 25, 255–267 CrossRef CAS.
- B. R. Pranzno, D. Cruickshank, M. Coruzzi, M. R. Caira and R. Bettini, J. Pharm. Sci., 2010, 99, 3731–3742 CrossRef PubMed.
-
https://www.ccdc.cam.ac.uk/structures/search?sid=ConQuest&coden=001078&year=2007&pid=ccdc:668711&aulast=Alhalaweh
.
-
SMART, version 5.625 and SHELX-TL, version 6.12, BrukerAXS Inc., Madison, Wisconsin, 2000 Search PubMed.
-
PLATON, A Multipurpose Crystallographic Tool, ed. A. L. Spek, Utrecht University, Utrecht, The Netherlands, 2002 CrossRef CAS; A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
-
X-Seed, Graphical interface to SHELX-97 and POV-Ray, Program for Better Quality of Crystallographic Figures, ed. L. J. Barbour, University of Missouri-Columbia, Columbia, Missouri, 1999 Search PubMed.
-
https://www.ccdc.cam.ac.uk/Community/csd-community/freemercury/
.
- M. C. Etter and J. C. Mcdonald, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- M. C. Etter and J. C. Macdonald, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- J. Chen, Z. Wang, C. Wu, S. Li and T. Lu, CrystEngComm, 2012, 14, 6221–6229 RSC.
- K. D. Paula, G. E. Camí, E. V. Brusau, G. E. Narda and J. Ellena, J. Pharm. Sci., 2013, 102, 3528–3538 CrossRef PubMed.
- J. Chen and T. Lu Chin, J. Chem., 2013, 31, 635–640 CAS.
- G. L. B. de Araujo, F. F. Ferreira, C. E. S. Bernardes, J. A. P. Sato, O. M. Gil, D. L. A. Faria, R. Loebenberg, S. R. Byrn, D. D. M. Ghisleni, N. A. Bou-Chacra, T. J. A. Pinto, S. G. Antonio, H. G. Ferraz, D. Zemlyanov, D. S. Gonçalves and M. E. M. Piedade, Cryst. Growth Des., 2018, 18, 2377–2386 CrossRef CAS.
- P. M. Bhatt and G. R. Desiraju, Chem. Commun., 2007, 2057–2059 RSC.
- M. K. Mishra, U. Ramamurty and G. R. Desiraju, J. Am. Chem. Soc., 2015, 137, 1794–1797 CrossRef CAS PubMed.
- T. Gelbrich and M. B. A. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448–460 RSC.
-
R. M. Silverstein, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., 6th edn, 2002, pp. 71–143 Search PubMed.
- R. K. Gilpin and W. Zhou, Vib. Spectrosc., 2005, 37, 53 CrossRef CAS.
- S. Karki, T. Friščić, L. Fábián and W. Jones, CrystEngComm, 2010, 12, 4038–4041 RSC.
- J. F. Remenar, M. L. Peterson, P. W. Stephens, Z. Zhang, Y. Zimenkov and M. B. Hickey, Mol. Pharmaceutics, 2007, 4, 386–400 CrossRef CAS PubMed.
- V. André, A. Fernandes, P. P. Santos and M. T. Duarte, Cryst. Growth Des., 2011, 11, 2325–2334 CrossRef.
- F. G. Vogt, J. S. Clawson, M. Strohmeier, A. J. Edwards, T. N. Pham and S. A. Watson, Cryst. Growth Des., 2009, 9, 921–937 CrossRef CAS.
- P. Sanphui, G. Bolla, A. Nangia and V. Chernyshev, IUCrJ, 2014, 1, 136–150 CrossRef CAS PubMed.
- L. Zhao, M. P. Hanrahan, P. Chakravarty, A. G. DiPasquale, L. E. Sirois, K. Nagapudi, J. W. Lubach and A. J. Rossini, Cryst. Growth Des., 2018, 18, 2588–2601 CrossRef CAS.
- Z. J. Li, Y. Abramov, J. Bordner, J. Leonard, A. Medek and A. V. Trask, J. Am. Chem. Soc., 2006, 128, 8199–8210 CrossRef CAS PubMed.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- B. Sarma, N. K. Nath, B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2009, 9, 1546–1557 CrossRef CAS.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
-
https://www.chemaxon.com/marvin/sketch/index.php (dated 05-04-2018).
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC.
- N. M. Blaton, O. M. Peeters and C. J. De Ranter, Cryst. Struct. Commun., 1980, 9, 181–186 CAS.
- K. D. Paula, G. E. Cami, E. V. Brusau, G. E. Narda and J. Ellena, J. Pharm. Sci., 2013, 102, 3528–3538 CrossRef PubMed.
- E. V. Brusau, G. E. Cami, G. E. Narda, S. Cuffini, A. P. Ayala and J. Ellena, J. Pharm. Sci., 2008, 97, 542 CrossRef CAS PubMed.
- K. Sepassi, G. S. Nichol and S. H. Yalkowsky, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5172–o5173 CrossRef CAS.
- K. K. Turgunov, A. G. Tozhiboev and B. Tashkhodzhaev, J. Struct. Chem., 2005, 46, 342 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1834924–1834929. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01311j |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Ashwini
Nangia
*ab and
Ashwini
Nangia