
Superbroad near-infrared photoluminescence covering the second biological window achieved by bismuth-doped oxygen-deficient gadolinium oxide
Kai Zhanga,
Jing-Shan Houa,
Bo-Mei Liub,
Yang Zhoub,
Zi-Jun Yongb,
Li-Na Lic,
Hong-Tao Sun*b and
Yong-Zheng Fang*a
aSchool of Materials Science and Engineering, Shanghai Institute of Technology, Shanghai 201418, China. E-mail: fyz1003@sina.com
bCollege of Chemistry, Chemical Engineering and Materials Science, State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Soochow University, Suzhou 215123, China. E-mail: timothyhsun@gmail.com
cSynchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
First published on 3rd August 2016
Abstract
In this work, we demonstrated that bismuth-doped oxygen-deficient gadolinium oxides, produced through a low-temperature topochemical reduction strategy using CaH2 as a solid-state reducing agent, show superbroad near-infrared photoluminescence covering the second biological window. Structural analyses confirm that the topochemical reduction treatment of bismuth-doped gadolinium oxides creates oxygen vacancies in the gadolinium oxide matrix, which changes the coordination of Bi and makes it situate in a defective environment. Given the tantalizing superbroad near-infrared emission from the reduced phases, it is anticipated that this novel system may find applications for in vitro/in vivo near-infrared fluorescence imaging in the second biological window.
Introduction
Near-infrared (NIR) light in the biological windows I (650–950 nm) and II (1000–1350 nm) possesses much higher tissue penetration capability than UV or visible light and is superior for in vivo optical imaging.1–3 NIR light in the II biological window could provide more efficient tissue penetration relative to that in the I biological window due to the substantial reduction of tissue autofluorescence and scattering.4–6 Up to date, NIR-luminescent materials emitting in the I window have been widely explored, and a broad range of systems have been reported.2,7–11 In sharp contrast, material systems luminescing in the II biological window are very limited. Only several kinds of systems including carbon nanotubes,12 Bi-doped aluminosilicate nanoparticles,13 Ag2Te quantum dots,14 Ag2S quantum dots15 have been reported over the past decade. Therefore, it is highly desirable to develop novel materials that can display NIR photoluminescence (PL) in the II biological window.Rare-earth (RE) doped NIR-luminescent phosphors have always been one of the most attractive members in the NIR phosphor family. They have been well developed as a new class of luminescent biomarkers that have become promising alternatives to organic fluorophores and quantum dots for applications in biological assays and medical imaging, due to particular characteristics of large Stokes shifts, high resistance to photobleaching, and high penetration depth and temporal resolution.16–18 However, these phosphors either rely on RE ions to achieve the NIR emission which usually show narrow excitation and NIR emission bands (such as Y2O3:Er3+,19 SrF2:Nd3+,20 and Y3Al2Ga3O12:Nd–Ce–Cr21), or demonstrate NIR emission that cannot cover the biological window II.22 These disadvantages hinder their application for in vitro/in vivo near-infrared fluorescence imaging. Therefore, it is highly desirable to develop novel phosphors which employ non-RE luminescent centers and display broad NIR PL that can cover the biological window II. Among the commonly used emission centers, bismuth, the heaviest stable element in the periodic table, owns diverse oxidation states, has become a promising optically active centre in a wide range of applications, such as visible and NIR photonics.23–33 It is well known that the unsheltered outer electrons of bismuth ion can be easily influenced by the crystal field environment. Therefore, it is important to tune the spectroscopic characteristics of Bi through rational selection of host materials, or tailor their coordination environments by a particular chemical method. We have recently achieved the shift of the PL emission from visible in Y2−xBixO3 to the NIR in Y2−xBixO3−z by using an efficient low-temperature topochemical reduction strategy.34 However, it is still of great significant to explore new bismuth-activated materials by using this powerful soft chemical route for broad applications, such as in vitro/in vivo NIR fluorescence imaging in the biological window II. Furthermore, combining the advantageous NIR emission feature with other characteristics in one material system might favour the development of a new class of multifunctional biomarkers.
Gadolinium oxide (Gd2O3) is a promising host matrix for optical materials because of its good chemical durability, thermal stability, and low phonon energy.35–37 To date, rare earth (RE) doped bulk Gd2O3 or Gd2O3 nanoparticles have been well studied, which have shown attractive PL properties and have been widely used in phosphors,38–42 television tubes,43 biological fluorescent labelling,44–46 MRI contrast36,45,47,48 and in vitro/in vivo optical imaging.35,49–53 Additionally, bismuth-doped Gd2O3 have also been reported to obtain broad visible PL,54,55 or to act as a sensitizer of other active centers to achieve enhanced emission.56,57 However, most of these materials show emission in the visible spectral range. In view of tantalizing prospect of Gd-containing materials for MRI, developing Gd2O3-based systems that are capable of emitting in the II biological window will present new avenue for their even broader application such as multifunctional biomarkers.
In this work, we address this issue by using a Bi-doped oxygen-deficient gadolinium oxide that is produced by low-temperature topochemical reduction using CaH2 as a mild reducing agent. Although the low-temperature topotactic reduction has been widely employed to prepare new magnetic phases with unusual cation coordination environments and oxidation states,58–62 its application for the synthesis of optically functional materials has been carried out only in recent several years.34,63 In this contribution, we find that the topochemical reduction of Bi-doped Gd2O3 yields a new class of luminescent materials demonstrating superbroad NIR PL that can cover the II window, owing to the creation of NIR-luminescent [BiOx] (x < 7). The appearance of oxygen vacancies in the matrix results in the coordination of Bi from distorted [BiO7] octahedra in the precursor to [BiO7−z] (z < 7) polyhedra in reduced phases. Such an evolution leads to the emergence of superbroad NIR PL in reduced phases. Thermogravimetric (TG) analysis, Raman spectroscopy, X-ray absorption spectra together with the visible and NIR PL spectra were taken to help us understand the unusual luminescent behaviours observed in Bi-doped oxygen-deficient gadolinium oxides. It is anticipated that the material system developed here could find applications for bioimaging in the II biological window.
Experimental
Sample preparation
Bi3+-Doped Gd2O3 were prepared by a co-precipitation method. Appropriate amounts of Gd(NO3)3·6H2O (Aladdin, 99.99%) and Bi(NO3)3·5H2O (Sinopharm Chemical Reagent Co., Ltd, 99%) were dissolved in hot deionized water with diluted nitric acid under stirring. All cations were co-precipitated by adding an excess of oxalic acid. The as-received precipitate was filter washed by distilled water and C2H5OH for several times to remove impurity anions. After being dried at 110 °C for 12 h in a vacuum oven, the precursor precipitate was calcined in air under temperatures of 1350 °C for 4.5 h. The precursor was mixed with two mole equivalents of CaH2 in a nitrogen-filled glovebox (O2 and H2O < 0.1 ppm). The mixtures were then sealed in an evacuated Pyrex tube and heated at 450 °C for different duration. Residual CaH2 and CaO produced during the reduction were washed out with an NH4Cl/methanol solution, and then dried in a vacuum oven at 50 °C to get the final samples. The samples were denoted St, where t is the treatment duration in hours.Characterizations
The phase purity of the samples was characterized and evaluated by powder X-ray diffraction (XRD) analysis on a Bruker D8 Advance diffractometer (Cu-Kα radiation). TG measurement was performed using a Netzsch STA 449C thermal analyser equipped with a Balzers ThermoStar GSD 300T mass spectrometer. Approximately 50 mg of the sample was loaded into an alumina crucible and heated at 10 K min−1 from 20 to 800 °C. The oxygen with a flow rate of 40 ml min−1 was used as the carrier gas for TG measurements. Raman spectra were recorded at room temperature with a Horiba Scientific Xplora Plus Raman spectrometer with an excitation wavelength of 638 nm from a He–Cd laser with a power of 1.25 mW. The X-ray absorption spectra of the Gd LIII edge for the precursor, S2, S12 and S96 samples were obtained in the transmission mode at the BL14W1 beam line of the Shanghai Synchrotron Irradiation Facility with stored electron energy of 3.5 GeV and ring currents of 200 mA. PL spectra were recorded with an FLS 980 spectrofluorometer (Edinburgh Instruments Ltd.) equipped with photomultipliers (Hamamatsu, R5509-73 and R928).Results and discussion
XRD measurements were carried out to confirm the phase purity and structural properties of the precursor and the reduced samples. It is well known that Gd2O3 has two polymorphs: the monoclinic phase (B type, space group, C2/m) and the cubic phase (C type, space group, Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ). Gd2O3 can be either cubic or monoclinic at room temperature.64 Previous results confirmed that both phases can co-exist under particular preparation condition.65 As shown in Fig. 1, the precursor consists of both monoclinic (JCPDS no. 42-1465) and cubic (JCPDS no. 43-1014) Gd2O3 phases, and the B type monoclinic polymorph is more dominant. Interestingly, we find that after undergoing CaH2 treatment, the resulting products demonstrate similar XRD patterns to the precursor, implying that the crystalline structure of gadolinium oxide retains well. As is known, Gd atom is seven-coordinated in the monoclinic Gd2O3, and is six-coordinated in the cubic structure. Since the radius of Gd3+ (CN = 6, r = 0.938 Å) is similar to that of Bi3+ (CN = 6, r = 1.03 Å),66 Bi3+ could randomly occupy the Gd3+ sites in the host structure.
). Gd2O3 can be either cubic or monoclinic at room temperature.64 Previous results confirmed that both phases can co-exist under particular preparation condition.65 As shown in Fig. 1, the precursor consists of both monoclinic (JCPDS no. 42-1465) and cubic (JCPDS no. 43-1014) Gd2O3 phases, and the B type monoclinic polymorph is more dominant. Interestingly, we find that after undergoing CaH2 treatment, the resulting products demonstrate similar XRD patterns to the precursor, implying that the crystalline structure of gadolinium oxide retains well. As is known, Gd atom is seven-coordinated in the monoclinic Gd2O3, and is six-coordinated in the cubic structure. Since the radius of Gd3+ (CN = 6, r = 0.938 Å) is similar to that of Bi3+ (CN = 6, r = 1.03 Å),66 Bi3+ could randomly occupy the Gd3+ sites in the host structure.
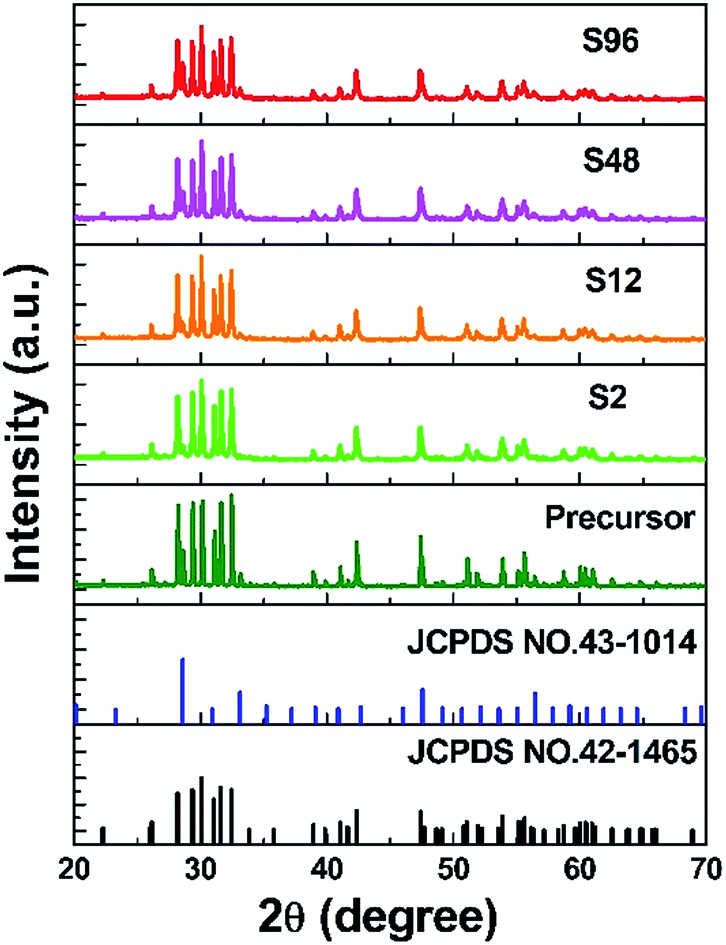 | ||
| Fig. 1 XRD patterns of the precursor and the reduced samples. | ||
In order to investigate the effect of the low temperature treatment on the composition change of gadolinium oxide, TG measurements were taken in an oxygen atmosphere. Typically, approximately 50 mg of the sample was loaded into an alumina crucible and heated at 10 K min−1 from 20 to 800 °C using oxygen with a flow rate of 40 ml min−1 as the carrier gas. If the topochemical reduction of the precursor using CaH2 could create oxygen defects in the structure, a weight increase is expected to appear in the reduced phases when they are heated to a certain temperature under the oxygen atmosphere. That is, the reduced phase can convert back to the phase without oxygen vacancies. Fig. 2 shows the TG curves of the reduced phases taken under flowing O2. Generally, three features are observed for these curves. The first step of weight loss from room temperature to approximately 360 °C was attributed to the removal of adsorbed water. A noticeable weight increase was observed in the second step from ca. 360 °C to ca. 580 °C due to a backfilling of the oxygen. Another weight loss stage was observed in the third step after the sample was heated to ca. 600 °C, are presumed to be caused by the decomposition of the CaCO3 and/or Ca(HCO3)2 by-products, as also observed in other cases.34 Based on TG data, we could roughly determine the content of oxygen vacancies in the S2, S12, S48 and S96 samples to be 0.089%, 0.09%, 0.124%, 0.186% and 0.216% (wt%), respectively. Obviously, the amount of oxygen vacancies increases with the increased harshness of the thermal treatment.
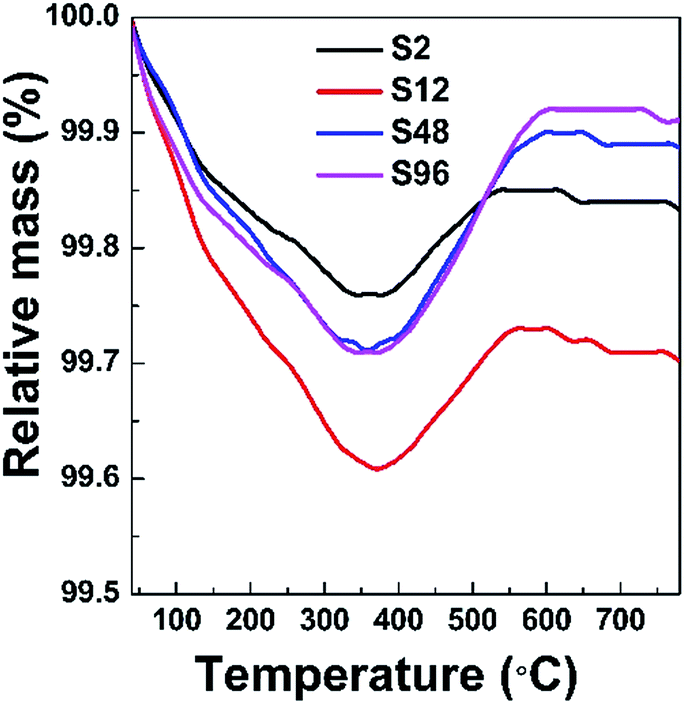 | ||
| Fig. 2 TG curves of the reduced phases taken under flowing O2. | ||
Raman spectroscopy is a very powerful tool for characterizing materials because it is an in situ and non-destructive method. Up to now, Gd2O3 has been studied theoretically and experimentally by Raman spectroscopy.67,68 To examine the influence of CaH2 treatment on the structure of Bi-doped gadolinium oxide, we further took Raman spectra. As show in Fig. 3, the precursor shows multiple sharp peaks similar to the undoped Gd2O3.67,68 It is obvious that these peaks cannot be totally attributed to one phase of Gd2O3. The scattering peaks at 361 and 442 cm−1 can be assigned to the combination of Fg and Eg modes of the cubic Gd2O3, while the others stem from the monoclinic phase of Gd2O3.67,68 This is in a good agreement with the XRD result. Interestingly, two distinct differences were observed in the Raman spectra of the reduced phases with respect to the precursor. First, the widths of Raman peaks were significantly broadened for all reduced samples (Fig. 3 and the inset in Fig. 3). Second, the scattering peaks assigned to both the monoclinic and cubic phases were affected by the thermal treatment. The peaks at 361 and 442 cm−1 almost disappear for the reduced phases, strongly evidencing that the structure of the cubic phase was seriously destroyed when treated by CaH2. In contrast, the scattering peaks assigned to the monoclinic phase merge in the range of 370–425 cm−1. All these facts indicate that the topochemical extraction of oxygen from the gadolinium oxide can seriously influence the short-range order of the systems, although the long-range order could maintain well. That is, the thermal treatment increases the disorder of the matrix, and doped Bi ions in the reduced phases can be viewed to situate in a defective environment (Fig. 4).
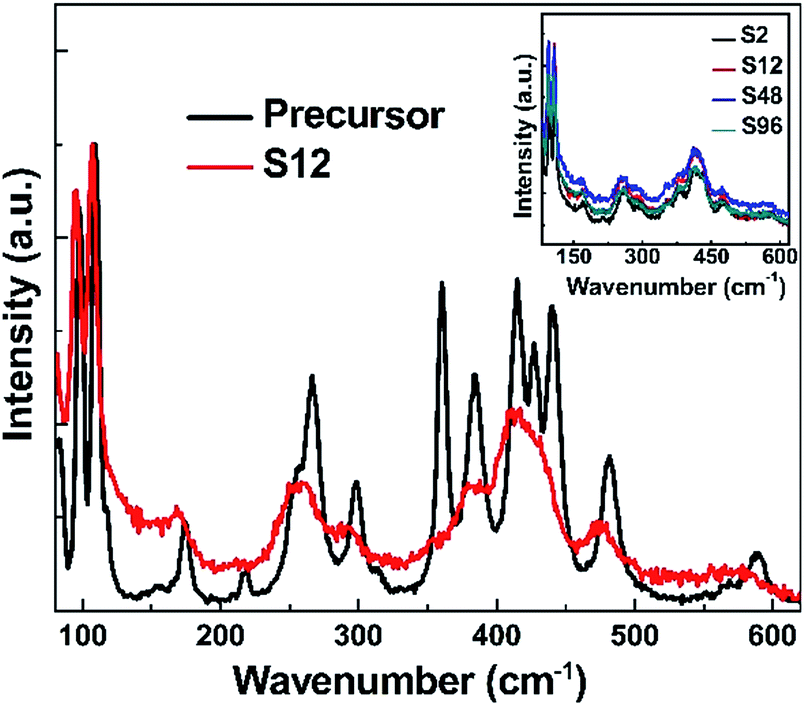 | ||
| Fig. 3 Raman spectra of the precursor and the S12 samples at room temperature (λexc. = 638 nm). The inset shows the Raman spectra of all the reduced samples. | ||
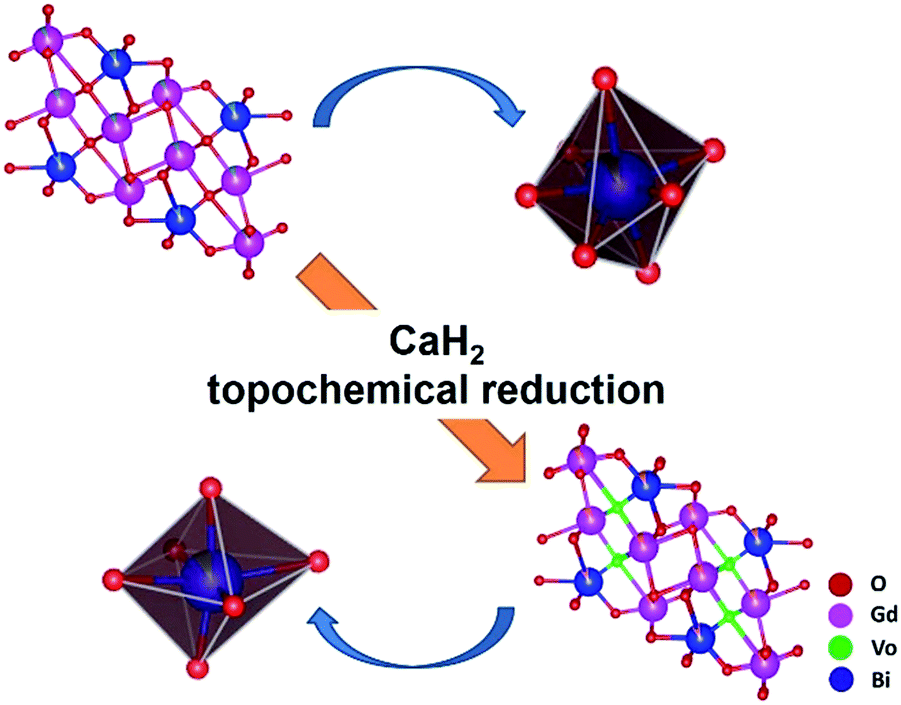 | ||
| Fig. 4 Schematic demonstration of structural evolution from Bi-doped Gd2O3 to the reduced phase through the topochemical reduction reaction strategy. | ||
As is well known, X-ray absorption spectroscopy is a powerful technique for more reliable information about the average oxidation states and the site-symmetry changes around the absorbing atoms. Unfortunately, due to the strong absorption of X-ray signal from Bi by Gd, we cannot obtain a sound X-ray absorption fine structure (XAFS) spectrum, thus making it impossible to analyze the oxidation state as well as the coordination environment of Bi in this system. We then turn our attention to the XAFS analysis of Gd in the precursor and the reduced phases. The Gd LIII-edge X-ray absorption near-edge structure (XANES) spectra are shown in Fig. 5. Remarkably, we find that the intensity of the Gd LIII-edge white line increases monotonically with the increase of treatment duration. It is well known that the Gd LIII-edge XANES corresponds to the transition from 2p3/2 to 5d. The increased peak intensity of white lines implies a greater number of unoccupied Gd 5d state being available for Gd 2p electrons to be excited to, which can be attributed to the increased ionic character of the Gd–O bond in the reduced phases.69 This becomes understandable if we take account of the created oxygen vacancies in the reduced phases. As a result of oxygen deintercalation, the coordination polyhedra of Gd can be influenced, resulting in the occurrence of under-coordinated Gd–O units and/or distorted Gd–O polyhedra. Such a change provides a chance for a decrease in sharing of electrons between the Gd 5d and O 2p states, resulting in more unoccupied Gd 5d states. Since Bi ions occupy the Gd sites, we thus conclude that the topochemical treatment could also alter the coordination environment of Bi present.
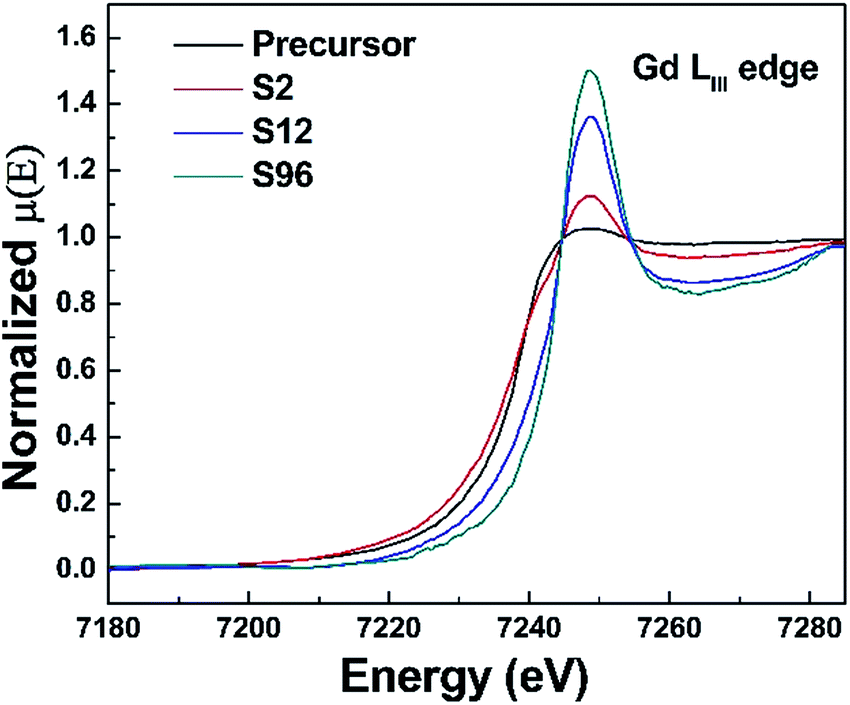 | ||
| Fig. 5 Gd LIII-edge XANES spectra of the precursor, S2, S12 and S96 samples. | ||
As is well known, Bi-doped materials can demonstrate ultraviolet, visible and infrared PL emission, and the emission range is intimately connected with the oxidation states as well as the local coordination environments of Bi.23–33 We thus employ the PL spectroscopy for monitoring the influence of oxygen vacancies on the photophysical properties of the systems studied here. Fig. 6a shows the PL excitation (PLE) spectra of the precursor and reduced samples monitored at 478 nm. A broad excitation band consisting of three peaks centred at 311, 340, 364 nm was observed. When excited by the 340 nm light, all the samples show a blue emission ranging from 420 nm to 650 nm with the maximum at 478 nm (Fig. 6b). This broad band is mainly ascribed to the characteristic 3P1 → 1S0 transition of Bi3+ ions. PLE spectra of the reduced samples monitored at 1025 nm NIR light were shown in Fig. 6c, indicating that our sample can response to the excitation from ca. 300 nm to ca. 800 nm. No NIR emission was detected in the precursor (Fig. 6d), while superbroad NIR emission ranging from ca. 800 nm to ca. 1600 nm emerges for all reduced samples. This indicates that the NIR PL is not from structural defects of Gd2O3, but intimately associated with the oxygen vacancies created. Since Bi3+ ions coordinated with seven oxygen atoms in the monoclinic phase can only show visible PL, the occurrence of NIR emission in reduced phases thus can be connected with defective Bi–O polyhedra. Among all samples, the S12 shows the strongest NIR PL, suggesting that the content of oxygen vacancies is of paramount importance for the NIR emission. This can be explained by the fact that the PL intensity is dictated by several parameters including the amount, quantum efficiency, and absorption cross section of the emitter. Importantly, we find that the treated samples except the S96 display stronger visible emission than the precursor. We propose that such a topochemical treatment may alter the local environments of Bi3+, thus making it even more efficient for visible emission. Given the salient enhancement of visible emission and the occurrence of NIR emission, we conclude that the treatment of the precursor at moderate conditions can yield new systems with significantly increased emission efficiency. Note that the existence of too many oxygen vacancies can weaken the visible emission owing to the decreased amount of Bi3+ (Fig. 6b). The two-dimensional excitation–emission graph of the S12 sample displayed in Fig. 7 clearly shows that the S12 sample possesses superbroad NIR emission centred at 1025 nm and with an optimal excitation band at 490 nm and a weak band at 660 nm. To the best our knowledge, this is the first report on the observation of NIR emission from Bi-doped Gd-containing system among Bi-activated phosphors. We think that further optimization of the emission properties of this system or developing other classes of Bi-doped Gd-containing oxygen-deficient materials by using the topochemical reduction reaction strategy may open the door for the development of a series of multimodal biomarkers that may find practical applications.
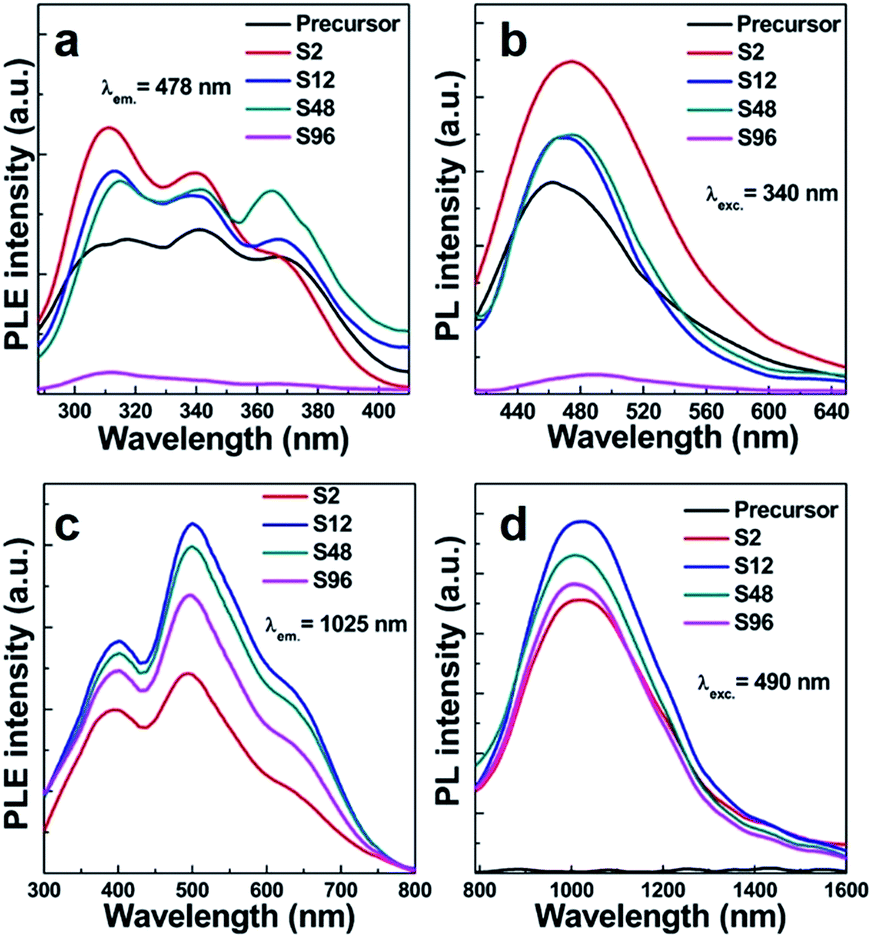 | ||
| Fig. 6 (a) PLE (λem. = 478 nm) and (b) PL (λexc. = 340 nm) spectra of the precursor and reduced samples in visible light region; (c) PLE (λem. = 1025 nm) and (d) NIR PL (λexc. = 490 nm) spectra of the precursor and reduced samples. | ||
 | ||
| Fig. 7 Two-dimensional excitation–emission graph of the S12 sample. | ||
Conclusions
In summary, we have successfully developed a class of NIR-emitting phosphor via low-temperature topochemical reduction of Bi-doped gadolinium oxide using CaH2 as a solid-state reducing agent. XRD, TG, Raman spectra, as well as the XAFS spectra were employed to help us unambiguously resolve structural evolution before and after thermal treatment. This strategy allows us to readily create oxygen vacancies of the host, and simultaneously retain the original crystalline phase. The existence of oxygen vacancies caused the change of the coordination environment of Bi, resulting in the occurrence of superbroad NIR PL ranging from 800 to 1400 nm. In light of the tantalizing superbroad near-infrared emission from the reduced phases, it is anticipated that this novel system may find applications for in vitro/in vivo near-infrared fluorescence imaging in the second biological window. We also anticipate that more systems with outstanding luminescent properties can be developed by using the topochemical reduction reaction strategy.Acknowledgements
Kai Zhang and Jing-Shan Hou have equally contributed to this work. This work is financially supported by the National Natural Science Foundation of China (Grant No. 51472162, and 11574225), Jiangsu Specially Appointed Professor program (SR10900214), Natural Science Foundation of Jiangsu Province for Young Scholars (BK20140336), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (TP2014062), and the Youth Project of National Natural Science Fund (11405256). We appreciate the staff at the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility for XAFS measurements. We also thank the staff at the 1W2B beamline at the Beijing Synchrotron Radiation Facility for XAFS measurements.Notes and references
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93 CrossRef CAS PubMed.
- Y. He, Y. Zhong, Y. Su, Y. Lu, Z. Jiang, F. Peng, T. Xu, S. Su, Q. Huang, C. Fan and S.-T. Lee, Angew. Chem., Int. Ed., 2011, 50, 5694 Search PubMed.
- P. Vijayaraghavan, C.-H. Liu, R. Vankayala, C.-S. Chiang and K. C. Hwang, Adv. Mater., 2014, 26, 6689 CrossRef CAS PubMed.
- M.-F. Tsai, S.-H. G. Chang, F.-Y. Cheng, V. Shanmugam, Y.-S. Cheng, C.-H. Su and C.-S. Yeh, ACS Nano, 2013, 7, 5330 CrossRef CAS PubMed.
- S.-M. Lee, H. Park and K.-H. Yoo, Adv. Mater., 2010, 22, 4049 CrossRef CAS PubMed.
- H. Liu, D. Chen, L. Li, T. Liu, L. Tan, X. Wu and T. Tang, Angew. Chem., Int. Ed., 2011, 50, 891 CrossRef CAS PubMed.
- K.-T. Yong, I. Roy, H. Ding, E. J. Bergey and P. N. Prasad, Small, 2009, 5, 1997 CrossRef CAS PubMed.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016 CrossRef CAS PubMed.
- R. Weissleder, C. Tung, U. Mahmood and A. Bogdanov Jr., Nat. Biotechnol., 1999, 17, 375 CrossRef CAS PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626 CrossRef CAS PubMed.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS PubMed.
- K. Welsher, S. P. Sherlock and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8943 CrossRef CAS PubMed.
- H.-T. Sun, J. Yang, M. Fujii, Y. Sakka, Y. Zhu, T. Asahara, N. Shirahata, M. Ii, Z. Bai, J.-G. Li and H. Gao, Small, 2011, 7, 199 CrossRef CAS PubMed.
- C. Chen, X. He, L. Gao and N. Ma, ACS Appl. Mater. Interfaces, 2013, 5, 1149 CAS.
- G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris, Q. Wang and H. Dai, Angew. Chem., Int. Ed., 2012, 51, 9818 CrossRef CAS PubMed.
- G. K. Das and T. T. Y. Tan, J. Phys. Chem. C, 2008, 112, 11211–11217 CAS.
- G. Wang, Q. Peng and Y. Li, Acc. Chem. Res., 2011, 44, 322–332 CrossRef CAS PubMed.
- G. Ajithkumar, B. Yoo, D. E. Goral, P. J. Hornsby, A.-L. Lin, U. Ladiwala, V. P. Dravid and D. K. Sardar, J. Mater. Chem. B, 2013, 1, 1561–1572 RSC.
- K. Soga, K. Tokuzen, K. Tsuji, T. Yamano, N. Venkatachalam, H. Hyodo and H. Kishimoto, Proc. SPIE, 2010, 7598, 759807 CrossRef.
- I. Villa, A. Vedda, I. X. Cantarelli, M. Pedroni, F. Piccinelli, M. Bettinelli, A. Speghini, M. Quintanilla, F. Vetrone, U. Rocha, C. Jacinto, E. Carrasco, F. S. Rodriguez, A. Juarranz, B. Rosal, D. H. Ortgies, P. H. Gonzalez, J. G. Sole and D. J. Garcia, Nano Res., 2015, 8, 649–665 CrossRef CAS.
- J. Xu, S. Tanabe, A. D. Sontakke and J. Ueda, Appl. Phys. Lett., 2015, 107, 081903 CrossRef.
- X.-W. Zhang, Z. Zhao, X. Zhang, D. B. Cordes, B. Weeks, B. Qiu, K. Madanan, D. Sardar and J. Chaudhuri, Nano Res., 2015, 8, 636–648 CrossRef CAS.
- H.-T. Sun, J. Zhou and J. Qiu, Prog. Mater. Sci., 2014, 64, 1 CrossRef CAS.
- A. G. Okhrimchuk, L. N. Butvina, E. M. Dianov, N. V. Lichkoca and K. N. Boldyrev, Opt. Lett., 2008, 33, 2182 CrossRef CAS PubMed.
- H.-T. Sun, A. Hosokawa, Y. Miwa, F. Shimaoka, M. Fujii, M. Mizuhata, S. Hayashi and S. Deki, Adv. Mater., 2009, 21, 3694 CrossRef CAS.
- H.-T. Sun, Y. Matsushita, Y. Sakka, N. Shirahata, M. Tanaka, Y. Katsuya, H. Gao and K. Kobayash, J. Am. Chem. Soc., 2012, 134, 2918 CrossRef CAS PubMed.
- H.-T. Sun, Y. Sakka, N. Shirahata, H. Gao and T. Yonezawa, J. Mater. Chem., 2012, 22, 12837 RSC.
- A. A. Veber, A. N. Romanov, O. V. Usovich, Z. T. Fattakhova, E. V. Haula, V. N. Korchak, L. A. Trusov, P. E. Kazin, V. B. Sulimov and V. B. Tsvetkov, Appl. Phys. B, 2012, 108, 733 CrossRef CAS.
- V. G. Plotnichenko, V. O. Sokolov, D. V. Philippovskiy, I. S. Lisitsky, M. S. Kouznetsov, K. S. Zaramenskikh and E. M. Dianov, Opt. Lett., 2013, 38, 362 CrossRef CAS PubMed.
- P. Boutinaud, Inorg. Chem., 2013, 52, 6028 CrossRef CAS PubMed.
- G. Blasse, A. Meijerink and J. Zuidema, J. Phys. Chem. Solids, 1994, 55, 171 CrossRef CAS.
- A. N. Romanov, A. A. Veber, D. N. Vtyurina, Z. T. Fattakhova, E. V. Haula, D. P. Shashkin, V. B. Sulimov, V. B. Tsvetkov and V. N. Korchak, J. Mater. Chem. C, 2015, 3, 3592 RSC.
- O. Laguta, H. EI Hamzaoui, M. Bouazaoui, V. B. Arion and I. Razdobreev, Optica, 2015, 2, 663 CrossRef CAS.
- B.-M. Liu, Z.-G. Zhang, K. Zhang, Y. Kuroiwa, C. Moriyoshi, H.-M. Yu, C. Li, L.-R. Zheng, L.-N. Li, G. Yang, Y. Zhou, Y.-Z. Fang, J.-S. Hou, Y. Matsushita and H.-T. Sun, Angew. Chem., Int. Ed., 2016, 55, 4967 CrossRef CAS PubMed.
- G. Tian, Z. Gu, X. Liu, L. Zhou, W. Yin, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, J. Phys. Chem. C, 2011, 115, 23790 CAS.
- N. M. Maalej, A. Qurashi, A. A. Assadi, R. Maalej, M. N. Shaikh, M. Ilyas and M. A. Gondal, Nanoscale Res. Lett., 2015, 10, 1 CrossRef CAS PubMed.
- G.-X. Liu, R. Zhang, Q.-L. Xiao, S.-Y. Zou, W.-F. Peng, L.-W. Cao, R. Zhang and J.-X. Meng, Opt. Mater., 2011, 34, 313 CrossRef CAS.
- C. Louis, R. Bazzi, M. A. Flores, W. Zhang, K. Lebbou, O. Tillement, B. Mercier, C. Dujardin and P. Perriat, J. Solid State Chem., 2003, 173, 335 CrossRef CAS.
- R. Bazzi, M. A. Flores, C. Louis, K. Lebbou, W. Zhang, C. Dujardin, S. Roux, B. Mercier, G. Ledoux, E. Bernstein, P. Perriet and O. Tillement, J. Colloid Interface Sci., 2004, 273, 191 CrossRef CAS PubMed.
- L. Li, H. K. Yang, B. K. Moon, B. C. Choi, J. H. Jeong and K. H. Kim, Mater. Chem. Phys., 2010, 119, 471 CrossRef CAS.
- T. Selvalakshmi, S. Sellaiyan, A. Uedono and A. C. Bose, RSC Adv., 2014, 4, 34257 RSC.
- A. Jain, G. A. Hirata, M. H. Farias and F. F. Castillon, Nanotechnology, 2016, 27, 065601 CrossRef PubMed.
- G.-X. Liu, G.-Y. Hong and D.-X. Sun, J. Colloid Interface Sci., 2004, 278, 133 CrossRef CAS PubMed.
- H.-C. Lu, G.-S. Yi, S.-Y. Zhao, D.-P. Chen, L.-H. Guo and J. Cheng, J. Mater. Chem., 2004, 14, 1336 RSC.
- R. M. Petoral, Jr., F. Soderlind, A. Klasson, A. Suska, M. A. Fortin, N. Abrikossova, L. Selegard, P.-O. Kall, M. Engstrom and K. Uvdal, J. Phys. Chem. C, 2009, 113, 6913 Search PubMed.
- S. Lechevallier, P. Lecante, R. Mauricot, H. Dexpert, J. Dexpert-Ghys, H.-K. Kong, G.-L. Law and K.-L. Wong, Chem. Mater., 2010, 22, 6153 CrossRef CAS.
- J. L. Bridot, A. C. Faure, S. Laurent, C. Riviere, C. Billotey, B. Hiba, M. Janier, V. Josserand, J. L. Coll, L. Vander Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS PubMed.
- M. Ahren, L. Selegard, A. Klasson, F. Soderlind, N. Abrikossova, C. Skoglund, T. Bengtsson, M. Engstrom, P.-O. Kall and K. Uvdal, Langmuir, 2010, 26, 5753 CrossRef CAS PubMed.
- J. L. Bridot, A. C. Faure, S. Laurent, C. Riviere, C. Billotey, B. Hiba, M. Janier, V. Josserand, J. L. Coll, L. V. Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS PubMed.
- G. Tian, Z.-J. Gu, X.-X. Liu, L.-J. Zhou, W.-Y. Yin, L. Yan, S. Jin, W.-L. Ren, G.-M. Xing, S.-J. Li and Y.-L. Zhao, J. Phys. Chem. C, 2011, 115, 23790 CAS.
- E. Hemmer, H. Takeshita, T. Yamano, T. Fujiki, Y. Kohl, K. Low, N. Venkatachalam, H. Hyodo, H. Kishimoto and K. Soga, J. Mater. Sci.: Mater. Med., 2012, 23, 2399 CrossRef CAS PubMed.
- L.-J. Zhou, Z.-J. Gu, X.-X. Liu, W.-Y. Yin, G. Tian, L. Yan, S. Jin, W.-L. Ren, G.-M. Xing, W. Li, X.-L. Chang, Z.-B. Hu and Y.-L. Zhao, J. Mater. Chem., 2012, 22, 966 RSC.
- S.-K. Sun, L.-X. Dong, Y. Cao, H.-R. Sun and X.-P. Yan, Anal. Chem., 2013, 85, 8436 CrossRef CAS PubMed.
- Y. Zou, L. Tang, J.-L. Cai, L.-T. Lin, L.-W. Cao and J.-X. Meng, J. Lumin., 2014, 153, 210 CrossRef CAS.
- H. Fukada, K. Ueda, J.-i. Ishino, T. Miyata and T. Minami, Thin Solid Films, 2010, 518, 3067 CrossRef CAS.
- G. Liu, Y. Zhang, J. Yin and W.-F. Zhang, J. Lumin., 2008, 128, 2008 CrossRef CAS.
- X.-T. Wei, Y.-H. Chen, X.-R. Cheng, M. Yin and W. Xu, Appl. Phys. B: Lasers Opt., 2010, 99, 763 CrossRef CAS.
- M. A. Hayward, M. A. Green, M. J. Rosseinsky and J. Sloan, J. Am. Chem. Soc., 1999, 121, 8843 CrossRef CAS.
- M. A. Hayward, E. J. Cussen, J. B. Claridge, M. Bieringer, M. J. Rosseinsky, C. J. Kiely, J. Blundell, I. M. Marshall and F. L. Pratt, Science, 2002, 295, 1882 CrossRef CAS PubMed.
- M. A. Hayward, Chem. Mater., 2005, 17, 670 CrossRef CAS.
- Y. Tsujimoto, C. Tassel, N. Hayashi, T. Watanabe, H. Kageyama, K. Yoshimura, M. Takano, M. Ceretti, C. Ritter and W. Paulus, Nature, 2007, 450, 1062 CrossRef CAS PubMed.
- Y. Kobayashi, O. J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami, A. Kitada, M. Ohkura, S. Hosokawa, Z. Li, K. Hayashi, Y. Kusano, J. Kim, N. Tsuji, A. Fujiwara, Y. Matsushita, K. Yoshimura, K. Takegoshi, M. Inoue, M. Takano and H. Kageyama, Nat. Mater., 2012, 11, 507 CrossRef CAS PubMed.
- G. K. Behrh, H. Serier-Brault, S. Jobic and R. Gautier, Angew. Chem., Int. Ed., 2015, 54, 11501 CrossRef PubMed.
- F.-X. Zhang, M. Liang, J.-W. Wang, U. Becker and R.-C. Ewing, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 064114 CrossRef.
- R. K. Tammrakar, D. P. Bisen and N. Brahme, J. Radiat. Res. Appl. Sci., 2014, 7, 550 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- J. Zarembowitch, J. Gouteron and A. M. Lejus, J. Raman Spectrosc., 1980, 9, 263 CrossRef CAS.
- C. Le Luyer, A. Garcia-Murillo, E. Bernstein and J. Mugnier, J. Raman Spectrosc., 2003, 34, 234 CrossRef CAS.
- E. R. Aluri and A. P. Grosvenor, Phys. Chem. Chem. Phys., 2013, 15, 10477 RSC.
| This journal is © The Royal Society of Chemistry 2016 |