
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of γ-alkylidenebutenolides isolated from Melodorum fruticosum: the role of the propylidene-type side chain structure on anti-melanogenic activity†
Katsuki
Takashima
 a,
Yoshiaki
Manse
b,
Riko
Suzuki
a,
Nanami
Masuda
a,
Yuki
Fukuda
a,
Risa
Fukuda
a,
Shinsuke
Marumoto
c,
Fumihiro
Ishikawa
a,
Toshio
Morikawa
*b and
Genzoh
Tanabe
*ab
a,
Yoshiaki
Manse
b,
Riko
Suzuki
a,
Nanami
Masuda
a,
Yuki
Fukuda
a,
Risa
Fukuda
a,
Shinsuke
Marumoto
c,
Fumihiro
Ishikawa
a,
Toshio
Morikawa
*b and
Genzoh
Tanabe
*ab
aFaculty of Pharmacy, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
bPharmaceutical Research and Technology Institute, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
cJoint Research Centre, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
First published on 8th April 2025
Abstract
The first structure–activity relationship study on γ-alkylidenebutenolides [(4Z)- and (4E)-6,7-dihydroxyhepta-2,4-dien-4-olide 6-monobenzoate] (4Z, 3 and 4E, 4) and 7-monobenzoate (4Z, 1 and 4E, 2) which are potent melanogenesis inhibitors (IC50 = 0.3–2.9 μM) isolated from the medicinal plant Melodorum fruticosum, was carried out using the related analogous (7–21). The inhibitory activities of the (4Z)- and (4E)-6,7-dideoxylated compounds (7) completely disappeared, while those of the 6-oxo-7-hydroxy-type compounds (4Z, 20 and 4E, 21) were significantly attenuated compared with those of 1–4. By contrast, the 6,7-dihydroxylated compounds (4Z, 8 and 4E, 9; IC50 = 5.8–1.5 μM) showed inhibitory activities similar to those of 1–4, suggesting that the two hydroxyl groups at positions C6 and C7 on the side chain play an essential role in the onset of potent inhibitory activity. The inhibitory activities of the 6,7-diacylated analogues (10–19, acyl group = Ac, Piv, or Bz; IC50 = 0.7–3.3 μM) were also nearly identical to those of the 6- or 7-monobenzoates (1, 3, and 4), regardless of the geometry of the double bond. However, acylation of the strongest inhibitor (4E, 2; IC50 = 0.3 μM) at position C6, reduced the activity by approximately 1/3 to 1/5. This result suggests the importance of the cooperative role of the acyl moiety at position C7 and the hydroxyl group at position C6 in the E-configured double bond. The total synthesis of the natural products melodorinone A (20) and melodorinone B (21) was also achieved during the synthesis of the γ-alkylidenebutenolide analogues.
Introduction
The ring system of 2,5-dihydrofuran-2-one [furan-2(5H)-one], also known as Δα,β-butenolide, is an important core shared by a number of natural products.1 A large number of Δα,β-butenolide derivatives with an exocyclic double bond at the γ-position have been found in nature. These derivatives are called γ-alkylidenebutenolides, and many of them have been reported to exhibit beneficial biological activities. For example, the simplest γ-alkylidenebutenolide, protoanemonin (A), exhibits various biological activities, such as cytotoxic, antibacterial, and antifungal activities.2–7 Bioactive γ-alkylidenebutenolides with more complex structures such as enhydrolide (B),8 piptocarfine A (C),9 and EBC-329 (D),10 exhibit antibacterial, cytotoxic, and antiproliferative activities, respectively, and cryptoconcatone I (E),11 and aspergone B (F),12 inhibit nitric oxide production and α-glucosidase activity, respectively (Fig. 1). Therefore, these natural products have attracted considerable attention as important sources of new therapeutic and physiological agents. In this context, the total synthesis13–17 and development of synthetic methods18–20 for these compounds have been actively investigated by many organic and bioorganic chemists. | ||
| Fig. 1 Chemical structures of representative natural γ-alkylidenebutenolides. | ||
Melodorinol (1), isomelodorinol (2), fruticosinol (3), and isofruticosinol (4) are isolated from the medicinal plant Melodorum fruticosum (also known as devil's tree or white cheesewood), which is endemic to Vietnam, Laos, Cambodia, and Thailand,21 and have a hepta-2,4-dien-4-olide (also called γ-propylidene-Δα,β-butenolide) structure belonging to the γ-alkylidenebutenolide family. The chiral HPLC analysis of optically pure (S)- and (R)-butenolides [(S)-1–(S)-4 and (R)-1–(R)-4], independently synthesised in our laboratory from D- and L-ribose, respectively, revealed that all isolates (1–4) from this plant are partially racemic mixtures predominantly containing the S-isomer (∼66% ee).22 Additionally, as part of our ongoing search for bioactive constituents from folk medicinal plants, we demonstrated that both enantiomers of butenolides 1–4 are potent melanogenesis inhibitors, regardless of their stereochemistry at position C6. (R)-2 and (S)-2 (IC50 = 0.29–0.39 μM) are particularly active compounds, exhibiting inhibitory activities that are 400–500 times more potent than that of arbutin (5, IC50 = 174 μM), a standard skin-lightening agent with potent melanin production inhibitory activity (Fig. 2). Therefore, these γ-alkylidenebutenolides can be considered ideal seeds for melanogenesis inhibitors.
 | ||
| Fig. 2 Chemical structures of γ-alkylidenebutenolides isolated from Melodorum fruticosum and arubutin, a reference standard for skin-whitening. *Melanogenesis inhibitory activity (IC50 value, μM). | ||
Melanin is a pigment that determines the colour of human hair, eyes, and skin. Normal melanin pigmentation is involved in many beneficial functions, such as UV shielding, photocarcinogenesis inhibition, and vitamin D3 biosynthesis.23 Abnormal melanin pigmentation causes serious dermatological issues, such as senile lentigines, freckles, melasma, and other melanin hyperpigmentations. Therefore, controlling melanogenesis may be an important approach for treating pigmentation-related disorders. Some phenomena associated with melanin accumulation may also be correlated with the development of Parkinson's disease, Alzheimer's disease, and melanoma.24,25 Thus, melanogenesis regulators are required for the chemical-based functional analysis of central nervous system diseases and malignancies. However, synthetic access to melanogenesis inhibitors and our understanding of their mode of action are currently limited.
As a continuation of our research project, we designed and synthesised relevant γ-alkylidenebutenolide-based analogues and evaluated their anti-melanogenic inhibitory activity to gain new insights into the design of better melanogenesis inhibitors. The synthesised materials were (a) compounds 6 and 7, which are the main carbon skeletons of natural products 1–4, (b) diols (S)-8 and (S)-9, (c) both enantiomers of diesters (S)-10–(S)-19 and (R)-10–(R)-19, and (d) ketones 20 and 21. This study is the first to examine the structure–activity relationship underlying the inhibition of melanogenesis based on the structure of γ-alkylidenebutenolides originating from M. fruticosum. Furthermore, we also report the first synthesis of ketones 20 (melodrinone A) and 21 (melodrinone B), which are cytotoxic natural products also isolated from M. fruticosum (Fig. 3).
 | ||
| Fig. 3 Designed analogues 6–21. | ||
Results and discussion
Compounds 6 and 7 were synthesised as shown in Scheme 1. According to the method reported by Jefford et al., 2-(trimethylsilyloxy)furan (22) was alkylated with 1-iodopropane in the presence of silver trifluoromethanesulfonate to afford 6,26 albeit in low yield. The TMSOTf-induced aldol reaction of the furan derivative 22 with propanal afforded 23 as a diastereomeric mixture (erythro/threo ratio = ca. 1/4). The subsequent E1cb elimination of the hydroxyl group of 23 with acetic anhydride (Ac2O) under basic conditions afforded 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 as a mixture of E- and Z-isomers (E/Z ratio = ca. 2/3).
27 as a mixture of E- and Z-isomers (E/Z ratio = ca. 2/3).
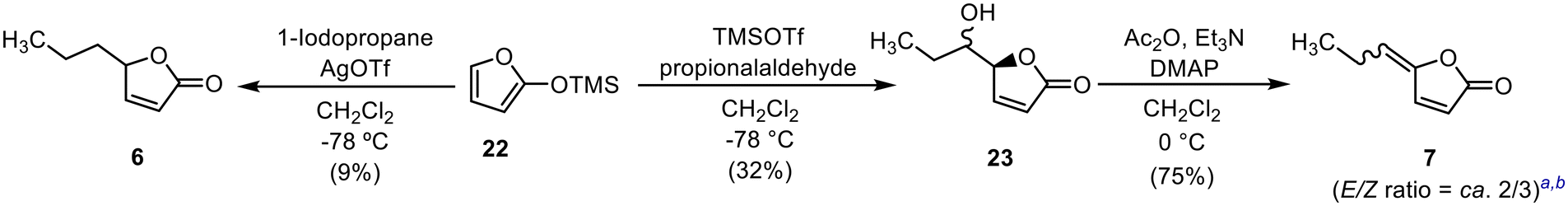 | ||
| Scheme 1 Synthesis of analogues 6 and 7: athe stereochemistry of the E- and Z-isomers was confirmed by NOESY experiments; bthe E/Z ratio was estimated by 1H NMR analysis. | ||
Next, the dihydroxylated compounds (S)-8 and (S)-9 were synthesised from a known α,β-unsaturated ester 25 prepared from D-ribose (24) according to the procedure22 used to synthesise natural products 3 and 4 (Scheme 2). The secondary hydroxyl group of 25 was protected with a TBDPS group to quantitatively afford silyl ether 26, which was then heated in aqueous acetic acid at 100 °C to afford the corresponding butenolide 27 in 44% yield. The IR spectrum of 27 showed two characteristic absorption bands at higher wavenumbers (1786 and 1747 cm−1) than the single band of its precursor 26 (1720 cm−1), thus supporting the formation of the α-unsubstituted Δα,β-butenolide ring. The HMBC correlation between the signals corresponding to H-4 and C-1, as shown in Scheme 2, confirmed these findings. The 1H NMR spectrum of 27 showed two signals at δH 6.03 and 7.21, which were attributed to the α and β protons of the Δα,β-butenolide ring, respectively, with a J2–3 value of 5.8 Hz, indicating a cis-configuration of the olefin moiety. Subsequently, 27 was subjected to an E1cb elimination reaction by treatment with benzoic anhydride under basic conditions to afford (Z)-γ-alkylidenebutenolide 28 and its E-isomer 29 in 25% and 57% yields, respectively. The stereochemistries of the newly introduced exo-double bonds in 28 and 29 were determined to be cis and trans, respectively, based on nuclear Overhauser effect spectroscopy (NOESY) experiments, as shown in Scheme 2. Finally, desilylation of 28 and 29 using tetrabutylammonium fluoride (TBAF) in the presence of excess acetic acid (3 equiv.) in THF afforded S-8 and S-9 in 97% and 94% yields, respectively. S-8 and S-9 showed the same NOESY correlations as their corresponding materials (28 and 29), supporting the stereochemistries of the exo-double bonds depicted in Scheme 2.
 | ||
| Scheme 2 Synthesis of dihydroxylated analogues (8 and 9). | ||
(S)-Melodorinol [(S)-1], (S)-isomelodorinol [(S)-2], (S)-fruticosinol [(S)-3], and (S)-isofruticosinol [(S)-4] were synthesised from D-ribose according to our method22 and then further converted into the corresponding diesters [(S)-10–(S)-19] in good yields by reaction with acylating reagents, such as Ac2O, pivaloyl chloride (PivCl), and benzoyl chloride (BzCl). The enantiomers [(R)-1–(R)-4] of (S)-1–(S)-4 were similarly synthesised from L-ribose (L-24) and converted into the corresponding acetates [(R)-13 and (R)-18] and pivaloates [(R)-14 and (R)-19]. Finally, the secondary hydroxyl groups in (S)-1 and (S)-2 were oxidised with Dess–Martin periodinane to give 20 and 21 in 82 and 86% yields, respectively. The 1H and 13C NMR properties of synthesised 20 and 21 were in good agreement with those of natural melodorinone A (20)28 and melodorinone B (21) (Scheme 3).28
 | ||
| Scheme 3 Synthesis of diesters [(S)-10–(S)-19 and (R)-10–(R)-19] and total synthesis of melodorinones (20 and 21). | ||
With all the target compounds in hand, we examined their melanogenesis inhibitory activities in theophylline-stimulated B16 melanoma 4A5 cells (Table 1 and ESI Tables S3 and S4†). First, the inhibitory activities of Δαβ-butenolides 6 and 7 were evaluated to determine the correlation between the carbon skeletons of 1–4 and their activity. Compound 6, a potential Michael acceptor, showed no inhibitory activity even at concentrations as high as 100 mM. Similarly, γ-propylidene-Δα,β-butenolide (7), the main carbon skeleton of 1–4, exhibited no inhibitory activity, suggesting that oxygen functionalization of the side chain, as observed in the structures of 1–4, is required for the onset of strong inhibitory activity (entries 17 and 18 vs. 1, 2, 19, and 20). Bioassays of the diols (S)-8 and (S)-9 demonstrated that they were both significantly more potent inhibitors than 7, a nonhydroxylated butenoide, with IC50 values of 5.8 and 1.5 μM, respectively (entries 3 and 22 vs. 18). These results confirm the validity of our prediction of the importance of oxygen functionalisation. Next, the activity of the diol S-8, which has a Z-configured double bond, was compared with those of the corresponding mono-O-benzoates [(S)-1 and (S)-3]. As shown in Table 1, O-benzoylation of the hydroxyl group at either position 6 or 7 of the side chain led to slightly increased inhibitory activity (entries 3 vs. 1 and 2). A comparison of the activities of the E-isomer [(S)-9] of (S)-8 and its corresponding benzoates [(S)-2 and (S)-4] revealed that O-benzoylation at position C6 did not lead to an increase in inhibitory activity; interestingly, however, O-benzoylation at position C7 strengthened this activity by one order of magnitude (entries 22 vs. 19 and 20).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | R1 | R2 | IC50 (μM) | Entry | Compound | R1 | R2 | IC50 (μM) |
| 1 | S-Melodorinol (S-1) | H | Bz | 2.7, (lit.22 2.5) | 19 | S-Isomelodorinol (S-2) | H | Bz | 0.2922 |
| 2 | S-Fruticosinol (S-3) | Bz | H | 1.522 | 20 | S-Isofruticosinol (S-4) | Bz | H | 0.8022 |
| 3 | S-8 | H | H | 5.8 | 22 | S-9 | H | H | 1.5 |
| 4 | S-10 | Ac | Bz | 0.67 | 23 | S-15 | Ac | Bz | 1.4 |
| 5 | S-11 | Piv | Bz | 0.92 | 24 | S-16 | Piv | Bz | 0.88 |
| 6 | S-12 | Bz | Bz | 2.0 | 25 | S-17 | Bz | Bz | 1.5 |
| 7 | S-13 | Bz | Ac | 2.9 | 26 | S-18 | Bz | Ac | 0.89 |
| 8 | S-14 | Bz | Piv | 1.1 | 27 | S-19 | Bz | Piv | 2.0 |
| 9 | R-Melodorinol (R-1) | H | Bz | 2.922 | 28 | R-Isomelodorinol (R-2) | Bz | Bz | 0.3922 |
| 10 | R-Fruticosinol (R-3) | Bz | H | 1.522 | 29 | R-Isofruticosinol (R-4) | H | H | 1.022 |
| 11 | R-10 | Ac | Bz | 1.0 | 30 | R-15 | Ac | Bz | 1.2 |
| 12 | R-11 | Piv | Bz | 0.82 | 31 | R-16 | Piv | Bz | 1.8 |
| 13 | R-12 | Bz | Bz | 3.3 | 32 | R-17 | Bz | Bz | 2.4 |
| 14 | R-13 | Bz | Ac | 3.3 | 33 | R-18 | Bz | Ac | 1.1 |
| 15 | R-14 | Bz | Piv | 1.2 | 34 | R-19 | Bz | Piv | 0.89 |
| 16 | Melodorinone A (20) | 71 | 35 | Melodorinone B (21) | 33 | ||||
| 17 | 6 | >100 | 36 | Arbutin (5) | 17422 | ||||
| 18 | 7 | >100 | |||||||

The above results motivated us to evaluate the effect of introducing a benzoyl group to the side chains of potent inhibitors [(S)-1–(S)-4]. In addition to the benzoyl group, the substitution effects of the relatively polar Ac group and bulky Piv group were also evaluated. Among the five Z-type diesters [(S)-10–(S)-14], (S)-10 exhibited a slight increase in inhibitory activity (entry 4, IC50 = 0.67 μM), but the potencies of the other compounds [(S)-11–(S)-14] remained nearly identical (entries 5–8, IC50 = 2.9–0.92 μM) to those of their parent compounds [(S)-1 and (S)-3]. The corresponding E-type diesters [(S)-15–(S)-19] also showed good inhibitory activities, with IC50 values ranging from 2.0 to 0.89 μM (entries 23–27). Thus, diacylation of the side chain did not lead to enhancements in inhibitory activity. On the contrary, the inhibitory activities of (S)-15, (S)-16 and (S)-17 were unexpectedly reduced to one-third to one-fifth that of the most potent inhibitor [(S)-2], indicating that acylation of (S)-4 at position C7 is not a suitable structural modification. Additionally, the 10 6R-diesters [(R)-10–(R)-14 and (R)-15–(R)-19] showed inhibitory activities similar to those of the corresponding 6S-diesters [(S)-10–(S)-14 and (S)-15–(S)-19]. The enantiomers of the natural products (1–4) showed the same tendency as the diesters, suggesting that the stereoconfiguration of C6, regardless whether the substituent is –OH or –OCOR, plays a minor role in its activity. Melodrinone A (20) and melodrinone B (21) exhibited melanogenesis inhibitory activity with IC50 values of 71 and 33 μM, respectively. The IC50 of 20 was one order of magnitude lower than that of the corresponding alcohols [(S)-1 and (R)-1] while that of 21 was two orders of magnitude lower than that of the corresponding alcohols [(S)-2 and (R)-2] (entries 1 and 9 vs. 16, entries 19 and 28 vs. 35), suggesting that the presence of an oxygen atom with an sp3 hybridisation orbital at position C6 is an important factor in the onset of potent inhibitory activity for this group of compounds.
Conclusions
In summary, we synthesised 26 analogues of natural γ-alkylidenebutenolides (1–4) and achieved the first total synthesis of melodorinone A (20) and melodorinone B (21). Evaluation of the inhibitory activity of all synthetics suggested that γ-alkylidenebutenolides require an E-configured side chain to express strong melanogenesis inhibitory activity. The importance of the cooperative role of the acyl moiety at position C7 and the oxygen atom with an sp3 hybrid orbital at position C6 was also highlighted. We confirmed that the stereochemistry of the C6 centre bearing the oxygen functional group did not contribute to the onset of potent inhibitory activity. Further studies on the structure–activity relationships of γ-alkylidenebutenolides with potential melanogenesis inhibitory activity are ongoing in our laboratory.Experimental section
Chemistry
5-Propyfuran-2(5H)-one (6). A mixture of 2-(trimethylsilyloxy)furan (22, 2.0 mL, 12.2 mmol), 1-iodopropane (1.54 mL, 15.9 mmol), silver trifluoroacetate (4.1 g, 15.9 mmol), and CH2Cl2 (20 mL) was stirred in the dark at −78 °C for 5 h. The reaction mixture was filtered and the filtrate was condensed in vacuo. The residue was purified by column chromatography (n-hexane/ether = 10/1) to give the title compound26 (6, 133 mg, 1.1 mmol, 9%) as a pale yellow oil. 1H-NMR (500 MHz, CDCl3) δ: 0.97 (3H, t, J = 7.4, CH2CH2CH3), 1.46–1.52 (2H, m, CH2CH2CH3), 1.61–1.69/1.71–1.79 (each 1H, m, CH2CH2CH3), 5.06 (1H, dddd, J = 7.4, 5.5, 1.8, 1.6, H-5), 6.11 (1H, dd, J = 5.6, 1.8, H-4), 7.48 (1H, dd, J = 5.6, 1.6, H-2). 13C-NMR (125 MHz, CDCl3) δ: 13.7 (CH2CH2CH3), 18.3 (CH2CH2CH3), 35.1 (CH2CH2CH3), 83.2 (C-5), 121.3 (C-3), 156.3 (C-4), 173.1 (C-2). HRMS (ESI) m/z: [M + Na]+ calcd for C7H10O2Na, 149.0573; found, 149.0574.
5-(1-Hydroxypropyl)furan-2(5H)-one (23). A mixture of 2-(trimethylsilyloxy)furan (29, 158 mg, 1.0 mmol), propionaldehyde (80 μL, 1.2 mmol), TMSOTf (40 μL, 0.20 mmol), and CH2Cl2 (3.0 mL) was stirred at −78 °C for 3 h. The reaction was quenched with 10% hydrochloric acid (1.0 mL) and the resulting mixture was extracted with CH2Cl2 (5 mL × 3). The extract was washed with brine (5 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 10/1) to give the title compound 23
29 (45 mg, 0.32 mmol, 32%) as a ca. 1:4 mixture of erythro and threo isomers. The HRMS (ESI) spectrum of the mixture of erythro- and threo-23 showed two peaks due to [M + Na]+ ions at m/z 165.0524 (for calcd for C7H10O3Na, 165.0522).
Compound erythro-23 (NMR data extracted from the spectrum of a mixture of erythro- and threo-23): 1H-NMR (800 MHz, CDCl3) δ: 1.06 (3H, t, J = 7.6, CHCH2CH3), 1.54–1.73 (2H, m, CHCH2CH3), 2.44 (1H, br s, OH), 3.79 (1H, br td, J = ca. 8.5, 5.0, H-6), 4.98 (1H, ddd, J = 5.0, 2.1, 1.5, H-5), 6.19 (2H, dd, J = 6.0, 2.1, H-3), 7.56 (1H, dd, J = 6.0, 1.5, H-4). 13C-NMR (200 MHz, CDCl3) δ: 9.9 (CHCH2CH3), 26.2 (CHCH2CH3), 72.8 (C-6), 85.8 (C-5), 122.7 (C-3), 153.7 (C-4), 173.0 (C-2).
Compound threo-23 (NMR data extracted from the spectrum of a mixture of erythro- and threo-23): 1H-NMR (800 MHz, CDCl3) δ: 1.05 (3H, t, J = 7.6, CHCH2CH3), 1.60–1.70 (2H, m, CHCH2CH3), 2.35 (1H, br s, OH), 3.70 (1H, br td, J = ca. 8.2, 4.6, H-6), 5.02 (1H, ddd, 4.6, 2.1, 1.6, H-5), 6.19 (2H, dd, J = 6.0, 2.1, H-3), 7.48 (1H, dd, J = 6.0, 1.6, H-4). 13C-NMR (200 MHz, CDCl3) δ: 9.9 (CHCH2CH3), 26.3 (CHCH2CH3), 73.0 (C-6), 85.9 (C-5), 122.6 (C-3), 153.8 (C-4), 173.0 (C-2).
E1cb elimination reaction of erythro- and threo-23. A mixture of erythro- and threo-23 (200 mg, 1.4 mmol), Et3N (0.58 mL, 4.2 mmol), Ac2O (0.27 mL, 2.8 mmol), DMAP (17 mg, 0.14 mmol), and CH2Cl2 (10 mL) was stirred at room temperature (rt) for 18 h. The reaction mixture was poured into cold water (20 mL) and extracted with CH2Cl2 (7 mL × 3). The extract was successively washed with aqueous NaHCO3 (7 mL) and brine (7 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/ether = 10/1) to give a ca. 2
:3 mixture of E- and Z-5-propylidenefuran-2(5H)-one27 [hepta-2,4-diene-4-olide (E- and Z-7, 132 mg, 1.06 mmol, 75%)]. The HRMS (ESI) spectrum of the mixture of E- and Z-7 showed two peaks due to [M + Na]+ ions at m/z 125.0598 (calcd for C7H9O2, 125.0597).
Compound E-7 (NMR data extracted from the spectrum of a mixture of E- and Z-7): 1H-NMR (800 MHz, CDCl3) δ: 1.13 (3H, t, J = 7.5, H-7), 2.32 (2H, dq, J = 8.3, 7.5, H-6), 5.78 (1H, ddd-like, J = ca. 8.3, 1.8, 0.8, H-5), 6.19 (1H, dd, J = 5.6, 1.8, H-2), 7.64 (1H, dd, J = 5.6, 0.8, H-3); 13C-NMR (200 MHz, CDCl3) E-20: δ: 14.4 (C-7), 20.1 (C-6), 118.2 (C-5), 120.1 (C-2), 139.3 (C-3), 149.5 (C-4), 170.0 (C-1).
Compound Z-7 (NMR data extracted from the spectrum of a mixture of E- and Z-7): 1H-NMR (800 MHz, CDCl3) δ: 1.11 (3H, t, J = 7.6, H-7), 2.43 (2H, quin, J = 7.6, 0.4, H-6), 5.29 (1H, br t-like, J = ca.7.6, H-5), 6.14 (1H, dd, J = 5.3, 0.6, H-2), 7.33 (1H, br d-like, J = ca. 5.3, 0.4, H-3); Z-20: δ: 13.4 (C-7), 19.9 (C-6), 119.0 (2C, C-2 and C-5), 143.6 (C-3), 149.2 (C-4), 170.1 (C-1).
Silylation of (2Z)-7-O-(tert-butyldiphenylsilyl)-2,3-dideoxy-4,5-O-isopropylidene-D-ribohept-2-enoate (25). A mixture of 25
22 (3 g, 6.28 mmol), AgNO3 (3.1 g, 18.8 mmol), TBDPSCl (4.9 mL, 18.8 mmol), pyridine (3.0 mL, 37.7 mmol), and CH3CN (20 mL) was stirred in the dark at rt for 2.5 h. After the reaction mixture was diluted with EtOAc (30 mL), the resulting suspension was filtered through Celite, and the filter cake was washed with EtOAc. The combined filtrate and washings were successively washed with ice-cold 10% aqueous sulfuric acid (20 mL × 2) and brine (20 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 50/1) to give (2Z)-6,7-bis-O-(tert-butyldiphenylsilyl)-2,3-dideoxy-4,5-O-isopropylidene-D-ribohept-2-enoate (26, 4.5 g, 6.20 mmol, 99%) as a colourless oil. [α]25D −68.4 (c = 0.96, CHCl3). IR (neat): 1720, 1589, 1473, 1427, 1381, 1222, 1111, 1072 cm−1. 1H NMR (800 MHz, CDCl3) δ: 0.96/1.07 [each 9H, s, (CH3)3CSi], 1.35/1.41 [each 3H, s, (CH3)2C], 3.48 (2H, dd, J = 11.1, 4.0, H-7a), 3.61 (3H, s, CO2CH3) 3.76 (2H, dd, J = 11.1, 6.2, H-7b), 3.91 (1H, ddd, J = 7.2, 6.2, 4.0, H-6), 4.62 (1H, dd, J = 7.2, 2.9, H-5), 5.54 (1H, dd, J = 11.5, 1.3, H-2), 5.58 (1H, ddd, J = 8.4, 2.9, 1.3, H-4), 5.83 (1H, dd, J = 11.5, 8.4, H-3), 7.24–7.72 (20H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.1/19.3 [(CH3)3CSi], 24.5/26.5 [(CH3)2C], 26.8(2C)/26.93(2C)/26.96(2C) [(CH3)3CSi], 51.4 (CO2CH3), 65.9(C-7), 73.0 (C-4), 73.8 (C-6), 80.0 (C-5), 108.5 [(CH3)2C], 120.7 (C-2), 127.75/127.81/129.85/129.89/135.5/135.6 (each 2C, d, arom.), 132.8/132.3 (each 2C, s, arom.), 145.8 (C-3), 165.6 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C43H54O6NaSi2, 745.3351; found, 745.3347.
Lactonisation of the Bis-O-TBDP ester (26). A mixture of 26 (7.5 g, 10.3 mmol), acetic acid (66 mL), and H2O (11 mL) was heated at 100 °C for 2 h. After being cooled, the reaction mixture was poured into cold water (300 mL). The resulting mixture was neutralised with sodium hydrogen carbonate and extracted with ethyl acetate (100 mL × 3). The extract was washed with brine (150 mL) and condensed in vacuo to give a pale yellow oil (7.4 g). The residue was purified by column chromatography (n-hexane/EtOAc = 100/1) to give (4S,5S,6R)-6,7-bis-O-(tert-butyldiphenylsilyl)-5-hydroxy-2-hepten-4-olide (27, 3.0 g, 4.56 mmol, 44%) as a colourless oil. [α]25D −62.6 (c = 0.78, CHCl3). IR (neat): 1786, 1747, 1589, 1464, 1427, 1392, 1361, 1080 cm−1. 1H NMR (800 MHz, CDCl3) δ: 1.04/1.05 [each 9H, s, (CH3)3C], 2.77 (1H, d, J = 5.3, OH), 3.80 (1H, dd, J = 10.9, 2.9, H-7a), 3.84 (1H, dd, J = 10.9, 5.1, H-7b), 3.84–3.88 (2H, m, H-6 and H-5), 5.13 (1H, ddd, J = 5.6, 1.9, 1.6, H-4), 6.03 (1H, dd, J = 5.8, 1.9, H-2), 7.21 (1H, dd, J = 5.8, 1.6, H-3), 7.27–7.62 (20H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.1/19.2 [(CH3)3CSi], 26.8/27.0 [each 3C, (CH3)3CSi], 65.0 (C-7), 72.8 (C-5), 73.7 (C-6), 83.1 (C-4), 122.1 (C-2), 127.7/127.8/127.9/128.0/129.9/129.96/130.00/130.2/135.5/135.6/135.7/135.8 (d, arom.), 132.4/132.5/132.6/133.1 (s, arom.), 154.4 (C-3), 172.8 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C39H46O5NaSi2, 673.2276; found, 673.2771.
E1cb elimination reaction of the butenolide (27). A mixture of 27 (3.0 g, 4.60 mmol), Et3N (1.6 mL, 11.4 mmol), DMAP (567 mg, 4.65 mmol), Bz2O (0.53 mL, 5.6 mmol), and CH2Cl2 (10 mL) was stirred at rt for 0.5 h. The reaction mixture was poured into cold water (30 mL) and extracted with CH2Cl2 (10 mL × 3). The extract was washed with brine (15 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 10/1 → 5/1) to give (4Z,6S)-6,7-bis-O-(tert-butyldiphenylsilyloxy)-2,4-heptadiene-4-olide (28, 728 mg, 1.15 mmol, 25%) and its E-isomer 29 (1.66 g, 2.62 mmol, 57%).
Z-Isomer ( 28 ): [α]25D −26.4 (c = 0.72, CHCl3). IR (neat): 1789, 1763, 1670, 1469, 1427, 1361, 1056 cm−1. 1H NMR (800 MHz, CDCl3) δ: 1.01/1.07 [each 9H, s, (CH3)3C], 3.68 (1H, dd, J = 10.3, 5.1, H-7a), 3.77 (1H, dd, J = 10.3, 5.1, H-7b), 4.98 (1H, dt, J = 8.9, 5.1, H-6), 5.11 (1H, ddd, J = 8.9, 0.7, 0.3, H-5), 6.00 (1H, dd, J = 5.4, 0.7, H-2), 7.02 (1H, dd, J = 5.4, 0.3, H-3), 7.26–7.65 (20H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.16/19.20 [(CH3)3C], 26.7/26.9 [(CH3)3C], 67.7 (C-7), 69.2 (C-6), 116.3 (C-5), 120.0 (C-2), 143.3 (C-3), 127.4/127.5/127.6/129.6/129.7/129.8/135.59/135.61/135.8/135.9 (each 2C, d, arom.), 133.17/133.27/133.30/133.7(s, arom.), 148.6 (C-4), 169.2 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C39H44O4NaSi2, 655.2670; found, 655.2663.
E-Isomer ( 29 ) [α]25D −54.5 (c = 0.90, CHCl3). IR (neat): 1782, 1762, 1678, 1470, 1427, 1107 cm−1. 1H NMR (800 MHz, CDCl3) δ: 0.88/0.95 [each 9H, s, (CH3)3C], 3.62 (1H, dd, J = 10.1, 6.6, H-7a), 3.80 (1H, dd, J = 10.1, 4.9, H-7b), 4.43 (1H, ddd, J = 9.3, 6.6, 4.9, H-6), 5.66 (1H, ddd, J = 9.3, 1.8, 0.7, H-5), 5.91 (1H, dd, J = 5.6, 1.8, H-2), 6.81 (1H, dd, J = 5.6, 0.7, H-3), 7.25–7.61 (20H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.1/19.2 [(CH3)3C], 26.7/26.8 [(CH3)3C], 67.6 (C-7), 70.1 (C-6), 115.5 (C-5), 120.2 (C-2), 127.7(6C)/129.78/128.88/129.95/135.5/135.6/135.7/135.9 (each 2C, d, arom.), 132.94(2C)/133.1/133.2 (s, arom.), 140.1 (C-3), 150.6 (C-4), 169.4 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C39H44O4NaSi2, 655.2670; found, 655.2667.
Desilylation of the Z-isomer (28). A mixture of 28 (63 mg, 99 μmol), acetic acid (17 μL, 0.30 mmol), 1 M solution of TBAF in THF (0.25 mL, 0.25 mmol), and THF (1 mL) was stirred at rt for 1 h. The reaction mixture was condensed in vacuo, and the residue was purified by column chromatography (n-hexane/EtOAc = 1/5) to give (4Z,6S)-6,7-dihydroxy-2,4-heptadien-4-olide30–33 (S-8, 15 mg, 96 μmol, 97%) as a colourless microcrystalline solid. Mp. 65–66 °C, [α]24D +12.5 (c = 0.5, MeOH). Lit.30 a pale yellow solid, Mp. 87–89 °C, [α]22D +53.1 (c = 0.42, acetone). Lit.31 a colourless liquid [α]32D +12 (c = 0.5, MeOH). Lit.32 a colourless viscous liquid, [α]29D +8 (c = 0.6, MeOH). Lit.33 colourless oil, [α]D +13 (c = 0.17, CHCl3). IR (KBr): 1786, 1755, 1674, 1558, 1400, 1303, 1122, 1076, 1030 cm−1. 1H NMR (800 MHz, CD3OD) δ: 3.58 (2H, d, J = 5.6, H-7a and H-7b), 4.72 (1H, dt, J = 8.7, 5.6, H-6), 5.41 (1H, d, J = 8.7, H-5), 6.30 (1H, d, J = 5.4, H-2), 7.65 (1H, d, J = 5.4, H-3). 13C NMR (200 MHz, CD3OD) δ: 66.6 (C-7), 68.6 (C-6), 116.5 (C-5), 121.1 (C-2), 145.9 (C-3), 151.4 (C-4), 171.4 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C7H8O4Na, 179.0315; found, 179.0314.
Desilylation of the E-isomer (29). Following the method similar to that used for the preparation of S-8, 29 (230 mg, 0.36 mmol) was desilylated with 1 M solution of TBAF in THF (0.91 mL, 0.91 mmol) in THF (5 mL) in the presence of acetic acid (62 μL, 1.09 mmol). Work up and column chromatographic purification (n-hexane/EtOAc = 1/5) gave (4E,6S)-6,7-dihydroxy-2,4-heptadien-4-olide34 (S-9, 53 mg, 0.94 mmol, 94%) as a colourless oil. [α]25D −30.6 (c = 0.32, CHCl3). IR (neat): 1786, 1755, 1670, 1554, 1419, 1300, 1188, 1126, 1072, 1033 cm−1. 1H NMR (800 MHz, CD3OD) δ: 3.54 (1H, dd, J = 11.2, 5.5, H-7a), 3.60 (1H, dd, J = 11.2, 5.8, H-7b), 4.54 (1H, ddd, J = 8.3, 5.8, 5.5, H-6), 5.77 (1H, ddd, J = 8.3, 1.8, 0.7, H-5), 6.32 (1H, dd, J = 5.6, 1.8, H-2), 8.04 (1H, dd, J = 5.6, 0.7, H-3). 13C NMR (200 MHz, CD3OD) δ: 66.9 (C-7), 69.4 (C-6), 116.4 (C-5), 121.6 (C-2), 143.0 (C-3), 151.9 (C-4), 171.5 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C7H8O4Na, 179.0315; found, 179.0312.
(4Z,6S)-6-Acethyloxy-7-benzoyloxy-2,4-heptadien-4-olide [(S)-acetylmelodorinol (S)-10]. To a stirred solution of (S)-melodorinol (S-1, 78 mg, 0.30 mmol) in pyridine (2.0 mL) was added acetic anhydride (57 μL, 0.60 mmol) at 0 °C, and the resulting mixture was stirred at rt for 4 h. The reaction mixture was poured into cold water (6 mL) and extracted with EtOAc (5 mL × 3). The extract was successively washed with 10% aqueous H2SO4, (6 mL), aqueous NaHCO3 (6 mL), and brine (6 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 10/1) to give the title compound30–32,35 [(S)-10, 79 mg, 0.26 mmol, 87%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of EtOAc and MeOH to give (S)-10 as colourless needles. Mp. 75–76 °C, [α]27D +54.0 (c = 1.0, CHCl3). Lit.30 colourless crystal, 78–80 °C (from iPrOH/n-hexane), [α]22D +41.0 (c = 0.33, CHCl3). Lit.31 [α]22D +32.5 (c = 2, CHCl3). Lit.32 a white solid, 78–80 °C (ether/n-hexane), [α]27D +31.9 (c = 0.23, CHCl3). Lit.35 a colourless liquid, [α]25D +42.3 (c = 0.7, CHCl3). IR (KBr): 1782, 1744, 1724, 1678, 1600, 1562, 1373, 1273, 1176, 1026 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 2.11 [3H, s, C(O)CH3], 4.53 (1H, dd, J = 11.7, 6.0, H-7a), 4.57 (1H, dd, J = 11.7, 4.0, H-7b), 5.34 (1H, d, J = 8.0, H-5), 6.15 (1H, ddd, J = 8.0, 6.0, 4.0, H-6), 6.29 (1H, d, J = 5.4, H-2), 7.38 (1H, d, J = 5.4, H-3), 7.44–7.48 (2H, m, arom.), 7.56–7.61 (1H, m, arom.), 8.01–8.05 (2H, m, arom.). 13C-NMR (125 MHz, CDCl3) δ: 20.9 [C(O)CH3], 64.6 (C-7), 67.2 (C-6), 108.9 (C-5), 121.6 (C-2), 128.4(2C)/129.7(2C)/133.3 (d, arom.), 129.5 (s, arom.), 143.3 (C-3), 150.6 (C-4), 166.0 [C(O)Ph], 168.5 (C-1), 169.8 [C(O)CH3]. HRMS (ESI) m/z: [M + Na]+ calcd for C16H14O6Na, 325.0683; found, 325.0678.
In a similar manner, (R)-1 (105 mg, 0.40 mmol) gave (R)-10 (102 mg, 0.34 mmol, 84%). [α]27D −52.9 (c = 0.98, CHCl3), lit.30 [α]27D −38.8 (c = 1.25, CHCl3). 1H and 13C-NMR spectral data of (R)-10 agreed well with those of the corresponding antipode [(S)-10].
(4Z,6S)-7-Acethyloxy-6-benzoyloxy-2,4-heptadien-4-olide [(S)-13]. Following the procedure used for the preparation of (S)-10, isofruticosinol [(S)-3, 158 mg, 0.61 mmol] was converted to the title compound [(S)-13, 162 mg, 0.54 mmol, 88%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-13 as colourless needles. Mp. 85–86 °C. [α]25D −166.9 (c = 0.60, CHCl3). IR (KBr): 1793, 1778, 1751, 1685, 1566, 1450, 1365, 1285, 1103 cm−1; 1H-NMR (800 MHz, CDCl3) δ: 2.08 [3H, s, C(O)CH3], 4.45 (1H, dd, J = 11.8, 4.1, H-7a), 4.48 (1H, dd, J = 11.8, 6.4, H-7b), 5.37 (1H, ddd, J = 8.1, 0.8, 0.4, H-5), 6.20 (1H, ddd, J = 8.1, 6.4, 4.1, H-6), 6.29 (1H, ddd, J = 5.5, 0.8, H-2), 7.38 (1H, dd, J = 5.5, 0.4, H-3), 7.45–7.48 (2H, m, arom.), 7.57–7.60 (1H, m, arom.), 8.03–8.05 (2H, m). 13C-NMR (200 MHz, CDCl3) δ: 20.7 [C(O)CH3], 64.1 (C-7), 67.9 (C-6), 108.9 (C-5), 121.6 (C-2), 128.5(2C)/129.8(2C)/133.4 (d, arom.), 129.5 (s, arom.), 143.3 (C-3), 150.7 (C-4), 165.4 [C(O)Ph], 168.5 (C-1), 170.5 [C(O)CH3]. HRMS (ESI) m/z: [M + Na]+ calcd for C16H14O6Na, 325.0683; found, 325.0683.
In a similar manner, (R)-3 (132 mg, 0.51 mmol) gave (R)-13 (142 mg, 0.47 mmol, 93%). [α]25D +169.2 (c = 0.70, CHCl3); 1H and 13C-NMR spectral data of (R)-13 agreed well with those of the corresponding antipode [(S)-13].
(4E,6S)-6-Acethyloxy-7-benzoyloxy-2,4-heptadien-4-olide [(S)-15]. Following the procedure used for the preparation of S-10, isomerodorinol [(S)-2, 480 mg, 1.8 mmol] was converted to the title compound35 [(S)-15, 427 mg, 1.4 mmol, 76%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-15 as colourless needles. Mp. 61–63 °C. [α]27D −18.3 (c = 0.94, CHCl3), lit.35 a white solid, [α]27D −11.2 (c = 0.5, CHCl3). IR (KBr): 1789, 1759, 1724, 1674, 1600, 1369, 1273, 1114, 1029 cm−1. 1H-NMR (500 MHz, CDCl3) δ: 2.10 [3H, s, C(O)CH3], 4.47 (1H, dd, J = 11.7, 6.9, H-7a), 4.53 (1H, dd, J = 11.7, 4.3, H-7b), 5.73 (1H, ddd, J = 10.0, 2.0, 0.8, H-5), 5.98 (1H, ddd, J = 10.0, 6.9, 4.3, H-6), 6.35 (1H, dd, J = 5.7, 2.0, H-2), 7.48–7.49 (2H, m, arom.), 7.90 (1H, dd, J = 5.7, 0.8, H-3), 7.89–7.92 (1H, m, arom.), 8.00–8.04 (2H, m, arom.). 13C-NMR (125 MHz, CDCl3) δ: 21.0 [C(O)CH3], 65.0 (C-7), 66.7 (C-6), 107.4 (C-5), 122.5 (C-2), 128.6(2C)/129.7(2C)/133.5 (d, arom.), 129.2 (s, arom.), 140.2 (C-3), 153.4 (C-4), 165.9 [C(O)Ph], 168.7 (C-1), 170.1 [C(O)CH3]. HRMS (ESI) m/z: [M + Na]+ calcd for C16H14O6Na, 325.0683; found, 325.0673.
In a similar manner, (R)-2 (171 mg, 0.66 mmol) gave (R)-15 (158 mg, 0.52 mmol, 79%). [α]27D +19.4 (c = 1.00, CHCl3). 1H and 13C-NMR spectral data of (R)-15 agreed well with those of the corresponding antipode [(S)-15].
(4E,6S)-7-Acetyloxy-6-benzoyloxy-2,4-heptadien-4-olide [(S)-18]. Following the procedure used for the preparation of (S)-10, (S)-isofruticosinol [(S)-4, 126 mg, 0.48 mmol] was converted to the title compound [(S)-18, 127 mg, 0.42 mmol, 87%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-18 as colourless needles. Mp. 81–82 °C. [α]25D −82.5 (c = 1.03, CHCl3). IR (KBr): 1789, 1747, 1712, 1636, 1558, 1269, 1176, 1068, 1026 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 2.08 [3H, s, C(O)CH3], 4.39 (1H, dd, J = 11.9, 6.8, H-7a), 4.42 (1H, dd, J = 11.9, 4.4, H-7b), 5.74 (1H, ddd, J = 10.0, 1.8, 0.7, H-5), 6.07 (1H, ddd, J = 10.0, 6.8, 4.4, H-6), 6.36 (1H, dd, J = 5.6, 1.8, H-2), 7.45–7.48 (2H, m, arom.), 7.58–7.61 (1H, m, arom.), 7.97 (1H, dd, J = 5.6, 0.7, H-3), 8.01–8.03 (2H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 20.7 [C(O)CH3], 64.6 (C-7), 67.3 (C-6), 107.3 (C-5), 122.6 (C-2), 128.6(2C)/129.7(2C)/133.6 (d, arom.), 129.6 (s, arom.), 140.3 (C-3), 153.6 (C-4), 165.6 [C(O)Ph], 168.6 (C-1), 170.5 [C(O)CH3]. HRMS (ESI) m/z: [M + Na]+ calcd for C16H14O6Na, 325.0683; found, 325.0683.
In a similar manner, (R)-isofruticosinol [(R)-4, 133 mg, 0.51 mmol] gave (R)-16 (147 mg, 0.49 mmol, 90%). [α]25D +83.8 (c = 1.04, CHCl3). 1H and 13C-NMR spectral data of (R)-16 agreed well with those of the corresponding antipode [(S)-16].
(4Z,6S)-6-Pivaloyloxy-7-benzoyloxy-2,4-heptadien-4-olide [(S)-11]. To a stirred solution of (S)-melodorinol [(S)-1, 154 mg, 0.59 mmol] in pyridine (1.0 mL) was added pivaloyl chloride (145 μL, 1.18 mmol) at 0 °C, and the resulting mixture was stirred at rt for 16 h. The reaction mixture was poured into cold water (20 mL) and extracted with EtOAc (10 mL × 3). The extract was successively washed with 10% aqueous H2SO4 (20 mL), aqueous NaHCO3 (20 mL), and brine (20 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 6/1) to give the title compound [(S)-11, 108 mg, 0.31 mmol, 53%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-11 as colourless needles. Mp. 84–85 °C. [α]23D +13.2 (c = 1.01, CHCl3). IR (KBr): 1774, 1747, 1732, 1674, 1458, 1276, 1149, 1122, 1037 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 1.20 [9H, s, C(CH3)3], 4.55 (1H, dd, J = 11.6, 4.3, H-7a), 4.57 (1H, dd, J = 11.6, 6.5, H-7b), 5.30 (1H, br d like, J = ca. 7.9, H-5), 6.11 (1H, ddd, J = 7.9, 6.5, 4.3, H-6), 6.28 (1H, dd like, J = 5.5, 0.5, H-2), 7.38 (1H, d, J = 5.5, H-3) 7.43–7.46 (2H, m, arom.), 7.56–7.68 (1H, m, arom.), 8.01–8.02 (2H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 27.0 [C(CH3)3], 38.8 [C(CH3)3], 64.4 (C-7), 67.2 (C-6), 109.1 (C-5), 121.5 (C-2), 128.5(2C)/129.8(2C)/133.5 (d, arom.), 129.5 (s, arom.), 143.3 (C-3), 150.6 (C-4), 165.6 [C(O)Ph], 168.5 (C-1), 177.3 [C(O)C(CH3)3]. HRMS (ESI) m/z: [M + Na]+ calcd for C19H20O6Na, 367.1152; found, 367.1150.
In a similar manner, (R)-1 (76 mg, 0.29 mmol) gave (R)-11 (58 mg, 0.17 mmol, 58%). [α]27D −11.6 (c = 0.95, CHCl3). 1H and 13C-NMR spectral data of (R)-11 agreed well with those of the corresponding antipode [(S)-11].
(4Z,6S)-6-Benzoyloxy-7-pivaloxy-2,4-heptadien-4-olide [(S)-14]. Following the procedure used for the preparation of (S)-pivaloylmerodorinol [(S)-11], (S)-fruticosinol [(S)-3, 103 mg, 0.40 mmol] was converted to the title compound [(S)-14, 124 mg, 0.36 mmol, 91%] as a colourless oil. [α]25D −149.0 (c = 0.92, CHCl3). IR (neat): 1786, 1751, 1728, 1681, 1454, 1315, 1265, 1107, 1026 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 1.17 [9H, s, C(CH3)3], 4.42 (1H, dd, J = 11.7, 4.2, H-7a), 4.48 (1H, dd, J = 11.7, 6.8, H-7b), 5.35 (1H, br d like, J = ca. 8.1, H-5), 6.24 (1H, ddd, J = 8.1, 6.8, 4.2, H-6), 6.30 (1H, d, J = 5.5, 0.6, H-2), 7.38 (1H, d, J = 5.5, H-3), 7.43–7.47 (2H, m, arom.), 7.56–7.60 (1H, m, arom.), 8.02–8.05 (2H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 27.1 [C(CH3)3], 38.9 [C(CH3)3], 63.8 (C-7), 68.0 (C-6), 109.0 (C-5), 121.7 (C-2), 128.5(2C)/129.7(2C)/133.3 (d, arom.), 129.5 (s, arom.), 143.2 (C-3), 150.8 (C-4), 165.3 [C(O)Ph], 168.5 (C-1), 177.9 [C(O)C(CH3)3]. HRMS (ESI) m/z: [M + Na]+ calcd for C19H20O6Na, 367.1152; found, 367.1144.
In a similar manner, (R)-3 (104 mg, 0.40 mmol) gave (R)-14 (119 mg, 0.35 mmol, 87%). [α]25D +148.6 (c = 0.88, CHCl3). 1H and 13C-NMR spectral data of (R)-14 agreed well with those of the corresponding antipode [(S)-14].
(4E,6S)-7-Benzoyloxy-6-pivaloyloxy-2,4-heptadien-4-olide [(S)-16]. Following the procedure used for the preparation of (S)-pivaloylmerodorinol [(S)-11], (S)-isomelodorinol [(S)-2, 91 mg, 0.35 mmol] was converted to the title compound [(S)-16, 102 mg, 0.30 mmol, 85%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc give the (S)-16 as colourless needles. Mp. 94–95 °C. [α]22D −35.6 (c = 0.70, CHCl3). IR (KBr): 1790, 1751, 1720, 1678, 1604, 1454, 1396, 1153, 1126, 1037 cm−1. 1H NMR (800 MHz, CDCl3) δ: 1.18 [9H, s, (CH3)3CO], 4.51 (1H, dd, J = 11.7, 6.9, H-7a), 4.53 (1H, dd, J = 11.7, 4.6, H-7b), 5.71 (1H, ddd, J = 10.1, 1.8, 0.8, H-5), 5.94 (1H, ddd, J = 10.1, 6.9, 4.6, H-6), 6.35 (1H, dd, J = 5.7, 1.8, H-2), 7.46 (2H, m, arom), 7.59 (1H, m, arom), 7.92 (1H, dd, J = 5.7, 0.8, H-3), 8.00 (2H, m, arom). 13C NMR (200 MHz, CDCl3) δ: 64.8 (C-7), 66.5 (C-6), 107.3 (C-5), 122.4 (C-2), 128.4/129.5/133.3 (d, arom.), 129.2 (s, arom.), 140.1 (C-3), 153.4 (C-4), 165.8 [C(O)Ph], 168.6 (C-1), 177.5 [9H, s, (CH3)3CO]. HRMS (ESI) m/z: [M + Na]+ calcd for C19H20O6Na, 367.1149; found, 367.1152.
In a similar manner, (R)-2, (121 mg, 0.46 mmol) gave (R)-16 (132 mg, 0.39 mmol, 83%). [α]22D 35.4 (c = 0.91, CHCl3); 1H and 13C-NMR spectral data of (R)-16 agreed well with those of the corresponding antipode [(S)-16].
(4E,6S)-6-Benzoyloxy-7-pivaloyloxy-2,4-heptadien-4-olide [(S)-19]. Following the procedure used for the preparation of (S)-pivaloylmerodorinol [(S)-11], (S)-isofruticosinol [(S)-4, 103 mg, 0.40 mmol] was converted to the title compound [(S)-19, 117 mg, 0.34 mmol, 85%] as a colourless oil. [α]25D −59.8 (c = 0.94, CHCl3). IR (neat): 1789, 1752, 1728, 1674, 1600, 1562, 1396, 1265, 1111, 1026 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 1.17 [9H, s, C(CH3)3], 4.39 (1H, dd, J = 5.2, 4.8, H-7a), 4.41 (1H, dd, J = 6.1, 4.8, H-7b), 5.75 (1H, ddd like, J = ca. 10.0, 1.8, 0.7, H-5), 6.09 (1H, ddd, J = 10.0, 6.1, 5.2, H-6), 6.36 (1H, dd, J = 5.6, 1.8, H-2), 7.44–7.48 (2H, m, arom.), 7.57–7.61 (1H, m, arom.), 7.98 (1H, br dd like, J = ca.5.6, 0.7, H-3), 7.99–8.02 (2H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 27.1 [C(CH3)3], 38.9 [C(CH3)3], 64.4 (C-7), 67.3 (C-6), 107.5 (C-5), 122.6 (C-2), 128.6(2C)/129.7(2C)/133.6 (d, arom.), 129.3 (s, arom.), 140.3 (C-3), 153.6 (C-4), 165.6 [C(O)Ph], 168.7 (C-1), 178.0 [C(O)C(CH3)3]. HRMS (ESI) m/z: [M + Na]+ calcd for C19H20O6Na, 367.1152; found, 367.1147.
In a similar manner, (R)-4 (97 mg, 0.37 mmol) gave (R)-19 (102 mg, 0.30 mmol, 79%). [α]25D +60.9 (c = 1.01, CHCl3); 1H and 13C-NMR spectral data of (R)-19 agreed well with those of the corresponding antipode [(S)-19].
(4Z,6S)-6,7-Bis(benzoyloxy)-2,4-heptadiene-4-olide [(S)-12]. To a stirred solution of (S)-1 (97 mg, 0.37 mmol) in pyridine (2.3 mL) was added benzoyl chloride (65 μL, 0.56 mmol) at 0 °C, and the resulting mixture was stirred at rt for 0.5 h. The reaction mixture was poured into cold water (10 mL) and extracted with EtOAc (10 mL × 3). The extract was successively washed with 10% aqueous H2SO4 (10 mL), aqueous. NaHCO3 (10 mL), and brine (10 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 3/1) to give the title compound32,34 [(S)-12, 104 mg, 0.28 mmol, 89%] as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-12 as colourless needles. Mp. 60–62 °C. [α]21D −83.4 (c = 1.00, CHCl3). Lit.32 [α]27D −60.8 (c = 1.00, CHCl3). Lit.34 a colourless oil, [α]27D −52.3 (c = 0.30, CHCl3). IR (KBr): 1782, 1750, 1717, 1685, 1559, 1316, 1267, 1177, 1098, 1087 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 4.69 (1H, dd, J = 11.7, 4.2, H-7a), 4.72 (1H, dd, J = 11.7, 6.2, H-7b), 5.45 (1H, br d like, J = ca. 8.2, H-5), 6.29 (1H, dd, J = 5.4, 0.5, H-2), 6.37 (1H, ddd, J = 8.2, 6.2, 4.2, H-6), 7.40 (1H, d, J = 5.4, H-3), 7.42–7.45 (4H, m, arom.), 7.55–7.58 (2H, m, arom.), 8.02–8.06 (4H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 64.6 (C-7), 68.0 (C-6), 108.9 (C-5), 121.6 (C-2), 128.5(4C)/129.7(2C)/129.8(2C)/133.3/133.4 (d, arom.), 129.4/129.5 (s, arom), 143.3 (C-3), 150.8 (C-4), 165.4/166.0 [C(O)Ph], 168.5 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C21H16O6Na 387.0839; found 387.0832.
In a similar manner, (R)-1 (102 mg, 0.39 mmol) gave (R)-12 (101 mg, 0.28 mmol, 71%). [α]21D +89.7 (c = 1.00, CHCl3). 1H and 13C-NMR spectral data of (R)-12 agreed well with those of the corresponding antipode [(S)-12].
(4E,6S)-7,6-Bis(benzoyloxy)-2,4-heptadiene-4-olide [(S)-17]. Following the procedure used for the preparation of (S)-12, isomelodorinol [(S)-2, 101 mg, 0.39 mmol] was converted to the title compound33 [(S)-17, 133 mg, 0.37 mmol, 94%] as colourless needles. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give (S)-17 as colourless needles. Mp. 69–70 °C, [α]21D −76.3 (c = 1.00, CHCl3), lit34 a pale yellow oil, [α]27D −24.9 (c = 0.30, CHCl3). IR (KBr): 1790, 1751, 1678, 1564, 1317, 1277, 1179, 1030 cm−1. 1H-NMR (800 MHz, CDCl3) δ: 4.64 (1H, dd, J = 11.9, 6.6, H-7a), 4.67 (1H, dd, J = 11.9, 4.4, H-7b), 5.84 (1H, ddd, J = 10.8, 1.8, 0.7, H-5), 6.21 (1H, ddd, J = 10.8, 6.6, 4.4, H-6), 6.36 (1H, dd, J = 5.7, 1.8, H-2), 7.42–7.46 (4H, m, arom.), 7.56–7.59 (2H, m, arom.), 8.02 (1H, dd, J = 5.7, 0.7, H-3), 8.01–8.03 (4H, m, arom.). 13C-NMR (200 MHz, CDCl3) δ: 65.1 (C-7), 67.4 (C-6), 107.4 (C-5), 122.6 (C-2), 128.5(2C)/128.5(2C)/129.7(2C)/129.7(2C)/133.4/133.6 (d, arom.), 129.2/129.3 (s, arom.), 140.3 (C-3), 153.7 (C-4), 165.6/166.0 [C(O)Ph], 168.6 (C-1). HRMS (ESI) m/z: [M + Na]+ calcd for C21H16O6Na 387.0839; Found 387.0839.
In a similar manner, (R)-2 (100 mg, 0.38 mmol) gave (R)-17 (136 mg, 0.37 mmol, 98%). [α]25D +76.8 (c = 1.00, CHCl3). 1H and 13C-NMR spectral data of (R)-17 agreed well with those of the corresponding antipode [(S)-17].
(4Z)-7-Benzoyloxy-6-oxo-2,4-heptadien-4-olide [melodorinone A (20)]. A mixture of (S)-1 (100 mg, 0.38 mmol), Dess–Martin periodinane (326 mg, 0.77 mmol), NaHCO3 (81 mg, 0.96 mmol), and CH2Cl2 (5.0 mL) was stirred at rt for 2.5 h. The reaction mixture was quenched with aqueous NaHCO3 (5 mL), and extracted with CH2Cl2 (5 mL × 3). The extract was washed with brine (10 mL) and condensed in vacuo. The residue was purified by column chromatography (n-hexane/EtOAc = 2/1) to give the title compound (81 mg, 0.31 mmol, 82%) as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc give the title compound (20) as a microcrystalline solid. Mp. 134–136 °C, lit.28 colourless bulky crystals, Mp. 136–138 °C. IR (KBr): 1788, 1730, 1705, 1649, 1491, 1379, 1178, 1014 cm−1. 1H-NMR (600 MHz, CDCl3) δ: 5.40 (2H, s, H-7ab), 5.71 (1H, s, H-5), 6.52 (1H, d, J = 5.6, H-2), 7.47 (2H, t, J = 7.2, arom.), 7.55 (1H, d, J = 5.6, H-3), 7.59 (1H, t, J = 7.2, arom.), 8.11 (2H, d, J = 7.2, arom.). 13C-NMR (150 MHz, CD3Cl3) δ: 69.3 (C-7), 107.3 (C-5), 124.2 (C-2), 128.4/129.9/133.3 (d. arom.), 129.3 (s, arom.), 145.0 (C-3), 155.9 (C-4), 165.8 [C(O)Ph], 166.9 (C-1), 190.8 (C-6). HRMS (ESI) m/z: [M + K]+ calcd for C14H10O5K 297.0160; found 297.0151.
(4E)-7-Benzoyloxy-6-oxo-2,4-heptadien-4-olide [melodorinone B (21)]. Following the procedure used for the preparation of 20, (S)-2 (176 mg, 0.68 mmol) was converted to the title compound (21, 150 mg, 0.58 mmol, 86%) as a colourless microcrystalline solid. For analytical purposes, a small portion was purified by recrystallization from a mixture of n-hexane and EtOAc to give 21 as colourless needles. Mp. 143–145 °C. Lit.28 colourless bulky crystals, Mp. 139–140 °C. IR (KBr): 1784, 1730, 1703, 1678, 1628, 1410, 1313, 1111, 1095, 1014, cm−1. 1H-NMR (600 MHz, CDCl3) δ: 5.01 (2H, s, H-7ab), 6.35 (1H, br s like, H-5), 6.52 (1H, d, J = 5.5, H-2), 7.49 (2H, t, J = 7.2, arom.), 7.62 (1H, t, J = 7.2, arom.), 8.11 (2H, d, J = 7.2, arom.), 8.33 (1H, d, J = 5.5, H-3). 13C-NMR (150 MHz, CDCl3) δ: 69.2 (C-7), 103.1 (C-5), 125.9 (C-2), 128.9(2C)/129.9(2C)/133.7 (d, arom.), 128.8 (s, arom.), 142.7 (C-3), 160.5 (C-4), 165.8 [C(O)Ph], 167.4 (C-1), 190.0 (C-6). HRMS (ESI) m/z: [M + Na]+ calcd for C14H10O5Na 281.0420; found 281.0414.
Biology
An aliquot (100 μL) of the lysate was transferred to a 96-well microplate, and the optical density of each well was measured with a microplate reader (SH-9000, CORONA) at 405 nm (reference: 655 nm). The test compound was dissolved in DMSO, and the final concentration in the medium was 0.1%. The rates of melanin production were corrected based on the viability of melanoma cells.
Inhibition (%) was calculated using the following formula, where A and B indicate the optical density of vehicle-treated and test compound-treated groups, respectively, and C indicates cell viability (%) (vide infra).
| Inhibition (%) = [(A − B)/A]/(C/100) × 100 |
IC50 values were determined graphically on figures including only non-toxic concentrations of compounds.
Cell viability (%) was calculated using the following formula, where A and B indicate the optical density of vehicle-treated and test compound-treated groups, respectively.
| Cell viability (%) = B/A × 100 |
Author contributions
Conceptualization, GT and TM; Synthesis and structural analysis, KT, RS, NM, YF, RF, and SM; biological experiments and evaluation, FI, YM; writing – original draft preparation, KT and FI; review and editing, GT and TM. The manuscript was reviewed and approved by all authors.Data availability
All data associated with this study have been included in either the manuscript or in the ESI† associated with the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study was supported by a Grant-in-Aid for scientific research from the Japan Society for the Promotion of Science [JSPS (KAKENHI), 22K06541 (G. T.)]. The authors thank financial supports extended by the “Antiaging” Project for Private Universities (G. T. and T. M.).References
- S. Chatterjee, R. Sahoo and S. Nanda, Org. Biomol. Chem., 2021, 19, 7298 RSC
.
- A. G. Villegas-Paneda and M. T. Ramirez-Apan, Rev. Colomb. Quim., 2020, 49, 13 CrossRef
- H. Baer, M. Holden and B. Seegal, J. Biol. Chem., 1946, 162, 65 CrossRef CAS
- D. Mares, Mycopathologia, 1987, 98, 33 CrossRef PubMed
- M. L. Martin, L. S. Román and A. Domínguez, Planta Med., 1990, 56, 66 CrossRef CAS PubMed
- H. Minakata, H. Komura, K. Nakanishi and T. Kada, Mutat. Res., 1983, 116, 317 CAS
- R. Erickson, Science, 1948, 108, 533 CrossRef CAS PubMed
- S. Felder, S. Kehraus, E. Neu, G. Bierbaum, T. F. Schaeberle and G. M. Koenig, ChemBioChem, 2013, 1363 CrossRef CAS PubMed
- P. Cowall, J. M. Cassady, C.-J. Chang and J. F. Kozlowski, J. Org. Chem., 1981, 46, 1108 CrossRef CAS
- L. A. Maslovskaya, A. I. Savchenko, E. H. Krenske, V. A. Gordon, P. W. Reddell, C. J. Pierce, P. G. Parsonsa and C. M. Williams, Chem. Commun., 2014, 50, 12315 RSC
- B. Yang, L. Kong, X. Wang, Y. Jhang, R. Li, M. Yang and J. Luo, J. Nat. Prod., 2016, 79, 196 CrossRef CAS
- F. Kong, C. Zhao, J. Hao, C. Wang, W. Wang, X. Huang and W. Zhu, RSC Adv., 2015, 5, 68852 RSC
- R. S. Thombal and V. H. Jadhav, Org. Biomol. Chem., 2015, 13, 9485 RSC
- Z. Ahmed and P. Langer, J. Org. Chem., 2004, 69, 3753 CrossRef CAS PubMed
- R. Acharyya, P. Pal, S. Chatterjee and S. Nanda, Org. Biomol. Chem., 2019, 17, 3552 RSC
- A. Ribaucourt and M. D. Hodgson, Org. Lett., 2016, 18, 4364 CrossRef CAS PubMed
- P. Pal and S. Nanda, Org. Lett., 2017, 19, 1164 CrossRef CAS PubMed
- P. M. Amin, A. Cui, S. Wang; and Z.-X. Yu, Org. Lett., 2021, 23, 5669 CrossRef CAS PubMed
- F. Liu and E. Negishi, J. Org. Chem., 1997, 62, 8591 CrossRef CAS PubMed
- C. Curti, G. Rassu, L. Battistini, G. Pelosi, A. Sartori, F. Zanardi, A. Lodola, M. Mor and G. Casiraghi, Org. Lett., 2011, 13, 4738 CrossRef CAS PubMed
- N. S. Engels, B. Waltenberger, S. Schwaiger, L. Huynh, H. Tran and H. Stuppner, J. Sep. Sci., 2019, 42, 3165 CrossRef CAS PubMed
- G. Tanabe, Y. Manse, T. Ogawa, N. Sonoda, S. Marumoto, F. Ishikawa, K. Ninomiya, S. Chaipech, Y. Pongpiriyadacha, O. Muraoka and T. Morikawa, J. Org. Chem., 2018, 83, 8250 CrossRef CAS PubMed
- N. G. Lindquist, Acta Radiol. Diagn., 1973, 325, 1 CAS
- T. Hasegawa, Int. J. Mol. Sci., 2010, 11, 1082 CrossRef CAS PubMed
- I. Tessari, M. Bisaglia, F. Valle, B. Samori, E. Bergantino, S. Mammi and L. Bubacco, J. Biol. Chem., 2008, 283, 16808 CrossRef CAS PubMed
- C. W. Jefford, A. W. Sledeski and J. Boukouvalas, J. Chem. Soc., Chem. Commun., 1988, 5, 364 RSC
- J. C. Carretero, S. D. Lombaert and L. Ghosez, Tetrahedron Lett., 1987, 28, 2135 CrossRef CAS
- C. Chaichantipyuth, S. Tiyaworanan, S. Mekaroonreung, N. Ngamrojnavanich, S. Roengsumran, S. Puthong, A. Petsom and T. Ishikawa, Phytochemistry, 2001, 58, 1311 CrossRef CAS PubMed
- D. W. Brown, M. M. Campbell, A. P. Taylor and X. A. Zhang, Tetrahedron Lett., 1987, 28, 985 CrossRef CAS
- X. Lu, G. Chen, L. Xia and G. Guo, Tetrahedron: Asymmetry, 1997, 8, 3067 CrossRef CAS
- C.-C. Shen, S.-C. Chou and C.-J. Chou, Tetrahedron: Asymmetry, 1996, 7, 3141 CrossRef CAS
- M. Pohmakotr, P. Tuchinda, P. Premkaisorn, A. Limpongpan and V. Reutrakul, Heterocycles, 1999, 51, 795 CrossRef CAS
- S. Samwela, S. J. M. Mdachia, M. H. H. Nkunyaa, B. N. Irungua, M. J. Moshib, B. Moultonc and B. S. Luisic, Nat. Prod. Commun., 2007, 2, 737–741 Search PubMed
- T. Khotavivattana, T. Khamkhenshorngpanuch, K. Rassamee, P. Siripong and T. Vilaivan, Tetrahedron Lett., 2018, 58, 2711 CrossRef
- S. Chatterjee, R. K. Acharyya, P. Pal and S. Nanda, Org. Biomol. Chem., 2022, 20, 2473 RSC
- F. Hu and P. F. Lesney, Cancer Res., 1964, 24, 1634 CAS
- T. Morikawa, Y. Nakanishi, K. Ninomiya, H. Matsuda, S. Nakashima, H. Miki, Y. Miyashita, M. Yoshikawa, T. Hayakawa and O. Muraoka, J. Nat. Med., 2014, 68, 539 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00449g |
| This journal is © The Royal Society of Chemistry 2025 |