
Cascade fractionation of poplar into xylose, glucan oligomers and less-condensed lignin via synergistic formic acid–LiBr molten salt hydrate pretreatment†
Weiyu
Xia
a,
Lupeng
Shao
 *a,
Chao
Wang
a,
Yu
Liu
a,
Xianhai
Zeng
c and
Feng
Xu
ab
*a,
Chao
Wang
a,
Yu
Liu
a,
Xianhai
Zeng
c and
Feng
Xu
ab
aKey Laboratory of Pulp and Paper Science & Technology of Ministry of Education, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250353, China. E-mail: shaolp@qlu.edu.cn
bBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing, 100083, China
cCollege of Energy, Xiamen University, Xiamen, 361102, China
First published on 14th January 2025
Abstract
Lignocellulose as an appealing renewable carbon resource holds great potential for producing high-value-added products. The implementation of an integrated pretreatment process that enables effective fractionation and targeted valorization is crucial for ensuring the feasibility of future biorefinery scenarios. In this study, a stepwise approach using formic acid (FA)–LiBr molten salt hydrate (LiBr MSH) pretreatment was successfully developed for producing high yields of xylose, glucose, glucan oligomers, and less-condensed lignin from poplar. The results showed that FA pretreatment removed the most hemicellulose, releasing a high yield (71.63%) of xylose at 160 °C for 1 h. Subsequent acidified LiBr MSH pretreatment at 100 °C for 0.5 h resulted in a remarkably high cellulose degradation rate of 84.55%, yielding 41.87% glucose and 42.68% glucan oligomers. Meanwhile, 95.57% purity of lignin with abundant uncondensed moieties (i.e., β-O-4 bonds, Hibbert's ketones) was obtained. The proposed integrated FA–LiBr MSH pretreatment exhibited great potential as an efficient deconstruction strategy in the current biorefinery scenario.
Green foundation1.Synergistic FA–MSH pretreatment is proposed to selectively degrade hemicellulose and cellulose into sugar products, simultaneously obtaining high-purity less-condensed lignin.2.71.63% of xylose, 41.87% of glucose, 42.68% of glucan oligomers, and 95.57% purity of lignin could be obtained under the optimal conditions. The fractionated lignin exhibited a β-O-4 bond content of 25% of the theoretical maximum yield, along with the presence of HK moieties. After three cycles of MSH, the cellulose conversion rate was sustained at 75%, while the lignin content remained relatively stable. 3.In future work, the focus will be on enhancing the efficiency of recycling and utilization of molten salt hydrates, as well as regulating the selective production of glucan oligomers with specific degrees of polymerization. |
Introduction
Lignocellulosic biomass, which consists hemicellulose, cellulose, and lignin, is the most abundant renewable carbon resource in nature.1,2 The selective conversion of renewable lignocellulosic biomass into value-added fuels and chemicals is considered a sustainable and promising solution to alleviate reliance on fossil resources, mitigate climate change, and achieve carbon neutrality in the near future.3,4 Currently, holistic integrated catalytic strategies have been proposed for biorefinery.5,6 The main objective of these processes is to achieve the overall valorization of lignocellulose at low operating costs through environmentally friendly routes.7 However, lignin as a structural support in lignocellulose wraps cellulose microfibrils with hemicellulose by hydrogen and covalent bonds, resulting in a highly recalcitrant performance, which restrains the decomposition of lignocellulose by chemicals and enzymes.8,9 Interestingly, facile and efficient pretreatment will play a crucial role in the integrated biorefinery industry because it can deconstruct lignocellulose, followed by the conversion of polysaccharides into soluble sugars and the extraction of lignin.10,11A series of pretreatment technologies, including physical,12 biological,13 acidic,14 alkaline,15 organosolv,16 and ionic liquid17 pretreatments, have been developed in recent years. Molten salt hydrate (MSH), a highly concentrated inorganic salt solution (Csalt ≥ 50 wt%), has a water-to-salt molar ratio close to the coordination number of the strongest hydrated cation.18,19 At this concentration, there is only water–ion interaction and almost no water–water or ion–ion interaction. The hydrated cations in MSH can polarize water molecules, making their protons acidic.20 The anions of the salt increase acidity by deshielding the protons and increasing the tendency to leave the water.21 MSH is similar to ionic liquid, but is significantly cheaper, and it can also swell cellulose and break hydrogen bonds. MSHs are (1) easy to prepare, (2) environmentally friendly due to their high boiling point and low vapor pressure and (3) less expensive than common ionic liquids.22 Because of these advantages, MSHs show broad application prospects in the field of cellulose saccharification.23,24
In particular, research relating to MSH pretreatment of biomass have been published in Green Chemistry during recent years. Saha et al. found that xylan underwent efficient saccharification to xylose with >90% yield within 20 min at a low temperature (85 °C) in acidified MSH (59 wt% LiBr, 0.05 mol L−1 H2SO4).21 Subsequently, Saha's group reported an effective strategy for the one-step depolymerization and saccharification of lignocellulose to obtain high sugar yields and less condensed isolated lignin.25 They demonstrated that a ZnBr2-based MSH system is effective for the direct conversion of poplar to 88% glucose and 89% xylose based on theoretical amounts of glucan and xylan, with lignin being the only solid product. However, despite the excellent performance of the MSH system in the hydrolysis of carbohydrates, glucose and xylose are difficult to separate and purify due to the high solubility of monosaccharides in MSH, which limits their subsequent utilization.26,27 Some measures have been adopted to overcome the problem of product separation. A biphasic reaction system was used to convert glucose to 5-hydroxymethylfurfural (5-HMF) in MSH with parallel extraction of HMF to an organic phase.21 Furthermore, glucan oligomers were generated through the hydrolysis of cellulose using MSH, followed by separation via anti-solvent precipitation or adsorption onto large-surface-area amorphous carbon.28–30
Compared with cellulose, which has a crystalline structure, hemicellulose with an amorphous structure has a lower degradation temperature and a wider degradation range, making it more economical for industrial applications.31 By initially hydrolyzing the hemicellulose in lignocellulose into monosaccharides, followed by treating the hydrolyzed residue with MSH, the cellulose in the residue is degraded into glucose or glucan oligomers, while lignin is isolated. This approach enables a reduction in raw material complexity at its source, enhances the selectivity of liquid products, and lowers the cost associated with subsequent product separation and purification. Organic acids with low acidity coefficients have the ability to break glycosidic bonds.32 Due to this peculiarity, organic acid pretreatment is considered a novel and efficient method for the clean fractionation of lignocellulosic biomass.33 Formic acid, which can be produced from biomass,34,35 has strong proton ionization ability.36 The separation yield of hemicelluloses improves because of the strong hydrogen ionization of formic acid. This is the key to its use in lignocellulose pretreatment. Formic-acid-assisted hot water extraction has been proven to be a promising technique for extracting hemicellulose from woody biomass.37
Hence, to realize the component separation and utilization of lignocellulose, a synergistic formic acid–MSH pretreatment biorefinery strategy was proposed to selectively degrade hemicellulose and cellulose into sugar products, simultaneously obtaining high-purity less-condensed lignin. First, formic acid pretreatment was performed to selectively degrade hemicellulose, which could reduce biomass recalcitrance. Subsequently, MSH pretreatment was used to treat the hydrolysis residue. The MSH was recovered for recycling using dialysis and rotary evaporation. The effects of various conditions on hemicellulose/cellulose removal efficiency and lignin fractionation efficiency were investigated. Chemical component analysis and characterization of raw materials and different pretreated substrates were conducted to evaluate the effect of pretreatment. Simultaneously, the recovered lignin samples were also evaluated using gel permeation chromatography (GPC) and nuclear magnetic resonance (NMR) techniques, and antioxidant performance was evaluated to better assess the selectivity of this synergistic process.
Experimental
Materials
Poplar was provided by a pulp mill in Shandong, China. The raw materials were ground using a pulverizer to obtain sawdust of 40–60 mesh after oven-drying at 60 °C for 24 h. The compositional analysis of poplar, performed following the National Renewable Energy Laboratory (NREL) method, showed 47.29 wt% of cellulose, 18.87 wt% of hemicellulose (4.25 wt% acetyl group), and 26.29 wt% of lignin. Formic acid (AR, 99%), lithium bromide (AR, 99%), pyridine d5, chloroform-d, N-hydroxy-5-norbornene-2,3-dicarboximide, DMSO-d6, chromium acetylacetonate, and high-performance liquid chromatography (HPLC) standards (glucose, xylose, and arabinose) were purchased from Aladdin Biotechnology Co. Ltd (Shanghai, China). 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane and sodium acetate were purchased from Sigma-Aldrich. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) (HPLC, ≥98.5%) and 2,6-di-tert-butyl-4-methylphenol (BHT) (AR) were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). All reagents were used directly without purification.Cascade fractionation of poplar via synergistic FA–MSH pretreatment
The whole process of cascade fractionation of poplar via synergistic FA–MSH pretreatment is shown in Fig. 1. The detailed experimental protocol can be outlined as follows. | ||
| Fig. 1 Schematic diagram of the overall process for the cascade fractionation of poplar. | ||
The FA pretreatment was conducted in a 75 mL pressure bottle reactor. In detail, 2 g (dry weight) of extractive-free poplar was immersed in 40 mL of FA solution (9–15 wt%). Then, the mixture was heated to the target temperature (140–160 °C) and kept constant for 0.5–1 h with magnetic stirring at 400 rpm. After the reaction, the pressure bottle reactor was immediately placed into ice water. The mixture was vacuum-filtered using a G3 filter. The filtrate was collected and stored at 4 °C for subsequent analysis of sugars and soluble lignin. The solid residue remaining in the filter was washed thoroughly with deionized water. The solid residue was dried at 60 °C for 24 h and then gravimetrically quantified.
MSH was prepared by dissolving 60 g of LiBr in 40 g of deionized water, forming MSH with 60 wt% LiBr. For MSH pretreatment, 0.6 g of solid residue (after FA pretreatment) or raw poplar was added into a pressure glass bottle with 9 mL of MSH (containing 0–40 mM HCl). The treatment was performed at a specific reaction temperature (90–110 °C) for 30 min with magnetic stirring at 400 rpm. After the reaction, the vial was removed from the heating block and quenched in ice water. The mixture in the vial was transferred into a 50 mL volumetric flask and diluted to scale with deionized water. Then, it was filtered using a preweighted G3 glass filter under vacuum. The filtrate was collected and stored at 4 °C for subsequent determination. All solids remaining in the flask were placed in the glass filter and thoroughly washed with deionized water, followed by drying and gravimetric quantification. In addition, the ash content in the residue was determined to correct for the insoluble lignin content.
The hydrolysate obtained after MSH pretreatment was dialyzed (Mw: 100 D) for 24 h. The liquid outside the dialysis bag was collected and concentrated using rotary evaporation. The concentrated solution was completely transferred and quantified. Based on the quantification data, an appropriate amount of LiBr was added to the solution, which was further utilized.
Analytical methods
 | (1) |
31P and 2D HSQC NMR results were recorded on a Bruker Avance NEO 600 instrument according to the reported ref. 38 and 39.
Assessment of antioxidant performance
The DPPH free radical scavenging assay of different lignin samples was performed using the method reported by An et al.40 Briefly, a lignin–methanol solution (0.001–0.1 mg mL−1, 3 mL) was combined with a DPPH–methanol solution (0.15 mmol L−1, 3 mL) for 30 min in the absence of light at room temperature. Subsequently, the absorbance of the mixture at 517 nm was measured using UV–vis spectroscopy. The commercial antioxidant BHT was used as a positive control. Each test was performed in triplicate. The DPPH free radical scavenging activities (RSA) of the samples were calculated with eqn (2): | (2) |
Results and discussion
Composition variations in FA pretreatment
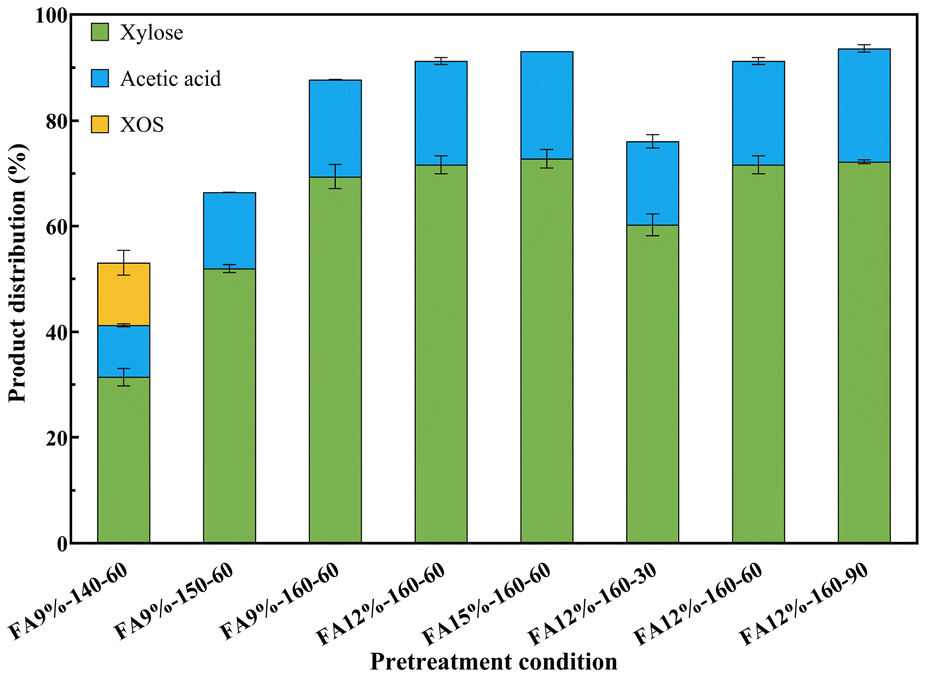
FA pretreatment of poplar was performed for selectively degrading hemicellulose. Fig. 2 shows the components of the solid residue obtained under different FA concentrations and reaction temperatures. The hemicellulose content gradually decreased from 8.35 to 2.23 wt% with increasing temperature at 9 wt% FA. In addition, when the temperature remained at 160 °C, there was a slight increase in the removal rate of hemicellulose with increasing FA concentration. Considering the impact of reaction time, the hemicellulose content decreased from 3.76 to 1.73 wt% as the reaction time was extended from 30 min to 60 min. When the reaction time was prolonged to 90 min, the hemicellulose content exhibited negligible variation. The degradation products of hemicellulose were mainly xylose and acetic acid. The product distribution of xylose and acetic acid in the hydrolysate during FA pretreatment is illustrated in Fig. 3. XOS was only detected in hydrolysates when poplar powder was treated with 9 wt% FA at 140 °C for 60 min. The corresponding yield of acetic acid ranged from 9.84% to 21.42% based on the dry weight of hemicellulose, and the yield of xylose ranged from 31.42% to 72.77%. It was observed that the yield of xylose exhibited a positive correlation with FA concentration, temperature, and reaction time. The hemicellulose content was found to be 1.73 wt% when poplar was treated with 12 wt% FA at 160 °C for a duration of 60 min. The levels of furfural and levulinic acid are shown in Fig. S1.† Even under the most extreme reaction conditions, less than 2% of hemicellulose was converted into furfural and levulinic acid.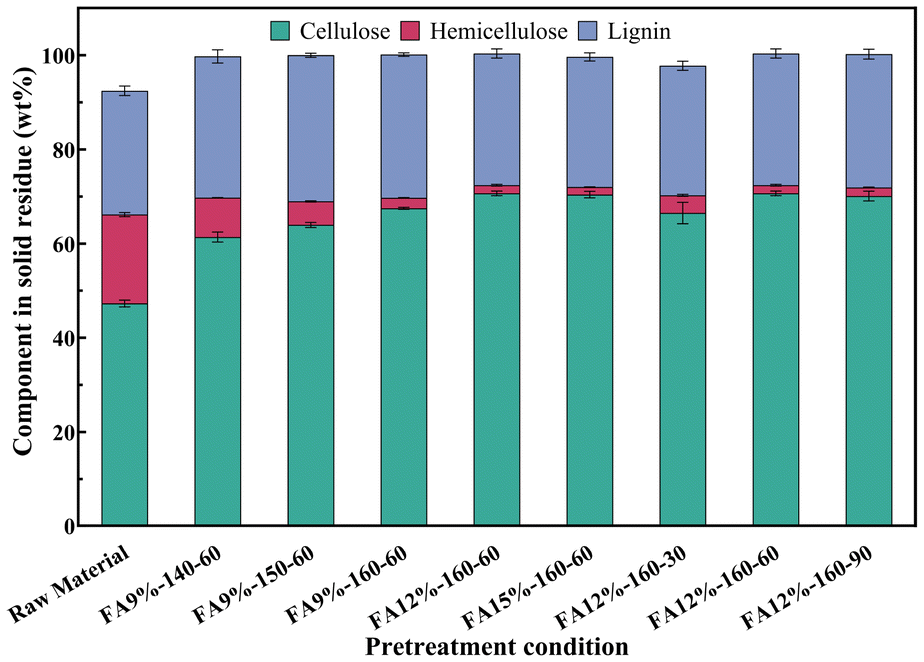 | ||
| Fig. 2 Chemical composition of raw material and FA-pretreated substrates. | ||
 | ||
| Fig. 3 Product distribution in the hydrolysate during FA pretreatment. | ||
The cellulose and lignin preserved in the solid after FA treatment were detected using the NREL standard method, and the results are shown in Fig. 4. Under identical FA concentration and reaction time, variations in temperature exhibited negligible influence on cellulose retention rate, consistently exceeding 95%. An increase in FA concentration was found to accelerate cellulose hydrolysis at the same temperature, as evidenced by a marginal decrease in cellulose retention rate. The cellulose retention rate remained at 96.53% even when subjected to the conditions of a formic acid concentration of 15 wt% and a reaction temperature of 160 °C, indicating that there was no significant degradation of cellulose during FA pretreatment of poplar. When the reaction time was prolonged from 30 min to 90 min, the retention rate of cellulose decreased from 98.45% to 95.64%. The retention rate of lignin increased from 70.59% to 74.77% accordingly.
 | ||
| Fig. 4 Cellulose and lignin retention rate during FA pretreatment. | ||
Taking into account the retention rates of lignin and cellulose together with the removal rate of hemicellulose, 160 °C, 60 min, and 12 wt% FA were selected as the optimized pretreatment conditions, which showed efficient hemicellulose removal of 91.24%, cellulose preservation of 96.78%, and lignin preservation of 86.54%. The solid residue part was used in the following experiments.
SEM and XRD characterization of the control and pretreated substrates
SEM technology was used to observe the morphological changes in poplar after FA pretreatment. As shown in Fig. S2,† the control poplar has a dense and flat surface morphology. By contrast, the FA-pretreated substrate showed a looser and rougher surface with wrinkles. The surface structure of lignocellulose exhibited signs of cracking while still maintaining an overall fibrous morphology. The change suggested that FA pretreatment could disrupt the surface structure of lignocellulose, leading to a looser fiber structure. This structural alteration was advantageous for enhancing the contact area between reagents and reaction substrates during subsequent MSH treatment, thereby facilitating more comprehensive reactions.As shown in Fig. S3,† both the control and pretreated substrates exhibited a cellulose type I structure, indicating the exceptional stability of the isolated crystalline cellulose in the solid part during FA pretreatment. In addition, the CrI values of cellulose in the control and pretreated substrates were 53.7% and 70.2%, respectively. The increase in CrI value after FA pretreatment could be attributed to the dissolution of hemicellulose and the amorphous region of cellulose by FA, whereas the crystalline structure of cellulose was well preserved. This was consistent with the previous finding of cellulose undergoing slight hydrolysis in formic acid solution.
Effect of reaction conditions on cellulose hydrolysis in MSH
After subjecting poplar to FA pretreatment under optimized reaction conditions, the solid residue obtained was subsequently hydrolyzed in MSH. Fig. 5 illustrates the yields of cellulose degradation products in the LiBr·3H2O system. In the absence of HCl, the cellulose degradation rate was limited to approximately 1%, thus precluding further discussion of this particular set of results. After treatment with the acidic LiBr·3H2O (20, 30, and 40 mM HCl) system, the cellulose conversion rate ranged from 45.36% to 84.55%, indicating that acidic LiBr·3H2O could effectively degrade cellulose in the residue. Under the same HCl concentration, increasing the temperature enhanced the cellulose conversion rate. At a concentration of 20 mM HCl, increasing the temperature from 90 °C to 100 °C resulted in an increase in glucan oligomer yield from 39.51% to 62.46%. However, as the temperature further increased to 110 °C, the oligosaccharide yield decreased to 38.94% and the glucose yield gradually increased from 5.58% to 43.53%. When the concentration of HCl was fixed at 30 mM, increasing the temperature led to a decrease in glucan oligomer yield from 52.41% to 14.46% and the glucose yield increased from 17.75% to 68.01%. The same trend was also observed in the distribution of products at 40 mM HCl. These findings suggested that higher temperature and HCl concentration facilitated the further degradation of glucan oligomers into glucose. | ||
| Fig. 5 Product distribution in the hydrolysate during LiBr–MSH pretreatment. | ||
Comparative analysis of hydrolysate between FA–MSH and direct MSH treatment
A control group experiment was conducted, where the poplar was directly treated with LiBr·3H2O according to a previous study41 (40 mM HCl, 110 °C, 30 min). The results demonstrated that the removal efficiency values for cellulose and hemicellulose were 85.32% and 89.04%, respectively. In the hydrolysate, the glucose yield was determined to be 65.72%, the glucan oligomer yield was 19.60%, the xylose yield reached 78.52%, and the acetic acid yield was 10.52%.The product distribution in the hydrolysate during FA–LiBr·3H2O pretreatment is illustrated in Fig. 6. The degradation products of hemicellulose were predominantly present in the hydrolysate during FA treatment, and the degradation products of cellulose were primarily observed in the hydrolysate after MSH treatment, thereby achieving the objective of segregating the cellulose and hemicellulose degradation products.
 | ||
| Fig. 6 Product distribution in the hydrolysate during FA–LiBr·3H2O pretreatment. In the region of FA treatment, A–T–t refers to the concentration of FA (wt%), reaction temperature (°C), and time (min), respectively. In the region of acidic LiBr–MSH treatment, T–A refers to the reaction temperature (°C) and the concentration of HCl (mM) in the LiBr·3H2O system. | ||
The cellulose degradation rate reached 84.55% via synergistic FA (12 wt% FA, 160 °C, 60 min) and LiBr·3H2O (30 mM HCl, 100 °C) pretreatment. Interestingly, both acid concentration and reaction temperature were reduced in the LiBr·3H2O system after using FA pretreatment compared with the reaction treating poplar directly with LiBr·3H2O (40 mM HCl, 110 °C). This indicated that the removal of hemicellulose contributed to the degradation of cellulose in MSH. From the data presented in Table S1,† it is evident that FA pretreatment can effectively remove hemicellulose at lower temperature than self-hydrolysis combined with MSH treatment. Furthermore, during the MSH treatment phase, it allows for the removal of a greater amount of cellulose under milder conditions. Compared to the sole application of MSH, although the yield of glucan oligomers decreases, there are inherent differences in the raw materials used, which complicates the issues faced in this study. Besides, the reaction conditions employed in this investigation are notably milder.
Physicochemical properties of isolated lignin
To investigate the impact of fractionation conditions on the lignin structure, we compared the structures of lignin samples obtained through FA–LiBr·3H2O treatment and direct LiBr·3H2O treatment with that of EMAL, which is commonly regarded as a representative of native lignin.
The average molecular weights (Mw and Mn) and the polydispersity index (PDI) of isolated lignin are shown in Table 1. Compared with EMAL, the molecular weights of lignin obtained through MSH treatment were reduced slightly. The weight-average molecular weight of lignin separated using the FA–LiBr·3H2O system (4922 g mol−1) was found to be comparable to that obtained with the direct LiBr·3H2O system (4972 g mol−1). The PDI of LFA/30 mM–100 °C was less than that of LC/40 mM–110 °C and EMAL, which indicates that the sample treated by the FA–LiBr·3H2O system exhibited a relatively narrow molecular distribution and FA–LiBr·3H2O treatment is competent for producing uniform lignin. Overall, the sample treated by the FA–LiBr·3H2O system with low molecular weight and homogeneous structure can be used as a precursor for value-added product processing.
| Lignin samples (La/b–c) | M w (g mol−1) | M n (g mol−1) | PDI (Mw/Mn) |
|---|---|---|---|
| La/b–c: a, conventional or formic acid; b, HCl concentration; and c, temperature. | |||
| EMAL | 5267 | 2015 | 2.61 |
| LC/40 mM–110 °C | 4972 | 1749 | 2.84 |
| LFA/30 mM–100 °C | 4922 | 1976 | 2.49 |
NMR analysis
The aliphatic-OH, phenolic-OH, and carboxylic-OH contents quantified by 31P-NMR measurement are shown in Table 2. Compared with EMAL, the aliphatic-OH content in lignin obtained after treatment with LiBr·3H2O was significantly reduced, potentially because of the dehydration reaction occurring during the cleavage of chemical bonds in lignin in the acidic LiBr·3H2O system. Among them, the aliphatic-OH content in LC/40 mM–110 °C was 1.77 mmol g−1, whereas it reached 2.92 mmol g−1 in LFA/30 mM–100 °C. This discrepancy could be attributed to the more rigorous reaction conditions of the LiBr·3H2O system compared with FA–LiBr·3H2O, where a higher acid concentration and temperature intensify the dehydration reaction. After treatment with the LiBr·3H2O system, there was a significant increase in the content of total phenolic hydroxyl groups in lignin, which could be attributed to the cleavage of aryl ether bonds. The content of condensed phenolic hydroxyl groups of LFA/20 mM–100 °C was lower than that of LC/40 mM–110 °C. Severe conditions during direct LiBr·3H2O treatment led to a lignin condensation reaction.| Sample | EMAL (mmol g−1) | LC/40 mM–110 °C (mmol g−1) | LFA/30 mM–100 °C (mmol g−1) |
|---|---|---|---|
| Aliphatic-OH | 3.46 | 1.77 | 2.92 |
| Condensed phenolic-OH | 0.02 | 0.44 | 0.21 |
| S–OH | 0.08 | 0.89 | 2.33 |
| G–OH | 0.32 | 0.40 | 0.83 |
| H–OH | 0.34 | 0.08 | 0.17 |
| –COOH | 0.31 | 0.30 | 0.59 |
| S–OH/G–OH | 0.24 | 2.22 | 2.82 |
| Total phenolic-OH | 0.74 | 1.37 | 3.32 |
For more insights into the structural information about the isolated lignin fractions, the 2D HSQC NMR analysis technique was conducted to examine the detailed chemical structure. The main visible subunits (i.e., S, G, and H) of lignin and the interunit linkages are presented in Table S3.† EMAL was selected to investigate the impact of the chosen pretreatment on the chemical substructures of lignin. The 2D HSQC NMR spectra of the lignin samples together with the main linkages and structural units are presented in Fig. 7. The relative abundance of various units in the lignin samples was estimated using semiquantitative analysis, as presented in Table 3.
 | ||
| Fig. 7 2D-HSQC NMR spectra of lignin samples and identified main structures. | ||
| Lignin samples | Linkages (% per 100 Ar) | ||
|---|---|---|---|
| β-O-4 | BD | HK | |
| EMAL | 63.44 | — | — |
| LC/40 mM–110 °C | — | 2.27 | — |
| LFA/30 mM–100 °C | 15.84 | — | 17.82 |
As shown in Fig. 7(a), EMAL had abundant β-O-4 (A) units, in agreement with reported studies.42 After LiBr·3H2O treatment, the signals of the β-O-4 structure were attenuated and some new chemical structures, including Hibbert's ketone (HK) and benzodioxane (BD) moieties, appeared. In particular, HK and BD are regarded as novel uncondensed lignin depolymerization products. The assignment of HK was made from the cross peaks at δC/δH of 44.5/3.67 ppm (α-position) and 67.1/4.19 ppm (γ-position), and the assignment of BD was made from the cross peak at δC/δH of 76.0/4.81 ppm.42 The signal of β-O-4 in LC/40 mM–110 °C disappeared, and the abundance of β-O-4 in LFA/30 mM–100 °C was 15.84%. The results indicated that FA–LiBr·3H2O treatment could effectively preserve a higher proportion of β-O-4 ether bonds in comparison to direct LiBr·3H2O treatment. An increased retention rate of β-O-4 ether bonds indicated that milder conditions (lower reaction temperature and reduced acidity) can effectively minimize the condensation of lignin, thereby preserving its natural structure more efficiently. Concurrently, we detected a higher signal for cellulose and xylan in Fig. 7(b1), which was consistent with the lignin purity observed in Table S2.†
Antioxidant properties of isolated lignin
To evaluate the antioxidant properties of the isolated lignin, a comparison was conducted with the commercial antioxidant BHT. The DPPH scavenging curves of lignin samples are presented in Fig. 8. It could be observed that LFA/30 mM–100 °C had the lowest IC50 value (1.4 μg mL−1) among all the test samples. The IC50 values of LC/40 mM–110 °C and EMAL were determined to be 2.2 and 3.2 μg mL−1, respectively, which were higher than the IC50 value of BHT (1.7 μg mL−1). A lower IC50 value represents higher antioxidant activity. Therefore, the results indicated that LFA/30 mM–100 °C exhibited superior antioxidant properties. Previous research43 has demonstrated that the presence of free phenolic hydroxyl groups is crucial for antioxidant activity. Together with the data in Table 3, we confirmed that the higher antioxidant properties of LFA/30 mM–100 °C could be attributed to the presence of a higher number of phenolic hydroxyl groups. By contrast, EMAL contained the highest number of aliphatic hydroxyl groups and the lowest number of phenolic hydroxyl groups, which resulted in it having the poorest antioxidant properties. The results above also indicate that LFA/30 mM–100 °C has potential applications as a commercial antioxidant. | ||
| Fig. 8 DPPH free radical scavenging capacity of isolated lignin samples. | ||
Biorefining mass balance
On the basis of the above results, the optimal conditions for FA–MSH pretreatment demonstrated the highest efficacy in deconstructing the various components of poplar biomass, including xylose, glucose, glucan oligomers, and lignin. The mass balance was performed using 100 kg of dried poplar feedstock under these optimal conditions. As shown in Fig. 9, 64.19 kg of solid was recovered after FA pretreatment, which contained 45.16 kg of glucan, 1.16 kg of xylan, and 17.87 kg of lignin. Simultaneously, 13.52 kg of xylose and 3.7 kg of acetic acid could be obtained in the filtrate. The result suggested that xylan was degraded after FA pretreatment. The subsequent MSH pretreatment yielded 18.91 kg of glucose and 19.27 kg of glucan oligomers that could be converted into fuels and chemicals. During the two-step treatment, FA treatment resulted in a significant lignin loss of 30.5 wt% (based on the lignin quality), primarily due to the dissolution of lignin in formic acid.39 In comparison, the lignin loss resulting from MSH treatment was nearly indiscernible. Finally, 17.08 kg of lignin (64.0 wt%) was isolated, which could be used for the preparation of antioxidant materials. The three main components of poplar feedstock were effectively deconstructed and converted. In summary, the FA–MSH pretreatment presented an upgraded approach for the high-value utilization of biomass feedstock. | ||
| Fig. 9 Mass balance of the proposed pretreatment process. | ||
Evaluation of the reuse of MSH
The recovered MSH was further utilized to treat the residues after FA treatment. Detailed data can be found in Table 4. After two cycles, the conversion rate of cellulose remained over 80%. In addition, we observed that the cellulose conversion rate decreased to less than 80% during the third cycle. Nevertheless, the lignin content during these cycles maintained a relatively constant level. These results showed that the MSH system had good reusability.| Sample | Glucose yielda (%) | Glucan oligomers yielda (%) | Lignin contentb (wt%) |
|---|---|---|---|
| a Based on the content of cellulose in the residues after FA treatment. b Based on the weight of the residues after FA treatment. | |||
| MSH fresh | 41.87 | 42.68 | 27.96 |
| 1st reuse | 43.49 | 40.83 | 26.06 |
| 2nd reuse | 42.27 | 38.46 | 27.28 |
| 3rd reuse | 37.86 | 37.32 | 28.39 |
Conclusions
In this study, a synergetic pretreatment process based on formic acid and LiBr molten salt hydrate was performed to efficiently generate xylose, glucose, glucan oligomers, and less-condensed lignin from poplar. It was found that 71.63% of xylose, 41.87% of glucose, 42.68% of glucan oligomers, and 95.57% purity of lignin could be obtained under the optimal conditions (FA pretreatment: 12 wt%, 160 °C, 1 h; LiBr·3H2O MSH pretreatment: 30 mM HCl, 100 °C, 30 min). The fractionated lignin exhibited a β-O-4 bond content of 25% of the theoretical maximum yield, along with the presence of HK moieties (as novel uncondensed lignin depolymerization products). This study provides a new stepwise method for the effective deconstruction of poplar into single components, which will facilitate the current highly available biorefinery.Author contributions
Weiyu Xia: data curation, formal analysis, investigation, methodology, software, writing – original draft; Lupeng Shao: conceptualization, investigation, project administration, resources, funding acquisition, writing – review & editing; Chao Wang: investigation, methodology, data curation; Yu Liu: project administration, methodology; Xianhai Zeng: data curation, funding acquisition, writing–review & editing; Feng Xu: project administration, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 22308177, 32371813, 32001275, U23A20123).References
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- X. Zhang, W. Zhang, F. Lei, S. Yang and J. Jiang, Bioresour. Technol., 2020, 309, 123385 CrossRef CAS PubMed.
- X. Cao, L. Shao, W. Huang, C. Wang, J. Mao, F. Xu and X. Zhang, J. Anal. Appl. Pyrolysis, 2021, 157, 105200 CrossRef CAS.
- B. Choe, S. Lee, H. Lee, J. Lee, H. Lim and W. Won, J. Cleaner Prod., 2021, 311, 127849 CrossRef CAS.
- K. Wang, S. Gao, C. Lai, Y. Xie, Y. Sun, J. Wang, C. Wang, Q. Yong, F. Chu and D. Zhang, Ind. Crops Prod., 2022, 187, 115366 CrossRef CAS.
- G. Velvizhi, K. Balakumar, N. P. Shetti, E. Ahmad, K. K. Pant and T. M. Aminabhavi, Bioresour. Technol., 2022, 343, 126151 CrossRef CAS.
- D. Sun, Z.-W. Lv, J. Rao, R. Tian, S.-N. Sun and F. Peng, Carbohydr. Polym., 2022, 281, 119050 CrossRef CAS.
- C. Zhao, Q. Shao and S. P. Chundawat, Bioresour. Technol., 2020, 298, 122446 CrossRef CAS PubMed.
- X. Yin, T. Cai, C. Liu, C. Huang, J. Wang, J. Hu, N. Li, J. Jiang and K. Wang, Chem. Eng. J., 2022, 437, 135408 CrossRef CAS.
- X.-J. Shen, J.-L. Wen, Q.-Q. Mei, X. Chen, D. Sun, T.-Q. Yuan and R.-C. Sun, Green Chem., 2019, 21, 275–283 RSC.
- R. Singh, R. Kumar, P. K. Sarangi, A. A. Kovalev and V. Vivekanand, Bioresour. Technol., 2022, 128458 Search PubMed.
- C. Wan and Y. Li, Biotechnol. Adv., 2012, 30, 1447–1457 CrossRef CAS PubMed.
- J. C. Solarte-Toro, J. M. Romero-García, J. C. Martínez-Patiño, E. Ruiz-Ramos, E. Castro-Galiano and C. A. Cardona-Alzate, Renewable Sustainable Energy Rev., 2019, 107, 587–601 CrossRef CAS.
- J. S. Kim, Y. Lee and T. H. Kim, Bioresour. Technol., 2016, 199, 42–48 CrossRef CAS PubMed.
- H. Sadeghifar, T. Wells, R. K. Le, F. Sadeghifar, J. S. Yuan and A. Jonas Ragauskas, ACS Sustainable Chem. Eng., 2017, 5, 580–587 CrossRef CAS.
- R. Alayoubi, N. Mehmood, E. Husson, A. Kouzayha, M. Tabcheh, L. Chaveriat, C. Sarazin and I. Gosselin, Renewable Energy, 2020, 145, 1808–1816 CrossRef CAS.
- H.-H. Emons, Electrochim. Acta, 1988, 33, 1243–1250 CrossRef CAS.
- S. Sen, J. D. Martin and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2013, 1, 858–870 CrossRef CAS.
- J. Duffy and M. Ingram, Inorg. Chem., 1978, 17, 2798–2802 CrossRef CAS.
- S. Sadula, O. Oesterling, A. Nardone, B. Dinkelacker and B. Saha, Green Chem., 2017, 19, 3888–3898 RSC.
- G. Gözaydın, S. Song and N. Yan, Green Chem., 2020, 22, 5096–5104 RSC.
- N. Rodriguez Quiroz, A. M. Norton, H. Nguyen, E. Vasileiadou and D. G. Vlachos, ACS Catal., 2019, 9, 9923–9952 CrossRef CAS.
- W. Deng, J. R. Kennedy, G. Tsilomelekis, W. Zheng and V. Nikolakis, Ind. Eng. Chem. Res., 2015, 54, 5226–5236 CrossRef CAS.
- S. Sadula, N. R. Quiroz, A. Athaley, E. O. Ebikade, M. Ierapetritou, D. G. Vlachos and B. Saha, Green Chem., 2021, 23, 1200–1211 RSC.
- Q. Liu, L. Zhou, D. Fan, M. Guan, Q. Ma, S. Li, X. Ouyang, X. Qiu and W. Fan, ACS Appl. Mater. Interfaces, 2021, 13, 52082–52091 CrossRef CAS PubMed.
- L. Zhou, Q. Liu, Q. Ma, M. Guan, X. Ouyang and X. Qiu, Cellulose, 2022, 1–13 Search PubMed.
- Q. Liu, Q. Ma, S. Sabnis, W. Zheng, D. G. Vlachos, W. Fan, W. Li and L. Ma, Green Chem., 2019, 21, 5030–5038 RSC.
- Q. Liu, S. Luo, W. Fan, X. Ouyang and X. Qiu, Green Chem., 2021, 23, 4114–4124 RSC.
- Q. Liu, L. Zhou, X. Xie, D. Fan, X. Ouyang, W. Fan and X. Qiu, Green Chem., 2022, 24, 8812–8819 RSC.
- A. Khodayari, W. Thielemans, U. Hirn, A. W. Van Vuure and D. Seveno, Carbohydr. Polym., 2021, 270, 118364 CrossRef CAS PubMed.
- X. Zhao, Y. Morikawa, F. Qi, J. Zeng and D. Liu, Bioresour. Technol., 2014, 151, 128–136 CrossRef CAS PubMed.
- B. Liu, L. Liu, B. Deng, C. Huang, J. Zhu, L. Liang, X. He, Y. Wei, C. Qin, C. Liang, S. Liu and S. Yao, Int. J. Biol. Macromol., 2022, 222, 1400–1413 CrossRef CAS PubMed.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Green Chem., 2008, 10, 612–615 RSC.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- C. Schuerch, J. Am. Chem. Soc., 1952, 74, 5061–5067 CrossRef CAS.
- W. M. Goldmann, J. Ahola, M. Mikola and J. Tanskanen, Bioresour. Technol., 2017, 232, 176–182 CrossRef CAS.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS.
- L. Shao, Q. Zhang, T. You, X. Zhang and F. Xu, Bioresour. Technol., 2018, 264, 238–243 CrossRef CAS PubMed.
- L. An, G. Wang, H. Jia, C. Liu, W. Sui and C. Si, Int. J. Biol. Macromol., 2017, 99, 674–681 CrossRef CAS.
- N. Li, X. Pan and J. Alexander, Green Chem., 2016, 18, 5367–5376 RSC.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, Green Chem., 2018, 20, 4224–4235 RSC.
- V. Ugartondo, M. Mitjans and M. P. Vinardell, Bioresour. Technol., 2008, 99, 6683–6687 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05494f |
| This journal is © The Royal Society of Chemistry 2025 |