
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low efficiency roll-off blue TADF OLEDs employing a novel acridine–pyrimidine based high triplet energy host†
Francesco
Rodella
 a,
Rishabh
Saxena
b,
Sergey
Bagnich
b,
Dovydas
Banevičius
c,
Gediminas
Kreiza
c,
Stavros
Athanasopoulos
d,
Saulius
Juršėnas
c,
Karolis
Kazlauskas
c,
Anna
Köhler
be and
Peter
Strohriegl
*ae
a,
Rishabh
Saxena
b,
Sergey
Bagnich
b,
Dovydas
Banevičius
c,
Gediminas
Kreiza
c,
Stavros
Athanasopoulos
d,
Saulius
Juršėnas
c,
Karolis
Kazlauskas
c,
Anna
Köhler
be and
Peter
Strohriegl
*ae
aMacromolecular Chemistry I, University of Bayreuth, Bayreuth 95440, Germany. E-mail: peter.strohriegl@uni-bayreuth.de
bSoft Matter Optoelectronics, University of Bayreuth, Bayreuth 95440, Germany
cInstitute of Photonics and Nanotechnology, Vilnius University, Saulėtekio av. 3, LT-10257 Vilnius, Lithuania
dDepartamento de Física, Universidad Carlos III de Madrid, Avenida Universidad 30, 28911 Leganés, Madrid, Spain
eBayreuth Institute of Macromolecular Research (BIMF), University of Bayreuth, Bayreuth 95440, Germany
First published on 26th November 2021
Abstract
The development of efficient blue emitter–host combinations is one of the biggest challenges in organic light-emitting diode (OLED) research. Host materials play a crucial role when it comes to enhancing the efficiency, improving the lifetime and decreasing the efficiency roll-off of the device. The need for new hosts is of prime importance, especially for blue phosphorescence and thermally activated delayed fluorescence (TADF) emitters, due to their high exciton energies. The hosts are less investigated than the emitters and require further progress. This work provides a new molecular strategy that combines an acridine derivative (donor) and pyrimidine moieties (acceptors) to obtain three novel host materials. This approach demonstrates that via careful selection of donor and acceptor units, it is possible to manage the properties of the host materials, obtaining at the same time superior thermal and morphological properties and high triplet energies up to 3.07 eV. The decrease of the conjugation in the acceptor unit was found to play a crucial role in increasing the triplet energy. The most promising host 1MPA was used to fabricate blue TADF OLEDs. Using a sky-blue emitter, we achieved electroluminescence at 491 nm and a maximum external quantum efficiency (EQE) of 13.6%, combined with a low efficiency roll-off, even beyond the practical brightness of 1000 cd m−1. The host 1MPA was also combined with a deep blue emitter to deliver a blue OLED with color coordinates of x = 0.16 and y = 0.18.
Introduction
Presently, organic light-emitting diodes (OLEDs) are expanding their boundaries and applications, such as in flat panel displays and smartphones, due to their significant advantages like facile preparation, cost-effectiveness, and energy efficiency.1,2 For these reasons, much effort is focused on developing this technology, both in academia and industry. Especially for display applications, commercial OLEDs contain stable and efficient green and red emitters based on iridium complexes.3 In contrast, blue emitters suffer from lacking simultaneous efficiency and stability.4,5 In the last few years, thermally activated delayed fluorescence (TADF) emitters seemed to be among the most promising candidates to solve this problem. Their capacity of harvesting triplet excited states can allow for an internal quantum efficiency of up to 100% through prompt and delayed fluorescence emission. Rapid thermally activated transfer of the excitation from the triplet to the singlet state, followed by efficient delayed fluorescence, avoids degradation processes that could otherwise occur when the excitation prevails for a long time in the triplet state. TADF OLEDs contain purely organic molecules, which avoid rare and costly noble metals and allow a broad range of molecular engineering. Aside from emitters, host materials play an essential role in obtaining the desired performance of the OLED. Emitters are frequently doped in a host matrix to decrease detrimental bimolecular effects caused by longer-lived triplet states.6 The dilution into the host is thus essential to enhance the lifetime, increase the efficiency, and reduce the efficiency roll-off of the device.7 The hosts, in general, must have high and balanced charge carrier mobility, good thermal and morphological stability, and chemical stability. They also must have high triplet energy to confine the excitons on the emitter, which is particularly challenging for blue-emitting OLEDs.8The majority of host materials adopt carbazole or less stable phosphine oxide groups.9 In this work, we present host molecules based on acridine and pyrimidine groups instead. Acridine is often used as a donor (D) in emitter molecules because of its rigidity and strong donating feature.10–13 Few cases report its use in hosts for red phosphorescence OLEDs.14,15 Furthermore, what brought our attention to the acridine group is that it has a higher triplet energy than carbazole (due to less conjugation).16 The pyrimidine unit is also a building block for TADF emitters due to its acceptor (A) properties.17–20 It is also used as a building block for hosts in phosphorescence OLEDs, with triplet energies of these hosts lying below 3 eV.21–23 Another use of pyrimidine is as an electron transport material (ETM).24
In this work, we report a strategy to obtain high triplet energy donor–acceptor host materials based on acridine and pyrimidine moieties. In particular, via careful molecular modification, it is possible to limit the conjugation of the acceptor pyrimidine, leading to a high triplet energy of more than 3 eV. To prove the strategy's applicability, we fabricated blue TADF OLEDs using the most promising host of the series (1MPA), expressing blue emission (from 461 nm to 493 nm) with low efficiency roll-off even beyond a practical brightness of 1000 cd m−2.
Experimental
Materials
9,9-Diphenyl-9,10-dihydroacridine; 2-chloro-4,6-diphenylpyrimidine; 4,6-dichloro-2-phenylpyrimidine; 4,6-dichloro-2-methylpyrimidine; palladium(II) acetate and tri(tert-butyl)phosphine (10 wt% in hexane) were purchased from abcr. Sodium tert-butoxide was purchased from TCI.Synthesis
10-(4,6-Diphenylpyrimidin-2-yl)-9,9-diphenyl-9,10-dihydroacridine (2PPA). 9,9-Diphenyl-9,10-dihydroacridine (1 g, 3 mmol, 1.1 eq.), 2-chloro-4,6-diphenylpyrimidine (727 mg, 2.73 mmol, 1 eq.), sodium tert-butoxide (340 mg, 3.54 mmol, 1.3 eq.), palladium(II) acetate (25 mg, 0.11 mmol, 0.04 eq.) and toluene (25 ml) were placed with a stirring bar in a Schlenk tube. The mixture was then degassed three times through freeze–pump–thaw cycles and the tube was backfilled with argon. Subsequently, tri(tert-butyl)phosphine(10 wt% in hexane) (0.07 ml, 0.22 mmol, 0.08 eq.) was added and the mixture was then refluxed for 12 h. The reaction mixture was extracted with water and the organic phase was dried over anhydrous sodium sulphate before evaporating the solvent with reduced pressure. Finally, the compound was purified by column chromatography on silica gel (20% ethyl acetate/hexane) and by train sublimation, obtaining 25% yield of pure product. 1H NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 8.01 (d, J = 7.5 Hz, 2H), 7.92 (d, J = 7.5 Hz, 4H), 7.45–7.42 (m, 7H), 7.39 (t, J = 7.5 Hz, 2H), 7.17 (t, J = 7.5 Hz, 2H), 7.05–6.99 (m, 6H), 6.96 (d, J = 7.0 Hz, 2H), 6.90 (d, J = 6.5 Hz, 4H). 13C NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 164.21, 160.05, 144.35, 142.55, 140.91, 137.73, 130.63, 130.27, 128.60, 128.04, 127.45, 127.28, 127.04, 126.32, 125.55, 124.07, 104.45, 58.62. EI-MS m/z [M]+: 564.10,10′-(2-Phenylpyrimidine-4,6-diyl)bis(9,9-diphenyl-9,10-dihydroacridine) (1PPA). 1PPA was synthesized following a similar procedure as described for 2PPA; using 9,9-Diphenyl-9,10-dihydroacridine (977 mg, 2.93 mmol, 2.2 eq.), 4,6-dichloro-2-phenylpyrimidine (300 mg, 1.33 mmol, 1 eq.), sodium tert-butoxide (320 mg, 3.33 mmol, 2.5 eq.), palladium(II) acetate (25 mg, 0.11 mmol, 0.08 eq.), tri(tert-butyl)phosphine (10 wt% in hexane) (0.07 ml, 0.22 mmol, 0.16 eq.) and toluene (25 ml). Yield = 28%. 1H NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 8.27 (m, 2H), 7.44–7.37 (m, 7H), 7.22 (t, J = 7.5 Hz, 4H), 7.16–7.10 (m, 12H), 7.07 (t, J = 7.5 Hz, 4H), 6.89 (d, J = 7.5 Hz, 4H), 6.82 (d, J = 7.5 Hz, 8H), 5.65 (s, 1H). 13C NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 162.68, 161.44, 144.60, 140.17, 140.15, 138.37, 130.40, 130.15, 128.93, 128.30, 128.17, 127.56, 126.63, 126.09, 123.74, 123.69, 91.1, 58.12. EI-MS m/z [M]+: 818.
10,10′-(2-Methylpyrimidine-4,6-diyl)bis(9,9-diphenyl-9,10-dihydroacridine) (1MPA). 1MPA was synthesized following a similar procedure as described for 2PPA; using 9,9-diphenyl-9,10-dihydroacridine (2.35 g, 7.05 mmol, 2.3 eq.), 4,6-dichloro-2-methylpyrimidine (0.50 g, 3.07 mmol, 1 eq.), sodium tert-butoxide (0.77 g, 8.01 mmol, 2.6 eq.), palladium(II) acetate (55 mg, 0.25 mmol, 0.08 eq.), tri(tert-butyl)phosphine(10 wt% in hexane) (0.15 ml, 0.49 mmol, 0.16 eq.) and toluene (25 ml). Yield = 58%. 1H NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 7.23 (d, J = 7.5 Hz, 4H), 7.18 (t, J = 7.5 Hz, 4H), 7.14–7.10 (m, 12H), 7.04 (t, J = 7.5 Hz, 4H), 6.87 (d, J = 7.5 Hz, 4H), 6.80 (d, J = 7.5 Hz, 8H), 5.60 (s, 1H), 2.43 (s, 3H). 13C NMR (CDCl3, 500 MHz, Me4Si): δ [ppm] 144.76, 140.07, 138.81, 130.34, 130.06, 129.01, 127.88, 127.55, 126.60, 126.28, 123.36, 122.31, 94.77, 57.92, 26.17. EI-MS m/z [M]+: 756.
Purification
The synthesized compounds 2PPA, 1PPA, and 1MPA were purified by train sublimation in a Carbolite split tube furnace HZS 12/450.Characterization
1H NMR and 13C NMR spectra were recorded on a Bruker Avance III HD (500 MHz), the chemical shifts were referred to chloroform-d3 (7.26 ppm), and the J values are given in Hz. MS spectra were obtained on a Finningan MAT 8500 using electron impact ionization.Thermal measurements
Thermogravimetric analysis (TGA) was conducted on a Mettler TGA/DSC3 with a heating rate of 10 K min−1 under a nitrogen flow. Differential scanning calorimetry was performed with a Mettler DSC3+ in pierced Al pans at 10 K min−1 under a nitrogen flow. The glass transition Tg was determined as the midpoint temperature of the step. Tm and Tr were determined as the peak temperature of the melting peak and recrystallization peak, respectively. These calculations were performed by Mettler STARe 15.00a software.Electrochemical measurements
Cyclic voltammetry measurements were carried out using a standard three electrode electrochemical micro-cell kit from Ametek Scientific Instruments, consisting of a Platinum wire counter electrode, a Platinum disk working electrode, and a Silver/Silver chloride reference electrode. A Gamry Interface 1010T served as the potentiostat. Electrochemistry grade solvent was used, and 100 mM tetrabutylammonium hexafluorophosphate was the supporting electrolyte. The solutions were thoroughly purged with dry nitrogen for 10 minutes before each measurement.Computational details
We have used density functional theory to obtain the ground state geometries of the compounds employing the M06-2X exchange–correlation functional in combination with a 6-31+G(d,p) atomic basis set. Geometry optimizations were successfully converged, and no imaginary frequencies were observed following vibrational frequency analysis. Singlet and triplet emission energies were further obtained by optimizing the structures with linear response time-dependent density functional theory at the M06-2X/6-31+G(d,p) level25 and within the Tamm–Dancoff approximation.26 Electron–hole natural transition orbitals (NTOs) were calculated to characterize the excited state transitions.27 All calculations were performed using the Gaussian 16 software package,28 and orbitals were visualized using the Avogadro molecular editor software.29 Charge transfer numbers ωCT with values in the range between 0 (for a Frenkel exciton) and 1 (for a complete charge transfer exciton) were computed by a transition density matrix analysis,30,31 based on defragmentation of the molecules in acridine and pyrimidine groups (D–A–D for 1PPA and 1MPA and D–A for 2PPA). Electron–hole correlation plots of the Ω matrix for the excitations were also constructed based on the fragments. The average electron–hole distance Δr, based on the charge centroids of the orbitals involved in the transition,32 was also computed as a complementary indicator to the nature of the states using Multiwfn software.33Photophysical measurements
For measurements in solution, all the compounds were dissolved in toluene or mTHF at a concentration of 6–9 × 10−5 M. Solutions were sonicated for 15 minutes after preparation. Absorption spectra were recorded using a Varian Cary 5000 spectrophotometer. Fluorescence spectra, phosphorescence spectra, and room temperature (RT) photoluminescence quantum yield (PLQY) were measured using a Jasco FP-8600 spectrofluorometer. For phosphorescence measurements, the detector unit was opened at a delay of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ms after excitation, and the signal was acquired for 50ms. The excitation wavelength for both fluorescence and phosphorescence was 300nm (unless specified in the text or figure). For the 77 K measurements, the sample or cuvette was immersed in liquid nitrogen. Values for the photoluminescence quantum yield of the molecules in solution were obtained using the Jasco FP-8600 spectrofluorometer equipped with an integrating sphere.
ms after excitation, and the signal was acquired for 50ms. The excitation wavelength for both fluorescence and phosphorescence was 300nm (unless specified in the text or figure). For the 77 K measurements, the sample or cuvette was immersed in liquid nitrogen. Values for the photoluminescence quantum yield of the molecules in solution were obtained using the Jasco FP-8600 spectrofluorometer equipped with an integrating sphere.
OLED fabrication and characterization
OLEDs were fabricated on pre-patterned indium tin oxide (ITO)-coated glass substrates (Kintec) with an ITO layer thickness of 100 nm and a sheet resistance of 15–20 Ω □−1. Prior to device fabrication, the substrates were cleaned by sonicating consecutively in detergent (Hellmanex II), distilled water, acetone, isopropyl alcohol, boiling distilled water, and finally treated with O2-plasma for 10 min. The substrates were then transferred into a vacuum evaporation chamber (Vacuum Systems and Technologies Ltd) integrated inside the nitrogen-filled glove box, where the stack of organic layers was deposited at a rate of 0.5–1 Å s−1 and a pressure <10−6 Torr. The doping of the emissive layer was accomplished by co-evaporating the host and guest materials from different evaporation sources at distinct rates for the desired doping concentration. Afterward, the samples were transported by the robotic arm to a metal deposition chamber without breaking the vacuum, which was followed by deposition of lithium fluoride (LiF) and aluminum (Al) layers at a rate of 0.2 and 2 Å s−1, respectively. The active area of the devices was 4 mm2 as defined by the ITO pattern and the shadow mask used for cathode deposition. The completed devices were removed from the vacuum chamber to the N2-filled glove box without exposure to air and encapsulated by using a glass cover and UV-curable epoxy KATIOBOND LP655 (DELO). A system consisting of a calibrated integrating sphere (ORB Optronix), a spectrometer PMA-11 (Hamamatsu), and a source-meter unit 2601A (Keithley) was utilized for evaluation of the electrical-optical properties of OLEDs such as current–voltage–luminance (I–V–L) characteristics, EQE and efficiency roll-off.Results and discussion
Design and synthesis
In this work, a strategy to increase the triplet energy of donor–acceptor molecules is shown. For this purpose, three novel host molecules were designed. As presented in previous work,34 by direct connection of donors (acridine and carbazole) and acceptors (triazine), it is possible to increase the triplet energy of donor–acceptor molecules compared to the case where the connection occurs via a conjugated bridge. In this work, acridine (donor) and three different pyrimidines (acceptors) with a different size of the conjugated system of the pyrimidine were chosen to obtain three materials of different conjugation. In particular, we aim to see a decrease in the conjugation of the acceptor moiety, a key strategy to reach a high-energy triplet (Fig. 1). 2PPA presents a donor–acceptor structure where the pyrimidine core is linked at positions 4 and 6 to two phenyl rings and at position 2 to one acridine. 1PPA has a donor–acceptor–donor (DAD) structure; i.e., the pyrimidine moiety is linked to just one phenyl ring (in position 2) while two acridines occupy positions 4 and 6. The last molecule of the series, 1MPA, still has the same DAD configuration, but the pyrimidine is substituted with a methyl group (in position 2) and two acridines (in positions 4 and 6). By replacing the phenyl ring with a methyl group, the conjugation of the acceptor is limited to the central pyrimidine ring. The products were synthesized using a general Buchwald-Hartwig protocol between the donor acridine and the chlorinated pyrimidine acceptor, which provides an efficient way for the formation of C–N bonds (Fig. 1). Furthermore, all products were purified via train sublimation to obtain highly pure compounds.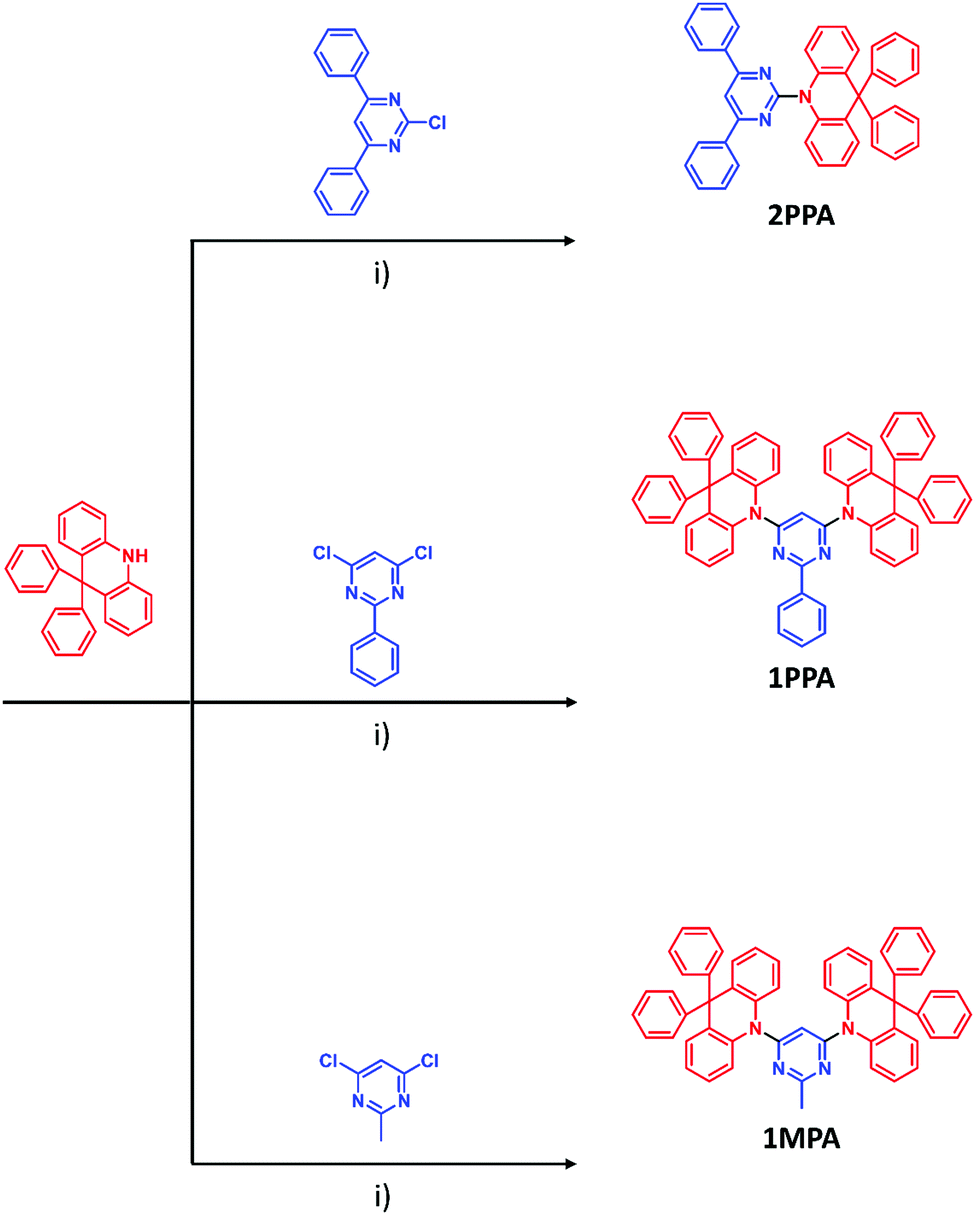 | ||
| Fig. 1 Synthesis route to the three host materials 2PPA, 1PPA and 1MPA. (i) Pd(Oac)2, P(tBu)3, NaOtBu, toluene, reflux, 12 h. | ||
Thermal and electrochemical analysis
The thermal properties of the compounds were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), as shown in Fig. 2a and b. DSC shows that all the compounds are morphologically stable beyond 100 °C with glass transitions (Tg) of 108 °C (2PPA), 150 °C (1PPA), and 138 °C (1MPA) and that recrystallization (Tr) upon heating occurs above 100 °C for 2PPA and above 200 °C for 1PPA and 1MPA, due to the addition of a second bulky acridine. The heating scan of 2PPA was obtained after the sample was melted and directly cooled with liquid nitrogen. This was necessary to obtain the amorphous material during the cooling process. The melting temperatures (Tm) are around 300 °C for 2PPA and 1PPA and 273 °C for 1MPA. | ||
| Fig. 2 Thermal and electrochemical properties of 2PPA, 1PPA and 1MPA. (a) Differential scanning calorimetry showing the second heating scan. (b) Thermogravimetric analysis. (c) Cyclic voltammograms (cathodic scan) measured in CH2Cl2 at room temperature with tetra-n-butylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte and a scan rate of 100 mV s−1. | ||
TGA data show high decomposition temperatures (Td) above 300 °C for 2PPA and above 400 °C for 1PPA and 1MPA. TGA also indicates that all the materials leave none or only minor residues which means that all three hosts completely evaporate already at normal pressure, which makes them ideal candidates for OLED fabrication via evaporation. Furthermore, throughout the series, it is possible to see that by adding a second acridine, the Tg, Tr, and Td values of the materials are improved. The melting points are instead similar, with a small decrease in 1MPA. These data confirm the high thermal stability of the materials and their potential applicability as hosts in OLEDs.
Furthermore, cyclic voltammetry of the three hosts was carried out to explore the electrochemical properties and to estimate the HOMO values (Fig. 2c). All the compounds show two quasi-reversible oxidation peaks. The first oxidation peak always appears slightly above 0.2 V vs. Ag/AgCl, and it corresponds to the oxidation of the acridine donor moiety. The HOMO values for the three compounds were estimated by the half-wave potential of the first oxidation potential, considering the solvent correction according to Gräf et al.35 The LUMO values were then calculated by adding the optical band gap to the HOMO values (Table 1).
| T g (°C) | T r (°C) | T m (°C) | T d (°C) | HOMO (eV) | LUMO (eV) | |
|---|---|---|---|---|---|---|
| 2PPA | 108 | 127, 147 | 305 | 359 | −5.12 | −2.00 |
| 1PPA | 150 | 229 | 304 | 417 | −5.11 | −1.71 |
| 1MPA | 138 | 222 | 273 | 413 | −5.10 | −1.55 |
Absorption and photoluminescence in solution
Absorption and photoluminescence studies of 2PPA, 1PPA, and 1MPA were performed in dilute toluene solutions to analyze their intrinsic photophysical properties, with a focus on determining their triplet state energies. Fig. 3a shows the room temperature absorption spectrum of 2PPA, 1PPA, and 1MPA. 2PPA exhibits a band attributed to CT absorption from around 3.2–3.9 eV. This assignment is based on the structureless absorption profile and comparatively low extinction of this particular band. 1PPA and 1MPA do not show any pronounced signatures of a CT state absorption. The absorption of 2PPA, 1PPA, and 1MPA differ significantly from the absorption of either donor or acceptor (Fig. S1a–c, ESI†). This indicates the existence of strong electronic interactions between the donor and acceptor units in these compounds.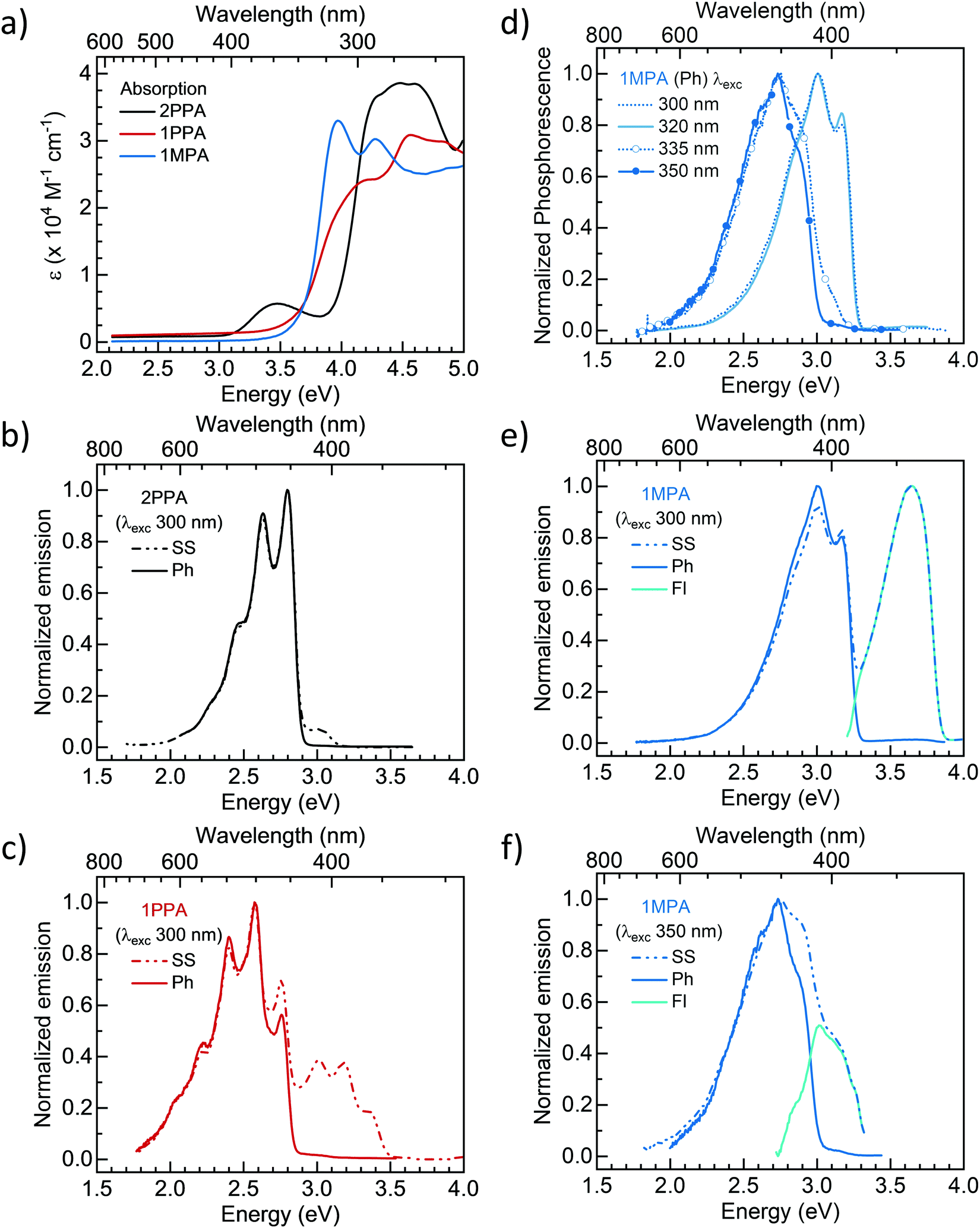 | ||
| Fig. 3 (a) The absorption spectra of 2PPA, 1PPA, and 1MPA in toluene solution at 300 K. (b) The emission spectra of 2PPA taken at 77 K in a toluene glass under steady-state conditions (SS), so that both fluorescence and phosphorescence show up, and taken with a delay of 150 ms after excitation and a gate width of 50 ms so that only phosphorescence (Ph) is visible. Excitation was at 300 nm. (c) The emission spectra were taken under the same conditions for 1PPA. (d) The phosphorescence (Ph) spectra of 1MPA in toluene glass at 77 K, taken for different excitation wavelengths as indicated. (e) Comparison of the Ph and SS spectra for excitation at 300 nm and (f) at 350 nm. Also indicated is the fluorescence spectrum obtained from subtracting the Ph from the SS. | ||
In order to estimate the triplet and singlet energies of the host materials, phosphorescence (Ph) and steady-state (SS) measurements were carried out at liquid nitrogen temperature (77 K). For 2PPA (Fig. 3b), the phosphorescence is structured with a 0–0 vibrational peak at 2.81 eV (442 nm). The steady-state spectrum is a combination of fluorescence and phosphorescence as the spectrum below 2.9 eV matches completely with the phosphorescence spectrum. Thus, the lowest singlet in 2PPA is a state exhibiting a structureless emission centered around 3 eV, with an onset at 3.15 eV (394 nm), with the lack of vibrational structure probably indicating a CT-character. In 1PPA, the lowest singlet and triplet states are both structured and have 0–0 peaks at 3.36 eV (369 nm) and 2.76 eV (449 nm), respectively, as shown in Fig. 3c. Thus, the T1 state is at a lower energy in 1PPA than in 2PPA. This can be accounted for by the nature of this state. The phosphorescence of 2PPA and 1PPA are similar to the ones of their respective pyrimidine acceptors (Fig. S2, ESI†), indicating that the triplet is localized on the pyrimidine acceptor moieties, 2PhPy and 1PhPy, in agreement with the quantum chemical calculations (vide infra). The triplet energy of the hosts therefore reflects the phosphorescence energies of the respective pyrimidine acceptors. The lower T1 energy in 1PPA results from the fact the phenyl ring is planar with the pyrimidine in 1PPA whereas there is a torsion in 2PPA, as evident in the quantum chemical calculations further below.
Due to the strong emission from the pyrimidine-based singlet state in 1PPA, we cannot discern whether there is also a CT singlet state at about 3 eV with a lower oscillator strength buried under it. When we measured the spectra with different excitation wavelengths between 300 nm and 350 nm, we found them to be independent of excitation wavelength (Fig. S3, ESI†).
The emission of 1MPA, in contrast, depends on the excitation wavelength, and the corresponding spectra are shown in Fig. 3d. As before, Ph and SS measurements were carried out at 77 K. For excitation at 300 nm and 320 nm (4.13 eV and 3.88 eV, respectively), we obtain a structured phosphorescence band with a 0–0 peak at 3.18 eV. However, when exciting at 335 nm and at 350 nm (3.70 eV and 3.54 eV), the emission band has a similar shape, yet it is shifted to lower energy, and it is broadened with a shoulder at 2.88 eV and an onset at 3.07 eV. To clarify the origin of these two bands, we performed photoluminescence excitation (PLE) spectra, and we also measured the phosphorescence of the acridine donor as well as the methylpyrimidine acceptor. These data are available in the ESI† (Fig. S4 and S5). For the higher energy phosphorescence, two assignments are principally possible, that is, to a locally excited (LE) state localized either on the acridine donor or on the pyrimidine acceptor. Based on the data in Fig. S4 and S5 and the arguments detailed in the ESI,† we tentatively assign the higher energy phosphorescence band to a 3LE state localized on the acridine donor, and we attribute the lower energy phosphorescence band to a triplet state with mixed CT–LE character, where the LE contribution would be again from the acridine.
The fluorescence in 1MPA shows the same excitation dependence. For excitation at 300 nm and 320 nm, we observe a broad band centered at about 3.6 eV (Fig. 3e), while for excitation at 335 nm and 350 nm, the fluorescence is centered at 3.0 eV (Fig. 3f). By comparison to the acridine fluorescence spectrum (Fig. S4, ESI†), the band at 3.6 eV is readily assigned to a 1LE state localized mainly on the acridine. The band at 3.0 eV, by virtue of its lower energy, must have some significant CT contribution.
We also determined the S1 and T1 energies of all compounds in neat films at 77 K and found them to be shifted to lower energies by 0.1 eV (see Fig. S6, ESI†). We attribute this rather moderate shift in the neat film to energy transfer within the DOS, which is known to be efficient in neat films, so that the emission comes from sites with lower energy within the DOS.
Fig. 4a summarizes the insights obtained on the energies and character of the excited states in 2PPA, 1PPA, and 1MPA based on the spectroscopic data. All energy values are taken from the onset of the emission spectra. It exemplifies the impact of reducing the conjugation length of the pyrimidine acceptor. While for 2PPA and 1PPA, the 3LE state localized on the 2-phenylpyrimidine (2PhPy) and 1-phenylpyrimidine (1PhPy) is the lowest triplet state with an onset at around 2.9 eV, this is no longer the case when the conjugation is reduced in methylpyrimidine (MePy). The triplet state for the units pyrimidine and methylpyrimidine have 0–0 peaks at 3.50 eV and 3.42 eV, respectively (see Nishi et al.36 and Fig. S5b, ESI†), i.e., well above the onset of phosphorescence for the acridine moiety (Fig. S4 and S5b, ESI†). As a result, in 1MPA, the lowest triplet state becomes one with a mixed CT–LE character, where the LE contribution is from the acridine unit. We also comment on the unusual observation of an excitation wavelength-dependent phosphorescence. This implies that when exciting into the acridine-based singlet LE state, the rates of intersystem crossing to its triplet state and phosphorescence can compete with the charge transfer required to form the CT-based singlet or triplet state. Evidently, the twists between the units slow down the charge transfer, which needs to take place through a bond.
 | ||
| Fig. 4 (a) Excited state energy level diagram for 2PPA, 1PPA, and 1MPA, based on the spectroscopic data, along with their structure (donor in red and acceptor in blue). The values are taken from the onset of the emission spectra. (b) TDA-DFT computed natural transition orbitals, emission energies, and charge transfer numbers for the lowest triplet state for 2PPA, 1PPA, and 1MPA at the M06-2X/6-31+G(d,p) level. Pyr represents pyrimidine acceptors and Acr the acridine donor. A side view is displayed in Fig. S8 (ESI†). | ||
Theoretical calculations
For a deeper understanding of the excited states, we turn our attention to the electronic structure calculations. We find that for all molecules, acridine units lose planarity, and their central ring adopts a distorted boat conformation both at the ground and excited state geometries. This boat conformation is in agreement with results on related compounds.37,38 At the same time, there is an intramolecular pi-stacking arrangement between the phenyl ring of one of the acridine donor units and the pyrimidine acceptor moiety (Fig. S7, ESI†). The obtained conformations suggest a strong electronic mixing between the donor and acceptor units, consistent with the observations made in the absorption spectra. The TDA-DFT calculated lowest excited singlet and triplet energies are in very good agreement with the fluorescence and phosphorescence spectroscopic data, respectively, as summarized in Table 2. Calculations support the above spectroscopic assignment on the nature of the emissive triplet states. Fig. 4b illustrates the spatial extent of the hole–electron NTO pairs for the optimized lowest triplet state of 2PPA, 1PPA, and 1MPA, along with the calculated charge transfer numbers ωCT. These transitions can be described by a single NTO pair. The calculated relaxed T1 state for 2PPA and 1PPA has a LE character and is localized on the pyrimidine unit, whereas the relaxed T1 state for 1MPA has a mixed CT–LE character, with the LE part on the acridine. An energy level diagram based on the spectroscopic data is depicted in Fig. 4a. The NTOs for fluorescence can be found in Fig. S7 (ESI†). The emissive singlet for 2PPA is localized on the pyrimidine, which is in contrast to the spectroscopic assignment of a CT state. This comes from the fact that the obtained gas-phase geometry from the TDA-DFT calculations leads to a distorted acridine. However, in the solid state, intermolecular interactions might favor pi–pi stacking configurations between the acridine units of the molecules and result in a twisted intramolecular CT state. For 1PPA, the obtained lowest singlet is at 3.41 eV and has a CT character. The lowest singlet state for 1MPA is at 3.13 eV, which displays a strong charge transfer character as depicted by the hole–electron pair natural transition orbitals and the electron–hole correlation plot of the Ω matrix (Fig. S9, ESI†) and has a weak oscillator strength f = 0.0037. Following optimization of the second lowest singlet state, S2, we find that it is located at 3.50 eV and has an oscillator strength f = 0.0134 (Fig. S7, ESI†). Although the charge transfer numbers obtained are similar for the two states, the electron–hole distance index Δr is larger for the S1 state (Δr = 3.26 Å) than for the S2 state (Δr = 2.07 Å), indicating a larger CT length for the S1 state. The hole NTO for S2 is delocalized across both acridine units. This could explain the experimentally observed excitation wavelength dependent emission described in the previous section. Given the relatively large energy difference between the S2 and S1 states and the fact that relaxation from the Franck–Condon state requires large conformational rearrangement, vibrational relaxation and internal conversion to S1 might compete with radiative emission from the S2 state leading to non-Kasha emission.39OLED properties
After having established that the acridine–pyrimidine based compound 1MPA is the one with the highest triplet energy (>3.0 eV) among the studied compounds, we employed it as a host for the fabrication of blue OLED. We combined it with the previously reported blue TADF emitters mPTC40 and OBA-O41 as dopants (Fig. S10e, ESI†). In the case of mPTC, a doping concentration of 12 wt% was used based on photoluminescence quantum yield measurements (Table S1, ESI†). For the emitter OBA-O we used several doping concentrations (5, 8, and 13 wt%) with a view to examine the emission wavelength tunability of the device. The choice of mPTC and OBA-O as TADF emitters was based on (i) their shallow HOMO levels (−5.12 and −5.15 eV, respectively), which were similar to that of 1MPA, (ii) their triplet energies being lower than that of the host to allow for triplet exciton confinement and (iii) their high photoluminescence quantum yields. OLEDs were fabricated using the following simple device architecture: ITO (100 nm)/TAPC (30 nm)/EML (x wt% mPTC and OBA-O in 1MPA, 30 nm)/TmPyPB (40 nm)/LiF (0.8 nm)/Al (100 nm). Here, ITO was used as a transparent anode, (1,1-bis[(di-4-tolylamino)phenyl]cyclohexane) TAPC and (1,3,5-tri(m-pyridin-3-ylphenyl)benzene) TmPyPB for hole and electron injection/transport, respectively, LiF/Al as the reflective cathode, whereas x denotes the doping concentration (in weight percentage) of the particular TADF emitter in the emissive layer (EML).The configuration and the energy level diagram of the device with the EML (12 wt% mPTC in 1MPA) are displayed in Fig. 5a. The main device characteristics are provided in Fig. 5b–d and Table 3. The OLED demonstrated a low turn-on voltage (Von) of 3.25 V, greenish-blue electroluminescence at 491 nm with the full width at half maximum (FWHM) of 75 nm resulting in Commission Internationale de L’Eclairage (CIE) coordinates of (0.2, 0.4). The maximum external quantum efficiency (EQEmax) was determined to be 13.6%, which is in good agreement with PLQY of mPTC in 1MPA (PLQY ≈ 70%, see Table S1 of the ESI†)40 if typical device outcoupling efficiency of about 20% is taken into account.42 The fabricated device exhibited low efficiency roll-off at practically useful brightness, i.e., EQE dropped down to 12.3% at the brightness of 100 cd m−2 and to 11.6% at 1000 cd m−2 (Fig. 5b). Notably, EQE remained above 10% up to the very high brightness of 5000 cd m−2. This behavior could be explained by the optimal device architecture and well-balanced electron and hole currents. The maximum luminance achieved by the device was nearly 44000 cd m−2. The properties of analogous devices based on the same host, yet the different TADF emitter OBA-O at various doping concentrations in the EML, are presented in Fig. S10 (ESI†) and Table 3. The OBA-O device with a similar doping concentration (13 wt%) as the previous one based on mPTC expressed comparable emission properties. Explicitly, peak emission wavelength, bandwidth, CIE color coordinates, and maximum EQE were found to be alike. On the other hand, the much steeper I–V and L–V curves in the OBA-O device indicated a significantly improved charge carrier mobility in the EML. Importantly, the reduced OBA-O concentration down to 5 wt% in the EML shifted the emission wavelength from greenish-blue (493 nm) to deep-blue (461 nm), demonstrating the potential of 1MPA to host deep-blue TADF emitters. The picture of the deep-blue emitting OLED is shown in the inset of Fig. S10 (ESI†). The reduced emitter concentration concomitantly caused a broadening of emission bandwidth (up to 94 nm) and an accelerated EQE roll-off. In OLEDs, EQE roll-off typically arises from exciton–polaron quenching and from exciton–exciton annihilation. The increased roll-off with lower emitter concentrations can arise from either effect. Since there is a barrier of about 1.15 eV for electron injection onto the host yet not for injection onto the emitter, the likely scenario is that electron transport proceeds by hopping between emitter molecules while hole transport occurs via the host, where hole injection is barrier-free. This results in a charge carrier imbalance that favors exciton–hole quenching. However, at low emitter concentrations percolative transport across emitting sites is no longer possible, so that the recombination zone narrows towards the electron injecting side. This decrease in recombination zone implies an increase of both exciton and charge carrier density, so that in addition to exciton–hole quenching there can also be a contribution from triplet–triplet annihilation. (see Fig. S10b, ESI†). We note that the dominant lifetime of delayed fluorescence in mPTC (2 μs) is shorter than that in OBA-O (9 μs), consistent with its lower susceptibility to exciton quenching processes (Fig. S12 and Table S2, ESI†). Furthermore, we fabricated an identical mPTC-based device where the 1MPA host is replaced with the benchmark host mCBP. In Fig. S11 (ESI†) we provide the comparison of OLED properties of the two identical devices differing only by the host material (1MPAvs. benchmark mCBP). Due to the lower LUMO of mCBP compared to 1MPA, many more electrons are being injected into the mCBP host resulting in significantly higher current densities flowing through the device, and subsequently, higher luminance attained at the same driving voltage. EL spectra of both devices utilizing the same mPTC dopant concentration (12 wt%) are found to be very similar. The device featuring benchmark mCBP host exhibits a higher EQEmax of 21% and similar efficiency roll-off as the other one based on the 1MPA host. Nevertheless, the obtained results show that high-triplet-energy host 1MPA is suitable for the fabrication of efficient blue TADF OLEDs, delivering low efficiency roll-off even beyond the practical brightness of 1000 cd m−2.
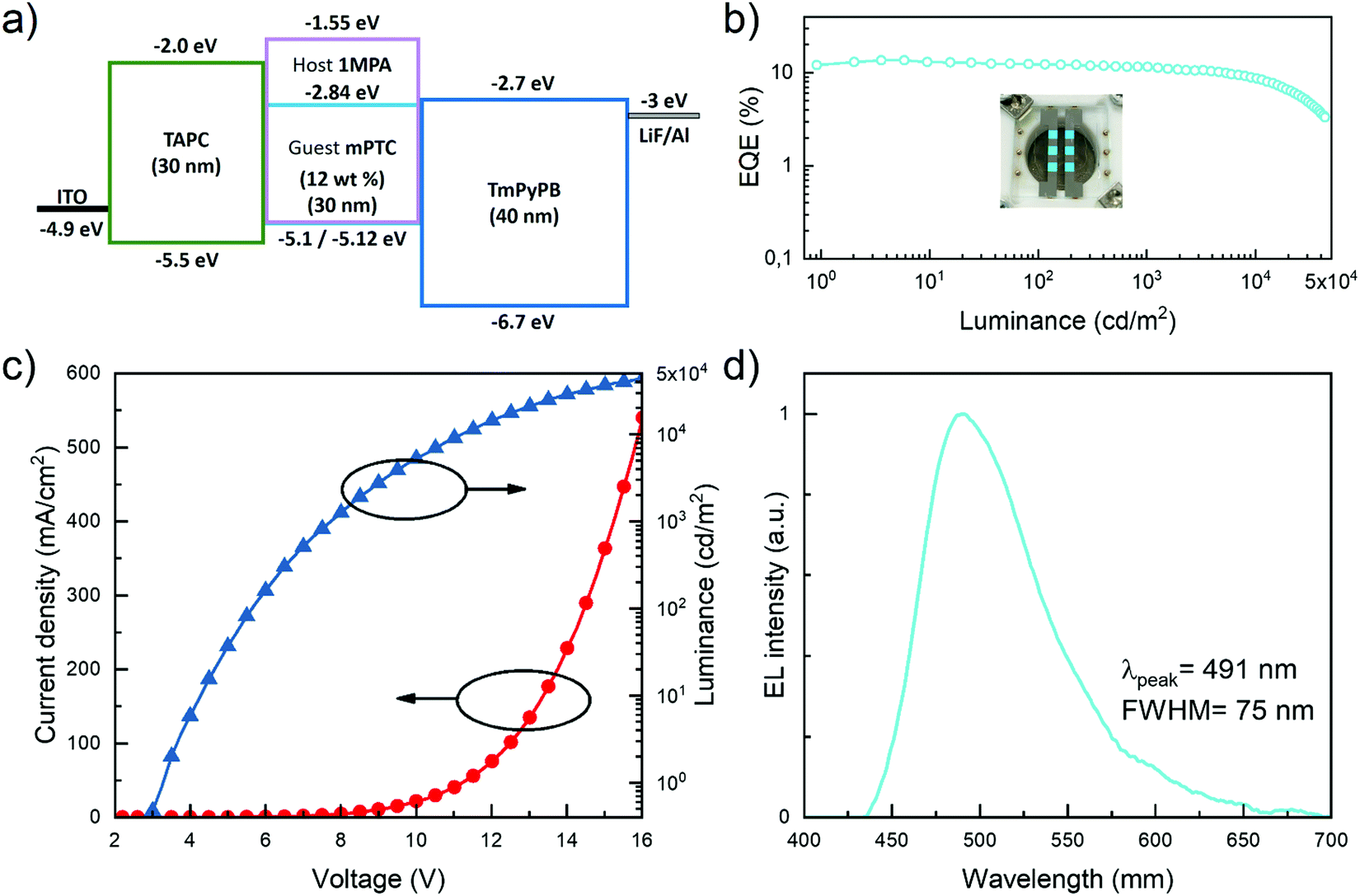 | ||
| Fig. 5 Main properties of the TADF OLED based on the 1MPA host doped with 12 wt% of mPTC. (a) Energy level diagram. (b) EQE vs. luminance. (c) Current density and luminance vs. applied voltage. (d) Electroluminescence spectrum with working device picture shown in the inset. | ||
| Emitter | V on (V) | EQEb (%) | L max (cd m−2) | CEmaxd (cd A−1) | LEmaxe (lm W−1) | λ max (nm) | FWHMg (nm) | CIE 1931h (x, y) |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at 1 cd m−2. b Maximum EQE/EQE at 100 cd m−2/EQE at 1000 cd m−2. c Maximum brightness. d Maximum current efficiency. e Maximum luminous efficiency. f Peak emission wavelength. g Full width at half maximum. h Commission Internationale de L'Eclairage color coordinates (x, y). | ||||||||
| mPTC (12 wt%) | 3.25 | 13.6/12.3/11.5 | 43911 |
33.3 | 27.5 | 491 | 75 | (0.2, 0.4) |
| OBA-O (13 wt%) | 2.75 | 14.2/11.9/8.9 | 30247 |
36.6 | 36.4 | 493 | 83 | (0.2, 0.38) |
| OBA-O (8 wt%) | 3.25 | 14.9/6.8/2.9 | 8449 | 26.2 | 24.5 | 475 | 93 | (0.16, 0.21) |
| OBA-O (5 wt%) | 3.5 | 10.6/3.5/1.6 | 5283 |
14.4 | 12.1 | 461 | 94 | (0.16, 0.18) |
Conclusions
In this work, we present three newly synthesized host molecules that adopt acridine and pyrimidine units. The bulky acridine provides superior thermal and morphological properties, further improved when two acridines are used, like in 1PPA and 1MPA. The donor and acceptor combination allows transporting both holes and electrons. Throughout the series, it is possible to observe a decrease in the conjugation of the acceptor moiety within the hosts. When the acceptor unit is limited to only one pyrimidine ring (1MPA host), the triplet energy is the highest, with a value of 3.07 eV. Through theoretical calculations and spectroscopy, we attribute its nature to a mixed CT–LE state. Finally, to demonstrate the potential of these molecules, 1MPA is employed as the host material to fabricate blue TADF OLEDs. Using the emitter mPTC we achieved a sky-blue OLED with electroluminescence at 491 nm and a maximum EQE of 13.6%, combined with a low roll-off. The host 1MPA is also shown to be suitable for deep-blue emitters like OBA-O, delivering blue OLEDs with color coordinates of x = 0.16 and y = 0.18.Author contributions
F. Rodella performed the synthesis and characterization of the host materials, as well as the thermal and electrochemical analysis. R. Saxena and S. Bagnich performed the photophysical studies. D. Banevičius and G. Kreiza fabricated OLED devices. S. Athanasopoulos was responsible for DFT calculations. S. Juršėnas, K. Kazlauskas, A. Köhler and P. Strohriegl supervised the experiments and corrected the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding through the EU Marie Skłodowska-Curie ITN TADFlife grant (GA. 812872). This work was also supported by Comunidad de Madrid (Spain) – multiannual agreement with UC3M (“Excelencia para el Profesorado Universitario” – EPUC3M14) – Fifth regional research plan 2016–2020 and by the Spanish Ministry of Science, Innovation and Universities (MICINN) through project RTI2018-101020-B-100. The research at Vilnius University was funded by the European Social Fund (project no. 09.3.3-LMT-K-712-01-0084) under grant agreement with the Research Council of Lithuania (LMTLT).References
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Small-Molecule Emitters with High Quantum Efficiency: Mechanisms, Structures, and Applications in OLED Devices, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- M. Y. Wong and E. Zysman-Colman, Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Recent progress in metal-organic complexes for optoelectronic applications, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- Y. Zhang, J. Lee and S. R. Forrest, Tenfold increase in the lifetime of blue phosphorescent organic light-emitting diodes, Nat. Commun., 2014, 5, 5008 CrossRef CAS PubMed.
- Z. Xu, B. Z. Tang, Y. Wang and D. Ma, Recent advances in high performance blue organic light-emitting diodes based on fluorescence emitters, J. Mater. Chem. C, 2020, 8, 2614–2642 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Recent advances in organic thermally activated delayed fluorescence materials, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Wang, J. H. Yun, L. Wang and J. Y. Lee, High Triplet Energy Hosts for Blue Organic Light-Emitting Diodes, Adv. Funct. Mater., 2020, 2008332 Search PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, Thermally activated delayed fluorescence blue dopants and hosts: from the design strategy to organic light-emitting diode applications, J. Mater. Chem. C, 2016, 4, 11355–11381 RSC.
- T. Chatterjee and K.-T. Wong, Perspective on Host Materials for Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes, Adv. Opt. Mater., 2019, 7, 1800565 CrossRef.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Sky-Blue Organic Light Emitting Diode with 37% External Quantum Efficiency Using Thermally Activated Delayed Fluorescence from Spiroacridine-Triazine Hybrid, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- I. E. Serdiuk, C. H. Ryoo, K. Kozakiewicz, M. Mońka, B. Liberek and S. Y. Park, Twisted acceptors in the design of deep-blue TADF emitters: crucial role of excited-state relaxation in the photophysics of methyl-substituted s -triphenyltriazine derivatives, J. Mater. Chem. C, 2020, 8, 6052–6062 RSC.
- T. Serevičius, R. Skaisgiris, I. Fiodorova, G. Kreiza, D. Banevičius, K. Kazlauskas, S. Tumkevičius and S. Juršėnas, Single-exponential solid-state delayed fluorescence decay in TADF compounds with minimized conformational disorder, J. Mater. Chem. C, 2021, 9, 836–841 RSC.
- X.-Y. Liu, F. Liang, Y. Yuan, L.-S. Cui, Z.-Q. Jiang and L.-S. Liao, An effective host material with thermally activated delayed fluorescence formed by confined conjugation for red phosphorescent organic light-emitting diodes, Chem. Commun., 2016, 52, 8149–8151 RSC.
- X.-Y. Liu, Y.-Y. Ma, W. Zhang, B. Song, L. Ding, M.-K. Fung and J. Fan, A Novel Linking Strategy of Using 9,10-Dihydroacridine to Construct Efficient Host Materials for Red Phosphorescent Organic Light-Emitting Diodes, Chemistry, 2018, 24, 11755–11762 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- B. Li, Z. Li, T. Hu, Y. Zhang, Y. Wang, Y. Yi, F. Guo and L. Zhao, Highly efficient blue organic light-emitting diodes from pyrimidine-based thermally activated delayed fluorescence emitters, J. Mater. Chem. C, 2018, 6, 2351–2359 RSC.
- K. Wu, T. Zhang, L. Zhan, C. Zhong, S. Gong, N. Jiang, Z.-H. Lu and C. Yang, Optimizing Optoelectronic Properties of Pyrimidine-Based TADF Emitters by Changing the Substituent for Organic Light-Emitting Diodes with External Quantum Efficiency Close to 25% and Slow Efficiency Roll-Off, Chemistry, 2016, 22, 10860–10866 CrossRef CAS PubMed.
- I. S. Park, J. Lee and T. Yasuda, High-performance blue organic light-emitting diodes with 20% external electroluminescence quantum efficiency based on pyrimidine-containing thermally activated delayed fluorescence emitters, J. Mater. Chem. C, 2016, 4, 7911–7916 RSC.
- H.-J. Park, S. H. Han and J. Y. Lee, Molecular Design of Thermally Activated Delayed-Fluorescent Emitters Using 2,2'-Bipyrimidine as the Acceptor in Donor-Acceptor Structures, Chem. – Asian J., 2017, 12, 2494–2500 CrossRef CAS PubMed.
- K. S. Son, M. Yahiro, T. Imai, H. Yoshizaki and C. Adachi, Blue organic electrophosphorescence diodes using diarylamino-substituted heterocyclic compounds as host material, J. Photopolym. Sci. Technol., 2007, 47–51 CrossRef CAS.
- S.-J. Su, C. Cai and J. Kido, RGB Phosphorescent Organic Light-Emitting Diodes by Using Host Materials with Heterocyclic Cores: Effect of Nitrogen Atom Orientations, Chem. Mater., 2011, 23, 274–284 CrossRef CAS.
- S.-J. Su, C. Cai and J. Kido, Three-carbazole-armed host materials with various cores for RGB phosphorescent organic light-emitting diodes, J. Mater. Chem., 2012, 22, 3447 RSC.
- R. Komatsu, H. Sasabe and J. Kido, Recent progress of pyrimidine derivatives for high-performance organic light-emitting devices, J. Photonics Energy, 2018, 8, 1 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 291–299 CrossRef CAS.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. G. Fox, Gaussian 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 84108 CrossRef CAS PubMed.
- F. Plasser and H. Lischka, Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, On the Metric of Charge Transfer Molecular Excitations: A Simple Chemical Descriptor, J. Chem. Theory Comput., 2013, 9, 3118–3126 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. Rodella, S. Bagnich, E. Duda, T. Meier, J. Kahle, S. Athanasopoulos, A. Köhler and P. Strohriegl, High Triplet Energy Host Materials for Blue TADF OLEDs-A Tool Box Approach, Front. Chem., 2020, 8, 657 CrossRef CAS PubMed.
- K. Gräf, M. A. Rahim, S. Das and M. Thelakkat, Complementary co-sensitization of an aggregating squaraine dye in solid-state dye-sensitized solar cells, Dyes Pigm., 2013, 99, 1101–1106 CrossRef.
- N. Nishi, R. Shimada and Y. Kanda, The Phosphorescence Processes in Pyrimidine and 2-Chloropymidine, Bull. Chem. Soc. Jpn., 1970, 41–46 CrossRef CAS.
- G. F. Makhaeva, S. V. Lushchekina, N. P. Boltneva, O. G. Serebryakova, E. V. Rudakova, A. A. Ustyugov, S. O. Bachurin, A. V. Shchepochkin, O. N. Chupakhin, V. N. Charushin and R. J. Richardson, 9-Substituted acridine derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors possessing antioxidant activity for Alzheimer's disease treatment, Bioorg. Med. Chem., 2017, 25, 5981–5994 CrossRef CAS PubMed.
- K. G. Thorat, R. P. Tayade and N. Sekar, Acridine-1, 8-diones – A new class of thermally stable NLOphores: Photophysical, (hyper)polarizability and TD-DFT studies, Opt. Mater., 2016, 62, 306–319 CrossRef CAS.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Breaking the Kasha Rule for More Efficient Photochemistry, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- D.-Y. Chen, W. Liu, C.-J. Zheng, K. Wang, F. Li, S. L. Tao, X.-M. Ou and X.-H. Zhang, Isomeric Thermally Activated Delayed Fluorescence Emitters for Color Purity-Improved Emission in Organic Light-Emitting Devices, ACS Appl. Mater. Interfaces, 2016, 8, 16791–16798 CrossRef CAS PubMed.
- D. Song, Y. Yu, L. Yue, D. Zhong, Y. Zhang, X. Yang, Y. Sun, G. Zhou and Z. Wu, Asymmetric thermally activated delayed fluorescence (TADF) emitters with 5,9-dioxa-13 b-boranaphtho[3,2,1-de]anthracene (OBA) as the acceptor and highly efficient blue-emitting OLEDs, J. Mater. Chem. C, 2019, 7, 11953–11963 RSC.
- N. C. Greenham, R. H. Friend and D. D. Bradley, Angular Dependence of the Emission from a Conjugated Polymer Light-Emitting Diode: Implications for efficiency calculations, Adv. Mater., 1994, 6, 491–494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03598c |
| This journal is © The Royal Society of Chemistry 2021 |