
Strategies for the synthesis of block copolymers with biodegradable polyester segments
Carlos
Diaz
and
Parisa
Mehrkhodavandi
 *
*
University of British Columbia, Department of Chemistry, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: mehr@chem.ubc.ca
First published on 18th January 2021
Abstract
The controlled synthesis of block copolymers offers great potential for the valorization of biodegradable polyesters, many of which can be bioderived. Combining polyester blocks with other oxygenated polymers through ring-opening polymerization (ROP) and copolymerization (ROCOP) reactions allows for the synthesis of new materials with tunable properties. In this review article, we describe recent advances in the synthesis of diblock and multiblock polyesters, as well as polyether and polycarbonate block copolymers bearing biodegradable polyester block segments. Due to the great diversity of oxygenated monomers available from petrochemical and biomass sources, a great number of polymerization strategies have been developed involving metal- and organo-catalysts with different degrees of control. This review aims to provide an overview of the strategies available for the synthesis of different block copolymers: from the more widespread sequential addition methods to the more rare systems displaying high degrees of kinetic control in a mixture of monomers or those with controlled switchable behavior.
 Carlos Diaz | Carlos Diaz obtained his M.Sc. in chemistry from Universidad de los Andes (Bogota, Colombia) in 2013. He joined the group of Parisa Mehrkhodavandi as a Ph.D. student in 2015 at the University of British Columbia (Vancouver, Canada). His research interests include the exploration of neutral and cationic Group 13 complexes in ring-opening polymerizations, and the controlled synthesis of block copolymers. |
 Parisa Mehrkhodavandi | Parisa Mehrkhodavandi is a Professor at the Department of Chemistry at the University of British Columbia, Vancouver, Canada. She completed her Ph.D. work at MIT (R. R. Schrock) and her postdoctoral work at Caltech (J. E. Bercaw) before starting her independent career at UBC in 2005. Prof. Mehrkhodavandi has garnered a number of awards including the Killam Research Fellowship and the Alexander von Humboldt Fellowship. Her research interests are focused on the cross section of inorganic chemistry, catalysis, polymer science, and green chemistry. |
1. Introduction
Synthetic polymers are ubiquitous in modern life, with applications ranging from every-day packaging and textiles to more specialized use in biomedicine and electronics. Overwhelmingly, these materials are derived from non-renewable petrochemicals. Despite using less than 5% of total oil and natural gas production,1 the global output of synthetic polymers is now well over 300 million tons a year and it is expected to keep growing in the foreseeable future.2 This has raised serious concerns with regards to the very low recyclability of most polymers and their bio-accumulation in various ecosystems.3 Thus, the development of synthetic polymer alternatives that include biobased and/or biodegradable building blocks is a pressing issue.Biobased polymers are macromolecules derived directly from biomass or generated from monomers derived from it.4 The large availability of functionalizable renewable resources has spurred research in more sustainable polymeric materials, many of which are biodegradable polyesters.5 The most common approaches for the utilization of biomass in synthetic polymers are: (i) isolation or chemical modification of natural polymers (e.g. poly(hydroxyalkanoates) from bacteria), (ii) modification of carbohydrates and triglycerides for the synthesis of new monomers (e.g. lactide, succinic acid) and (iii) direct extraction of monomers from natural oils (e.g. tulipalin A, limonene).6 Another important research direction looks at the use of carbon dioxide (a renewable resource) in a copolymerization with epoxides.7 Despite the great diversity of renewable feedstocks, currently, only 1% of all synthetic polymers produced yearly are biobased.8 Some of the biggest barriers to their more widespread commercialization are the higher costs and current limited production scale.9
Biodegradable polymers are macromolecules that experience degradation (i.e. lowering of their molar mass) by a wide variety of biological activities.10 While biodegradable polymers can be either derived from biomass or petrochemicals (i.e. not all biobased polymers are biodegradable and vice versa), nowadays a great proportion of the building blocks used for the synthesis of biodegradable polymers (Scheme 1) come from petrochemicals.11 In order for greener polymers (i.e. bioderived and/or biodegradable) to become more prevalent, they are expected to exhibit properties comparable to those of traditional plastics as well as show new and complementary properties that open the door to new applications. Controlled synthesis of copolymers including biobased and/or biodegradable components has shown promising results, as the diversity of functionalities from renewable resources can be used to tune thermal and mechanical properties of the macromolecules and often allow for post-functionalization.12 In particular, the use of block copolymers including polyester segments is attractive because of their tunable biodegradation, which has been applied in thermoplastic elastomers, polymeric polyols, phase compatibilizers, and polymer-drug conjugates.13
 | ||
| Scheme 1 Some oxygenated monomers employed for the synthesis of polyester, polyether and polycarbonate blocks. (A) Cyclic esters/lactones, (B) O-carboxyanhydrides (OCAs), C = cyclic carbonates; D = anhydrides and epoxides. | ||
Most commonly, the synthesis of block copolymers including polyester units involves the coupling of a polyester with another block through post-functionalization, or the use of a pre-made block as a macroinitiator for the polymerization of a monomer. Such approaches comprise several steps and have been applied with success to some copolymers.13b,14
In this review, we summarize some of the most recent methods for the one-pot synthesis of block copolymers bearing biodegradable polyester segments, covering both metal- and organocatalyst systems active in the ring-opening polymerization (ROP) and ring-opening copolymerization (ROCOP) of biobased and non-biobased monomers (Scheme 1). The synthesis of stereoblock polyester sequences is not included, as the topic has been reviewed earlier.15 Similarly, the combination of ROP and radical polymerization (RAFT or ATPR) for the synthesis of polyester–polyolefin copolymers has been recently reviewed and therefore is not included here.14a,16 For applications of block copolymers, the reader can refer to recent review articles covering their applications in materials and biomedical science.17
2. Synthesis of polyester–polyester blocks
The synthesis of polyester-only block copolymers offers some illustrative examples of how the mechanical and thermal properties of materials can be engineered. For instance, poly(lactic acid) (PLA); one of the most successful biobased polyesters produced on an industrial scale, displays high tensile strength and high Young modulus that make it suitable for replacing polyolefins in some packaging applications.18 However, its high intrinsic brittleness, low impact strength and thermal instability hinder its more widespread use.19 Toughening of PLA and the synthesis of PLA thermoplastic elastomers has been achieved successfully by forming block copolymers with polycaprolactone (PCL)20 and other biobased polyesters.21 The synthesis of those block copolymers can be achieved through isocyanate or amine coupling of two pre-formed polyesters (forming a urethane or amide linkage).22 In some cases, blocky architectures have been achieved using Novozym 435 (immobilized lipase B)23 or through sequential polycondensation methods in the presence of a transesterification agent.24 Due to a large variety of biobased diols and dicarboxylic acids available from biomass, polycondensation methods are an attractive route to a greater variety of block polyesters. However, they suffer from uncontrolled transesterification and frequently yield samples with large dispersities (Đ > 2.0).252.1. Synthesis of polyester–polyester block copolymers through the sequential feeding of monomers
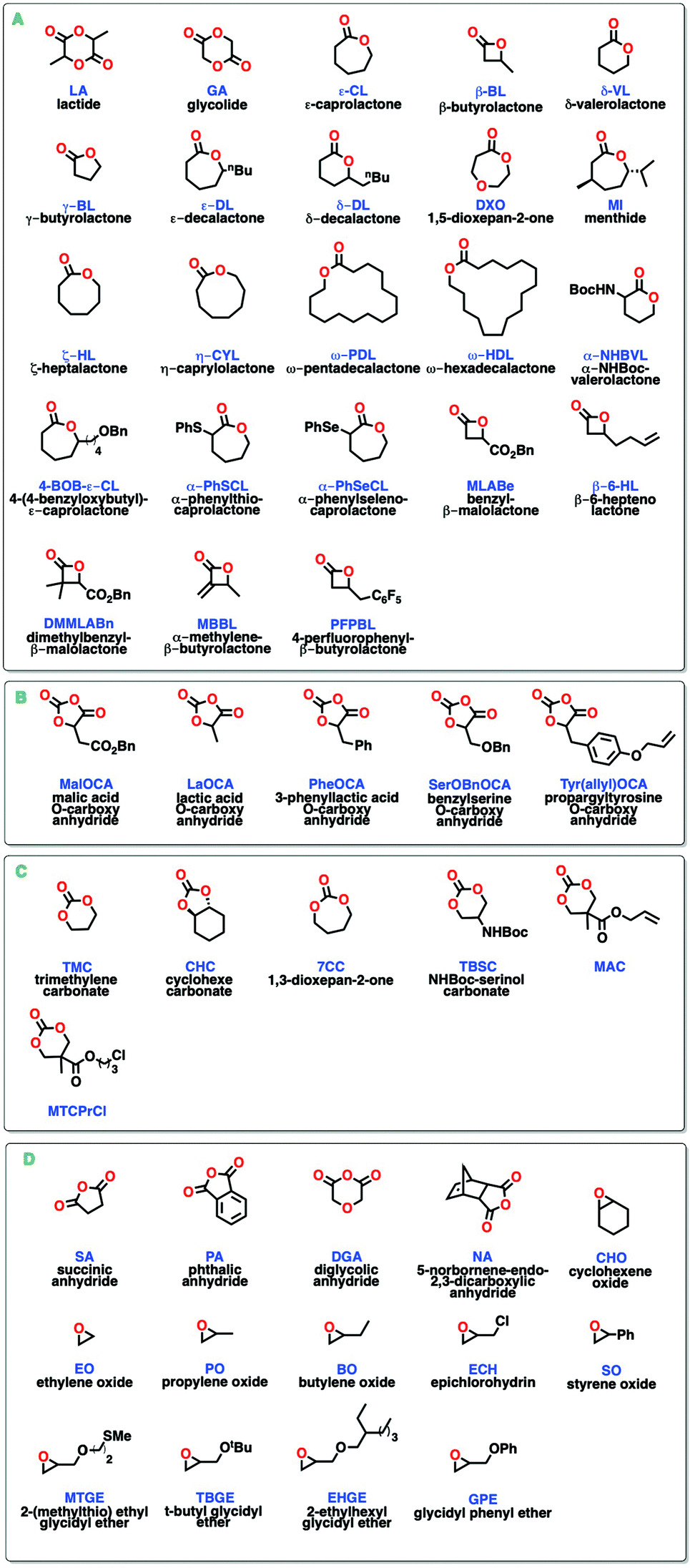
Most of the aluminum systems reported for ROP of cyclic esters are based on phenoxy-imine, salen- and salan-type alkyls or alkoxides that give access to different copolymer microstructures.34 Shaver and co-workers reported that in the presence of alcohol both salen- and salan-type aluminium alkyl (1 and 2, Scheme 2) complexes were active at room temperature and up to 2500 equivalents of ε-CL in an immortal fashion to yield high molecular weight PCL with low dispersities (Đ < 1.1).29b Substituted ε-caprolactones proved difficult to polymerize; particularly in the 2- or 6-position, as no polymerization was achieved with (4R,7S)-4-methyl-7-(1-methylethyl)oxepan-2-one (menthide, MI), even at higher temperatures. Substitution at the 4-position of the ring allowed for a relatively faster and more controlled polymerization at high temperatures, thus 10 equivalents of 4-(4-benzyloxybutyl)-ε-caprolactone (4-BOB-ε-CL) were polymerized at 70 °C in 2 hours. Cooling down the reaction mixture to room temperature followed by addition of 90 equivalents of ε-CL formed a diblock copolymer with monomodal distribution and low dispersity (Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 Da; Đ = 1.15), ruling out transesterification.29b Block copolymers of 4-BOB-ε-CL contain benzyloxy groups that can be deprotected to give free hydroxy groups and can also be used as initiators for the synthesis of brush copolymers. Using the same catalysts, this group also reported the successful copolymerization of L-LA and different alkyl substituted β-butyrolactones at 85 °C to make triblock copolymers with good control (Mn = 33700 Da; Đ = 1.16).35 β-butyrolactones are challenging monomers to polymerize due to poor ring-opening selectivity (where O-acyl or O-alkyl bond cleavage are possible) and the formation of crotonization side-products.36
600 Da; Đ = 1.15), ruling out transesterification.29b Block copolymers of 4-BOB-ε-CL contain benzyloxy groups that can be deprotected to give free hydroxy groups and can also be used as initiators for the synthesis of brush copolymers. Using the same catalysts, this group also reported the successful copolymerization of L-LA and different alkyl substituted β-butyrolactones at 85 °C to make triblock copolymers with good control (Mn = 33700 Da; Đ = 1.16).35 β-butyrolactones are challenging monomers to polymerize due to poor ring-opening selectivity (where O-acyl or O-alkyl bond cleavage are possible) and the formation of crotonization side-products.36
 | ||
| Scheme 2 Metal complexes for the block copolymerization of functionalized cyclic esters through sequential addition approach. Subsequent de-protection reactions form amphiphilic block copolymers. | ||
Zaitsev, Kostjuk and co-workers reported bulky iminophenolate aluminum alkoxides that were relatively controlled in the polymerization of ε-CL or rac-LA at high temperatures (but showed higher dispersities under monomer starved conditions).37 The fluorinated initiator (4, Scheme 3) allowed for a more controlled synthesis of block copolymer, first polymerizing ε-CL and then rac-LA under neat conditions at 130 °C with moderate molecular weight and dispersity (Mn = 13200 Da; Đ = 1.27). Dinuclear salan aluminium system in the presence of an alcohol (5, Scheme 3) has also been reported to give defined block copolymers of ε-CL and L-LA by sequential addition, but only by the initial polymerization of ε-CL followed by lactide.34e Matsubara and co-workers reported an aluminum isopropoxide complex (7, Scheme 3) bearing half-salen-type ligands that could polymerize first rac-LA and then ε-CL in a sequential manner to form block copolymers (Mn = 11500 Da; Đ = 1.24) at 70 °C in pyridine (to suppress dimerization of Al complexes).29c
 | ||
| Scheme 3 Metal complexes for the block copolymerization of lactide and ε-caprolactone through sequential addition. | ||
Pappalardo and co-workers employed a salicylaldiminate aluminum alkyl complex (9, Scheme 4) in the presence of alcohol for the block copolymerization of LA and glycolide (GA) through sequential addition method.38 Under the reaction conditions studied (xylenes, 130 °C), analysis by 13C{1H} NMR spectroscopy showed only homosequence triads, supporting the formation of a block copolymer through a living mechanism (with good agreement between experiment and theoretical molecular weight). Interestingly, switching to the simultaneous addition of monomers formed either blocky-structures or totally random copolymers depending on the reaction conditions used (solution or bulk polymerization). Previously, the same group reported a very similar aluminum alkyl complex active in the block copolymerization of rac- or L-LA with ε-CL through a sequential addition method in toluene at 70 °C.34f
 | ||
| Scheme 4 Salicylaldiminato aluminum alkyl complex for the block-copolymerization of lactide and glycolide through sequential addition. | ||
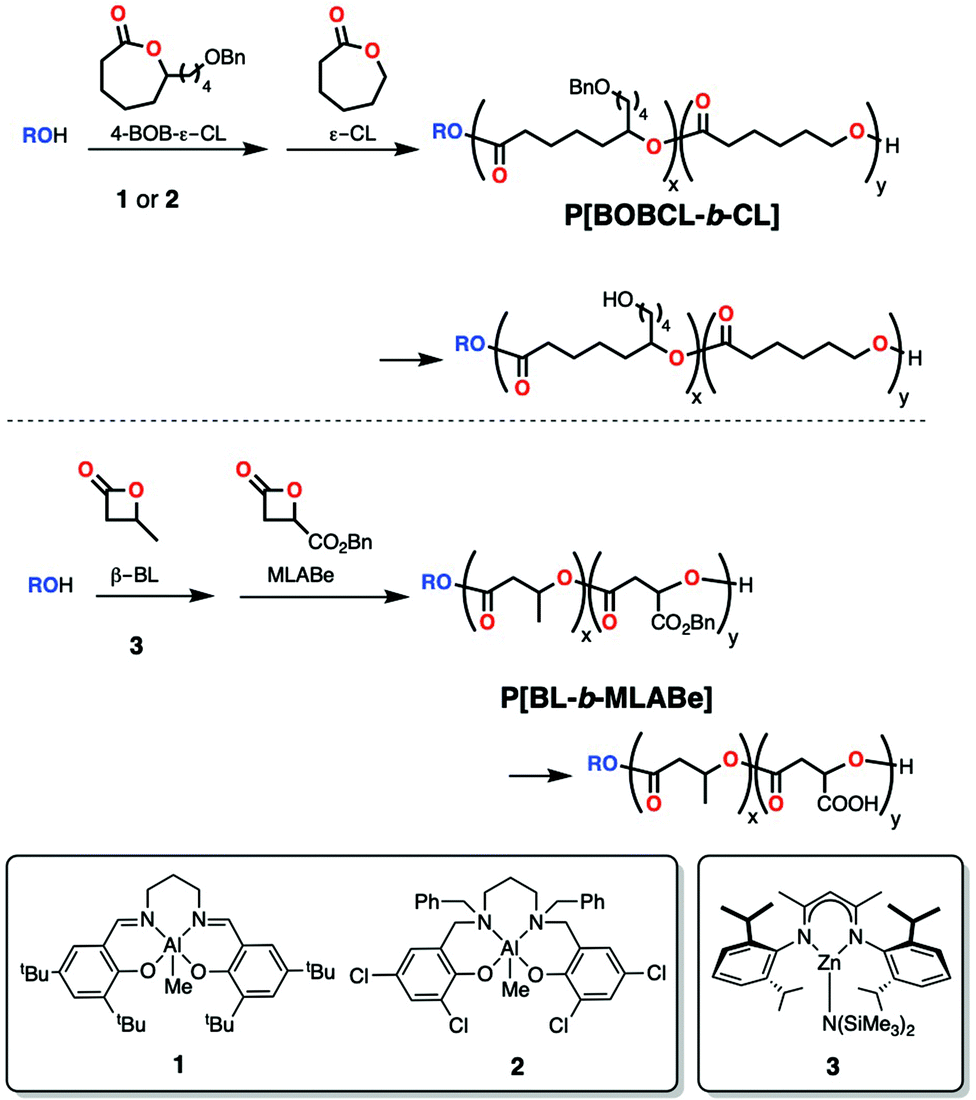
Zinc and indium initiators have been reported for the sequential block copolymerization of L- or D-LA with other cyclic esters. In particular, phosphinophenolate zinc alkoxide complexes reported by Dagorne, Avilés and co-workers were well-behaved initiators for the synthesis of PCL-b-PLLA, but not when lactide was polymerized first, as it's been observed for most aluminum systems (6, Scheme 3).39 The molecular weights obtained with this system were higher than the theoretical values suggesting low initiation efficiency (Mn = 22830 Da; Đ = 1.05). Guillaume and co-workers reported a zinc β-diketiminate amide complex (3, Scheme 2) that, in the presence of alcohol, was capable of copolymerizing challenging rac-β-butyrolactone (β-BL) with benzyl-β-malolactone (MLABe) in a sequential manner (only when BBL was polymerized first).40 Later, Chen, Tong and co-workers employed a lactate version of this catalyst (10, Scheme 5) for the ROP of different enantiopure O-carboxyanhydrides (OCAs) in a sequential manner to form diblock copolymers with controlled molecular weight and isotactic microstructure (no epimerization).41 One of these monomers, L-3-penyllactic acid-O-carboxyanhydride (L-PheOCA) was polymerized in a sequential manner with L-LA to form triblock copolymers with excellent control over the molecular weight (Mn = 32300 Da; Đ = 1.05). Due to their easy synthesis from natural amino acids, OCAs have become attractive monomers for the synthesis of polyesters with functionalizable side groups and therefore tunable physicochemical properties.42
 | ||
| Scheme 5 Zinc β-diketiminate complex active in the block-copolymerization of enantipure OCAs and lactide without epimerization. | ||
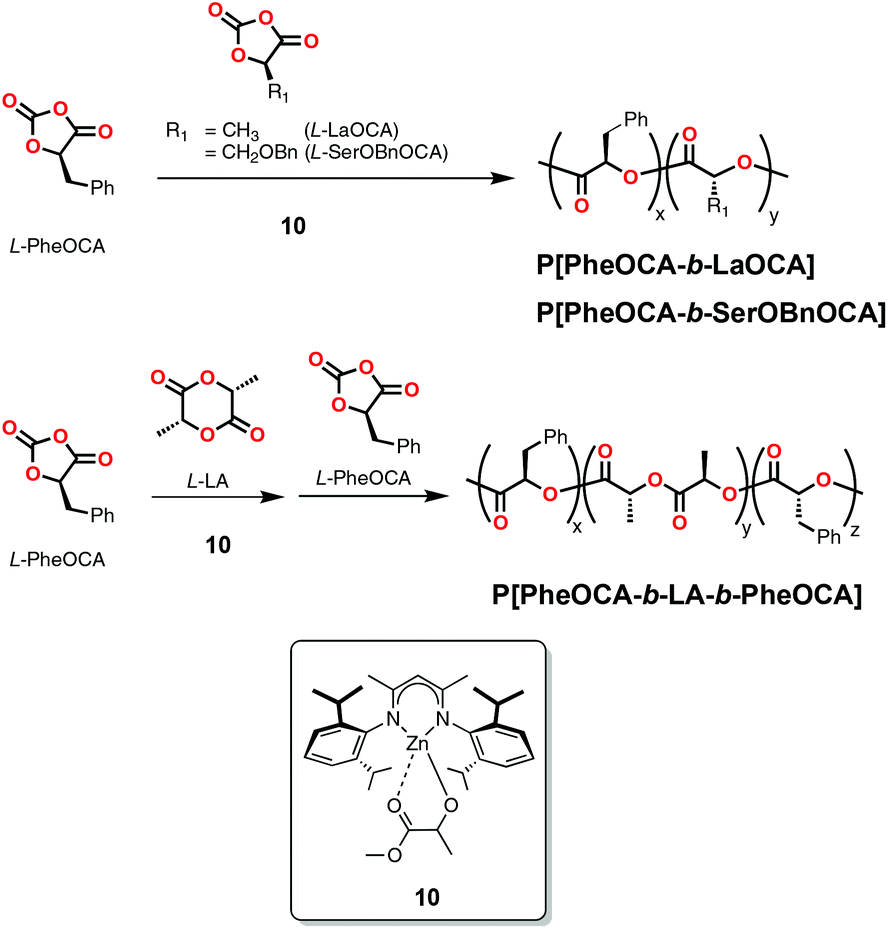
Mehrkhodavandi and co-workers reported the application of a very active dinuclear indium catalyst (11, Scheme 6) in the synthesis of triblock copolymers of L- and D-LA with rac-β-BL.31b The high molecular weight copolymers (Mn = 138000 Da; Đ = 1.22) were made through three consecutive additions and behaved as thermoplastic elastomers with improved mechanical properties over block copolymers of lactide isomers.31b Later, the same group reported another indium catalyst (12, Scheme 6) supported by a salan ligand that could polymerize LA in an immortal fashion to make linear and star block-copolymers when exposed to air and moisture. With this system, diblock copolymers of LA and β-BL were synthesized at room temperature with good control (Mn = 48600 Da; Đ = 1.01).31c
 | ||
| Scheme 6 Indium alkoxide complexes active in the block-copolymerization of lactide and butyrolactone through sequential addition. | ||
Using Hillmyer and Tolman's simple system43 for the syndio-selective polymerization of rac-LA (indium trichloride, triethylamine and an alcohol), Martin-Vaca, Bourissou and coworkers reported the copolymerization of ε-CL and ε-decanolactone (ε-DL) (Scheme 1) to diblock copolymers with good control (Mn up to 22000 Da; Đ = 1.24) and showed no evidence of transesterification regardless of the order of monomer addition.44
Transition metal complexes have been used with success in the sequential block-copolymerization of lactide and other cyclic esters. Interestingly, a titanium(IV) amidinate complex (8, Scheme 3) in the presence of an alcohol was reported to synthesize both PCL-b-PLA and PLA-b-PCL through sequential addition of ε-CL and L-LA (Mn = 12200 Da; Đ = 1.41). Block microstructure was confirmed by the predominance of CL–CL and LA–LA sequences in 13C{1H} NMR analysis.33a Also using group 4 metals, Jones and co-workers reported bimetallic zirconium and hafnium trisphenolate alkoxide complexes for the sequential polymerization of rac-β-BL and rac-LA to make both PHB-b-PLA (Mn = 38900 Da; Đ = 1.33) and PLA-b-PHB (Mn = 41700 Da; Đ = 1.09).45
Over the past decade, the ROP of macrolactones has attracted a lot of interest as a result of their long aliphatic backbone that resembles low density poly ethylene (LDPE),46 with the added advantage of a higher biodegradability imparted by the ester group. Despite their limited degradation under physiological conditions, they can be biocompatible47 and their copolymerization with smaller monomers can tune their physical properties for different applications.26d,48 In 2015, Duchateau and co-workers reported a salen aluminum alkyl complex (13, Scheme 7) capable of copolymerizing in a sequential manner ω-pentadecalactone (ω-PDL) and L-LA in p-xylene at 100 °C to high molecular weight block copolymer with moderate dispersity (Mn = 144000 Da; Đ = 1.50).49 DSC analysis confirmed block microstructure, with two clear melting transitions. Interestingly, block copolymers could only be made if ω-PDL was polymerized first, but not through reverse order of addition, demonstrating the lack of reactivity of a secondary alkoxide group towards macrolactone ROP. Previously, it was found that polymerization of ω-PDL with ε-CL with the same catalyst formed only random copolymers due to uncontrolled transesterification, while tridentate iminophenolate complexes of calcium and zinc (14 and 15, Scheme 7) were capable of forming such block copolymers in toluene at 100 °C (Mn not reported).50 In contrast, the analogous tridentate iminophenolate aluminum complex (bearing the same ligand framework) only produced random copolymers by sequential addition due to extensive transesterification.51
 | ||
| Scheme 7 Aluminum, zinc and calcium complexes for the block-copolymerization of ω-pentadecalactone and lactones through sequential addition. | ||
Later, Lu and coworkers reported the copolymerization of the racemic functionalized lactone α-methylene-β-butyrolactone (rac-MBBL) with rac-β-BL using two different catalysts (16 and 17, Scheme 8).52 Due to the specific stereoinduction exerted by each catalyst (the aluminum complex formed syndio-rich PMBBL block and the yttrium complex formed syndio-rich PHB/PBL block), different diblock semicrystalline copolymers with distinct thermal properties were formed.
 | ||
| Scheme 8 Aluminum and yttrium complexes for the block-copolymerization of butyrolactones through sequential addition. | ||
 | ||
| Scheme 9 Some common organocatalysts active in the ROP of cyclic esters. | ||
Some of the most common bases, 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0] dec-5-ene (TBD) and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) (Scheme 9) have been reported by Guillaume and coworkers to synthesize block copolymers of rac-β-BL and benzyl-β-malolactone (MLABe) with relatively high molecular weight (Mn = 73500 Da; Đ = 1.44), regardless of the order of monomer addition.40 In comparison, zinc β-diketiminate amide complex (3) and alcohol system reported by the same group could only form blocks under these conditions only if β-BL was polymerized first (Scheme 2). As there was no chain-transfer agent (i.e. alcohol) in these polymerizations using organocatalysts, the organic bases were covalently attached to the polymer chain end. The use of TBD afforded polymers with slightly reduced dispersities compared to DBU and BEMP, but high monomer concentration conditions were needed in order to inhibit transesterification, which has been reported with TBD.55 Block copolymers including benzyl-β-malolactone (MLABe) units (Scheme 1) have shown promising drug-delivery activity through the hydrolysis of its benzyl groups, affording amphiphilic block PMLA-b-PHB that can assemble into nanoparticles.56
Using bases in the presence of an alcohol can afford a higher degree of control to the block copolymerization through the formation of alkoxides with tunable basicity. In 2014, Hadjichristidis and coworkers reported that phosphazene base t-BuP2 (Scheme 9) in the presence of various aliphatic and aromatic alcohols polymerized ε-CL with excellent control of the molecular weight and subsequently copolymerized L-LA to form PCL-b-PLLA with excellent control of the molecular weight and low dispersities (Mn = 14900 Da; Đ = 1.15).57 Using TBD in the presence of benzyl alcohol, He and coworkers achieved the polymerization of α-substituted δ-valerolactones (bearing thioether-amino and thioether-alkyl chains of different lengths) in a sequential manner to form block copolymers (Scheme 10) with good control (Mn = 5458 Da; Đ = 1.18).58 This block copolymer showed applicability in gene delivery, with higher efficiency compared to its random counterpart and excellent biocompatibility compared to common non-viral carriers. Later, Li and coworkers used a new cyclic phosphazene base CTPB (Scheme 9) in the presence of benzyl alcohol for the sequential ROP of γ-butyrolactone (γ-BL) and L-LA (Scheme 10) to form block copolymers with relatively good control (Mn up to 20500 Da; Đ = 1.62). This was the first report of a block copolymer of γ-BL,59 whose unfavorable ROP thermodynamics require low polymerization temperatures, giving products with relatively high dispersities.60
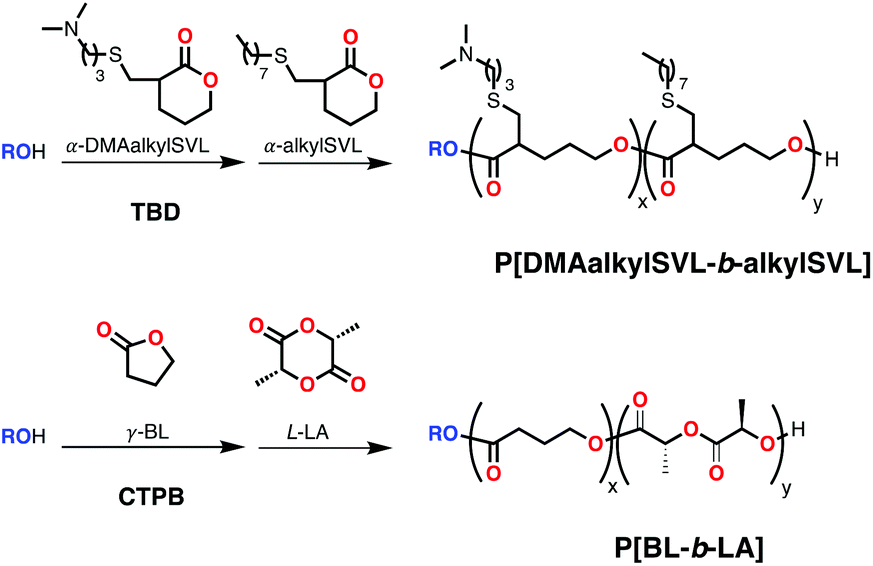 | ||
| Scheme 10 Block copolymerization of cyclic esters using organobases and alcohol through sequential addition. | ||
Using organic acids in the presence of an alcohol can also lead to controlled polymerizations through an activated monomer mechanism. In 2014 Martín-Vaca, Bourissou and coworkers applied this strategy for the polymerization of rac-β-BL using triflic acid and n-pentanol or 1,4-butanediol followed by the polymerization of ε-CL to give diblock or triblock copolymers with medium molecular weight (Mn = 6670 Da; Đ = 1.21 for diblock).61 Other organic acids such as diphenyl phosphate (DPP) have been the synthesis of block copolymers, in particular for δ-valerolactone (δ-VL) and ε-CL regardless of the monomer addition order (Scheme 11).62 Years later, Guo and coworkers employed commercially available dibutyl phosphate and 3-phenyl-1-propanol under industrial-relevant conditions (neat, 180 °C) to form block copolymers PCL-b-PVL with medium molecular weight and remarkably low dispersities (Mn up to 5340 Da; Đ = 1.11).63
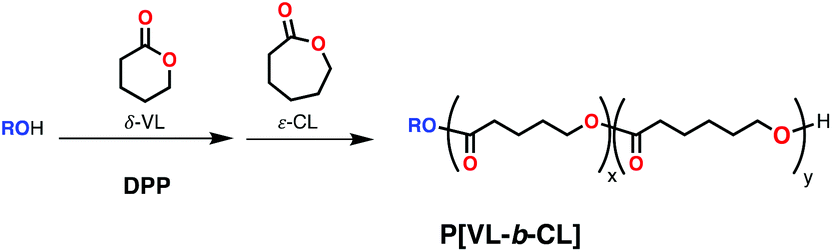 | ||
| Scheme 11 Block copolymerization of δ-VL and ε-CL using organic acid diphenyl phosphate (DPP) and alcohol through sequential addition. | ||
Some organocatalyst systems have also been applied to the block copolymerization of macrolactones to give high molecular weight products. In 2015, Dubois, Todd and coworkers reported that TBD in the presence of an alcohol was capable of polymerizing ω-PDL and L-LA in a sequential manner (Scheme 7): first in bulk at 100 °C, then at 25 °C in CHCl3 to prevent transesterification in the lactide polymerization step (Mn = 107910 Da; Đ = 1.11). The block nature of the copolymer was corroborated by 1H, 13C{1H} and DOSY NMR spectroscopy.64 More recently, Liu, Li and coworkers adopted a similar strategy for the synthesis of PPDL-b-PLA (Mn = 37800 Da; Đ = 1.73) using cyclic trimeric phosphazene base (CTPB) in toluene at 80 °C without evidence of scrambling by transesterification.65
Recently Hadjichristidis and coworkers reported a dual organocatalyst approach for the synthesis of block copolymers using ω-PDL or ω-hexadecalactone (ω-HDL) and smaller lactones δ-VL or ε-CL.66 The system involved the use of strong phosphazene base t-BuP4 (Scheme 9) with alcohol for the polymerization of the macrolactone in toluene at 80 °C, followed by neutralization using an equimolar amount of protic acid diphenyl phosphate (DPP) and the addition of weaker phosphazene base t-BuP2 with the smaller lactone to avoid transesterification (Scheme 12). This catalyst-switch strategy produced block copolymers PPDL-b-PCL, PPDL-b-PVL, PHDL-b-PCL and PHDL-b-PVL (Mn up to 83300 Da; Đ = 1.92).
 | ||
| Scheme 12 Catalyst-switch strategy for the block-copolymerization of cyclic esters through sequential addition of monomers. | ||
Another example of a dual organocatalyst approach was reported by Kakuchi and coworkers (Scheme 12).67 By switching from diphenyl phosphate (DPP) activation in the presence of an alcohol to 4-(dimethylamino)pyridine (DMAP) activation, they achieved the synthesis of PVL-b-PLLA (Mn = 11600 Da; Đ = 1.16) and PCL-b-PLLA (Mn = 12600 Da; Đ = 1.12) in a sequential, one-pot procedure. Also, 1,5-dioxepan-2-one (DXO) was polymerized with this system to form PDXO-b-PLLA (Mn = 12100 Da; Đ = 1.08). PDXO is a highly amorphous polymer and more prone to hydrolysis compared to PCL or PLA.68 A similar strategy was used by Li, Guo and coworkers, who reported the synthesis of block copolymers PVL-b-PLLA (Mn = 12200 Da; Đ = 1.09) and PCL-b-PLLA (Mn = 13800 Da; Đ = 1.06).69 Using a reverse switch, Lin, Li and coworkers could polymerize first rac-LA (employing benzyl alcohol and DBU) followed by a 2-substituted δ-VL (using an excess of DPP) to yield amphiphilic block copolymers.70
Exploiting synergistic effects between different types of organocatalysts (an electrophile and a nucleophile) has opened the door to some of the most active and controlled systems capable of sequential copolymerization of different cyclic esters at room temperature. For example, Dove and coworkers applied a thiourea/amine system with an alcohol for the rapid block copolymerization of substituted glycolide and L-LA (Mn up to 24160 Da; Đ = 1.07).71 Similarly, Li, Liu and coworkers reported a thiourea/CTPB system with different alcohols to give in a matter of minutes PVL-b-PCL (Mn = 12700 Da; Đ = 1.16).72
Following the pioneering contributions of Amgoune,74 Bourissou,74 Dove,73a Naumann75 and coworkers, where inactive DMAP or a N-heterocyclic olefin (NHO) were rendered active in the polymerization of LA, ε-CL or ω-PDL with high activity and control in the presence of magnesium or zinc compounds, Zhang and his group reported the synthesis of block polyesters of ε-CL and δ-VL in a living fashion to high molecular weight copolymers (Scheme 13) and excellent control (Mn = 72100 Da; Đ = 1.15 for triblock) using Al(C6F5)3 and NHO (Scheme 9) as a Lewis pair.76 The mechanism of polymerization involved an initiation step by the nucleophilic attack of the NHO base to a monomer unit activated by the Lewis acid. This forms a propagating imidazolium-enolate group (Scheme 13) that is controlled in further ROP and remains as chain-end group in the block copolymers.76
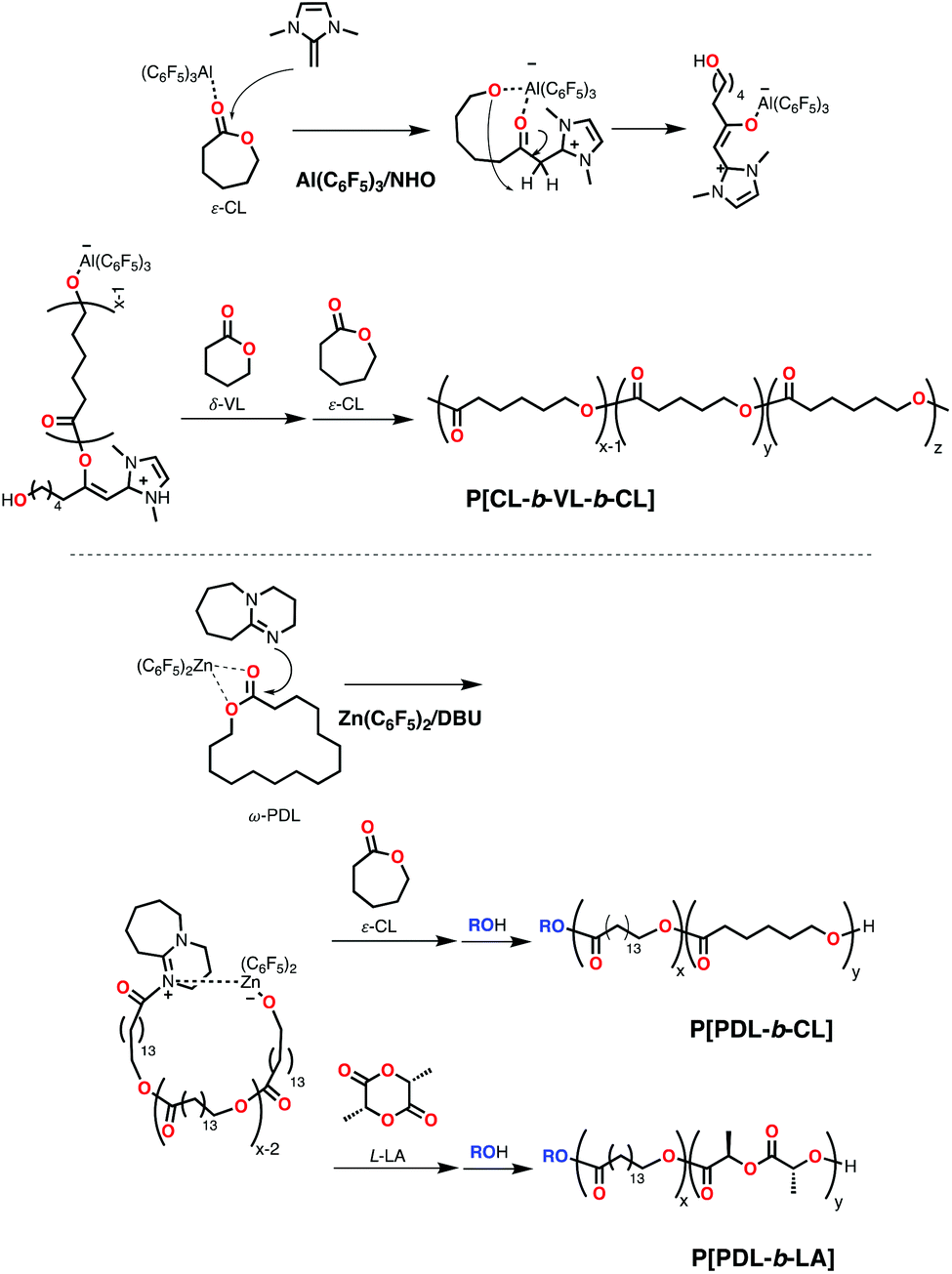 | ||
| Scheme 13 Mechanism of ROP of cyclic esters using different Lewis pairs for the synthesis of block copolymers through sequential addition. | ||
Recently, Li and coworkers reported the block copolymerization of ω-PDL/ε-CL (Mn = 79900 Da; Đ = 1.90) and ω-PDL/LA (Mn = 87900 Da; Đ = 1.80) through sequential addition using Zn(C6F5)2 and DBU (Scheme 9) in the presence of an alcohol (Scheme 13).77 The mechanism proceeds similarly to the previous system with initial dual activation of the monomer by the Lewis acid and base to form a propagating alkoxide species active in sequential copolymerization (Scheme 13), but in this case the close interaction of the ion pair allows for the control synthesis of cyclic copolymers in the absence of alcohol. When an alcohol is added, linear diblock structures could be obtained (Scheme 13). However, randomization of the block structure was detected in the case of PPDL-b-PCL after prolonged reaction times under the conditions studied (xylene, 110 °C). Neither PPDL-b-PLA nor cyclic PPDL-b-PCL (prepared without alcohol addition) showed evidence of scrambling by transesterification under these conditions.77
2.2. Synthesis of polyester–polyester block copolymers through simultaneous feeding of monomers
Simultaneous synthesis of block copolymers that follow the same ROP mechanism can be challenging and requires that the different monomers have very different reactivities with a particular catalyst, implying that one gets consumed much faster than the other (kinetic control), but even in that case transesterification in the system can turn blocky structures into random copolymers. For example, polymerization of unsaturated macrolactones such as globalide (macrolactone) and 1,5-dioxepan-2-one (DXO) formed random copolymers in the presence of Novozym 435 as a catalyst (Candida Antarctica Lipase B immobilized). This scrambling occurred despite the different reactivity ratios of those two monomers.78Waymouth and coworkers addressed this issue in 2011, reporting Zwitterionic ROP as a strategy for gradient (block-like) copolymers in the simultaneous copolymerization of ε-CL and δ-VL with nucleophilic N-heterocyclic carbenes.79 Both linear and cyclic products (obtained in the presence and absence of alcohol respectively) contained a larger fraction of homo-dyads than hetero-dyads by 13C{1H} NMR spectroscopy compared to the control experiment with tin(II) octoate, corroborating the blocky-structure of the copolymers.
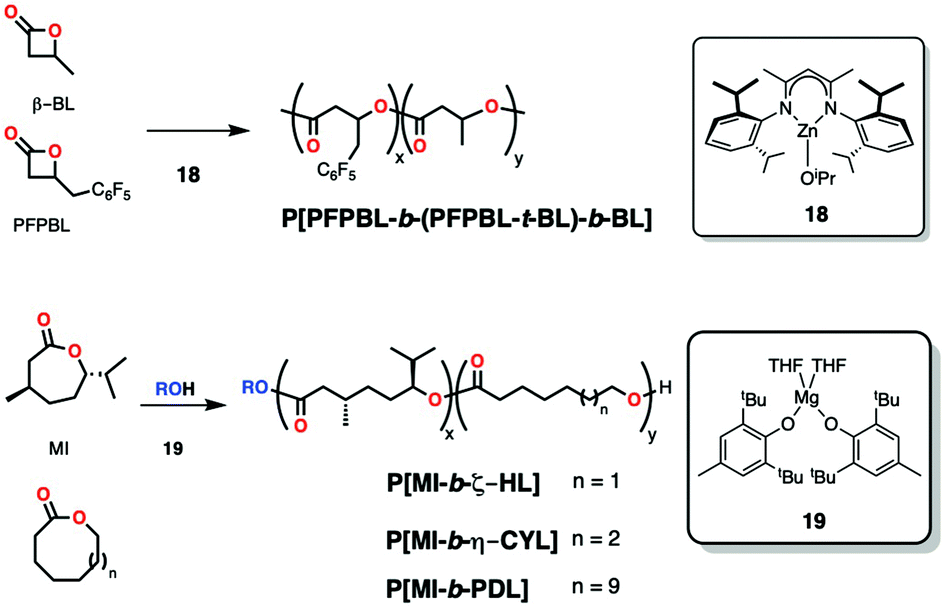 | ||
| Scheme 14 Zinc and magnesium complexes active in the synthesis of block and block-like copolymers through the simultaneous addition of monomers. | ||
Recently, Dove and coworkers reported a simple magnesium phenoxide complex (19, Scheme 14) for the block-copolymerization of 3,6-disubstituted lactone menthide (MI) and larger lactones with minimal ring-strain like ζ-heptalactone (ζ-HL, 8-membered ring) or η-caprylolactone (η-CYL, 9-membered ring) in toluene at 80 °C (Scheme 14). In a mixture of monomers, the magnesium complex polymerized the more reactive MI followed by the less strained lactone to give block-like macromolecules (Mn up to 18400 Da; Đ = 1.50), as evidenced by the presence of major homocoupling carbonyl diads in 13C{1H} NMR spectroscopy (with only a minor presence of heterocoupling diads). Previously, the same group had reported a similar degree of kinetic control in the polymerization of MI and ω-PDL (Scheme 14) as evidenced by conversion profiles and 13C{1H} NMR spectroscopy (Mn = 13800 Da; Đ = 2.41).26c Their rationale behind the kinetic control exhibited by this catalyst was that the initial formation of PMI block is “locked” against transesterification side reactions (due to the presence of methyl and isopropyl groups in each monomer unit) and once MI has been depleted, the second block starts forming and can only undergo transesterification mostly within itself. In contrast, reaction of MI with a more reactive lactone (such as ε-CL) forms completely random copolymers as the more reactive small monomer will react first and any incorporation of MI added at the chain end will rapidly be pushed to the middle of the chain by fast transesterification. Random copolymers in this case were evidenced by equal integration of homocoupling and heterocoupling diads in NMR spectroscopy.81
000 Da; Đ = 1.65), polymerizing first more sterically hindered MLABe and then rac-β-BL (despite homopolymerizations having very similar rates). In comparison, TBD and DBU only polymerized MLABe in the simultaneous addition, indicating inhibition in the polymerization of rac-β-BL, although no detailed mechanistic studies were presented.40
 | ||
| Scheme 15 Synthesis of block copolymers by different organocatalysts with simultaneous feeding of monomers. | ||
In 2013, Basko and coworkers reported another organocatalytic system capable of block-copolymerization with simultaneous feeding of monomers: triflic acid (TfOH, Scheme 9) in the presence of alcohol copolymerized rac-β-BL and L-LA in CH2Cl2 at room temperature through an activated monomer mechanism. The conversion profile showed that rac-β-BL was polymerized quickly with this system (in less than an hour) and L-LA was consumed following an induction period after full conversion of rac-β-BL (Scheme 15). Formation of a block copolymer (Mn = 3420 Da; Đ = 1.20) was corroborated by 13C{1H} NMR spectroscopy and thermo gravimetric analysis (TGA).82
Exploiting the previously reported O-alkyl cleavage ROP of β-lactones catalyzed by carboxylate salts,83 Coulembier and co-workers employed a bifunctional hydroxyl-carboxylic acid initiator in the presence of DBU to achieve dual activation of β-lactone 2,2-dimethylbenzyl-β-malolactone (DMMLABn) and lactide in the simultaneous addition of monomers (Scheme 15).84 The dual polymerization occurs first by an acid-base reaction between the carboxylic group in the initiator and DBU, forming a carboxylate group that polymerizes DMMLABn (with a carboxylate-propagating species). The carboxylate propagating species also works as an activator of the alcohol chain-end to polymerize LA (with an alcohol-propagating species). The resulting block copolymers (Mn = 12450 Da; Đ = 1.46), however suffered from transesterification and large dispersities when long reaction times were applied.
Diaconescu and coworkers exploited ligand conformational changes through redox switching for the block copolymerization of very similar monomers L-LA and ε-CL.88 The authors tested different zirconium and titanium alkoxide complexes supported by redox-active ferrocene-based ligands. Block copolymerization in a mixture of monomers was achieved by chemical redox switching using a titanium complex (20red, Scheme 16) that on its reduced form was active in LA polymerization and on its oxidized form (20ox, Scheme 16) was much more active for ε-CL polymerization, yielding PLA-b-PCL (Mn = 3230 Da; Đ = 1.12). Narrow dispersity indicated a controlled polymerization, but only low conversions of monomer were achieved (as an increase in reaction times led to a decrease in substrate selectivity). Formation of multiblock copolymers through several switches was not reported.88a
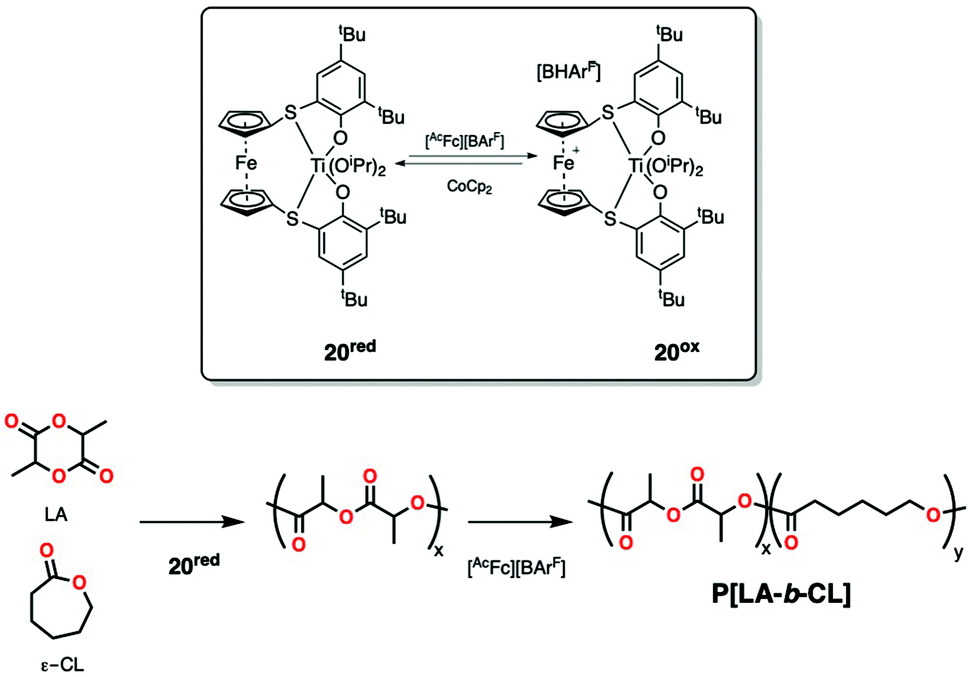 | ||
| Scheme 16 Redox-switchable system active in the one-pot block copolymerization of lactide and ε-caprolactone with simultaneous feeding of monomers. | ||
900 Da; Đ = 1.21). The ability of this catalyst to switch between two different polymerization cycles was rationalized by the very different rates of insertion of PA and ε-DL into the zinc alkoxide bond.89
Using a variation of this catalyst, a dinuclear zinc acetate complex (21, Scheme 17) Williams and coworkers reported the formation of block copolymers from mixtures of PA, CHO and ε-CL (Mn = 22500 Da; Đ = 1.46). DFT studies performed on this system revealed that selectivity results from lower activation barriers (lower for anhydride insertion) and more stable linkages during polymerization (more thermodynamically stable ester linkages from ROCOP).90 The same group also reported that commercially available chromium(III) salen complex (22, Scheme 17) was competent in both ROP of ε-DL and ROCOP of different anhydrides with CHO in the presence of an equimolar amount of bis(triphenylphosphoranylidene) imminium chloride (PPNCl) as a cocatalyst. Polymerizations were carried out under immortal conditions at 100 °C in toluene-d8 using 1,2-cyclohexanediol as a chain-transfer agent to yield low molecular weight triblock copolymers with good control (Mn = 4430 Da; Đ = 1.20). In a similar way to the bimetallic zinc complex 21, the chromium salen complex 22 showed high selectivity to alternating polyesters through ROCOP in a mixture of different monomers with high activity and no significant ROP activity. Once the anhydride was consumed catalyst switched to ROP of ε-DL. The large scope of anhydrides tested allowed for postfunctionalization through the thiol-ene reaction.91
 | ||
| Scheme 17 Catalysts active in the tandem ROCOP of anhydrides/epoxides and ROP of lactones for the synthesis of polyester block copolymers. | ||
More recently, Mazzeo and coworkers employed a bimetallic aluminum alkyl complex bearing salen-type ligands with dinaphthalene Schiff bases (23, Scheme 18) in the same reaction using PA, CHO and L-LA in toluene at 110 °C and in the presence of isopropyl alcohol and PPNCl as a cocatalyst. Formation of an alkoxide species in situ was active in ROCOP, but with a lower selectivity (81% polyester linkages) compared to William's systems (vide supra).90 Following ROP of lactide formed a block copolymer with high molecular weight (Mn = 61000 Da; Đ = 1.44).92
 | ||
| Scheme 18 Aluminum complex active in the tandem ROCOP of anhydrides/epoxides and ROP of lactide for the synthesis of polyester block copolymers. | ||
In contrast to metal-base systems, organocatalysts do not typically combine ROCOP and ROP in a controlled fashion for the synthesis of block copolymers from a mixture of monomers. One of rare example was reported independently by Zhao,93 Wang, Li and their coworkers94 and involved the use of a non-nucleophilic phosphazene base (t-BuP1) with alcohol at 60 or 100 °C for the polymerization of PA with different epoxides and rac-LA.93 As it was observed with the metal-based systems (vide supra), ROCOP of PA and epoxides took place first with high selectivity, followed by ROP of LA in a tandem mechanism giving diblocks, triblocks or pentablocks with very good control (Mn up to 31300 Da; Đ = 1.09).
3. Synthesis of polyether–polyester blocks
Polyester–polyether block copolymers can be synthesized by ROP of cyclic ethers and lactones. Although multiple metal-based initiators and organocatalysts have been reported to give a controlled polymerization of these molecules, very often a catalytic system optimal for the polymerization of one monomer is inactive or leads to uncontrolled polymerization of the other. Consequently, many of the synthetic procedures reported for the synthesis of polyester–polyether blocks require multiple steps involving different catalytic systems and the purification of intermediates. Some approaches include controlled polycondensation reactions between two preformed polymers95 and their linkage through post-functionalization (e.g. click reactions).96By far the most common synthetic strategy involves using commercially available polyethers such as polyethylene glycol/oxide (PEG/PEO), polytetramethylene oxide (PTMO), polypropylene oxide (PPO) or poloxamers as macroinitiators in the ROP of the cyclic ester in the presence of a suitable catalyst. In this fashion, block copolymers of LA,97 ε-CL,98 δ-VL,83b,99 β-BL,100 ω-PDL,101O-carboxyanhydrides (OCAs)102 and other functionalized monomers103 have been accessed. In particular, ROP of OCAs has attracted significant interest due to their straightforward synthesis from naturally abundant precursors and easy access to functionalized monomers.104 Other functionalized monomers based on ε-CL and LA have also been employed in their copolymerization with PEG macroinitiators to make diblock and triblock copolymers.105
The most widely used initiator for these polymerizations is tin(II) octoate, which works well with most common 6- or 7-membered rings in the presence of a hydroxy-capped polyethers, yielding polyester–polyether block copolymers with good control over molecular weight (dispersities as low as 1.1, given that a monodispersed macroinitiator is used). In order to copolymerize PEG and 4-membered lactones, Gillies and coworkers employed a salen aluminum alkyl complex 1 (Scheme 2) to polymerize challenging β-6-heptenolactone (β-6-HEL, Scheme 1) with excellent control in the presence of MeO-PEG (Mn = 2000 Da) forming amphiphilic diblocks (Mn = 12910 Da; Đ = 1.03) that could be subsequently functionalized through the thiol-ene reaction for application in drug delivery (Scheme 19).106 Another notable exception is the polymerization of OCAs. Using organocatalysts such as DMAP or 4-methoxypyridine, Dove and coworkers have reported the polymerization of OCAs derived from malic acid (L- or D-MalOCA) in the presence of MeO-PEG (Mn = 7500 Da) to give amphiphilic diblock copolymers (Mn = 9400 Da; Đ = 1.04) that form stable stereocomplexed micelles in solution.102b Polymerization of macrolactone ω-PDL or bioderived δ-decalactone (δ-DL) has also been achieved using organocatalyst TBD in the presence of diamino- or dihydroxy-capped PEG macroinitiator to give amphiphilic triblock copolymers.101,107 Other organocatalytic systems such as DBU,99 sparteine/thioureas108 and phosphazene base t-BuP257 have also been employed for the polymerization of cyclic esters in the presence of polyether macroinitiators. ROCOP of anhydrides and epoxides has also been performed in the presence of a polyether macroinitiator to make block copolymers. Recently, Xiao, Chen and coworkers employed phosphazene base t-BuP1 in the presence of MeO-PEG (Mn = 5000 Da) for the polymerization of 2-(methylthio)ethylglycidyl ether (MTG, Scheme 1) and PA to make an amphiphilic block copolymer (Mn = 12500 Da; Đ = 1.03) with redox-responsive properties for application in drug delivery (Scheme 19).109
 | ||
| Scheme 19 Synthesis of amphiphilic polyether–polyester block copolymers using PEG as a preformed block. Subsequent reactions lead to post-modification of the hydrophobic block in the copolymer. | ||
3.1. Synthesis of polyether–polyester blocks through the sequential feeding of monomers
The synthesis of amphiphilic polyester–polyether block copolymers has attracted a considerable attention as a route to novel drug and gene delivery vehicles with pH- or light-responsive properties.110 While PEG has been the polyether of choice in these copolymers due to its wide availability, low toxicity, solubility in water and favorable pharmacological properties,111 its use as a preformed block offers limited possibilities of functionalization. It is reasonable to expect a greater chemical diversity from more versatile synthetic methods. Combining the ROP of epoxides with other ring-opening polymerization and copolymerization reactions is an attractive strategy to access new polyester–polyether block copolymers with tunable properties.100 Da; Đ = 1.97) and PCHO-b-PLA (Mn = 11000 Da; Đ = 1.57) through the sequential feeding of CHO and ε-CL/L-LA. Interestingly, simultaneous feeding of the monomers afforded only the polyester even after prolonged reaction times.112 Employing more Lewis acidic titanium and zirconium isopropoxide complexes bearing 8-hydroxyquinoline ligands, Chand and coworkers reported formation of blocky structures with simultaneous feeding of t-butyl glycidyl ether (tBGE) (Scheme 1) and L-LA by a faster polymerization of the cyclic ester.113 No simultaneous feeding was attempted in this case.
More recently, Mehrkhodavandi and coworkers reported a cationic indium complex (24, Scheme 20) capable of polymerizing epoxides such as epichlorohydrin (ECH, Scheme 1) in a cationic mechanism and subsequently could polymerize rac-LA in a coordination-insertion mechanism to form block copolymers (Scheme 20).114 The rationale in this case was that cationic polymerization of the epoxides led to the formation of a polyether block with a cationic chain-end (active in epoxide polymerization) and an alkoxide chain-end bound to a neutral indium species (active in lactide polymerization). Some of the block copolymers prepared in this fashion exhibited increased ductility and stiffness compared to rac-lactide homopolymer of similar molecular weight.114 A disadvantage of this strategy was the lower-than-expected molecular weights based on the monomer to initiator ratios attributed to back biting and transesterification reactions.
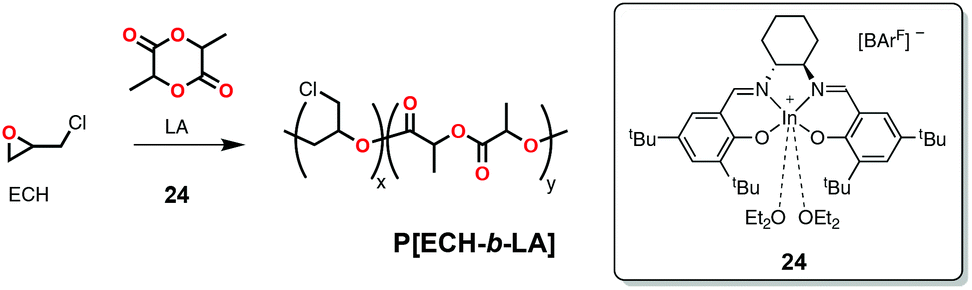 | ||
| Scheme 20 Cationic indium complex active in the block copolymerization of epichlorohydrin and lactide through the sequential addition of monomers. | ||
400 Da; Đ = 1.08) with the subsequent addition of ε-CL (Scheme 21).116 Later, Limbach and coworkers adapted this strategy using more stable NHC-CO2 precursors that could generate free NHCs in situ when heated to 120 °C reaching good catalytic activities in the formation of block copolymers in a sequential fashion (Scheme 21).117
 | ||
| Scheme 21 Catalysts active in the sequential polymerization of ethylene oxide and cyclic esters. | ||
Another initiator that shows controlled reactivity towards epoxides and cyclic esters is phosphazene t-BuP2 (Scheme 9). Hadjichristidis and coworkers reported its rather slow activity in the polymerization of EO (days) through activation of 3-phenyl-1-propanol or water in a pseudo-anionic mechanism. Subsequent addition of ε-CL formed block copolymers in a living fashion that allowed further addition of L-LA for multiblock synthesis (Mn = 4600 Da; Đ = 1.03), but only in that order as ε-CL couldn't be polymerized after L-LA (Scheme 21). In this case, the choice of phosphazene base is crucial as the use of the more basic t-BuP4 brings about extensive transesterification in the polyesters blocks and less basic t-BuP1 is not active in epoxide ROP.118
To improve upon their phosphazene polymerization system, Hadjichristidis and coworkers reported a catalyst-switch strategy using strong base t-BuP4 (Scheme 9) in the presence of an alcohol for the fast polymerization of epoxides, followed by an addition of protic diphenyl phosphate (DPP) in excess to quench the phosphazene base. After a couple of minutes, δ-VL or ε-CL were added to the reaction mixture and were polymerized by the remaining DPP in an activated monomer mechanism, forming block copolymers with excellent control. This was not only faster than the previously reported, but also showed living epoxide ROP and allowed the use of bulkier butylene oxide (BO) together with EO for the synthesis of triblock PBO-b-PEO-b-PCL (Mn = 15100 Da; Đ = 1.09).119 This methodology was later expanded to the block copolymerization of bulky epoxides BO or 2-ethylhexyl glycidyl ether (EHGE) (Scheme 1) with bulky, bioderived 5-substituted δ-valerolactones.120 As DPP is not a suitable initiator for LA polymerization, the strategy was modified by addition of a stoichiometric amount of DPP (to phosphazene base) after polymerization with t-BuP4. Subsequent addition of t-BuP2 and cyclic esters allowed the formation of triblock copolymers (Scheme 22) PSO-b-PCL-b-PLA (Mn = 13700 Da; Đ = 1.16) by the sequential polymerization of styrene oxide (SO), ε-CL and L-LA.121
 | ||
| Scheme 22 Catalyst-switch strategy for the block-copolymerization of epoxides and cyclic esters through the sequential addition of monomers. | ||
Similar catalyst-switch strategies have been reported with other organocatalysts. In 2016, Zhao and coworkers employed a base-to-base switch approach involving initial polymerization of EO by t-BuP4 in the presence of 1,4-butanediol followed by addition of stoichiometric 1,3-dicyclohexylthiourea (to phosphazene base). Deactivation of strong phosphazene-alkoxide complex led to the formation of a weaker thioureate anion and an alcohol in an equilibrium that could polymerize either L-LA or ε-CL. This base attenuation strategy allowed for the formation of triblock copolymers (Scheme 19) at room temperature with excellent control PLA-b-PEO-b-PLA (Mn = 9900 Da; Đ = 1.01) and PCL-b-PEO-b-PCL (Mn = 21100 Da; Đ = 1.11).122 The mechanism at play might be a dual activation of the monomer and the chain-end as previously described by Waymouth and coworkers in their very active and selective thiourea/alkoxide systems.123 Using activator 1,3-diphenylthiourea with higher acidity significantly affected the polymerization of ε-CL (but not of L-LA), giving very low conversions even at 40 °C. The even more acidic activator 1,3-bis[3,5-bis(trifluoromethyl)phenyl]thiourea (pKa 8.5, DMSO124) was not active in any cyclic ester polymerizations, evidencing that the basicity of the ureate plays a key role in the modulation of alkoxide strength. Guo and coworkers adopted a similar switching strategy polymerizing first glycidyl phenyl ether (GPE) through an anionic mechanism with tetrabutylammonium fluoride (TBAF) at 50 °C. Cooling down the reaction mixture to room temperature and adding a mixture of 1-(3,5-bis(trifluoromethyl)phenyl)-3-phenylthiourea and LA formed block copolymers with good control and no transesterification over extended periods of time (Mn = 7600 Da; Đ = 1.19). Not surprisingly, these optimal conditions favorable for LA could not form block copolymers with ε-CL.124
3.2. Synthesis of polyether–polyester blocks through simultaneous feeding of monomers
Cationic complex 24 (Scheme 20) as well as complex 25 (Scheme 23)125 were reported by Mehrkhodavandi and coworkers to form block copolymers from epoxide and cyclic ester mixtures through the coupling of cationic and coordination-insertion mechanisms (Scheme 23). The complexes could polymerize a mixture of epichlorohydrin (ECH) and rac-LA in 1 hour at 130 °C forming block copolymers with monomodal molecular weight distributions, but with a significantly lower-than-expected molecular weight (based on monomer to initiator ratios), even more when compared to the sequential addition method (vide supra).125 When comparing the two complexes, the authors found that complex 24 with the bigger and less-coordinating tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (BArF) as counterion yielded more controlled copolymerization (Mn = 64447 Da; Đ = 1.35) than 25 (Mn = 10130 Da; Đ = 2.67).
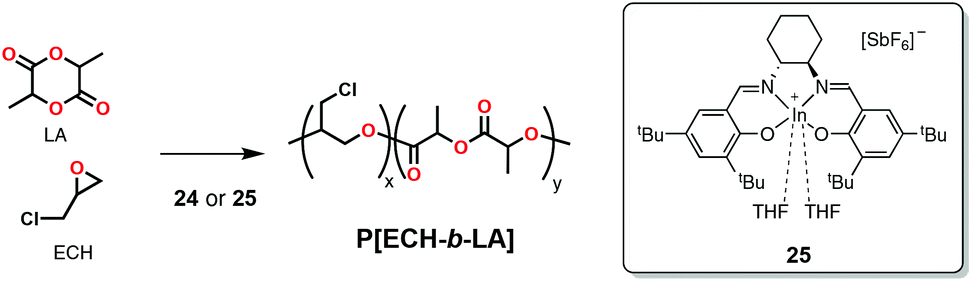 | ||
| Scheme 23 Cationic indium complex active in the block copolymerization of epichlorohydrin and lactide through the simultaneous addition of monomers. | ||
300 Da; Đ = 1.44). Reverse switch starting from the reduced version of the catalyst (26red, Scheme 24) was not as controlled due to side reactions of the added oxidant acetyl ferrocenium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate ([AcFc][BArF]) with CHO, but block copolymers with good control could still be formed through sequential addition PLA-b-PCHO-b-PLA (Mn = 16900 Da; Đ = 1.25).126 Byers and coworkers also reported a redox switching system based on a bis(imino)pyridine iron complex that could cycle between neutral iron(II) (27red, Scheme 24) and cationic iron(III) (27ox, Scheme 24) with orthogonal activity towards rac-LA and CHO. Interestingly, diblock copolymers (Mn up to 12500 Da; Đ = 1.40) could be formed with the system switching from both the oxidized and reduced forms, but always with some formation of PCHO homopolymer (which could be removed by selective precipitation). Block copolymers synthesized by the sequential addition of monomers had similar properties to those synthesized by simultaneous feeding, further confirming the selectivity of the system in a mixture of monomers.127 Some years later, the same group reported an adaptation of their iron system to make it work under electrochemical switches, obviating the need of adding sacrificial redox agents (Mn up to 40300 Da; Đ = 1.80).128
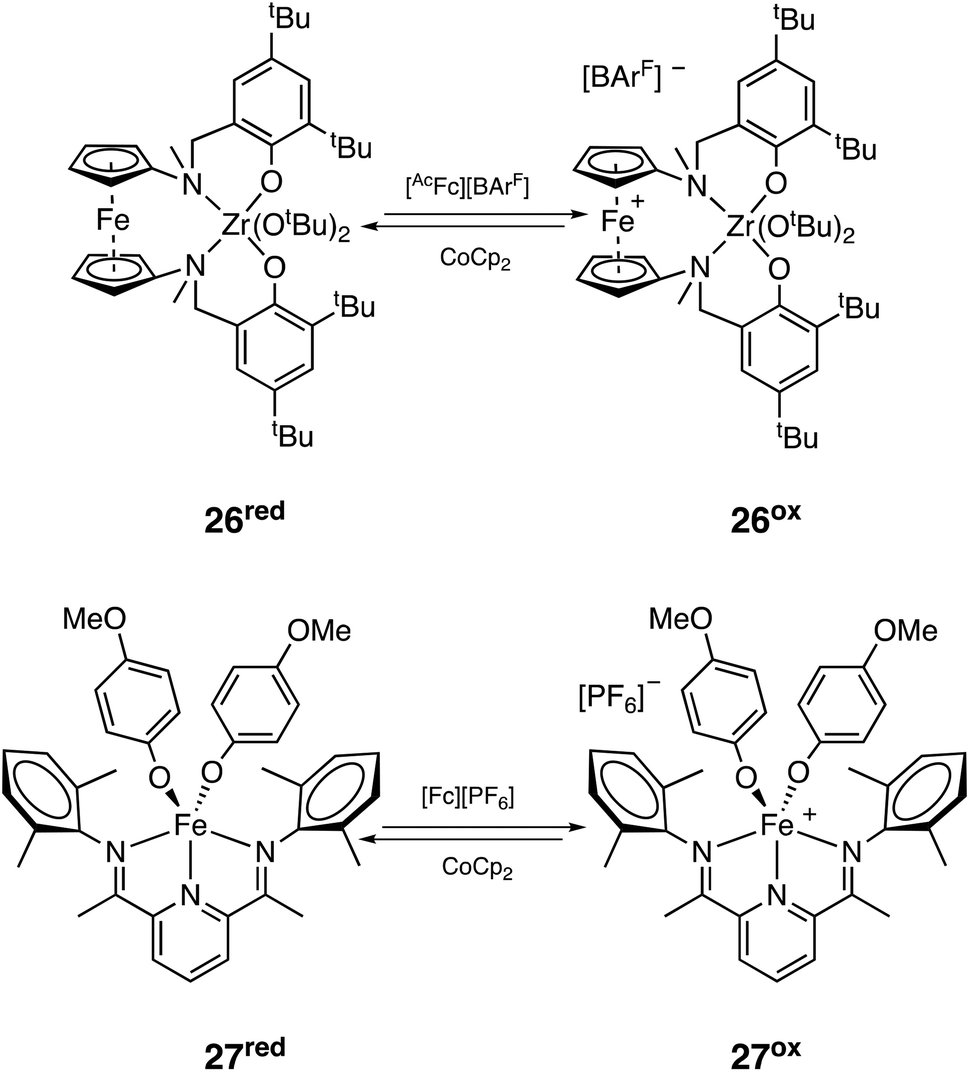 | ||
| Scheme 24 Redox-switchable system active in the copolymerization of cyclohexene oxide and lactide in a mixture of monomers. | ||
700 Da; Đ = 1.50).129 More recently, Williams and coworkers exploited a similar strategy for the well-controlled synthesis of multiblock polyester–polyether macromolecules.130 Employing a commercial salen chromium(III) chloride catalyst (22, Scheme 17) in the presence of PPNCl and alcohol as a chain-transfer agent, different substituted epoxides could be copolymerized with different anhydrides to give highly alternating polyesters (>95% polyester linkages). Once the anhydride was totally consumed the system could switch to a ROP mechanism in the presence of excess epoxide to make polyester–polyether block copolymers. Although the same system was reported earlier for the synthesis of polyester–polyester blocks in mixtures of anhydrides, CHO and ε-DL with no evidence of polyether formation (vide supra),91 here the use of monosubstituted epoxides (instead of CHO) provided a lower barrier for epoxide homopolymerization. Using 1,2-cyclohexanediol as a chain-transfer agent, the system could be switched several times between ROCOP and ROP by further additions of anhydride and epoxide. This very convenient chemical switch strategy allowed isolation of multiblock copolymers of up to 15 blocks (Mn = 17800 Da; Đ = 1.38), with the notable advantage of producing functionalized polymer structures with controlled molecular weights.130
Another promising strategy employs Lewis acid/base pairs for the synthesis of these block copolymers. Building up on their previous work,131 Wang, Li and coworkers applied the synergistic effect between phosphazene t-BuP2 and BEt3 for the ROCOP of PA and butylene oxide (BO) in the presence of a diol to give a linear polyester. Starting the polymerization with 2 equivalents of BEt3 per phosphazene equivalent and an excess of BO makes possible the switch to BO ROP after all anhydride has been consumed, forming the desired triblock copolymers with good control (Mn up to 113100 Da; Đ = 1.14). As the t-BuP2 phosphazene alone is weak to induce BO polymerization and also leads to significant transesterification in the polyester synthesis, the authors hypothesized that the addition of the Lewis acid serves two purposes: one being the tuning of alkoxide nucleophilicity and the other being epoxide activation for polymerization (through an activated monomer mechanism). This methodology was applied to different epoxides and anhydrides, maintaining excellent control.132 Also, Zhao and coworkers reported a similar system based on t-BuP1 and BEt3 for the block copolymerization in mixtures of PA and PO or EO. Due to the lower basicity of t-BuP1, polymerization trials required only 0.3–0.5 equivalents of BEt3 to form controlled blocks (Mn up to 117300 Da; Đ = 1.12). An excess of Lewis acid slowed down ROCOP and led to a premature epoxide polymerization before full anhydride consumption precluding clean block formation.133 This observation proved to be critical to later expand the scope of these systems (vide infra).
To further demonstrate the high versatility of the Lewis acid/base approach for switchable block copolymerizations in mixtures of monomers, Zhao, Ling and coworkers showed recently its applicability to the ROP of epoxides and cyclic esters. Using t-BuP2 phosphazene and BEt3 in different proportions, the catalytic system could switch from ROP of epoxides (excess of BEt3) to the ROP of δ-VL or ε-CL (excess of t-BuP2) in a clean manner and with no transesterification, by just adding Lewis acid or Lewis base. This elegant system could also form uncommon block copolymers of reversed sequence such as PCL-b-PEO and PEO-b-PCL-b-PEO, which are difficult to obtain with more traditional synthetic methods that employ preformed PEG/PEO. With multiple switches, a pentadecablocks could be formed with excellent control (Mn = 40000 Da; Đ = 1.07).134
4. Synthesis of polycarbonate–polyester blocks
Aliphatic polycarbonates are an attractive class of materials with tunable glass transition temperatures,135 high toughness136 and biodegradability.137 They can be synthesized from the ROP of 5-, 6- or 7-membered ring cyclic carbonates, many of which can be readily accessed from glycerol, a byproduct of biodiesel synthesis.138 Block copolymers containing polyester and polycarbonate segments are fully biodegradable materials that show improved toughness (in comparison to the polyester homopolymer) as well as improved stability and ductility.1394.1. Synthesis of polycarbonate–polyester blocks through polymerization of cyclic carbonates and cyclic esters
000 Da; Đ = 1.56) and triblock (Mn = 77150 Da; Đ = 1.50) copolymers.140 In the same study, it was shown that organocatalysts DMAP and BEMP (Scheme 9), as well as Al(OTf)3 were also active for this block copolymerization in the presence of an alcohol, but with lower reactivities compared to 3. Interestingly, the simultaneous addition of monomers still produced in some cases blocky-microstructures with different reactivity profiles for the different monomers (despite showing similar rates in the sequential addition): the zinc β-diketiminate complex 3 polymerized first and preferentially L-LA, while Al(OTf)3 first polymerized preferentially TMC.140 Polymerization with the simultaneous addition using organocatalyst TBD gave random copolymers.141 Later, the same group applied a similar sequential addition strategy (first cyclic carbonate ROP at 60 °C followed by lactone ROP at 100 °C) to accomplish the block copolymerization of rac- or R,R-trans-cyclohexene carbonate (CHC, Scheme 9) with L-LA (Mn up to 28000 Da; Đ = 1.7) using a diaminophenolate zinc alkyl complex in the presence of an alcohol (28, Scheme 25).142 Similar results could be obtained with organobase TBD or with yttrium amide Y[N(SiMe3)2]3, but with lower activities. Very importantly, no decarboxylation (a side-reaction that leads to the formation of ether linkages) was observed in any of the products. Copolymerization carried out in the reverse order (first LA, then CHC) afforded random copolymers via extensive transesterification, just as simultaneous feeding of monomers also yielded random copolymers.142
 | ||
| Scheme 25 Sequential polymerization of cyclic carbonates and lactide. | ||
As the order of addition of monomers is often an issue for these systems, formation of polyester–polycarbonate multiblock copolymers is not easily attainable. Recently, Diaconescu and coworkers reported the first system that allows multiple additions of L-LA and TMC to yield multiblock copolymers. The bimetallic zinc complex supported by heteroscorpionate ligands with pendant ferrocene groups (29, Scheme 25) could polymerize both monomers in a living fashion and support multiple addition of monomers to make tri, tetra and pentablock copolymers (Mn up to 58900 Da; Đ = 1.49) with monomodal molecular weight distributions and a characteristic diffusion coefficient by DOSY NMR spectroscopy.143
Exploiting significantly different rates of ROP of L-LA and functional cyclic carbonate (MTC-AE) (Scheme 9) with a highly isoselective aluminum complex (30, Scheme 25), Cui, Liu and coworkers reported the synthesis of blocky-copolymers with the simultaneous addition of monomers.144 In comparison, highly heteroselective complex 2 (Scheme 2) only produced random copolymers under the same conditions.
500 Da; Đ = 1.12) and PBL-PTMC (Mn = 7170 Da; Đ = 1.21) with good control using alcohols and DPP (Scheme 9) or significantly more acidic bis(4-nitro)DPP bearing electron withdrawing groups (Scheme 26).146 Block copolymers of TMC with LA could also be obtained in a similar fashion (using DPP/alcohol), but only with co-addition of DMAP during the LA polymerization step (as DPP/alcohol system alone does not polymerize LA).
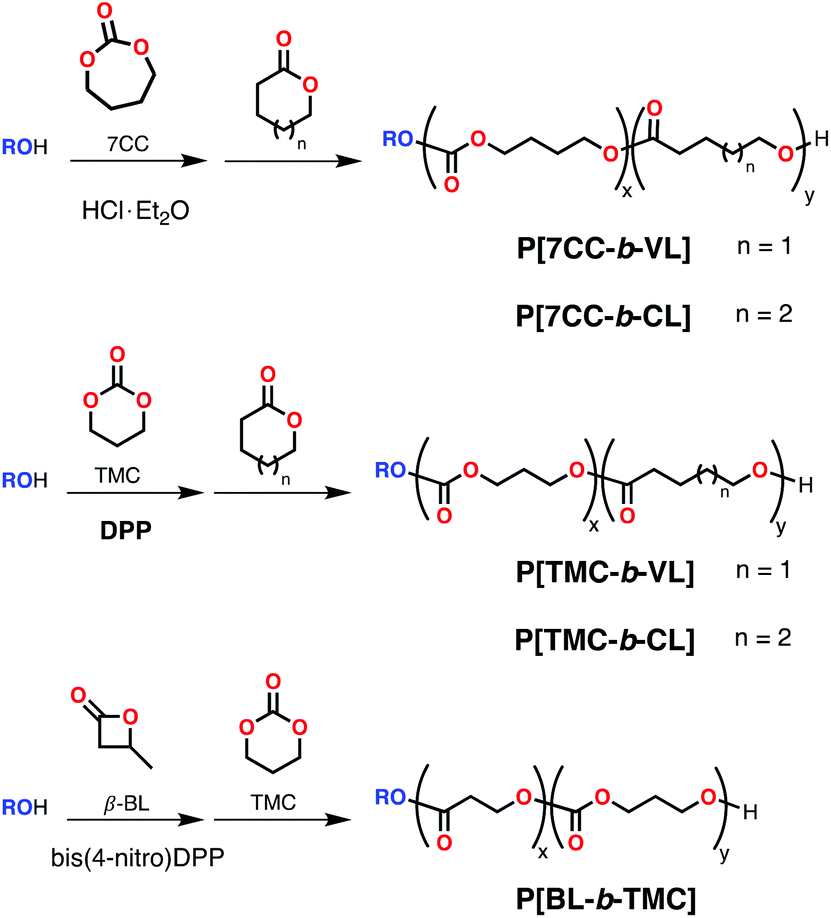 | ||
| Scheme 26 Sequential polymerization of cyclic carbonates and cyclic esters using organic and inorganic acids as initiators in the presence of alcohols. | ||
An important advantage of cyclic carbonates over cyclic esters is their greater diversity of functional side-groups available by chemical synthesis, requiring either 1,2- or 1,3-diols or an olefin, which can often come from bioderived sources.147 Such side-groups can impart hydrophilicity to polycarbonates or allow for incorporation of cargo molecules. In 2011, Song and coworkers reported the ROP of an azido-functionalized cyclic carbonate with L-LA using organobase DBU and an alcohol to make block and random copolymers that could later be reacted with different alkynes to install different functional groups in the polymer backbone.148
Following their reports on the synthesis of 5-functionalized 5-methyltrimethylene carbonates (MTCs),149 Hedrick and coworkers reported the synthesis of a triblock copolymer with a polycarbonate core bearing pending halide groups that could be post-functionalized to quaternary ammonium groups with antimicrobial activity. Block copolymer synthesis was achieved through the sequential polymerization of the MTC first, followed by addition of D-LA using a diol and a spartein/thiourea couple (Scheme 27).150 Alexander, Taresco and coworkers employed a trimethylene carbonate derived from serinol (tBSC) to form triblock amphiphilic copolymers PEG-b-PLA-b-tBSC using DBU and methyl PEG as a macroinitiator. The block copolymer could be deprotected and post-functionalized to bear a drug molecule by reaction of the free amine groups in the carbonate backbone (Scheme 27).151
 | ||
| Scheme 27 Block copolymerization of functionalized cyclic carbonates and lactide through sequential addition. | ||
Using TBD (Scheme 9) and an alcohol for the polymerization of ε-CL and a functionalized cyclic carbonate, Albertsson and coworkers reported a sequential polymerization involving switches in temperature.152 In this system, TBD/alcohol first polymerized ε-CL at 30 °C (only to low conversions to avoid transesterification) and then the reaction mixture was cooled to −40 °C, followed by addition of the cyclic carbonate. Under these conditions the cyclic carbonate was polymerized to full conversion in a matter of minutes and CL ROP was suppressed. Subsequent cycles of heating and cooling/addition of cyclic carbonate afforded multiblock copolymers.
4.2. ROP/ROCOP Systems for the synthesis of polycarbonate–polyester blocks involving carbon dioxide incorporation
Using waste CO2 as a reagent for the synthesis of polycarbonates is a powerful strategy with potential economic and environmental benefits.153 Alternating ROCOP of petrochemically- or bio-derived epoxides with CO2 offers a route to more sustainable homopolymers and copolymers through combination with other ROCOP and ROP mechanisms with exquisite control.154By far, the most common approach for the synthesis of polycarbonate–polyester blocks that incorporate CO2 in their backbone combines ROCOP of epoxides and CO2 with ROCOP of epoxides and anhydrides. In 2008, Coates and coworkers re-applied their previously reported zinc β-diketiminate acetate complex155 (31, Scheme 28) to the synthesis of block copolymers in a mixture of monomers. Using such complex, mixtures of diglycolic anhydride (DGA) or succinic anhydride (SA) with cyclohexene oxide (CHO) could be polymerized in the presence of CO2 to form block copolymers of perfectly-alternating polyesters and polycarbonate (Scheme 28) by means of kinetic resolution/control, i.e. anhydride/epoxide coupling having a much faster rate and therefore preceding CO2/epoxide coupling.156
 | ||
| Scheme 28 Metal catalysts (with and without cocatalysts) active in the tandem ROCOP of epoxides and anhydrides and ROCOP of epoxides and carbon dioxide. | ||
Independently, the Darensbourg157 and Duchateau158 research groups re-applied previously reported chromium(III) salen complex (22, Scheme 17) and phorphirine complex (32, Scheme 28) in the presence of different cocatalysts to the synthesis of block copolymers in mixtures of phthalic anhydride (PA) or other anhydrides with CHO/CO2 (Scheme 28) following a similar kinetic control profile. Later, analogous cobalt(III) salen complex and aluminum phorphirine complex (33, Scheme 28) would also be developed for similar block copolymerizations.159
Similarly, Williams and coworkers have applied their dizinc acetate catalyst (21, Scheme 17) to the polymerization of mixtures of PA/CHO and CO2 without the need for a cocatalyst.90 Using DFT and spectroscopic analysis to study the high degree of kinetic selectivity, the authors showed that the acetate catalyst initially reacts with an epoxide monomer to form an intermediate alkoxide species which is common to both ROCOP mechanisms. This alkoxide not only has a lower kinetic barrier for PA insertion, but also forms a thermodynamically more stable ester linkage that propagates to form the polyester block, which explains the more favored ROCOP of anhydride/epoxide. Once the PA has depleted, the alkoxide intermediate (with a preformed polyester block) can incorporate CO2 forming a polycarbonate block. Later, other dinuclear zinc- and magnesium-systems (homo- and heteronuclear) would be developed by the same group for similar block copolymerizations.160
A different approach for the synthesis of polycarbonate–polyester blocks involves combining ROP of cyclic esters with ROCOP of epoxides and CO2. This strategy is not only attractive because of the expanded monomer scope, but also because of the peculiarity of many of these systems for being directly switched on/off by the presence of CO2. For example, the dizinc acetate catalyst (21, Scheme 17) reported by Williams group has been reported to form block copolymers in a mixture of CHO and ε-CL through the formation of an intermediate alkoxide species (produced by insertion of one CHO into the Zn–OAc bond) that is active in both ROCOP and ROP cycles.90,161 In the presence of CO2 gas (approx. 1 bar), ROCOP takes precedence and shuts down completely ROP: this is due to the rapid insertion of CO2 into the metal alkoxide bond to form a Zn–carbonate bond active in ROCOP and inactive in ROP. In the absence of CO2, ROP takes over. Likely, this is due to the slower insertion of ε-CL into the metal–alkoxide bond to form a polycarbonate–polyester block. As has been shown previously, this bimetallic system shows unprecedented chemoselectivity in a mixture of epoxides, lactones, anhydrides and CO2 that allows the synthesis of polyester–polyester blocks and different polycarbonate–polyester blocks. This is attributed in part to the very different rates of insertion into the intermediate Zn–alkoxide bond, following the order PA (fastest) > CO2 > ε-CL (slowest).90 Other variations of this catalyst have been proposed with similar kinetic and switch effects.139c,162 For instance, in order to make block copolymers including bioderived ε-decanolactone (ε-DL, Scheme 1), an analogous zinc-magnesium heteronuclear complex was developed (34, Scheme 29). Using a two-step methodology in the presence of a bifunctional initiator (1,2-cyclohexanediol), triblock copolymers with high toughness and elongation at break were obtained.139c
 | ||
| Scheme 29 Metal catalysts (with and without cocatalaysts) used alone or in conjunction with organobases active in the coupling of ROP of cyclic esters with ROCOP of epoxides and carbon dioxide. | ||
Also following a multi-step approach, Rieger and coworkers employed a zinc β-diketiminate amidate complex (35, Scheme 29) for the synthesis of polyester–polycarbonate diblock and triblock copolymers from mixtures of β-BL and CHO.163 Interestingly, the ROP of the cyclic ester could also be switched off in this system by addition of CO2, but only at high pressures (40 bar). Low CO2 pressures (3 bar) in a mixture of monomers yielded totally random copolymers product of simultaneous ROP and ROCOP mechanisms.
In order to incorporate PLA blocks into polycarbonates, Darensbourg and coworkers reported a tandem catalytic approach involving a salen cobalt(III) complex (36, Scheme 29) with PPNTFA (PPN trifluoroacetate) as cocatalyst and organobase DBU.164 Initial polymerization of propylene oxide (PO) and CO2 using 36 and cocatalyst system yielded the initial polycarbonate block. Subsequent addition of water formed hydroxy end-groups, which could subsequently react with added LA/DBU to form triblock copolymers (Scheme 29). Other multicomponent systems have been reported for tandem ROCOP/ROP.165
5. Conclusions
With the constant development of new functionalized monomers and more selective catalytic systems, the controlled synthesis of block copolymers including biodegradable and/or bioderived components has experienced impressive advances in the last 20 years. The development of new synthetic methodologies has opened the door to new materials with tunable properties. While the most common synthetic approach is the sequential addition of monomers polymerizable by ROP or ROCOP methods, systems that can switch between different catalytic cycles by internal or external stimuli in a mixture of monomers offer the greatest advantage by yielding well-defined polymer sequences that can be tailored according to the reactivities of the different monomers (e.g. TMC > ε-CL > LA) or a specific application.In general, metal-catalysts tend to display higher activities and can exert different degrees of stereocontrol in polymerizations, but they typically require a lengthy synthesis and therefore are not always readily available. In contrast, relatively simple organocatalysts can show high activities, while being more readily accessible and ideal for applications where trace metals are undesirable. Combination of metal catalysis (or other Lewis acids for the matter) with organocatalysis has shown promising results in the synthesis of uncommon block copolymers with high activities and offers great potential in the future development of more versatile tools applicable to a greater arrange of monomers.
Similarly, combination of ROP and ROCOP mechanisms in one catalytic system (typically a metal complex) has opened the door to new block compositions with exquisite control. Future combination with other mechanisms of polymerization will certainly expand the applications of new block copolymer architectures.
Reducing number of synthetic and purification steps as well as energy consumption in accordance with green chemistry principles will simplify synthetic protocols, making novel block copolymers architectures more accessible for different applications. In particular, the biomedical field that relies heavily on poly(ethylene) glycol (PEG) can benefit greatly from more versatile synthetic protocols and the study of new amphiphilic block copolymer architectures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Council of Canada for support.Notes and references
- S. Spierling, V. Venkatachalam, H. Behnsen, C. Herrmann and H.-J. Endres, in Progress in Life Cycle Assessment, Springer, 2019, pp. 147–154 Search PubMed.
- (a) The Future of Petrochemicals: Towards More Sustainable Plastics and Fertilisers; ; (b) E. J. North and R. U. Halden, Rev. Environ. Health, 2013, 28, 1–8 CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS.
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CAS.
- (a) G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS; (d) G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS; (e) A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- (a) K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS; (b) F. L. Hatton, Polym. Chem., 2020, 11, 220–229 RSC; (c) K. Müller, C. Zollfrank and M. Schmid, Macromol. Mater. Eng., 2019, 304, 1800760 CrossRef; (d) K. G. Nair, A. Dufresne, A. Gandini and M. N. Belgacem, Biomacromolecules, 2003, 4, 1835–1842 CrossRef CAS.
- (a) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS; (b) S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC; (c) G. Trott, P. Saini and C. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef.
- M. Van den Oever, K. Molenveld, M. van der Zee and H. Bos, Bio-based and biodegradable plastics: facts and figures: focus on food packaging in the Netherlands, Wageningen Food & Biobased Research, 2017 Search PubMed.
- R. P. Babu, K. O'Connor and R. Seeram, Prog. Biomater., 2013, 2, 8 CrossRef.
- K. Horie, M. Baron, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Marechal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlidal, E. S. Wilks, W. J. Work, G. Allegra, M. Baron, A. Fradet, K. Hatada, J. He, M. Hess, K. Horie, A. D. Jenkins, J. I. Jin, R. G. Jones, J. Kahovec, T. Kitayama, P. Kratochvil, P. Kubisa, E. Marcechal, I. Meisel, W. V. Metanomski, G. Moad, W. Mormann, S. Penczek, L. P. Rebelo, M. Rinaudo, I. Schopov, M. Schubert, V. P. Shibaev, S. Slomkowskj, R. F. T. Stepto, D. Tabak, J. Vohlidal, E. S. Wilks, W. J. Work, K. Dorfner, M. J. Frechet, W. I. Harris, P. Hodge, T. Nishikubo, C. K. Ober, E. Reichmanis, D. C. Sherrington, M. Tomoi and D. Wohrle, Pure Appl. Chem., 2004, 76, 889–906 CAS.
- (a) S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS; (b) J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS.
- B. Imre and B. Pukánszky, Eur. Polym. J., 2013, 49, 1215–1233 CrossRef CAS.
- (a) Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC; (b) C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. Coelho, C. S. Freire and A. J. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC; (c) D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS; (d) F. R. Kersey, G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, ACS Nano, 2010, 4, 4989–4996 CrossRef CAS.
- (a) U. C. Palmiero, M. Sponchioni, N. Manfredini, M. Maraldi and D. Moscatelli, Polym. Chem., 2018, 9, 4084–4099 RSC; (b) V. S. Voet, G. O. A. van Ekenstein, N. L. Meereboer, A. H. Hofman, G. ten Brinke and K. Loos, Polym. Chem., 2014, 5, 2219–2230 RSC; (c) D. Chi, F. Liu, H. Na, J. Chen, C. Hao and J. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 9893–9902 CrossRef CAS; (d) K. L. Liu, J.-l. Zhu and J. Li, Soft Matter, 2010, 6, 2300–2311 RSC.
- (a) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (b) M. Kakuta, M. Hirata and Y. Kimura, J. Macromol. Sci., Polym. Rev., 2009, 49, 107–140 CAS; (c) X. Tang and E. Y.-X. Chen, Chem, 2019, 5, 284–312 CrossRef CAS.
- (a) I. Yildirim, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2018, 76, 111–150 CrossRef CAS; (b) D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef CAS.
- (a) X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS; (b) H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS; (c) H. Y. Ye, K. Y. Zhang, D. Kai, Z. B. Li and X. J. Loh, Chem. Soc. Rev., 2018, 47, 4545–4580 RSC.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS.
- H. Liu and J. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1051–1083 CrossRef CAS.
- (a) A. Amgoune, C. M. Thomas, T. Roisnel and J. F. Carpentier, Chem. – Eur. J., 2006, 12, 169–179 CrossRef CAS; (b) G. Zhang, G. L. Fiore, T. L. St. Clair and C. L. Fraser, Macromolecules, 2009, 42, 3162–3169 CrossRef CAS; (c) D. Cohn and A. H. Salomon, Biomaterials, 2005, 26, 2297–2305 CrossRef CAS; (d) M.-H. Huang, S. Li and M. Vert, Polymer, 2004, 45, 8675–8681 CrossRef CAS.
- (a) M. Ryner and A.-C. Albertsson, Biomacromolecules, 2002, 3, 601–608 CrossRef CAS; (b) J.-O. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- (a) X. Zeng, B. Wu, L. Wu, J. Hu, Z. Bu and B.-G. Li, Ind. Eng. Chem. Res., 2014, 53, 3550–3558 CrossRef CAS; (b) W. D. Li, J. B. Zeng, Y. D. Li, X. L. Wang and Y. Z. Wang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5898–5907 CrossRef CAS; (c) J.-B. Zeng, Y.-D. Li, W.-D. Li, K.-K. Yang, X.-L. Wang and Y.-Z. Wang, Ind. Eng. Chem. Res., 2009, 48, 1706–1711 CrossRef CAS; (d) B. Hazer, E. Akyol, T. Şanal, S. Guillaume, B. Çakmakli and A. Steinbuchel, Polym. Bull., 2019, 76, 919–932 CrossRef CAS.
- S. Dai, L. Xue, M. Zinn and Z. Li, Biomacromolecules, 2009, 10, 3176–3181 CrossRef CAS.
- (a) X. Lu, J.-B. Zeng, C.-L. Huang and Y.-Z. Wang, Ind. Eng. Chem. Res., 2012, 51, 8262–8272 CrossRef CAS; (b) C. H. Wilsens, J. M. Verhoeven, B. A. Noordover, M. R. Hansen, D. Auhl and S. Rastogi, Macromolecules, 2014, 47, 3306–3316 CrossRef CAS.
- T. Tang, T. Moyori and A. Takasu, Macromolecules, 2013, 46, 5464–5472 CrossRef CAS.
- (a) L. Jasinska-Walc, M. R. Hansen, D. Dudenko, A. Rozanski, M. Bouyahyi, M. Wagner, R. Graf and R. Duchateau, Polym. Chem., 2014, 5, 3306–3310 RSC; (b) L. Jasinska-Walc, M. Bouyahyi, A. Rozanski, R. Graf, M. R. Hansen and R. Duchateau, Macromolecules, 2015, 48, 502–510 CrossRef CAS; (c) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS; (d) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Macromolecules, 2015, 48, 950–958 CrossRef CAS.
- (a) A. Duda, Z. Florjanczyk, A. Hofman, S. Slomkowski and S. Penczek, Macromolecules, 1990, 23, 1640–1646 CrossRef CAS; (b) M. Sobczak, Polym. Bull., 2012, 68, 2219–2228 CrossRef CAS.
- (a) J. Jang, H. Park, H. Jeong, E. Mo, Y. Kim, J. S. Yuk, S. Q. Choi, Y.-W. Kim and J. Shin, Polym. Chem., 2019, 10, 1245–1257 RSC; (b) V. Arias, P. Olsén, K. Odelius, A. Höglund and A.-C. Albertsson, Polym. Chem., 2015, 6, 3271–3282 RSC; (c) C. Ba, J. Yang, Q. Hao, X. Liu and A. Cao, Biomacromolecules, 2003, 4, 1827–1834 CrossRef CAS; (d) J. Hao, J. Servello, P. Sista, M. C. Biewer and M. C. Stefan, J. Mater. Chem., 2011, 21, 10623–10628 RSC.
- (a) I. Taniguchi and N. G. Lovell, Macromolecules, 2012, 45, 7420–7428 CrossRef CAS; (b) J. P. MacDonald, M. Sidera, S. P. Fletcher and M. P. Shaver, Eur. Polym. J., 2016, 74, 287–295 CrossRef CAS; (c) K. Matsubara, K. Eda, Y. Ikutake, M. Dan, N. Tanizaki, Y. Koga and M. Yasuniwa, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2536–2544 CrossRef CAS.
- V. Simic, S. Pensec and N. Spassky, Macromol. Symp., 2000, 153, 109–121 CrossRef CAS.
- (a) I. Yu, T. Ebrahimi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Dalton Trans., 2015, 44, 14248–14254 RSC; (b) D. C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965–3974 CrossRef CAS; (c) T. Ebrahimi, D. C. Aluthge, B. O. Patrick, S. G. Hatzikiriakos and P. Mehrkhodavandi, ACS Catal., 2017, 7, 6413–6418 CrossRef CAS.
- (a) A. Kundys, A. Plichta, Z. Florjańczyk, A. Frydrych and K. Żurawski, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1444–1456 CrossRef CAS; (b) J. Bai, X. Xiao, Y. Zhang, J. Chao and X. Chen, Dalton Trans., 2017, 46, 9846–9858 RSC.
- (a) R. Lapenta, M. Mazzeo and F. Grisi, RSC Adv., 2015, 5, 87635–87644 RSC; (b) D. J. Gilmour, R. L. Webster, M. R. Perry and L. L. Schafer, Dalton Trans., 2015, 44, 12411–12419 RSC; (c) T. J. Whitehorne and F. Schaper, Can. J. Chem., 2014, 92, 206–214 CrossRef CAS.
- (a) N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132, 1750–1751 CrossRef CAS; (b) C. Kan and H. Ma, RSC Adv., 2016, 6, 47402–47409 RSC; (c) Z. Sun, R. Duan, J. Yang, H. Zhang, S. Li, X. Pang, W. Chen and X. Chen, RSC Adv., 2016, 6, 17531–17538 RSC; (d) T. Fuoco and D. Pappalardo, Catalysts, 2017, 7, 64 CrossRef; (e) Y. Wang and H. Ma, Chem. Commun., 2012, 48, 6729–6731 RSC; (f) D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056–6062 CrossRef CAS.
- J. MacDonald, M. Parker, B. Greenland, D. Hermida-Merino, I. Hamley and M. Shaver, Polym. Chem., 2015, 6, 1445–1453 RSC.
- J. F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS.
- K. V. Zaitsev, Y. A. Piskun, Y. F. Oprunenko, S. S. Karlov, G. S. Zaitseva, I. V. Vasilenko, A. V. Churakov and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1237–1250 CrossRef CAS.
- A. Meduri, T. Fuoco, M. Lamberti, C. Pellecchia and D. Pappalardo, Macromolecules, 2014, 47, 534–543 CrossRef CAS.
- C. Fliedel, V. Rosa, F. M. Alves, A. M. Martins, T. Avilés and S. Dagorne, Dalton Trans., 2015, 44, 12376–12387 RSC.
- C. d. G. Jaffredo, J.-F. o. Carpentier and S. M. Guillaume, Macromolecules, 2013, 46, 6765–6776 CrossRef CAS.
- R. Wang, J. Zhang, Q. Yin, Y. Xu, J. Cheng and R. Tong, Angew. Chem., Int. Ed., 2016, 55, 13010–13014 CrossRef CAS.
- (a) Y. Yu, J. Zou and C. Cheng, Polym. Chem., 2014, 5, 5854–5872 RSC; (b) B. Martin-Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef CAS; (c) A. Basu, K. R. Kunduru, J. Katzhendler and A. J. Domb, Adv. Drug Delivery Rev., 2016, 107, 82–96 CrossRef CAS.
- A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736–2737 RSC.
- S. Thongkham, J. Monot, B. Martin-Vaca and D. Bourissou, Macromolecules, 2019, 52, 8103–8113 CrossRef CAS.
- B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC.
- M. P. Pepels, R. A. Koeken, S. J. van der Linden, A. Heise and R. Duchateau, Macromolecules, 2015, 48, 4779–4792 CrossRef CAS.
- Y. Xiao, J. Pan, D. Wang, A. Heise and M. Lang, Biomacromolecules, 2018, 19, 2673–2681 CrossRef CAS.
- I. Van Der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS.
- M. P. Pepels, W. P. Hofman, R. Kleijnen, A. B. Spoelstra, C. E. Koning, H. Goossens and R. Duchateau, Macromolecules, 2015, 48, 6909–6921 CrossRef CAS.
- M. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS.
- M. Bouyahyi and R. Duchateau, Macromolecules, 2014, 47, 517–524 CrossRef CAS.
- Y.-C. Xu, W.-M. Ren, H. Zhou, G.-G. Gu and X.-B. Lu, Macromolecules, 2017, 50, 3131–3142 CrossRef CAS.
- (a) T. M. Hermans, J. Choi, B. G. Lohmeijer, G. Dubois, R. C. Pratt, H. C. Kim, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2006, 45, 6648–6652 CrossRef CAS; (b) S. H. Kim, F. Nederberg, L. Zhang, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Nano Lett., 2008, 8, 294–301 CrossRef CAS; (c) C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401–16403 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. Bouyahyi, M. P. Pepels, A. Heise and R. Duchateau, Macromolecules, 2012, 45, 3356–3366 CrossRef CAS.
- G. Barouti, K. Jarnouen, S. Cammas-Marion, P. Loyer and S. M. Guillaume, Polym. Chem., 2015, 6, 5414–5429 RSC.
- H. Alamri, J. Zhao, D. Pahovnik and N. Hadjichristidis, Polym. Chem., 2014, 5, 5471–5478 RSC.
- L. Song, A.-X. Ding, K.-X. Zhang, B. Gong, Z.-L. Lu and L. He, Org. Biomol. Chem., 2017, 15, 6567–6574 RSC.
- Y. Shen, J. Zhang, N. Zhao, F. Liu and Z. Li, Polym. Chem., 2018, 9, 2936–2941 RSC.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42 CrossRef CAS.
- A. Couffin, B. Martín-Vaca, D. Bourissou and C. Navarro, Polym. Chem., 2014, 5, 161–168 RSC.
- (a) K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS; (b) Y. Jin, Y. Ji, X. He, S. Kan, H. Xia, B. Liang, J. Chen, H. Wu, K. Guo and Z. Li, Polym. Chem., 2014, 5, 3098–3106 RSC; (c) K. Makiguchi, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3769–3777 CrossRef CAS.
- J. Liu, C. Zhang, Z. Li, L. Zhang, J. Xu, H. Wang, S. Xu, T. Guo, K. Yang and K. Guo, Eur. Polym. J., 2019, 113, 197–207 CrossRef CAS.
- R. Todd, S. Tempelaar, G. Lo Re, S. Spinella, S. A. McCallum, R. A. Gross, J.-M. Raquez and P. Dubois, ACS Macro Lett., 2015, 4, 408–411 CrossRef CAS.
- N. Zhao, C. Ren, Y. Shen, S. Liu and Z. Li, Macromolecules, 2019, 52, 1083–1091 CrossRef.
- V. Ladelta, J. D. Kim, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2018, 51, 2428–2436 CrossRef CAS.
- K. Makiguchi, S. Kikuchi, K. Yanai, Y. Ogasawara, S. i. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1047–1054 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- X. Wang, J. Liu, S. Xu, J. Xu, X. Pan, J. Liu, S. Cui, Z. Li and K. Guo, Polym. Chem., 2016, 7, 6297–6308 RSC.
- J. Bai, X. Tang, Y. Zhang, J. Lin and M. Li, RSC Adv., 2018, 8, 1905–1908 RSC.
- R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS.
- Y. Li, N. Zhao, C. Wei, A. Sun, S. Liu and Z. Li, Eur. Polym. J., 2019, 111, 11–19 CrossRef CAS.
- (a) S. Naumann, P. B. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS; (b) B. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade and R. M. Waymouth, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- E. Piedra-Arroni, C. Ladavière, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13306–13309 CrossRef CAS.
- P. Walther and S. Naumann, Macromolecules, 2017, 50, 8406–8416 CrossRef CAS.
- Q. Wang, W. Zhao, J. He, Y. Zhang and E. Y.-X. Chen, Macromolecules, 2017, 50, 123–136 CrossRef CAS.
- B. Wang, L. Pan, Z. Ma and Y. Li, Macromolecules, 2018, 51, 836–845 CrossRef CAS.
- I. Van Der Meulen, Y. Li, R. Deumens, E. A. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837–843 CrossRef CAS.
- E. J. Shin, H. A. Brown, S. Gonzalez, W. Jeong, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2011, 50, 6388–6391 CrossRef CAS.
- J. W. Kramer and G. W. Coates, Tetrahedron, 2008, 64, 6973–6978 CrossRef CAS.
- J. Wilson, S. Hopkins, P. Wright and A. Dove, ACS Macro Lett., 2016, 5, 346–350 CrossRef CAS.
- M. Basko, A. Duda, S. Kazmierski and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4873–4884 CrossRef CAS.
- (a) D. J. Coady, K. Fukushima, H. W. Horn, J. E. Rice and J. L. Hedrick, Chem. Commun., 2011, 47, 3105–3107 RSC; (b) T. Saito, Y. Aizawa, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Macromolecules, 2018, 51, 689–696 CrossRef CAS.
- B. Raeskinet, S. Moins, L. Harvey, J. De Winter, C. Henoumont, S. Laurent and O. Coulembier, Macromolecules, 2019, 52, 6382–6392 CrossRef CAS.
- (a) A. Sauer, J. C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima and J. Okuda, ChemCatChem, 2013, 5, 1088–1091 CrossRef CAS; (b) E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897–9899 RSC; (c) C. K. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS.
- O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 4964–4968 CrossRef CAS.
- (a) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS; (b) A. Lai, Z. C. Hern and P. L. Diaconescu, ChemCatChem, 2019, 11, 4210–4218 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef.
- F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213–1220 CrossRef CAS.
- H. Li, H. Luo, J. Zhao and G. Zhang, ACS Macro Lett., 2018, 7, 1420–1425 CrossRef CAS.
- H. Y. Ji, B. Wang, L. Pan and Y. S. Li, Angew. Chem., Int. Ed., 2018, 57, 16888–16892 CrossRef CAS.
- (a) A. Vassiliou, S. Papadimitriou, D. Bikiaris, G. Mattheolabakis and K. Avgoustakis, J. Controlled Release, 2010, 148, 388–395 CrossRef CAS; (b) A. Jäger, E. z. Jäger, Z. k. Syrová, T. Mazel, L. r. Kováčik, I. Raška, A. Höcherl, J. Kučka, R. Konefal and J. Humajova, Biomacromolecules, 2018, 19, 2443–2458 CrossRef; (c) Y. Chen, Y. Li, J. Gao, Z. Cao, Q. Jiang, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 490–501 CrossRef CAS; (d) S. Paszkiewicz, A. Szymczyk, D. Pawlikowska, I. Irska, I. Taraghi, R. Pilawka, J. Gu, X. Li, Y. Tu and E. Piesowicz, RSC Adv., 2017, 7, 41745–41754 RSC.
- (a) H. Wang, D. Tong, L. Wang, L. Chen, N. Yu and Z. Li, Polym. Degrad. Stab., 2017, 140, 64–73 CrossRef CAS; (b) S. Zhang, J. Xu, H. Chen, Z. Song, Y. Wu, X. Dai and J. Kong, Macromol. Biosci., 2017, 17, 1600258 CrossRef; (c) J. Babinot, E. Renard and V. Langlois, Macromol. Chem. Phys., 2011, 212, 278–285 CrossRef CAS.
- J. Li, S. Guo, M. Wang, L. Ye and F. Yao, RSC Adv., 2015, 5, 19484–19492 RSC.
- K. Yoon, H. C. Kang, L. Li, H. Cho, M.-K. Park, E. Lee, Y. H. Bae and K. M. Huh, Polym. Chem., 2015, 6, 531–542 RSC.
- C. L. Maikawa, A. Sevit, B. Lin, R. J. Wallstrom, J. L. Mann, A. C. Yu, R. M. Waymouth and E. A. Appel, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1322–1332 CrossRef CAS.
- (a) C. Chen, C. H. Yu, Y. C. Cheng, H. Peter and M. K. Cheung, Biomaterials, 2006, 27, 4804–4814 CrossRef CAS; (b) E. Oledzka, P. Sliwerska, M. Sobczak, B. Kraska, W. Kamysz, G. Nalecz-Jawecki and W. Kolodziejski, Macromol. Chem. Phys., 2015, 216, 1365–1375 CrossRef CAS; (c) C. Xu, I. Yu and P. Mehrkhodavandi, Chem. Commun., 2012, 48, 6806–6808 RSC.
- E. Tinajero-Díaz, A. Martínez-de Ilarduya, S. Muñoz-Guerra, M.-V. de-Paz and E. Galbis, Eur. Polym. J., 2018, 108, 380–389 CrossRef.
- (a) H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef CAS; (b) R. J. Pounder, H. Willcock, N. S. Ieong, K. Rachel and A. P. Dove, Soft Matter, 2011, 7, 10987–10993 RSC; (c) Y. Li, Y. Niu, D. Hu, Y. Song, J. He, X. Liu, X. Xia, Y. Lu and W. Xu, Macromol. Chem. Phys., 2015, 216, 77–84 CrossRef CAS; (d) Z. Zhang, L. Yin, C. Tu, Z. Song, Y. Zhang, Y. Xu, R. Tong, Q. Zhou, J. Ren and J. Cheng, ACS Macro Lett., 2013, 2, 40–44 CrossRef CAS.
- (a) Z. T. Hu, Y. Chen, H. H. Huang, L. X. Liu and Y. M. Chen, Macromolecules, 2018, 51, 2526–2532 CrossRef CAS; (b) L. Yu, M. Zhang, F. S. Du and Z. C. Li, Polym. Chem., 2018, 9, 3762–3773 RSC.
- Q. Yin, L. Yin, H. Wang and J. Cheng, Acc. Chem. Res., 2015, 48, 1777–1787 CrossRef CAS.
- (a) S. Cajot, P. Lecomte, C. Jérôme and R. Riva, Polym. Chem., 2013, 4, 1025–1037 RSC; (b) S. M. Garg, X.-B. Xiong, C. Lu and A. Lavasanifar, Macromolecules, 2011, 44, 2058–2066 CrossRef CAS; (c) H. Deng, J. Liu, X. Zhao, Y. Zhang, J. Liu, S. Xu, L. Deng, A. Dong and J. Zhang, Biomacromolecules, 2014, 15, 4281–4292 CrossRef CAS; (d) J. Yan, Z. Ye, H. Luo, M. Chen, Y. Zhou, W. Tan, Y. Xiao, Y. Zhang and M. Lang, Polym. Chem., 2011, 2, 1331–1340 RSC; (e) J. Zou, C. C. Hew, E. Themistou, Y. Li, C. K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS; (f) B. Surnar and M. Jayakannan, Biomacromolecules, 2013, 14, 4377–4387 CrossRef CAS.
- B. M. Raycraft, J. P. MacDonald, J. T. McIntosh, M. P. Shaver and E. R. Gillies, Polym. Chem., 2017, 8, 557–567 RSC.
- K. K. Bansal, D. Kakde, L. Purdie, D. J. Irvine, S. M. Howdle, G. Mantovani and C. Alexander, Polym. Chem., 2015, 6, 7196–7210 RSC.
- (a) S. J. Buwalda, A. Amgoune and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1222–1227 CrossRef CAS; (b) J. P. Tan, S. H. Kim, F. Nederberg, E. A. Appel, R. M. Waymouth, Y. Zhang, J. L. Hedrick and Y. Y. Yang, Small, 2009, 5, 1504–1507 CrossRef CAS.
- Y. Zhang, P. He, X. Liu, H. Zhang, H. Yang, C. Xiao and X. Chen, Eur. Polym. J., 2018, 107, 308–314 CrossRef CAS.
- (a) C. Zhang, L. Hao, C. M. Calabrese, Y. Zhou, C. H. J. Choi, H. Xing and C. A. Mirkin, Small, 2015, 11, 5360–5368 CrossRef CAS; (b) C. Jing, R. Wang, H. Ou, A. Li, Y. An, S. Guo and L. Shi, Chem. Commun., 2018, 54, 3985–3988 RSC; (c) J. Tian, L. Xu, Y. Xue, X. Jiang and W. Zhang, Biomacromolecules, 2017, 18, 3992–4001 CrossRef CAS.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS.
- F. Isnard, M. Carratù, M. Lamberti, V. Venditto and M. Mazzeo, Catal. Sci. Technol., 2018, 8, 5034–5043 RSC.
- S. Pappuru, D. Chakraborty, V. Ramkumar and D. K. Chand, Polymer, 2017, 123, 267–281 CrossRef CAS.
- C. Diaz, T. Tomković, C. Goonesinghe, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2020, 53, 8819–8828 CrossRef CAS.
- (a) E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS; (b) G. W. Nyce, T. Glauser, E. F. Connor, A. Möck, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046–3056 CrossRef CAS.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS.
- R. Lindner, M. L. Lejkowski, S. Lavy, P. Deglmann, K. T. Wiss, S. Zarbakhsh, L. Meyer and M. Limbach, ChemCatChem, 2014, 6, 618–625 CrossRef CAS.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 3750–3753 RSC.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 3814–3822 CrossRef CAS.
- J. Zhao and N. Hadjichristidis, Polym. Chem., 2015, 6, 2659–2668 RSC.
- H. Alamri and N. Hadjichristidis, Polym. Chem., 2016, 7, 3225–3228 RSC.
- Y. Xia, Y. Chen, Q. Song, S. Hu, J. Zhao and G. Zhang, Macromolecules, 2016, 49, 6817–6825 CrossRef CAS.
- X. Zhang, G. O. Jones, J. L. Hedrick and R. M. Waymouth, Nat. Chem., 2016, 8, 1047–1053 CrossRef CAS.
- Y. Liu, X. Wang, Z. Li, F. Wei, H. Zhu, H. Dong, S. Chen, H. Sun, K. Yang and K. Guo, Polym. Chem., 2018, 9, 154–159 RSC.
- C. Diaz, T. Ebrahimi and P. Mehrkhodavandi, Chem. Commun., 2019, 55, 3347–3350 RSC.
- S. M. Quan, X. Wang, R. Zhang and P. L. Diaconescu, Macromolecules, 2016, 49, 6768–6778 CrossRef CAS.
- A. B. Biernesser, K. R. Delle Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS.
- C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 20, 4789–4795 CrossRef CAS.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 1–9 CrossRef.
- H.-Y. Ji, X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 3963–3973 RSC.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- H. Li, G. He, Y. Chen, J. Zhao and G. Zhang, ACS Macro Lett., 2019, 8, 973–978 CrossRef CAS.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 CrossRef CAS.
- Y. Sasanuma and Y. Takahashi, ACS Omega, 2017, 2, 4808–4819 CrossRef CAS.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- (a) D. Pospiech, H. Komber, D. Jehnichen, L. Häussler, K. Eckstein, H. Scheibner, A. Janke, H. R. Kricheldorf and O. Petermann, Biomacromolecules, 2005, 6, 439–446 CrossRef CAS; (b) Z. Zhang, D. W. Grijpma and J. Feijen, Macromol. Chem. Phys., 2004, 205, 867–875 CrossRef CAS; (c) G. S. Sulley, G. L. Gregory, T. T. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS.
- W. Guerin, M. Helou, J.-F. Carpentier, M. Slawinski, J.-M. Brusson and S. M. Guillaume, Polym. Chem., 2013, 4, 1095–1106 RSC.
- W. Guerin, M. Helou, M. Slawinski, J.-M. Brusson, S. M. Guillaume and J.-F. Carpentier, Polym. Chem., 2013, 4, 3686–3693 RSC.
- A. K. Diallo, W. Guerin, M. Slawinski, J.-M. Brusson, J.-F. o. Carpentier and S. M. Guillaume, Macromolecules, 2015, 48, 3247–3256 CrossRef CAS.
- M. Abubekerov, J. Wei, K. R. Swartz, Z. Xie, Q. Pei and P. L. Diaconescu, Chem. Sci., 2018, 9, 2168–2178 RSC.
- X. F. Hua, X. L. Liu and D. M. Cui, Polym. Chem., 2019, 10, 4042–4048 RSC.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316–4320 CrossRef CAS.
- (a) K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772–1782 CrossRef CAS; (b) K. Makiguchi, T. Saito, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2032–2039 CrossRef CAS.
- (a) R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC; (b) F. de la Cruz-Martínez, M. M. de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. n. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef; (c) W. Guerin, M. Helou, M. Slawinski, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2014, 5, 1229–1240 RSC.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS.
- (a) D. P. Sanders, D. J. Coady, M. Yasumoto, M. Fujiwara, H. Sardon and J. L. Hedrick, Polym. Chem., 2014, 5, 327–329 RSC; (b) D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS.
- Y. Li, K. Fukushima, D. J. Coady, A. C. Engler, S. Liu, Y. Huang, J. S. Cho, Y. Guo, L. S. Miller and J. P. Tan, Angew. Chem., Int. Ed., 2013, 52, 674–678 CrossRef CAS.
- C. E. Vasey, A. K. Pearce, F. Sodano, R. Cavanagh, T. Abelha, V. C. Crucitti, A. B. Anane-Adjei, M. Ashford, P. Gellert and V. Taresco, Biomater. Sci., 2019, 7, 3832–3845 RSC.
- P. Olsen, K. Odelius, H. Keul and A. C. Albertsson, Macromolecules, 2015, 48, 1703–1710 CrossRef CAS.
- (a) A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS; (b) S. H. Lee, A. Cyriac, J. Y. Jeon and B. Y. Lee, Polym. Chem., 2012, 3, 1215–1220 RSC; (c) Y. Q. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS.
- (a) O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC; (b) Y. Li, Y. Y. Zhang, L. F. Hu, X. H. Zhang, B. Y. Du and J. T. Xu, Prog. Polym. Sci., 2018, 82, 120–157 CrossRef CAS; (c) Y. Y. Zhang, G. P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- (a) R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS; (b) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- (a) A. Bernard, C. Chatterjee and M. H. Chisholm, Polymer, 2013, 54, 2639–2646 CrossRef CAS; (b) Z. Y. Duan, X. Y. Wang, Q. Gao, L. Zhang, B. Y. Liu and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 789–795 CrossRef CAS.
- (a) P. K. Saini, C. Romain, Y. Q. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC; (b) A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS.
- D. J. Darensbourg and G. P. Wu, Angew. Chem., Int. Ed., 2013, 52, 10602–10606 CrossRef CAS.
- (a) C. Y. Hu, R. L. Duan, S. C. Yang, X. Pang and X. S. Chen, Macromolecules, 2018, 51, 4699–4704 CrossRef CAS; (b) G. P. Wu, D. J. Darensbourg and X. B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |