
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing functionalized nanodiamonds with hyaluronic acid–phospholipid conjugates for enhanced cancer cell targeting and fluorescence imaging capabilities†
Sofia
Sturari‡
ab,
Ilaria
Andreana‡
c,
Pietro
Aprà
 b,
Valeria
Bincoletto
d,
Joanna
Kopecka
e,
Lorenzo
Mino
df,
Beatrice
Zurletti
c,
Barbara
Stella
c,
Chiara
Riganti
e,
Silvia
Arpicco
*c and
Federico
Picollo
*abd
b,
Valeria
Bincoletto
d,
Joanna
Kopecka
e,
Lorenzo
Mino
df,
Beatrice
Zurletti
c,
Barbara
Stella
c,
Chiara
Riganti
e,
Silvia
Arpicco
*c and
Federico
Picollo
*abd
aDepartment of Physics, University of Torino, via P. Giuria 1, 10125 Torino, Italy. E-mail: federico.picollo@unito.it
bNational Institute of Nuclear Physics, Sect. Torino, via P. Giuria 1, 10125 Torino, Italy
cDepartment of Drug Science and Technology, University of Torino, via P. Giuria 9, 10125, Torino, Italy. E-mail: silvia.arpicco@unito.it
dNIS Inter-Departmental Centre, via G. Quarello 15/a, 10135 Torino, Italy
eDepartment of Oncology, University of Torino, Piazza Nizza 44, 10126 Torino, Italy
fDepartment of Chemistry, University of Torino, via P. Giuria 7, 10125 Torino, Italy
First published on 29th May 2024
Abstract
Nanomedicine aims to develop smart approaches for treating cancer and other diseases to improve patient survival and quality of life. Novel nanoparticles as nanodiamonds (NDs) represent promising candidates to overcome current limitations. In this study, NDs were functionalized with a 200 kDa hyaluronic acid–phospholipid conjugate (HA/DMPE), enhancing the stability of the nanoparticles in water-based solutions and selectivity for cancer cells overexpressing specific HA cluster determinant 44 (CD44) receptors. These nanoparticles were characterized by diffuse reflectance Fourier-transform infrared spectroscopy, Raman spectroscopy, and photoluminescence spectroscopy, confirming the efficacy of the functionalization process. Scanning electron microscopy was employed to evaluate the size distribution of the dry particles, while dynamic light scattering and zeta potential measurements were utilized to evaluate ND behavior in a water-based medium. Furthermore, the ND biocompatibility and uptake mediated by CD44 receptors in three different models of human adenocarcinoma cells were assessed by performing cytofluorimetric assay and confocal microscopy. HA-functionalized nanodiamonds demonstrated the advantage of active targeting in the presence of cancer cells expressing CD44 on the surface, suggesting higher drug delivery to tumors over non-tumor tissues. Even CD44-poorly expressing cancers could be targeted by the NDs, thanks to their good passive diffusion within cancer cells.
Introduction
Although it took approximately three decades from their first synthesis in the 1960s for nanodiamonds (NDs) to capture the interest of researchers, once they gained attention in the late 1990s, the fascination with these nanoparticles not only endured, but also steadily intensified.1 As a result, a wide variety of methods for their production has been developed, encompassing both top-down approaches, like detonation of carbon-containing explosives1,2 and grinding of High-Pressure High-Temperature (HPHT) microdiamond powders,3 and bottom-up techniques, such as Chemical Vapor Deposition (CVD)4 and synthesis from adamantane derivatives.5 Similarly, an extensive array of post-synthetic procedures, including treatments with microwave plasma,6,7 high-temperature thermal processes in controlled environments8–10 and wet chemistry-based strategies,11–13 have been established. This combined spectrum of synthesis methods and subsequent modifications enables the creation of NDs with diverse sizes, covering the 5–300 nm range, morphologies, and surface characteristics. These find applications in a rich landscape of fields, which includes tribology,14,15 nanocomposites,16,17 energy-related technologies,18,19 sensing20,21 and photonics,22,23 thanks to their unique combination of physical and chemical properties.In particular, due to their remarkable chemical stability and biocompatibility,24 NDs stand out in the biomedical field, where they have been investigated as drug delivery systems,25,26 additives in tissue scaffolds and surgical implants,27,28 biosensors29,30 and radiosensitizers.31–33 In this context, they have also been extensively employed as fluorescent cellular biomarkers,34–36 owing to their fluorescence properties arising from the presence of optically active lattice defects, such as Nitrogen-Vacancy (NV) centers, which show an intense photoluminescence in the red wavelength range when excited with a green laser.37 Notably, the emission from NV centers is highly stable and resistant to quenching or photobleaching,38–40 setting NDs apart from organic fluorophores, thus providing a significant advantage for their use in optical bioimaging. As a result, NDs have emerged as a promising tool for the visualization of cancer cells, relying on their preferential accumulation in tumor tissues, thanks to the Enhanced Permeation and Retention (EPR) effect.41
Moreover, to improve selectivity, thus enhancing ND uptake and internalization in cancer cells, active targeting can be accomplished through surface functionalization, which involves decorating the ND surface with specific molecules, acting as ligands that bind to overexpressed receptors on cancer cells or tumour vasculature.42 Examples of this approach, typically grounded in covalent bond formation with the functionalizing moiety, comprise linking with antibodies,43,44 vascular endothelial growth factor,45 transferrin,46 folic acid,47 and mannose.48
Another targeting agent that can be used for the functionalization of NDs is hyaluronic acid (HA). This is a natural ubiquitous polysaccharide with excellent biocompatibility and biodegradability in the body, which can be recognized by cluster determinant 44 (CD44) receptors,49,50 overexpressed on the surface of several tumour cells, such as breast, ovarian, liver and prostate cancer cells.51,52 Although the high specificity of HA towards CD44 receptors has been proved for a broad range of nanoparticles,53–55 research on the derivatization of NDs with HA has been limited. Yun et al. developed ND-based nanoparticles incorporating HA and a photosensitive molecule for photodynamic and photothermal tumour therapy, highlighting the selective ND uptake by HeLa cells due to the presence of HA.56 Han et al. successfully attached HA on fluorescent NDs, delving into particle capabilities for liver-targeted molecular imaging. Through both in vitro and in vivo experiments, they demonstrated specific delivery of HA–ND conjugates to liver cells. Their work also showed the safety, biocompatibility, and imaging capability of these HA conjugates assessed through in vivo fluorescence lifetime measurements.57 Both the works described are based on covalent bonded hyaluronic acid on a diamond surface obtained through advanced and complex chemical pathways. On the other hand, Cui et al. focused on developing ND-based theranostic platforms for triple-negative breast cancer treatment. They achieved ND functionalization through non-covalent bonding involving electrostatic interactions for coating NDs with a protamine sulphate layer. The proposed multi-step approach guarantees the adsorption of HA onto a protamine sulphate layer through charge complexation. In one study, curcumin and a photosensitive compound were encapsulated within the NDs,58 while another study involved ND loading with the anticancer drug doxorubicin through electrostatic interactions.59 In both cases, the developed nanosystems were characterized by uniform dimensions, high loading efficiency, excellent biocompatibility, and colloidal stability, showing prevalent localization in the tumour cells, thus evidencing the high potentiality of HA-functionalized NDs for the investigated application.58,59 Lastly, Chernysheva et al. recently confirmed that the adsorption of miramistin on NDs enhances the adsorption of HA. This exploration introduces an alternative strategy for ND functionalization with HA, suggesting potential advancements in the field of drug nanocarriers.60
Due to the promising results from the aforementioned studies, the present investigation aims to explore a novel and smarter system for functionalizing NDs with HA through a non-covalent approach. Here, we demonstrate, for the first time, the successful functionalization of NDs with a conjugate of HA and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (HA/DMPE). This process showcases the selective internalization of functionalized particles into cancer cells that overexpress CD44 receptors, highlighting the potential for tracking these particles due to the fluorescence properties of NDs.
The targeting efficacy of HA/DMPE conjugates towards CD44-overexpressing cancer cells has previously been reported by our group for carbon nano-onions61 and carbon nanotubes.62 Functionalization not only imparts cell specificity to the nanoconstructs, but also enhances their dispersion in aqueous media. In this study, NDs were functionalized with a 200 kDa HA/DMPE conjugate using a ND![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HA/DMPE ratio of 5:1. The characterization of the particles was conducted through diffuse reflectance Fourier transform infrared spectroscopy, Raman spectroscopy, and photoluminescence spectroscopy to gather information on the efficacy of the functionalization process. Scanning electron microscopy was employed to evaluate the size distribution of the dry particles, while Dynamic Light Scattering (DLS) and zeta potential measurements were utilized to evaluate the nanoparticles’ behaviour in a water-based medium.
:HA/DMPE ratio of 5:1. The characterization of the particles was conducted through diffuse reflectance Fourier transform infrared spectroscopy, Raman spectroscopy, and photoluminescence spectroscopy to gather information on the efficacy of the functionalization process. Scanning electron microscopy was employed to evaluate the size distribution of the dry particles, while Dynamic Light Scattering (DLS) and zeta potential measurements were utilized to evaluate the nanoparticles’ behaviour in a water-based medium.
Furthermore, we assessed the ND biocompatibility and uptake mediated by CD44 receptors in three different models of human adenocarcinoma cells by performing cytofluorometric assay and confocal microscopy.
Results and discussion
ND sample preparation
The NDs considered in this study are MSY 0–0.1 purchased from Pureon and are characterized by different surface features. The samples were labelled as NDAnn, NDOx, and NDH, according to the various thermal treatments they underwent, which are detailed in the Experimental section and in Fig. S1 in the ESI.† NDAnn were subjected solely to an annealing process under nitrogen flow. On the other hand, NDOx underwent annealing followed by oxidation in air, whereas NDH experienced a sequential treatment involving annealing, oxidation and hydrogenation under hydrogen flux. The annealing and thermal processes ending with hydrogenation were intended to promote surface hydrophobicity by fostering the formation of C–H terminations,63,64 while post-annealing oxidation was performed to endow the ND surface with hydrophilic character through the establishments of oxygenated moieties.65All the samples were non-covalently functionalized with a HA/DMPE conjugate. The conjugate was obtained by linking the phospholipid to HA via an amidic bond in the presence of a soluble carbodiimide derivative to give a product in which the phospholipid amino group is randomly linked to the carboxylic residues of HA.62,66 After the dispersion of NDs in water for 90 min of bath sonication, the HA/DMPE conjugate previously solubilized in water was added and bath sonicated for further 90 min (Fig. S2†). The NDs were purified by centrifugation and a carbazole assay was performed to detect any amount of the non-covalently bound conjugate. The colorimetric assay confirmed that all the HA/DMPE was adsorbed onto the ND surface. Upon functionalization, NDAnn, NDOx, and NDH were respectively labelled as HA–NDAnn, HA–NDOx, and HA–NDH, as also reported in the Experimental section and in Fig. S1.†
ND characterization
Prior to evaluating ND behaviour in the cellular environment, to gain insights into the physical and chemical properties of both non-functionalized and hyaluronated samples, comprehensive material characterization was performed employing Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy, Raman spectroscopy, Scanning Electron Microscopy (SEM), Dynamic Light Scattering (DLS) and photoluminescence (PL) spectroscopy.To investigate sample's surface chemistry, assessing the success of thermal processes in the modifications of the ND surface and the effectiveness of the non-covalent functionalization with the HA-based conjugate, a DRIFT spectroscopy study was performed (Fig. 1a).
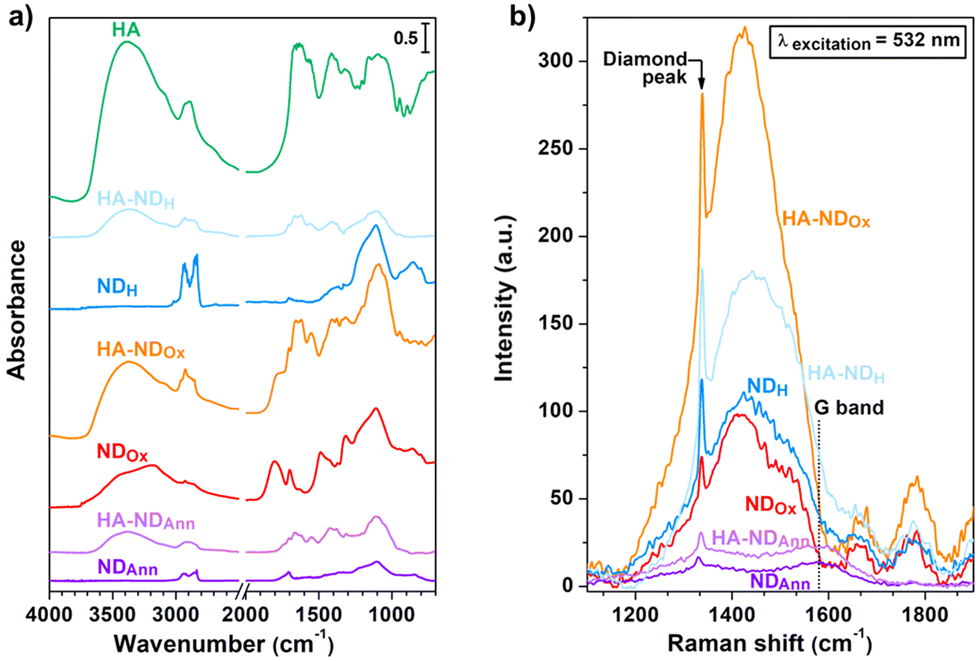 | ||
| Fig. 1 (a) DRIFT spectra of the NDs and HA. (b)NDs Raman spectra obtained after baseline subtraction. | ||
The DRIFT spectrum of NDAnn reveals distinctive features in the 3000–2800 cm−1 spectral range, corresponding to the asymmetric and symmetric stretching of C–H bonds.67 On the other hand, no bands related to ν(O–H) stretching of the surface adsorbed water and hydroxyl groups appear between 3700 cm−1 and 3000 cm−1.68 Furthermore, only a faint signal at 1705 cm−1 can be detected in the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) region.69 These findings underscore the efficacy of the annealing treatment in forming C–H moieties on the NDAnn surface, thus imparting it a hydrophobic behaviour. In contrast, the DRIFT spectrum of NDOx is characterized by a broad ν(O–H) band, along with a prominent and complex signal at 1800 cm−1, which is attributed to the CO stretching in various surface functionalities, encompassing esters, carboxylic acids, acid anhydrides and lactones.69,70 Notably, the feature at 1705 cm−1 and the signals of C–H stretching are also present in the DRIFT spectrum of NDOx. However, the intensity of the former is higher in the case of NDOx compared to NDAnn, while the opposite holds true for the latter. All such observations highlight that the oxidation process successfully resulted in the establishment of surface oxygenated moieties, thus rendering the surface of NDOx highly hydrophilic. This is further supported by the appearance of a series of new bands in the spectral region between 1500 cm−1 and 1300 cm−1, ascribed to epoxide and ketone vibrations,69,70 in addition to the visible shoulder at 1630 cm−1, attributed to the water bending mode.64,71 The DRIFT spectrum of NDH is similar to the one of NDAnn, being characterized by the absence of the ν(O–H) bands, as well as the presence of the main signals due to oxygen-containing moieties at 1800 cm−1 and in the 1500–1300 cm−1 range. Remarkably, it can be noted that the signals in the C–H stretching range are enhanced for NDH with respect to the DRIFT spectrum of NDAnn. The previously discussed features reveal the effectiveness of the hydrogenation process in forming surface C–H bonds, thus giving to the NDH surface a hydrophobic character.
O) region.69 These findings underscore the efficacy of the annealing treatment in forming C–H moieties on the NDAnn surface, thus imparting it a hydrophobic behaviour. In contrast, the DRIFT spectrum of NDOx is characterized by a broad ν(O–H) band, along with a prominent and complex signal at 1800 cm−1, which is attributed to the CO stretching in various surface functionalities, encompassing esters, carboxylic acids, acid anhydrides and lactones.69,70 Notably, the feature at 1705 cm−1 and the signals of C–H stretching are also present in the DRIFT spectrum of NDOx. However, the intensity of the former is higher in the case of NDOx compared to NDAnn, while the opposite holds true for the latter. All such observations highlight that the oxidation process successfully resulted in the establishment of surface oxygenated moieties, thus rendering the surface of NDOx highly hydrophilic. This is further supported by the appearance of a series of new bands in the spectral region between 1500 cm−1 and 1300 cm−1, ascribed to epoxide and ketone vibrations,69,70 in addition to the visible shoulder at 1630 cm−1, attributed to the water bending mode.64,71 The DRIFT spectrum of NDH is similar to the one of NDAnn, being characterized by the absence of the ν(O–H) bands, as well as the presence of the main signals due to oxygen-containing moieties at 1800 cm−1 and in the 1500–1300 cm−1 range. Remarkably, it can be noted that the signals in the C–H stretching range are enhanced for NDH with respect to the DRIFT spectrum of NDAnn. The previously discussed features reveal the effectiveness of the hydrogenation process in forming surface C–H bonds, thus giving to the NDH surface a hydrophobic character.
Before delving into the DRIFT analysis of the functionalized NDs, examination of HA alone was carried out with the purpose to determine distinctive markers for assessing the presence of the molecule on the surface of the functionalized samples. The HA spectrum is characterized by a broad band centred at 3390 cm−1, associated with stretching vibrations of hydrogen bonded O–H. This is accompanied by a weak shoulder at ca. 3100 cm−1, assigned to N–H vibrations, and by signals in the 3000–2800 cm−1 range, due to ν(C–H).72 Additionally, several complex bands appear in the 1750–1500 cm−1 spectral region, assigned to amides I and II and the vibrations of various carbonyls and carboxyls.73 Moreover, the broad signal extending between 1200 cm−1 and 1000 cm−1 gathers C–O stretching vibrations in alcohols and the antisymmetric C–O–C stretching in glycosidic groups.73
The comparative analysis between the DRIFT spectra of the hyaluronated NDs and those of the non-functionalized samples reveals discernible alterations in the shape of the characteristic bands of the latter upon derivatization. Substantial modifications are indeed evident in the C–H stretching bands when comparing HA–NDAnn with NDAnn and HA–NDH with NDH. Similarly, relevant changes can be observed in the O–H and CO stretching signals when examining HA–NDOx with respect to NDOx. The spectral comparison between the functionalized NDs with the NDs prior to derivatization also displays the appearance of additional features in the former. Such extra bands are the same in HA–NDAnn, HA–NDOx and HA–NDH and correspond to the signals detected for HA, which have been discussed above. Notably, ν(O–H) signals appear in HA–NDAnn and HA–NDH, which are not present in NDAnn and NDH. Moreover, C–H stretching bands are more pronounced in the DRIFT spectrum of HA–NDOx with respect to the one of NDOx. Further confirmation of the correlation between the DRIFT features of ND samples and successful functionalization is evident in Fig. S3,† where a series of DRIFT spectra collected at decreasing HA/DMPE:ND ratios (1:10 and 1:15) are presented. As expected, the intensity of the bands attributed to HA decreases for the nanoparticles functionalized with a lower amount of the conjugate. These observations suggest the effective anchoring of HA to the nanodiamond surface.
With the aim of examining the structures of various samples and monitoring any potential structural perturbations to the NDs arising from the introduction of functionalization, Raman spectroscopy was performed and the corresponding results are presented in Fig. 1b. The Raman spectra of the samples all exhibit the first-order Raman peak of diamond,74 while the G-band is observable only in the Raman spectra of NDAnn and HA–NDAnn, appearing as a weak signal at 1580 cm−1.74 The latter observation indicates the formation of some graphite during the annealing treatment, suggesting at the same time its removal through oxidation and hydrogenation processes, as also documented in prior studies.64,65 Notably, the Raman spectra of NDOx, HA–NDOx, NDH, and HA–NDH reveal a signal at 1405 cm−1 overlapped with a broad shoulder, along with additional features at above 1600 cm−1. These signals are not related to Raman scattering; instead, they arise from the photoluminescence of NV centers (further details concerning the photoluminescence properties of NDs are presented in the discussion of Fig. 4, displaying the photoluminescence spectra of the samples). Specifically, the feature at 1405 cm−1, corresponding to 575 nm on the wavelength scale, is associated with the zero-phonon line of NV0 centers, while the other peaks are phonon replicas due to the interaction between phonon states and electronic transitions.74 It can be observed that the spectral features of NDAnn, NDOx and NDH are conserved in the Raman spectra of their functionalized counterparts, namely HA–NDAnn, HA–NDOx and HA–NDH, meaning that the association of HA does not induce structural alterations to the NDs, not changing the diamond and graphite contents of the original particles.
To explore the characteristics of the ND samples in terms of median particle size, SEM and DLS analyses were carried out. SEM was employed to gain insights into the size distribution of the NDs in their dry state, as well as for analyzing their morphology. On the other hand, DLS characterized the particle dimensions in an aqueous environment, thus offering complementary information to the ones collected through SEM micrographs, in addition to the data regarding dispersibility and stability in solution, which are crucial parameters for the application of the NDs in cellular context.
The size distributions of NDs obtained from SEM images are shown in Fig. 2a–c for the non-functionalized particles and in Fig. 2d–f for the functionalized ones. Median size diameters (Dmed) for NDAnn, NDOx, and NDH are respectively 53 nm, 55 nm and 53 nm with a corresponding standard deviation (σ) of 10 nm, 10 nm and 9 nm. These values align consistently with each other and with the dimensions specified by the producer (Dmed = 50 nm ± 10 nm), indicating no appreciable influence of thermal treatments on the ND dimensions. Median size diameter values of the functionalized particles are 54 nm for HA–NDAnn, 55 nm for HA–NDOx and 52 nm for HA–NDH with a standard deviation equal to 10 nm, 10 nm and 8 nm, respectively. Notably, such values closely mirror those of their non-hyaluronated equivalents. Fig. 2 also shows details from the SEM micrographs of NDs. The pictures demonstrate that all the NDs exhibit irregular and jagged geometries, a distinctive feature preserved across both non-functionalized and functionalized samples. The collective results from both shape analysis and size distribution thus show that the functionalization has essentially no discernible impact on the geometry and the dimensions of the NDs in their dry state.
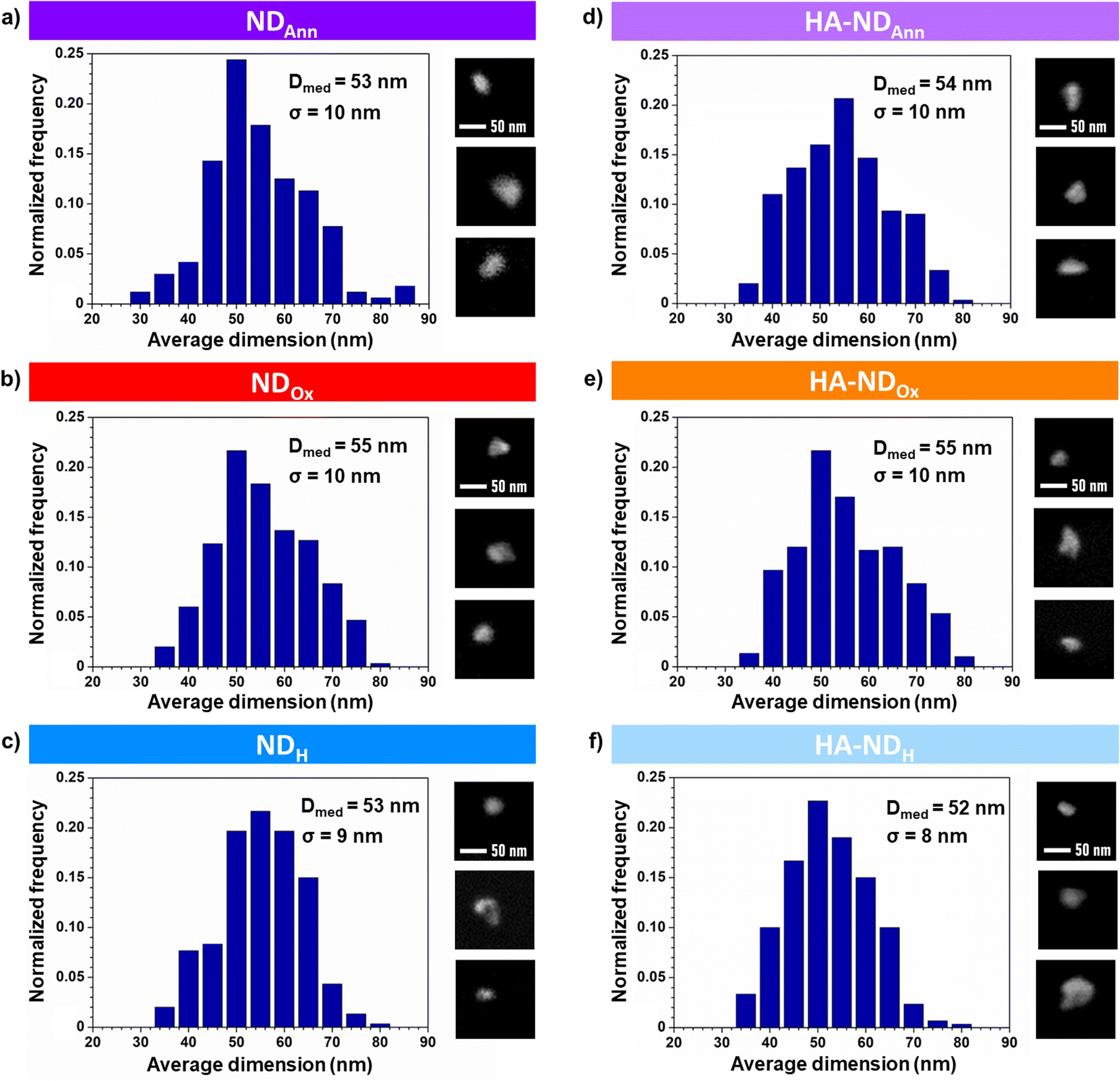 | ||
| Fig. 2 Size distributions of the NDs derived from SEM images. Particle size histograms were constructed based on 300 NDs from 3–4 micrographs, except for NDAnn, where the distribution was derived from 168 NDs. Details from SEM micrographs showing ND particles are also presented. (a) NDAnn, (b) NDOx, (c) NDH, (d) HA–NDAnn, (e) HA–NDOx, and (f) HA–NDH. | ||
The median particle size and zeta potential of NDs and HA–NDs dispersed in water were analyzed by DLS after sonicating the prepared solution for 180 minutes using a sonicator bath at a frequency of 40 kHz. After aggressive sonication, the median diameter of the bare NDs closely matches the primary particle size, confirming that a negligible quantity of aggregates is present in the solution. This is a crucial aspect, as these ND solutions represent an intermediate step for the preparation of hyaluronated ones. Only NDH, however, was confirmed to be less dispersible in water due to the hydrophobic nature of the hydrogenation termination, as demonstrated by the higher diameter and PDI compared to other bare NDs. As shown in Table 1, after modification with the HA/DMPE conjugate, the hydrodynamic median diameter of NDs (NDAnn and NDOx) does not change, but the zeta potential was more negative, due to the HA carboxyl groups, indicating the successful modification of the outer surface of the NDs.
| Sample | Median diameter (nm ± S.D.) | Polydispersity index (PDI) | Zeta potential (mV ± S.D.) |
|---|---|---|---|
| NDAnn | 59 ± 4 | 0.180 | −22.6 ± 0.3 |
| HA–NDAnn | 59 ± 1 | 0.166 | −33.7 ± 0.9 |
| NDOx | 59 ± 4 | 0.122 | −37.5 ± 3.1 |
| HA–NDOx | 59 ± 5 | 0.153 | −45.8 ± 0.5 |
| NDH | 80 ± 12 | 0.214 | −20.1 ± 0.7 |
| HA–NDH | 69 ± 6 | 0.119 | −34.9 ± 0.3 |
The colloidal stability of the NDs is an important parameter for their biomedical applications, thus the stability of HA–NDs in RPMI 1640 + 10% FBS and water was evaluated. The size and zeta potential of the samples remained stable over 72 hours, indicating good stability in both water and cell medium (data not shown), while the unconjugated sample precipitated at the bottom of the container after a few hours. NDs in the presence of unconjugated 200 kDa HA were also prepared as a reference to compare the effect of the HA/DMPE conjugate with the one of the free HA in water. Fig. 3 shows the different behaviours of NDs in water immediately after dispersion and over one month at room temperature. HA–NDs were well-dispersed and stable without aggregation and any differences in median diameter and zeta potential values. On the other hand, NDs in the presence of unconjugated HA showed aggregation after one month.
 | ||
| Fig. 3 Representative images of the NDs obtained immediately after dispersion (a–c) and after one month (d–f): (a and d) unconjugated HA–NDOx; (b and e) NDOx; and (c and f) HA–NDOx. | ||
These results confirm that the HA/DMPE conjugate confers excellent dispersibility and stability to ND preparations.
In order to investigate the photoluminescence (PL) properties of the NDs, playing a key role in their visualization in cells, the samples were analyzed by means of PL spectroscopy. The PL spectrum was collected also for the sole conjugate to assess its potential contributions to a reduction of particle intrinsic fluorescence through any quenching effect. PL spectra of the samples are presented in Fig. 4, where the wavelength range highlighted by the black-contoured rectangle corresponds to the region reported in Fig. 1b in Raman values, since it displays features not only due to PL effects, but also arising from Raman scattering, as examined above (see the discussion of Fig. 1b). The PL spectra of NDs are all characterized by a broad band extending from 600 nm to 780 nm, which can be ascribed to the superposition of the phonon sidebands of NV0 and NV− centers, covering the 600–750 nm and the 650–750 nm wavelength range, respectively.37 In the PL spectra of NDOx, HA–NDOx, NDH, and HA–NDH, signals corresponding to the zero-phonon lines of both types of NV centers are discernible. In particular, the one at 638 nm is associated with NV− centers, while the one at 575 nm, falling in the contoured region and corresponding to 1405 cm−1 on the Raman shift scale, is ascribed to NV0 centers,37 as already discussed above. Observing together the PL spectra of the non-functionalized NDs, it can be noted that the fluorescence of NDOx is increased compared to NDAnn, and NDH exhibits even higher fluorescence than NDOx. Such a trend is likely due to the elimination of graphitic phases formed upon annealing through oxidation and combined oxidation + hydrogenation processes, as also suggested by the analysis of Raman spectra. Graphitic phases have indeed been reported to have a quenching effect on the PL of NV centers75 and hence their reduced presence should enhance the fluorescence of the NDs.
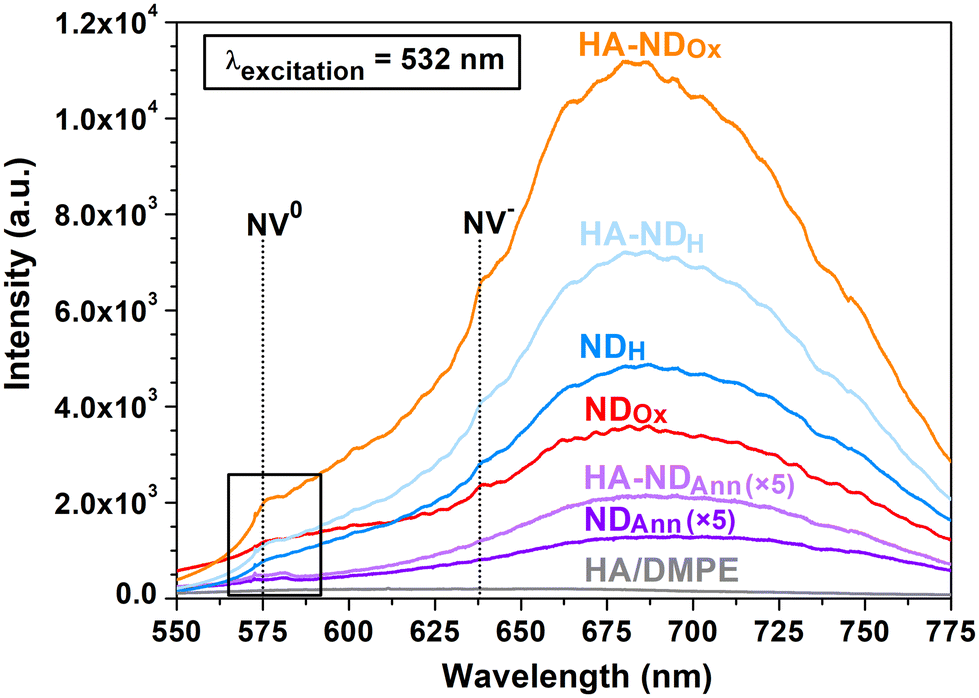 | ||
| Fig. 4 Photoluminescence spectra of the NDs and the HA/DMPE conjugate. The PL spectra of NDAnn and HA–NDAnn are multiplied by a factor of five. The black rectangle identifies the region highlighted in Fig. 1b. | ||
A noteworthy observation for HA–NDAnn, HA–NDOx, and HA–NDH is the presence of luminescence, comparable to or even increased compared to that of their non-functionalized counterparts. The influence of the HA-based conjugate on optical spectral features can be deemed insignificant, as demonstrated by the photoluminescence spectrum of the pure conjugate, which exhibits no detectable new PL bands or peaks. The key finding emerging from the analysis of PL spectra is thus the preservation of particle fluorescence upon association, indicating that NDs continue to be well-suited agents for cellular imaging applications.
Taken together, all these data suggest the efficacy of our approach, which offers distinctive advantages over others reported in the literature for coating NDs with HA. In fact, these require some steps to obtain modified NDs, either involving the formation of a covalent bond between the ND-functional groups and HA or a two-step assembly strategy through the preliminary modification of NDs with protamine sulfate or miramistin. In contrast, our method streamlines the HA anchoring process, offering a more straightforward route for decorating NDs with HA, regardless of their different starting surface terminations.
ND cytotoxicity
First, we compared the cytotoxicity of different formulations of non-functionalized (NDAnn, NDH, NDOx) and HA-functionalized NDs (HA–NDAnn, HA–NDH and HA–NDOx) across three pairs of cancer cell lines of pancreatic, breast and lung origin, which are three of the most common and aggressive cancer types. Each pair contained one cell line with low levels of CD44 and the second one with high levels of the receptor, respectively (Fig. S4†), to allow a better comparison between the effects of plain and HA-functionalized NDs. Dose- and time-dependent experiments suggested a good biocompatibility of all types of NDs across the different cancer cell lines. Indeed, even at the highest concentration (20 μg ml−1) and at the most prolonged time (72 h), the reduction in cell viability was not >30% (see Fig. 5 and Fig. S5†). This experimental set demonstrates that NDs in all their formulations do not exert an appreciable cytotoxicity, therefore they could be proposed as safe carriers of chemotherapeutic drugs as future development. The not significant differences in terms of cytotoxicity between the cancer cell lines of different origins and histotypes within the same tumor type indicate that the good biocompatibility of ND was not cell line- or tumor-dependent.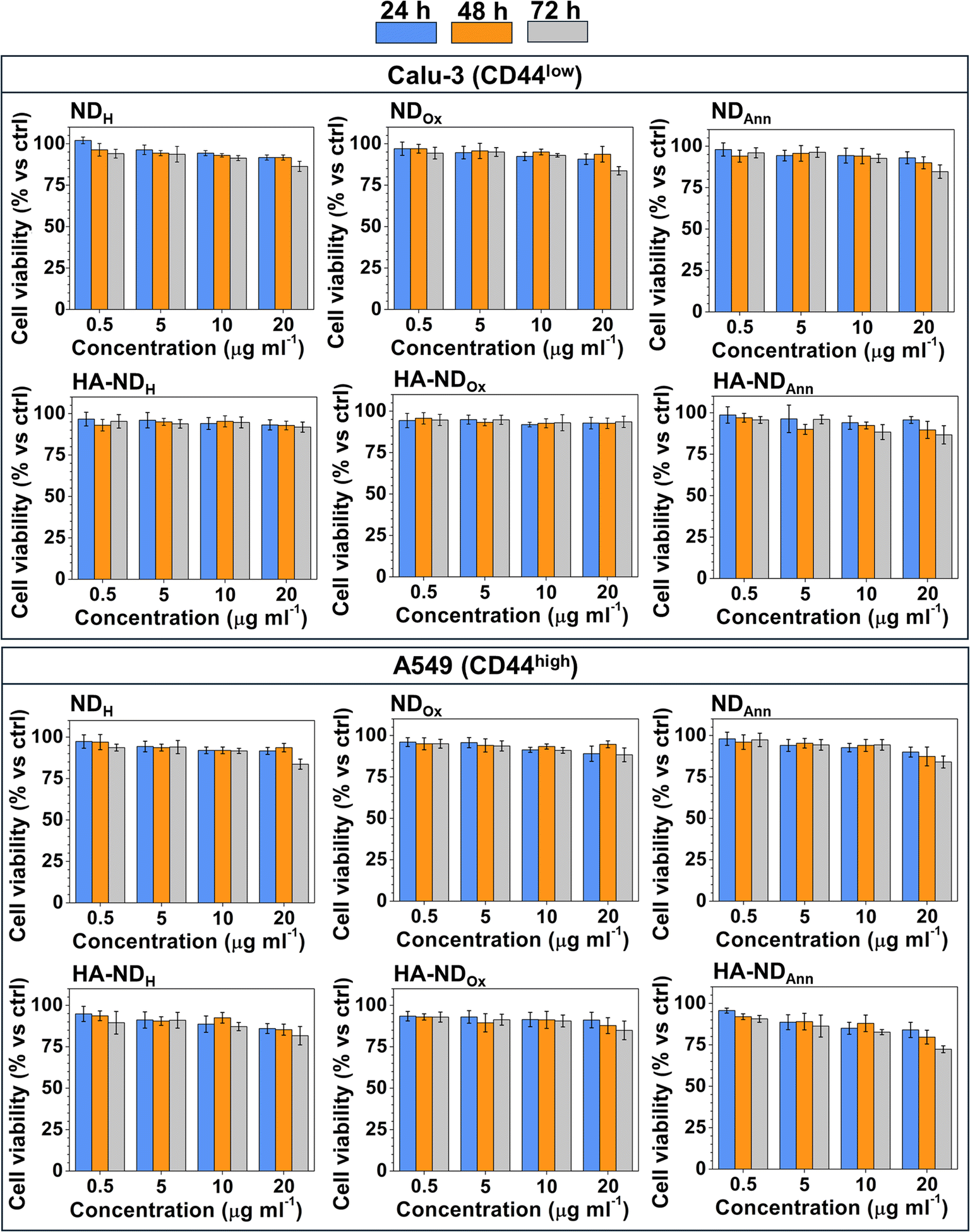 | ||
| Fig. 5 Cell viability at 24, 48 and 72 h in human non-small cancer cell lines, Calu-3 and A549, incubated with NDAnn, NDH, NDOx, HA–NDAnn, HA–NDH and HA–NDOx at the indicated concentration by a chemiluminescence-based assay in triplicate (n = 3 independent experiments). Data are means + SD. | ||
ND uptake
We measured the intracellular uptake of different ND formulations at the same non-cytotoxic concentrations, exploiting the intrinsic fluorescence of NDs. By the fluorimetric analysis of the intracellular fluorescence, we observed a time- and dose-dependent uptake of NDs. For non-decorated NDs, the intracellular retention at 24 h followed this rank order: NDAnn ≥ NDH ≥ NDOx, independently from the surface level of CD44 (Fig. 6).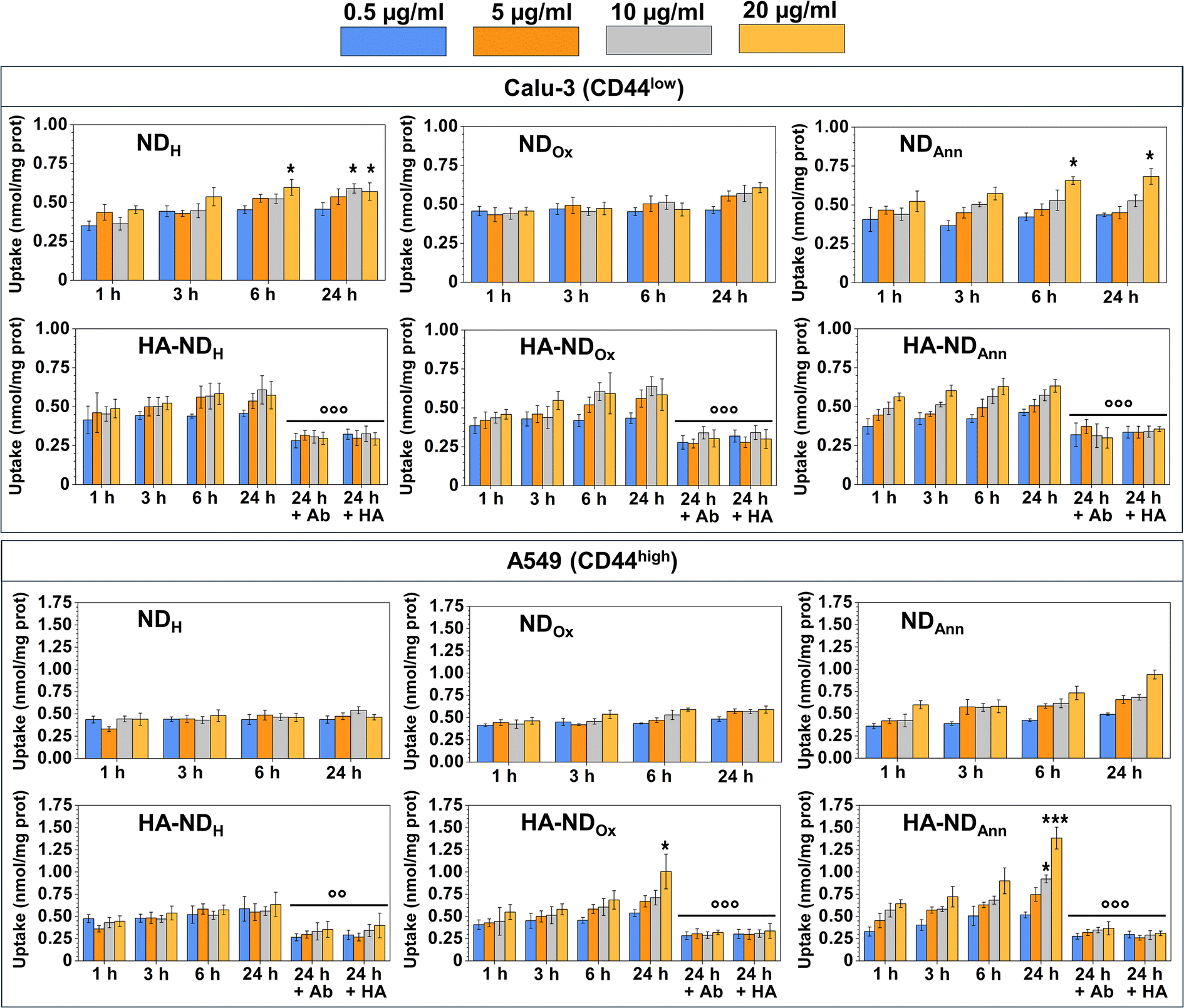 | ||
| Fig. 6 Uptake of NDAnn, NDH, NDOx, HA–NDAnn, HA–NDH and HA–NDOx at the indicated concentrations after 1, 3, 6, and 24 h in human non-small cancer cell lines, Calu-3 and A549, by a fluorimetry-based assay in triplicate (n = 3 independent experiments). When indicated, an anti-CD44 neutralizing antibody (Ab, diluted 1/100) or 100 μM hyaluronic acid (HA) was co-incubated. Data are the means + SD. *p < 0.05, ***p < 0.001: ND fluorescence vs. cell autofluorescence; ○○p < 0.01, ○○○p < 0.001: ND fluorescence in cells treated with Ab/HA vs. ND fluorescence in the cells without Ab/HA. | ||
The slow time- and dose-dependent increase, without significant differences in the uptake between the different ND formulations, is indicative of a passive endocytosis of NDs in different cell types. The presence of the negative charge of the oxygenated groups in NDOx, deprotonated at the physiological pH, may render the passive diffusion of the NDs less efficient; indeed, in Calu-3, PANC-1 and MDA-MB-231 cells, NDOx was less taken up by the cells. The different behaviours may be explained by the different patterns of surface charge, due to glycolipids and glycoproteins, that can make the entry of NDs more difficult because of electrostatic repulsion events. In low-CD44 surface level cells, we observed a comparable yet slightly enhanced uptake of HA–NDs compared to their non-functionalized counterparts. This phenomenon could be attributed to the general facilitation of cellular uptake conferred by ND surface coatings, as for instance observed in the case of zwitterionic moieties.76 However, the observed increase also hints at the role of HA in promoting NDs internalization through interaction with CD44 receptors by facilitating receptor-mediated endocytotic pathways. Although the endocytosis of HA-functionalized NDs was dependent on CD44-mediated endocytosis, as demonstrated by a significant reduction in the uptake in the presence of a CD44-blocking antibody or an excess of HA, the impact of the CD44-mediated route in the endocytosis of NDs seems equivalent to the impact of an unspecific endocytosis (Fig. S6†). Interestingly, in CD44-highly expressing A549 cells, the uptake of HA–NDAnn and HA–NDOx was higher than the uptake of the corresponding non-functionalized NDs, indicating that in this cell model the endocytosis triggered by CD44 plays a preponderant role. Only HA–NDH did not show a significantly higher uptake than NDH; it is known that different tumors express different CD44 isoforms: the presence of a specific variant instead of another may facilitate or hamper the interaction between CD44 and HA–NDs. Therefore, a preliminary analysis of the amount and variants of CD44 could be useful in determining the type of tumor that is the most suitable target for HA–NDs instead of bare NDs.
To prove that our NDs are good tools to label the CD44-positive cells, we analyzed the intracellular distribution of NDOx and HA–NDOx in A549 cells by confocal microscopy (Fig. 7). The intracellular accumulation was time dependent and appeared faster with NDOx than with HA–NDOx, in line with the different kinetics of entry dependent on the endocytosis route (unspecific versus receptor-mediated endocytosis). NDOx reached indeed the maximum accumulation already after 3 h (data not shown), while HA–NDOx reached a comparable accumulation after 6 h. At 24 h, the amount was similar for both NDOx and HA–NDOx, indicating a likely saturation in the uptake mechanisms. Interestingly, the main site of intracellular localizations was the nuclear and the perinuclear regions. Such distribution may suggest that NDs could be potentially good candidates as carriers of chemotherapeutic drugs that should be directed to the nucleus to exert their cytotoxic potential as gemcitabine, anthracyclines, taxanes, and platinum derivatives. These results may have a translational potential since these types of drugs are indeed the first-line treatment in pancreatic, breast and lung cancers, the three types of cancers analyzed in this work.
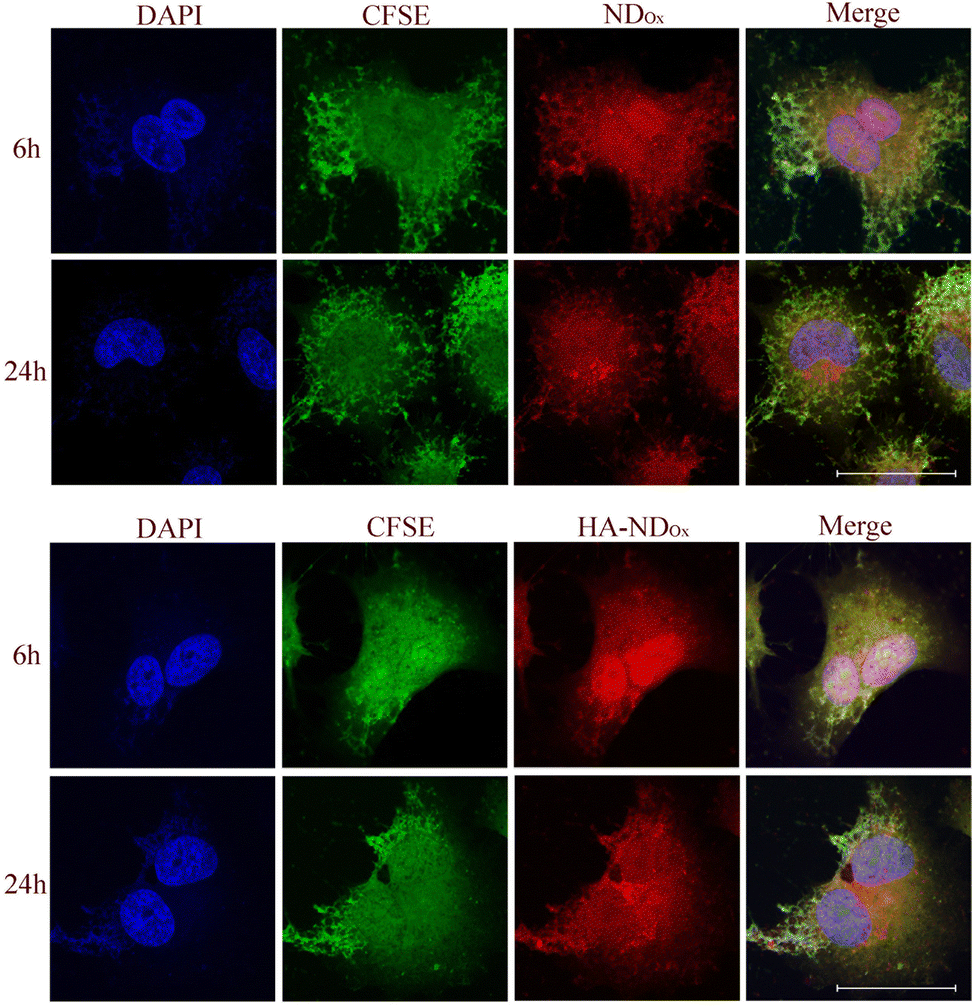 | ||
| Fig. 7 Representative microscopy images of A549 cells incubated with 100 μg ml−1 NDOx and HA–NDOx for the indicated time. Photographs were taken with a Leica SP8 confocal microscope using a 60× objective. The scale bar is 50 μm. | ||
Conclusions
In the present work, we reported a new approach for the functionalization of NDs with HA based on non-covalent anchoring of the molecule on the particle surface. This was accomplished by exploiting a HA/DMPE conjugate, which was attached to NDs with different surface chemistry, obtained by performing annealing, oxidation and hydrogenation thermal treatments on commercial diamond nanocrystals. The ND samples were comprehensively characterized both before and after the functionalization in terms of surface chemistry, structure, size and optical properties.DRIFT spectroscopy evidenced distinct changes in the surface functional groups induced on the NDs by thermal treatments, thus allowing us to prove the efficacy of each process in modifying ND surface chemistry. On the other hand, the comparison between the DRIFT spectra of non-functionalized NDs and their hyaluronated counterparts, showing significant alterations in the ND spectral features upon derivatization, confirmed the successful anchoring of HA to the ND surface.
Functionalized nanoparticles exhibited enhanced dispersibility and stability in water while preserving the fluorescence properties of nitrogen vacancy colour centers. Biocompatibility assays across pancreatic, breast, and lung cancer cell lines indicated the safety of both non-functionalized and HA-functionalized ND formulations with no relevant differences observed among the cell lines of different origins and histotypes within the same tumor type. Fluorimetric analysis revealed the time- and dose-dependent uptake of NDs, primarily through passive endocytosis across various cell types, with HA-functionalized NDs showing similar uptake patterns in CD44-rich cells dependent on CD44-mediated endocytosis. Confocal microscopy further supported ND potential to target the nuclear regions of cancer cells expressing CD44, underscoring the promising potential of NDs as efficacious drug delivery nanocarriers, particularly for targeting pancreatic, breast, and lung cancers. The observed time-dependent intracellular accumulation, with preferential localization in the nuclear and perinuclear regions, not only elucidates the kinetics of ND uptake via different endocytosis pathways, but also highlights their suitability as vehicles for chemotherapeutic agents requiring nuclear targeting, thus suggesting the translational potential of these systems in cancer therapy.
Overall, we observed that NDs are safe nanoparticles that can be taken up by different cancer types. The presence of CD44 on the cancer cell surface offers the possibility of using HA-functionalized NDs that have the advantage of an active targeting, implying a higher delivery of the drug to the tumors over the non-tumor tissues. Also, CD44-poorly expressing cancers, however, could be easily targeted by the NDs, thanks to their good passive diffusion within cancer cells.
The next steps will be the application of NDs as drug nanocarriers loaded with chemotherapeutic drugs. Improving the drug delivery into pancreatic, breast and lung cancer, where chemoresistance is well documented at the clinical level,77–79 is still an open challenge. Our approach can represent a step forward in this direction.
Experimental
ND sample preparation
The employed NDs are MSY 0–0.1 with a nominal median diameter of (50 ± 10) nm purchased from Pureon. Realized by means of grinding of HPHT diamond single crystals, they are classified as type Ib, having a nominal concentration of single substitutional nitrogen between 10 ppm and 100 ppm. NDs were processed with different thermal treatments in a tubular furnace to modify their surface, as represented in the scheme of Fig. S1 in the ESI.† NDs were subjected to an annealing process under nitrogen flow, which was performed at 800 °C for 2 h. This had the purpose of standardizing the ND surface through the elimination of surface functionalities, concurrently graphitizing amorphous carbon components present on particle surface without damaging the diamond core. After the treatment with nitrogen, on a part of the annealed powders, an air oxidation process for 36 h at 500 °C was performed with the aim of purifying NDs from graphitic layers thanks to the selective removal of sp2 carbon, while simultaneously decorating at the surface with oxygen-containing functional groups. A batch of the NDs subjected to the oxidation was further processed via a hydrogenation treatment under hydrogen flow for 3 h at 850 °C to promote the formation of hydrogen termination on the surface.Preparation of HA/DMPE
The 200 kDa hyaluronic acid–1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (HA/DMPE) conjugate and the fluoresceinamine-labelled HA/DMPE conjugate (f-HA/DMPE) were synthesized as previously described.62Non-covalent functionalization of NDs
The HA/DMPE conjugate was used for the non-covalent functionalization of NDs (HA–NDs). NDs were dispersed in MilliQ® water at a concentration of 0.5 mg ml−1 and bath sonicated for 90 min at 25 °C. Then, the amount of HA/DMPE to give a 5:1 NDs:HA/DMPE weight ratio was added to the ND suspension and bath sonicated for 90 min at 25 °C.
To remove the possible excess of HA/DMPE, the suspension was centrifuged at 11000 rpm for 5 min and the percentage of HA on the supernatant was determined by the carbazole assay.80
The same experimental procedure was used for the functionalization of NDAnn with the f-HA/DMPE conjugate for further cellular uptake experiments.
ND characterization
R, where R represents the measured reflectance.
For the analysis of size distribution, the ImageJ program was utilized. The dimensions of the nanocrystals were determined from the micrographs by manually tracing the contours of the projected areas of the NDs, considering only well-separated particles. Assuming a spherical nanoparticle shape, the obtained data were converted back to diameter values and particle size histograms were generated using an appropriate binning technique.
:10 dilution in MilliQ® water.
The surface charge was investigated via zeta potential measurement at 25 °C using the Smoluchowski equation and the Zetasizer Pro after 1:10 dilution of the suspensions in MilliQ® water. Each value reported is the average of three measurements.
The colloidal stability of functionalized NDs was assessed after 1:5 dilution in RPMI 1640 + 10% fetal bovine serum (FBS). The particle size and zeta potential were measured after 30 min, 24, 48 and 72 h incubation at 37 °C by diluting 1:2 in MilliQ® water. The colloidal stability of functionalized NDs dispersed in MilliQ® water was also evaluated at room temperature and the dispersions in water were also photographed immediately after sonication and over a 30-day period.
Author contributions
Conceptualization: SA and FP; formal analysis: LM; investigation: SS, IA, PA, VB, JK and BZ; methodology: BS, SA, FP and CR; resources: BS, SA, FP and CR; supervision: SA and FP; validation: SS, IA, PA, VB, JK and BZ; writing – original draft: SS, CR and IA; writing – review and editing: SS, IA, PA, VB, JK, LM, BS, BZ, CR, SA and FP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project AURORA of Italian Institute of Nuclear Physics (INFN, to PA), Italian Ministry for University and Research (MIUR)—University of Torino, “Fondi Ricerca Locale (ex-60%)” and Italian Associations for Cancer Research (AIRC; ID21480, IG29750 to CR). This work was also supported by the coordinated research project “Sub-cellular imaging and irradiation using accelerator-based techniques” of the International Atomic Energy Agency (IAEA, CRP F11024 to FP).Notes and references
- V. V. Danilenko, Phys. Solid State, 2004, 46, 595–599 CrossRef CAS.
- A. M. Panich, A. I. Shames, D. Mogilyansky, S. D. Goren and V. Y. Dolmatov, Diamond Relat. Mater., 2020, 108, 107918 CrossRef CAS.
- J.-P. Boudou, P. A. Curmi, F. Jelezko, J. Wrachtrup, P. Aubert, M. Sennour, G. Balasubramanian, R. Reuter, A. Thorel and E. Gaffet, Nanotechnology, 2009, 20, 235602 CrossRef PubMed.
- M. Frenklach, W. Howard, D. Huang, J. Yuan, K. E. Spear and R. Koba, Appl. Phys. Lett., 1991, 59, 546–548 CrossRef CAS.
- M. Alkahtani, J. Lang, B. Naydenov, F. Jelezko and P. Hemmer, ACS Photonics, 2019, 6, 1266–1271 CrossRef CAS.
- J.-R. Bertrand, C. Pioche-Durieu, J. Ayala, T. Petit, H. A. Girard, C. P. Malvy, E. Le Cam, F. Treussart and J.-C. Arnault, Biomaterials, 2015, 45, 93–98 CrossRef CAS.
- D. P. Mitev, A. M. Alsharabasy, L. Morrison, S. Wittig, C. Diener and A. Pandit, Front. Bioeng. Biotechnol., 2021, 9, 1–18 Search PubMed.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- Ashek-I-Ahmed, L. Gines, S. Mandal, C.-Y. Song, O. A. Williams, M. N. Sarmiento and C.-L. Cheng, ACS Omega, 2019, 4, 16715–16723 CrossRef CAS.
- S. Sturari, V. Varzi, P. Aprà, A. Britel, N.-H. Amine, G. Andrini, E. Corte, G. Tomagra, L. Mino, P. Olivero and F. Picollo, Surf. Interfaces, 2023, 38, 102831 CrossRef CAS.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS PubMed.
- C. Li, X. Zhang, E. F. Oliveira, A. B. Puthirath, M. R. Neupane, J. D. Weil, A. G. Birdwell, T. G. Ivanov, S. Kong, T. Gray, H. Kannan, A. Biswas, R. Vajtai, D. S. Galvao and P. M. Ajayan, Carbon, 2021, 182, 725–734 CrossRef CAS.
- F. Picollo, L. Mino, A. Battiato, S. Ditalia Tchernij, J. Forneris, K. Martina, M. Sacco, S. Tagliapietra, E. Vittone, P. Olivero and A. Barge, Diamond Relat. Mater., 2019, 91, 22–28 CrossRef CAS.
- C.-C. Chou and S.-H. Lee, Wear, 2010, 269, 757–762 CrossRef CAS.
- I. Neitzel, V. Mochalin, J. A. Bares, R. W. Carpick, A. Erdemir and Y. Gogotsi, Tribol. Lett., 2012, 47, 195–202 CrossRef CAS.
- Y. Zhang, J. R. Choi and S.-J. Park, Composites, Part A, 2017, 101, 227–236 CrossRef CAS.
- Y. Zhang, K. Y. Rhee, D. Hui and S.-J. Park, Composites, Part B, 2018, 143, 19–27 CrossRef CAS.
- Y. Song, H. Li, L. Wang, D. Qiu, Y. Ma, K. Pei, G. Zou and K. Yu, Chem. Commun., 2016, 52, 10497–10500 RSC.
- H. Wang and Y. Cui, Carbon Energy, 2019, 1, 13–18 CrossRef.
- Y. Wu, M. N. A. Alam, P. Balasubramanian, A. Ermakova, S. Fischer, H. Barth, M. Wagner, M. Raabe, F. Jelezko and T. Weil, Nano Lett., 2021, 21, 3780–3788 CrossRef CAS PubMed.
- N. Norouzi, A. C. Nusantara, Y. Ong, T. Hamoh, L. Nie, A. Morita, Y. Zhang, A. Mzyk and R. Schirhagl, Carbon, 2022, 199, 444–452 CrossRef CAS.
- H. Zhang, X. Chen and Z. Yin, Adv. Quantum Technol., 2021, 4, 2000154 CrossRef CAS.
- M. Radulaski, J. L. Zhang, Y. K. Tzeng, K. G. Lagoudakis, H. Ishiwata, C. Dory, K. A. Fischer, Y. A. Kelaita, S. Sun, P. C. Maurer, K. Alassaad, G. Ferro, Z. X. Shen, N. A. Melosh, S. Chu and J. Vučković, Laser Photonics Rev., 2019, 13, 1–14 Search PubMed.
- K. van der Laan, M. Hasani, T. Zheng and R. Schirhagl, Small, 2018, 14, 1–17 CrossRef PubMed.
- S. Chauhan, N. Jain and U. Nagaich, J. Pharm. Anal., 2020, 10, 1–12 CrossRef PubMed.
- Y. Tian, A. C. Nusantara, T. Hamoh, A. Mzyk, X. Tian, F. Perona Martinez, R. Li, H. P. Permentier and R. Schirhagl, ACS Appl. Mater. Interfaces, 2022, 14, 39265–39273 CrossRef CAS PubMed.
- Y. Zamani, A. Zareein, L. Bazli, R. NasrAzadani, B. P. Mahammod, S. Nasibi and A. M. Chahardehi, J. Compos. Compd., 2020, 2, 215–227 Search PubMed.
- M. Adel, P. Keyhanvar, I. Zare, Z. Tavangari, A. Akbarzadeh and M. Zahmatkeshan, J. Drug Delivery Sci. Technol., 2023, 90, 105130 CrossRef CAS.
- G. Petrini, G. Tomagra, E. Bernardi, E. Moreva, P. Traina, A. Marcantoni, F. Picollo, K. Kvaková, P. Cígler, I. Pietro Degiovanni, V. Carabelli and M. Genovese, Adv. Sci., 2022, 9, 2202014 CrossRef PubMed.
- Y. Wu and T. Weil, Adv. Sci., 2022, 9, 2200059 CrossRef CAS.
- R. Grall, H. Girard, L. Saad, T. Petit, C. Gesset, M. Combis-Schlumberger, V. Paget, J. Delic, J. C. Arnault and S. Chevillard, Biomaterials, 2015, 61, 290–298 CrossRef CAS PubMed.
- M. Kurzyp, H. A. Girard, Y. Cheref, E. Brun, C. Sicard-Roselli, S. Saada and J. C. Arnault, Chem. Commun., 2017, 53, 1237–1240 RSC.
- V. Varzi, E. Fratini, M. Falconieri, D. Giovannini, A. Cemmi, J. Scifo, I. Di Sarcina, P. Aprà, S. Sturari, L. Mino, G. Tomagra, E. Infusino, V. Landoni, C. Marino, M. Mancuso, F. Picollo and S. Pazzaglia, Int. J. Mol. Sci., 2023, 24, 16622 CrossRef CAS PubMed.
- Y. Y. Hui, C. L. Cheng and H. C. Chang, J. Phys. D: Appl. Phys., 2010, 43, 374021 CrossRef.
- N. Mohan, C. S. Chen, H. H. Hsieh, Y. C. Wu and H. C. Chang, Nano Lett., 2010, 10, 3692–3699 CrossRef CAS.
- W. W. W. Hsiao, Y. Y. Hui, P. C. Tsai and H. C. Chang, Acc. Chem. Res., 2016, 49, 400–407 CrossRef CAS.
- M. W. Doherty, N. B. Manson, P. Delaney, F. Jelezko, J. Wrachtrup and L. C. L. Hollenberg, Phys. Rep., 2013, 528, 1–45 CrossRef CAS.
- J. I. Chao, E. Perevedentseva, P. H. Chung, K. K. Liu, C. Y. Cheng, C. C. Chang and C. L. Cheng, Biophys. J., 2007, 93, 2199–2208 CrossRef CAS PubMed.
- C.-C. Fu, H.-Y. Lee, K. Chen, T.-S. Lim, H.-Y. Wu, P.-K. Lin, P.-K. Wei, P.-H. Tsao, H.-C. Chang and W. Fann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 727–732 CrossRef CAS PubMed.
- S.-J. Yu, M.-W. Kang, H.-C. Chang, K.-M. Chen and Y.-C. Yu, J. Am. Chem. Soc., 2005, 127, 17604–17605 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- D. H. Jariwala, D. Patel and S. Wairkar, Mater. Sci. Eng., C, 2020, 113, 110996 CrossRef CAS PubMed.
- B. M. Chang, H. H. Lin, L. J. Su, W. Der Lin, R. J. Lin, Y. K. Tzeng, R. T. Lee, Y. C. Lee, A. L. Yu and H. C. Chang, Adv. Funct. Mater., 2013, 23, 5737–5745 CrossRef CAS.
- D. Terada, S. Sotoma, Y. Harada, R. Igarashi and M. Shirakawa, Bioconjugate Chem., 2018, 29, 2786–2792 CrossRef CAS.
- M. D. Torelli, A. G. Rickard, M. V. Backer, D. S. Filonov, N. A. Nunn, A. V. Kinev, J. M. Backer, G. M. Palmer and O. A. Shenderova, Bioconjugate Chem., 2019, 30, 604–613 CrossRef CAS.
- M.-F. Weng, S.-Y. Chiang, N.-S. Wang and H. Niu, Diamond Relat. Mater., 2009, 18, 587–591 CrossRef CAS.
- B. Zhang, Y. Li, C. Fang, C. Chang, C. Chen, Y. Chen and H. Chang, Small, 2009, 5, 2716–2721 CrossRef CAS PubMed.
- K. Kvakova, M. Ondra, J. Schimer, M. Petrik, Z. Novy, H. Raabova, M. Hajduch and P. Cigler, Adv. Funct. Mater., 2022, 32, 1–14 CrossRef.
- G. Mattheolabakis, L. Milane, A. Singh and M. M. Amiji, J. Drug Targeting, 2015, 23, 605–618 CrossRef CAS PubMed.
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Adv. Drug Delivery Rev., 2016, 97, 204–236 CrossRef CAS PubMed.
- C. Chen, S. Zhao, A. Karnad and J. W. Freeman, J. Hematol. Oncol., 2018, 11, 1–23 CrossRef.
- M. Hassn Mesrati, S. E. Syafruddin, M. A. Mohtar and A. Syahir, Biomolecules, 2021, 11, 1850 CrossRef CAS PubMed.
- J. Kim, M. Moon, D. Kim, S. Heo and Y. Jeong, Polymers, 2018, 10, 1133 CrossRef PubMed.
- Y. Liang, Y. Wang, L. Wang, Z. Liang, D. Li, X. Xu, Y. Chen, X. Yang, H. Zhang and H. Niu, Bioact. Mater., 2021, 6, 433–446 CAS.
- M. C. de Paula, S. G. Carvalho, A. L. P. Silvestre, A. M. dos Santos, A. B. Meneguin and M. Chorilli, Carbohydr. Polym., 2023, 320, 121257 CrossRef CAS PubMed.
- T. H. Yun, G. Ahn, I. Choi, Y. Bae, K. Hwang, S. Kang and S. Choi, Polym. Adv. Technol., 2020, 31, 2990–2998 CrossRef CAS.
- H. H. Han, H. Kang, S.-J. Kim, R. Pal, A. T. N. Kumar, H. S. Choi and S. K. Hahn, RSC Adv., 2021, 11, 23073–23081 RSC.
- X. Cui, X. Deng, Z. Liang, J. Lu, L. Shao, X. Wang, F. Jia, Z. Pan, Q. Hu, X. Xiao, Y. Wu and W. Sheng, Biomater. Sci., 2021, 9, 3838–3850 RSC.
- X. Cui, Z. Liang, J. Lu, X. Wang, F. Jia, Q. Hu, X. Xiao, X. Deng, Y. Wu and W. Sheng, Nanoscale, 2021, 13, 13375–13389 RSC.
- M. G. Chernysheva, A. V. Sinolits, V. S. Votyakova, A. G. Popov and G. A. Badun, Mendeleev Commun., 2022, 32, 501–503 CrossRef CAS.
- M. d'Amora, A. Camisasca, A. Boarino, S. Arpicco and S. Giordani, Colloids Surf., B, 2020, 188, 110779 CrossRef.
- S. Arpicco, M. Bartkowski, A. Barge, D. Zonari, L. Serpe, P. Milla, F. Dosio, B. Stella and S. Giordani, Front. Chem., 2020, 8, 1–12 CrossRef PubMed.
- N. S. Xu, J. Chen and S. Z. Deng, Diamond Relat. Mater., 2002, 11, 249–256 CrossRef CAS.
- P. Aprà, L. Mino, A. Battiato, P. Olivero, S. Sturari, M. C. Valsania, V. Varzi and F. Picollo, Nanomaterials, 2021, 11, 2740 CrossRef PubMed.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, J. Am. Chem. Soc., 2006, 128, 11635–11642 CrossRef CAS.
- C. Surace, S. Arpicco, A. Dufaÿ-Wojcicki, V. Marsaud, C. Bouclier, D. Clay, L. Cattel, J.-M. Renoir and E. Fattal, Mol. Pharm., 2009, 6, 1062–1073 CrossRef CAS PubMed.
- C. Barzan, L. Mino, E. Morra, E. Groppo, M. Chiesa and G. Spoto, ChemCatChem, 2017, 9, 4324–4327 CrossRef CAS.
- L. Mino, Á. Morales-García, S. T. Bromley and F. Illas, Nanoscale, 2021, 13, 6577–6585 RSC.
- T. Petit and L. Puskar, Diamond Relat. Mater., 2018, 89, 52–66 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CrossRef CAS.
- L. Mino, C. Negri, R. Santalucia, G. Cerrato, G. Spoto and G. Martra, Molecules, 2020, 25, 4605 CrossRef CAS PubMed.
- R. Gilli, M. Kacuráková, M. Mathlouthi, L. Navarini and S. Paoletti, Carbohydr. Res., 1994, 263, 315–326 CrossRef CAS PubMed.
- K. Haxaire, Y. Maréchal, M. Milas and M. Rinaudo, Biopolymers, 2003, 72, 10–20 CrossRef CAS PubMed.
- A. M. Zaitsev, Optical Properties of Diamond, Springer, 2001 Search PubMed.
- B. R. Smith, D. Gruber and T. Plakhotnik, Diamond Relat. Mater., 2010, 19, 314–318 CrossRef CAS.
- A. Sigaeva, V. Merz, R. Sharmin, R. Schirhagl and A. Krueger, J. Mater. Chem. C, 2023, 11, 6642–6650 RSC.
- M. Ashrafizadeh, K. Luo, W. Zhang, A. Reza Aref and X. Zhang, Environ. Res., 2024, 240, 117443 CrossRef CAS PubMed.
- K. Bhise, N. S. Gavande and A. K. Iyer, Drug Discovery Today, 2023, 28, 103761 CrossRef CAS PubMed.
- P. Malik, R. Rani, R. Solanki, V. H. Patel and T. K. Mukherjee, Explor. Targeted Anti-Tumor Ther., 2023, 850–895 CAS.
- T. Bitter and H. M. Muir, Anal. Biochem., 1962, 4, 330–334 CrossRef CAS PubMed.
- M. E. Cano, D. Lesur, V. Bincoletto, E. Gazzano, B. Stella, C. Riganti, S. Arpicco and J. Kovensky, Carbohydr. Polym., 2020, 248, 116798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00932k |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |