
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the synthesis of LDH using the urea method: morphology and intercalated anion control†
Alessandro Di Michele a,
Elisa Boccalonb,
Ferdinando Costantinob,
Maria Bastianinic,
Riccardo Vivani*d and
Morena Nocchetti*d
a,
Elisa Boccalonb,
Ferdinando Costantinob,
Maria Bastianinic,
Riccardo Vivani*d and
Morena Nocchetti*d
aDepartment of Physics and Geology, University of Perugia, Via Pascoli, 006123 Perugia, Italy. E-mail: alessandro.dimichele@unipg.it
bDepartment of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di sotto, 8, 006123 Perugia, Italy. E-mail: elisa.boccalon@unipg.it; ferdinando.costantino@unipg.it
cProlabin & Tefarm S.r.l., Via dell'Acciaio 9, 06134 Perugia, Italy. E-mail: maria.bastianini@prolabintefarm.com
dDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 006123 Perugia, Italy. E-mail: riccardo.vivani@unipg.it; morena.nocchetti@unipg.it
First published on 3rd July 2024
Abstract
Layered double hydroxides (LDHs) are a class of layered solids applied in many application fields. The study of synthetic methods able to control the interlayer composition and morphology of LDH is an open issue. The urea method, which exploits the thermal decomposition of urea, is known for yielding highly crystalline LDH in the carbonate form. This form is highly stable and, to replace carbonate ions with more easily exchangeable anions, a second step is required. In this work, we modified the urea method to obtain MgAl and ZnAl LDH in the chloride or nitrate form through a one-step synthesis. The effects of the urea/(Al + M(II)) molar ratio (R), reaction time and metal salt concentrations were deeply investigated. We found that LDH in chloride and nitrate forms can be prepared from solutions of metal salts not exceeding 1 M by adjusting R and maintaining the reaction time at 48 hours. The morphology of these products was found to depend on the R value and on the metal salts used in the synthesis. A high R value and nitrate salts favoured the formation of sand-rose crystals, while chloride salts induced the formation of plate-like crystals. The crystal growth mechanism and the parameters influencing the morphology are discussed with reference to ZnAl LDH by monitoring the synthesis over time.
Introduction
Layered double hydroxides (LDHs) are a fascinating class of lamellar solids characterized by the general formula [M(II)1−xM(III)x(OH)2]x+[Ax/nn−·mH2O], where M(II) and M(III) are divalent and trivalent metal cations, respectively, and An− are anionic species.1–3 x can be defined as the M(III)/[M(II) + M(III)] molar ratio in the solid, and its value is generally in the 0.20–0.33 range. The LDH properties can be tuned by changing the nature of metal cations and of the intercalated anions, thus enabling their use in various application areas such as catalysis, biomaterials,1,4–6 energy storage and conversion,7 polymer composites,8 and environmental pollution control.9–11The design of materials for a specific application requires not only the preparation of compounds with an appropriate chemical composition, but also with suitable textural properties (i.e., surface area, micro- and/or meso-porosity), particle size, thickness and morphology, and selected intercalated anions.12 In this connection, the development of new synthetic methods to obtain LDH with suitable dimensions and morphology of crystallites, and a desired intercalated anion, represents an open challenge to increase the potentiality of these materials. The most common synthetic route used to prepare LDH is the so-called co-precipitation method. This procedure was first investigated by Miyata13 and consists of the simultaneous precipitation of the metal cations in the hydroxide form, following the slow addition of a basic solution, such as NaHCO3/Na2CO3 buffer (precipitation at constant pH value) or NaOH solution (precipitation at variable pH). Furthermore, the precipitation may be carried out at a low or high supersaturation degree according to the solution concentration and the rate of addition of the precipitating reagent. After complete precipitation, the solid is aged in the mother solution for a certain time, ranging from a few hours to several days. Poorly crystalline samples are generally obtained, unless a low degree of supersaturation and a relatively high temperature are maintained during the precipitation.1 Aggregates of microcrystals with a plate-like morphology are commonly formed. However, LDHs are obtained in the carbonate form, except when the synthesis is carried out under a nitrogen atmosphere. In this case, LDHs in the chloride or nitrate form can be prepared.
Syntheses that include separate nucleation (formation of seeds) and aging (crystal growth) steps (SNAS)14–16 or the precipitation in reverse micellae17–20 were proposed to obtain LDH nanocrystals having a dimension of 50–250 nm and a plate-like morphology. These methods allow the insertion of transition metals,21 such as Ir3+ and lanthanides22,23 (i.e., Eu3+, Yb3+, Tb3+ and Nd3+) in the brucite structure. The type of intercalated interlayer anion depends on the surfactant used to prepare the reverse micellae: when an anionic surfactant, like dodecylsulphate, is used, it normally remains intercalated in the precipitated LDH and it is very difficult to replace it, whereas, in the presence of a cationic surfactant, like cetyltrimethylammonium bromide, the LDHs are intercalated with bromide, an easily exchangeable anion.19
The sol–gel route allows the fabrication of LDH with the desired x value and different combinations of metal ions.24 Recently, a new sol–gel method was proposed in which the LDH gel precursor was calcined and the mixed metal oxides obtained were then rehydrated in water to reconstitute the LDH structure.25,26 This method produced nanoscale LDH with a high specific surface area, although the crystallinity remained poor and there was no control over the intercalated anions. The crystallinity can be improved by subjecting the sample to further treatments like microwave irradiation,27 sonication or hydrothermal treatments.
Other preparative routes, sometimes referred to as homogeneous precipitation, use hexamethylenetetramine (HMT),28,29 or urea30–32 that, upon hydrolysis, generates ammonia. The hydrolysis of HMT and urea at a high temperature (60–100 °C) increases the pH value, inducing the precipitation of a large amount of metal hydroxides. These methods provide well-defined hexagonal microcrystals with the required stoichiometry and generally lead to the formation of LDH in the carbonate form. Rosette-like LDH microcrystals in the carbonate form were obtained by a reduction of HMT (or urea) concentration,29 whereas the substitution of carbonate with chloride and perchlorate anions resulted in materials having suitable anion-exchange and water permeability characteristics.33 Flower-like MgAl–CO3 were obtained by a solvothermal method in a mixed solution of ethylene glycol and water using urea as a precipitating reagent.34 A systematic decreasing of the urea concentration in a solution of nitrate salts of zinc and aluminum was used to prepare LDH directly in the nitrate form.35
Table 1 shows the synthetic conditions used by several authors to prepare LDH via the urea method such as the urea/(Al + M(II)) molar ratio (R), reaction time, concentration of the metal salts and temperature. Noteworthy is the prevalent formation of LDH in carbonate form despite the different synthetic conditions used. In addition, from the literature data, the comprehension of the parameters driving the synthesis towards the formation of LDH with anions other than carbonate is not straightforward.
| Salt | R | M(II) | Reaction time (h) | [M(II) + M(III)] | Phase | T (°C) | Ref. |
|---|---|---|---|---|---|---|---|
a The synthesis was carried out in the presence of 1 M NH4NO3.b Solvent: ethylene glycol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 9:1.c In a closed vessel. :H2O = 9:1.c In a closed vessel. |
|||||||
| NO3 | 0.58–4 | Mg | 24 | 0.15 | CO3 | 140c | 29 |
| NO3 | 3–10 | Mg | 18–48 | 1 | CO3 | 90–100 | 31 |
| NO3 | 2.5 | Mg | 20 | 0.5 | CO3/NO3 | 90 | 36 |
| NO3 | 7 | Zn | 24 | 0.15 | CO3 | 100 | 37 |
| NO3 | 4 | Mg | 96 | 0.01–0.5 | CO3 | 90–120c | 38 |
| NO3 | 3.3–3.5a | Zn | 10–124 | 0.5 | NO3 | 90 | 35 |
| NO3 | 5b | Mg | 6 | — | CO3 | 160c | 34 |
| NO3 | 2.3–9.2 | Mg, Zn, Ni | 30 | 0.9 | CO3 | 95 | 39 |
| Cl | 2–3.5 | Mg | 24 | 0.5 | CO3 | 120c | 40 |
| Cl | 4 | Zn | 24 | 0.6 | Cl | 100 | 41 |
The direct synthesis of LDH with easily exchangeable anions is very advantageous for applications that imply ion-exchange reactions.
Therefore, in this work, the effect of various synthetic parameters was investigated with the aim of obtaining LDH in the nitrate or chloride form directly in a one pot synthesis. The evolution of the samples over time was investigated to determine the crystalline phases, the composition and the morphology of the solids. Interestingly, different morphologies were obtained according to the salt type, chloride or nitrate, used.
Experimental
Materials
Urea (percent purity, 98+%), Zn(NO3)2·6H2O (percent purity, 98%), ZnO (percent purity 99.5+%), Mg(NO3)2·6H2O (percent purity, 98%), MgCl2·6H2O (percent purity, 98+%), Al(NO3)3·9H2O (percent purity, 98+%) and AlCl3·6H2O (percent purity, 98%) were purchased from Thermo Fisher Scientific and were used without further purification. All the other reagents were supplied by Sigma-Aldrich.Synthesis of ZnAl and MgAl LDHs with different urea/(Al + M(II)) molar ratios
ZnAl LDHs were prepared starting either from a solution of nitrate or chloride metal salts. For the preparation of zinc chloride solution, ZnO was used. Specifically, to prepare a solution of 0.5 M of ZnCl2, 40.7 g of ZnO were suspended in 500 mL of deionized water and concentrated hydrochloric acid was added under stirring to dissolve almost all the solid (80 mL). The complete dissolution of ZnO was performed by adding, under stirring, 1 M HCl and monitoring the pH of the solution that has to be in the range of 4.2 to 4.3. Finally, the solution was brought to 1 L with deionized water adjusting the pH to the value reported above.Volumes of 0.5 M aqueous solutions of Zn(NO3)2·6H2O and Al(NO3)3·9H2O solutions were mixed in order to obtain an Al/(Al + Zn) molar ratio in solution (y in the following) equal to 0.30. Finally, solid urea was added to the previous solution in order to reach a urea/(Al + Zn) molar ratio (R in the following) equal to 3, 1.8, and 1.2 (the samples were labelled as: ZAN/3, ZAN/2, and ZAN/1). The synthesis was carried out under magnetic stirring at reflux temperature in an open flask for 24 h. Additional samples with R = 1.8 and 1.2 were precipitated from a solution containing 0.5 M KNO3; these samples were named ZAN/2 s and ZAN/1 s.
The previous procedure was repeated by using 0.5 M aqueous solutions of ZnCl2 and AlCl3·6H2O and R = 1.2 (ZACl/1).
MgAl LDH were prepared starting from 0.5 M aqueous solutions of Mg(NO3)2·6H2O and Al(NO3)3·9H2O (or MgCl2·6H2O and AlCl3·6H2O) and R = 1.8, samples named MAN/2 and MACl/2 were obtained. The syntheses were carried out under magnetic stirring, at reflux temperature and in an open flask for 48 h.
The solids were separated by centrifugation (5000 rpm for 10 min), washed with water until the neutrality of the water (at least 3 × 100 mL) and dried under vacuum at 75% of relative humidity (RH).
The reaction time and the amount of salts and urea used to prepare about 5 g of the LDH described in this paragraph are reported in Table S1.†
Synthesis of ZnAl and MgAl LDHs with different concentrations of metal salts
The synthesis of ZAN/1 and MAN/2 were carried out by using concentrations of metal salts equal to 1, 2, and 2.5 M and refluxing the solution in an open vessel for 48 h. The recovered samples were named ZAN/1-M and MAN/2-M (with M = 1, 2, and 2.5). The solids were separated by centrifugation (5000 rpm for 10 min), washed with water until the neutrality of the water (at least 3 × 100 mL) and dried under vacuum at 75% RH.The volumes of metal solutions used to prepare about 5 g of the LDH described in this paragraph are reported in Table S2.†
Synthesis of ZAN/1, ZACl/1 and ZnAlCO3 at different reaction times
ZnAlCO3 was prepared as previously reported.30 Briefly, volumes of 0.5 M Zn(NO3)2·6H2O and Al(NO3)3·9H2O aqueous solutions were mixed in order to obtain an y value equal to 0.30 and solid urea was added in order to obtain R = 3. The volumes of metal solutions used to prepare about 5 g of ZnAlCO3 are equal to those used for ZAN/3, Table S1.† The final solution was kept under magnetic stirring at reflux temperature an open flask for 72 h.The syntheses of ZAN/1, ZACl/1 and ZnAlCO3 were repeated and the precipitating solutions were maintained at reflux temperature in an open flask for 5 days. Small samples were taken at set times of 0, 2, 4, 6, 20, 24, 28, 48, 120 hours after the beginning of the precipitation process, which was assumed to start when the mixed solution became cloudy (t = 0). The mother liquors were filtered, and the pH was monitored by a pH-meter provided with a glass electrode.
Each sample was separated by centrifugation (5000 rpm for 10 min), washed with water until the neutrality of the water (at least 3 × 30 mL) and dried under vacuum at 75% RH.
Analytical procedures
PXRD patterns were recorded using a Bruker D8 Advance diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) in Bragg–Brentano geometry, equipped with a Lynxeye XE-T position sensitive detector, Cu Kα radiation operating at 40 kV and 40 mA, with a step size of 0.0170° 2θ, and a step scan of 20 s. The phase identification was performed by using the Bruker DIFFRAC.EVA V5 software equipped with COD database. Quantitative phase analyses were performed with the Rietveld method with the help of the GSAS-II42 and Bruker-AXS TOPAS V6.0 software packages.Metal analyses were performed by ICP with a Varian Liberty Series spectrometer. 20 mg of ZnAl LDH, or MgAl LDH, was first dissolved in concentrated HNO3 and de-ionized water until a final volume of 100 mL. The solution, properly diluted, was analyzed by ICP.
The morphology of the samples was investigated with a transmission electron microscope (TEM, Philips 208) and with a scanning electron microscope (SEM, FEG LEO 1525). For the TEM images, a small drop of the aqueous dispersion was deposited on a copper grid precoated with a Formvar film and then evaporated in air at room temperature. SEM micrographs were collected after depositing the dry samples on a stub and sputter coating them with chromium (8 nm of thickness). Measurements were performed with an acceleration voltage of 15 kV and a working distance from 3 to 1.5 mm using an in-lens detector. The images were collected at different magnifications, from ×10 K to ×100 K. The elemental mapping of the samples was carried out by using energy-dispersive X-ray spectroscopy (EDX) supported with a field emission scanning electron microscope. Measurements were performed with an acceleration voltage of 15 kV and a working distance of 8 mm using a secondary electron detector. The images were collected at different magnifications, from ×10 K to ×80 K.
Nitrogen adsorption and desorption isotherms were performed using a Micromeritics ASAP 2010 analyser. Prior to the analysis, the samples were degassed overnight under vacuum at 80 °C. Brunauer–Emmett–Teller (BET) analysis of the data was used to calculate the specific surface area.
FT-IR spectra of the different samples, dispersed in KBr pellets, were recorded at room temperature using a Bruker IFS113V spectrometer. Typically, each spectrum was obtained at a resolution of 1 cm−1 in the 370–5000 cm−1 spectral region.
Results and discussion
The urea method was properly modified to prepare ZnAl and MgAl LDH directly in nitrate or chloride form. With this aim, some synthetic parameters, i.e. the urea/(Al + M(II)) molar ratio, the reaction time and the concentration of the metal salts, were finely tuned. In the following, the effect of each parameter on the composition and morphology of ZnAl LDH will be mainly discussed. Some preliminary results of MgAl LDH systems will be also outlined.Effect of the urea/(Al + Zn) molar ratio
Syntheses of ZnAl LDH were carried out by using the nitrate salts of the metals and varying R. The total concentration of metal ions was set at 0.5 M, the y value was kept at 0.30 and the reflux time was 24 h. Table 2 shows the R value, the presence of added KNO3, the Al/(Al + Zn) molar ratio in the solid (F), and the phases formed as detected by PXRD. From these data it is clear that the R value affects the anionic form of the LDH obtained: high R values favour the formation of the carbonate form, while low R values tend to produce the nitrate or chloride form, depending on the metal salt used for the synthesis. Furthermore, the presence of added KNO3 does not seem to affect this trend.The compounds consisting of a single phase at PXRD analysis were characterized for their Al content; the found F values fell in the 0.2–0.33 range, which is typical of a pure LDH phase. Rietveld refinements of these phases (entries 1, 5, and 6 in Table 2) based on the literature structural model43 confirmed our attribution. Furthermore, additional quantitative analyses based on the Rietveld method, devoted to the determination of the amount of the amorphous fraction in the samples, were performed. For this aim, weighed amounts of crystalline corundum, as the internal standard, were added to the above samples and the fraction of the amorphous phase was determined as reported in ref. 44. The results showed that the three samples contained a negligible amount, less than 3% w/w, of the amorphous phase, and therefore they can be considered as pure crystalline phases (see the ESI† for details of the Rietveld refinement results). For these samples it is clear that the F values coincide with x in their formula.
These experiments show that when R = 1.2, ZnAlNO3 is obtained as a pure crystalline phase after 24 h of reaction (sample ZAN/1, entry 5 in Table 2). Moreover, it was found that these synthetic conditions produce pure ZnAlCl starting from the chloride salts of the metals (sample ZACl/1, entry 6 in Table 2).
Effect of the concentration of metal salts
It was also interesting to study the effect of the concentration of metal salts on the formation of LDH containing anions other than carbonate. Increasing the metal concentration opens up the possibility to prepare large quantities of LDH in small volumes of solution. In this connection, the phases formed in the synthesis of ZnAlNO3 were investigated at increasing concentrations of metal nitrate salts. Fig. 1 shows the PXRD patterns of the solids obtained by using Zn(II) and Al(III) solutions at different concentrations and keeping fixed the R value (as reported for ZAN/1) and the reaction time at 48 h.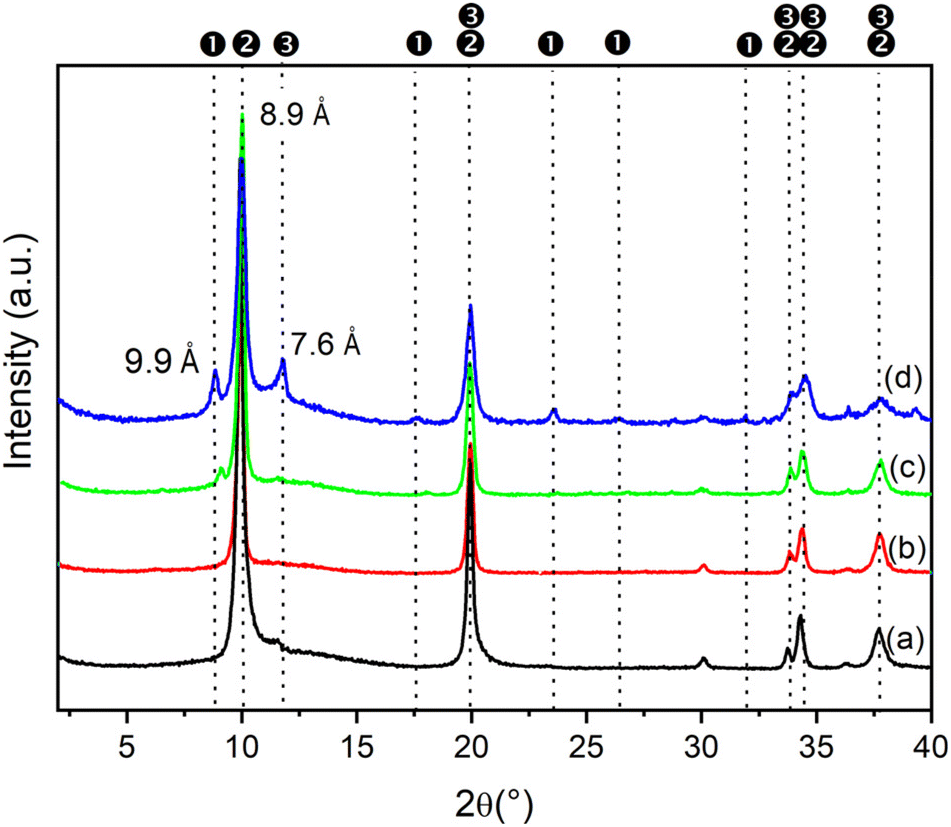 | ||
| Fig. 1 PXRD patterns of ZnAl LDH synthesized by using Zn(NO3)2·6H2O and Al(NO3)3·9H2O salts with concentrations: 0.5 M (a); 1 M (b); 2 M and (c); 2.5 M (d). R = 1.2, reaction time 48 h at a reflux temperature. ❶ Zn5(OH)8(NO3)2·2H2O, ❷ ZnAlNO3, and ❸ ZnAlCO3. | ||
ZnAl compounds precipitated from solutions with the concentration of metals ranging from 0.5 to 1 M consisted of a single phase with an interlayer distance of 8.9 Å that can be attributed to the nitrate phase. In contrast, when the metal concentration exceeded 2 M, reflections of a new phase (the first reflection at 9.9 Å) appeared next to the nitrate phase at PXRD analysis. By comparison with the literature data, this phase can be attributed to Zn5(OH)8(NO3)2·2H2O (COD ID: 2106442) and could not be converted back into the LDH phase even when the reaction time was increased (data not shown).
The composition of the samples consisting of a pure phase was determined by ICP and is shown in Table 3. For metal salt concentrations equal to 0.5 M and 1 M (samples ZAN/1 and ZAN/1-1), the F values, 0.29 and 0.33, respectively, are typical of an LDH phase, and are close to the aluminum molar fraction in solution (y). Summarizing the results of these experiments, the best conditions to obtain a pure crystalline ZnAlNO3 phase with a high yield were: ion concentration 1 M, R = 1.2, time 48 h under reflux.
The effect of the concentration of metals was also evaluated in terms of the surface area of the pure LDH samples. Table 3 shows the surface areas of the solids degassed at 80 °C overnight. Changes in the synthetic parameters did not lead to major modifications in the measured surface areas.
Mechanism of ZnAl LDH formation: effect of the reaction time on the sample composition and morphology
Fig. 2 shows the evolution over the time of the PXRD patterns of the solids obtained during the synthesis of ZnAlCO3, ZAN/1 and ZACl/1. The moment the solution turned into a white colloidal dispersion was set as 0 h. The three series show very different behaviors.
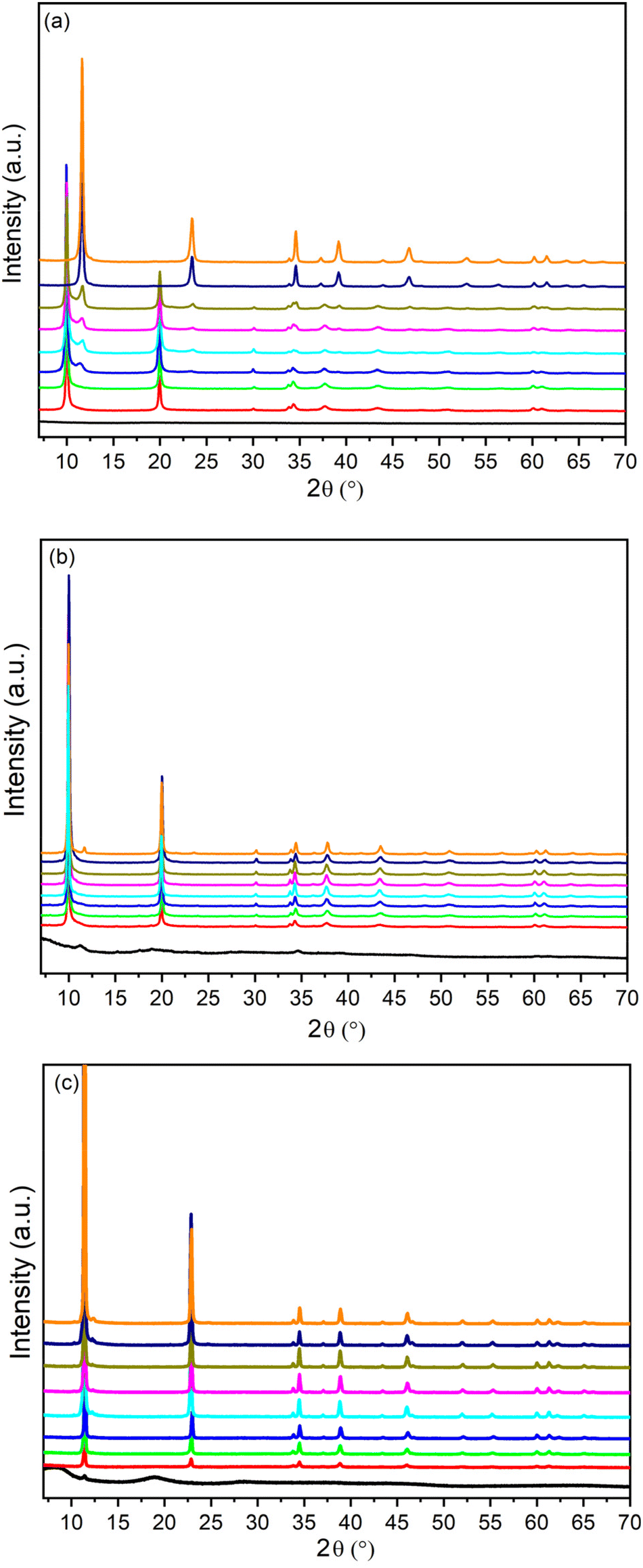 | ||
| Fig. 2 PXRD of ZnAlCO3 (R = 3) (a); ZAN/1 (b) and ZACl/1 (c) samples recovered at the increasing times (0 h, black; 2 h, red line; 4 h, green line; 6 h, blue line; 20 h, cyan line; 24 h, pink line; 28 h, olive line; 48 h, navy line; 120 h, orange line). Reaction conditions: concentration of metal ions of 0.5 M, y = 0.30, and at a reflux temperature. | ||
For as concerns the synthesis of ZnAlCO3 (starting from nitrate salts, and R = 3, Fig. 2a), the first precipitate (time 0) consisted of an amorphous material (black line in Fig. 2a), while the diffraction peaks characteristic of LDH in the nitrate form (d = 8.9 Å) appeared after 2 hours and became sharper over time, indicating an increase in crystallinity. Starting from about 6 h of reaction, a second basal peak at d = 7.6 Å, that was attributed to the LDH in the carbonate form, appeared. The intensity of this peak increased, while that relating to the nitrate form decreased over time, indicating that first, LDH precipitated by incorporating the nitrate anions of the starting metal salts, then the pristine nitrate phase gradually converted into the carbonate one as the carbonate concentration in solution increased due to the degradation of urea. The a and b lattice parameters of nitrate and carbonate phases were found in good agreement with the literature data43 over time, indicating that very likely, the molar fraction of the metals remained unchanged, as also confirmed by ICP analysis (Table 4).
| Reaction time (h) | F (±0.01) | ||
|---|---|---|---|
| ZnAlCO3 | ZAN/1 | ZACl/1 | |
| 0 | 0.84 | 0.82 | 0.91 |
| 2 | 0.35 | 0.58 | 0.70 |
| 4 | 0.33 | 0.45 | 0.42 |
| 6 | 0.33 | 0.41 | 0.33 |
| 20 | 0.33 | 0.35 | 0.32 |
| 24 | 0.33 | 0.32 | 0.32 |
| 28 | 0.33 | 0.31 | 0.32 |
| 48 | 0.33 | 0.33 | 0.32 |
| 120 | 0.33 | 0.33 | 0.33 |
When the synthesis conditions of ZAN/1 were used, a small peak ascribed to the carbonate phase appeared only after 120 h of reaction (Fig. 2b). However, in the first 6 h, the F value, obtained by ICP, was found to be much greater than 0.33, the characteristic x value of an LDH phase, suggesting the presence of aluminum-rich amorphous phases. The pure nitrate phase was observed between 24 and 48 h (Table 4).
Fig. 2(c) shows the time evolution of the PXRD patterns for the ZACl/1 system. In this case, the pure chloride phase (d = 7.76 Å) was obtained and maintained throughout the duration of the synthesis. As with ZAN/1, after 120 hours of reaction, a very low reflection appears, due to the carbonate phase. The broadening of diffraction peaks was found to reduce with time, suggesting a progressive increase in crystal size and/or a reduction in lattice defects in the sample.
At time 0, the precipitate was completely amorphous and the high F value suggests the presence of aluminum rich phases, that were present up to 4 h of reaction. After 6 h, F reached the value of 0.33 and the unit cell parameters were consistent with literature data,43 indicating the formation of a pure LDH phase. By prolonging the contact of the precipitate with the mother liquor, both unit cell parameters and F remained unchanged and only an improvement of the crystallinity of the sample occurred. These results confirm that the urea method could be used to obtain LDH with good crystallinity and containing anions other than carbonate in the interlayer region directly from the synthesis.
In order to get more information on the evolution of the samples during the synthesis, FT-IR spectra were collected (Fig. 3). In the first precipitate (0 h) of all the samples, three broad bands centred at 966, 740 and 584 cm−1 were detected and can be attributed to AlOH deformations and AlOH translations, attesting to the presence of amorphous oxohydroxides of alumina.45 Moreover, multiple bands are observed in the 1300–1500 cm−1 region that can be assigned to both nitrate (at around 1390 cm−1) and carbonate anions (between 1450 and 1300 cm−1). The position of the carbonate bands depends on the environment surrounding the anions.46 The bending mode of hydrogen bonded water (ν2) with carbonate and nitrate is displayed at 1650 cm−1 in Fig. 3(a) and (b); this mode is shifted towards lower wavenumbers, about 1635 cm−1, in the sample precipitated from the chloride salts (Fig. 3(c)) given the contribution of weak hydrogen bonds between the water molecules and the chloride anions.1
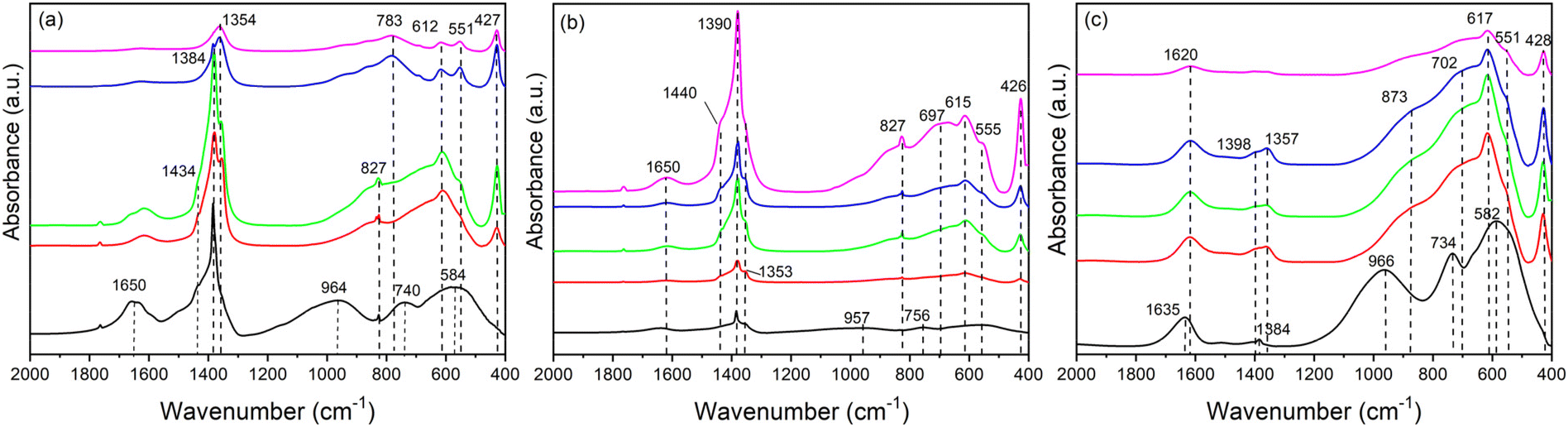 | ||
| Fig. 3 FT-IR spectra of ZnAlCO3 (R = 3) (a); ZAN/1 (b) and ZACl/1 (c) samples recovered at increasing times (0 h, black line; 6 h, red line; 24 h, green line; 48 h, blue line; 120 h, magenta line). Reaction conditions: concentration of metal ions 0.5 M, y = 0.30, and at a reflux temperature. | ||
Over time, in the 800–400 cm−1 region, the bands of amorphous aluminum compounds are replaced by several Al–OH and Zn–OH translation modes,47 positioned at about 427, 551, 612 and 697 cm−1, that suggest the formation of LDH as observed by PXRD.
The region 1300 to 1500 cm−1 of FT-IR spectra provides information on the composition of the interlayer region during the synthesis. Regarding the ZnAlCO3 samples recovered after 6–24 h of reaction, this region (Fig. 3(a)) reveals the presence of three superimposed bands, one sharp and two broad. The first is ascribable to the ν3 mode of nitrate (1384 cm−1) and the others to the ν3 mode of carbonate that is split in two bands at 1354 and 1434 cm−1. This splitting is due to a decrease in the symmetry of the carbonate ion upon the interactions with water molecules.48,49 Moreover, the samples also show the ν2 mode of nitrate50 at 827 cm−1. From the initial stage (time 0), the intense adsorption band of nitrate (1384 cm−1) is progressively replaced by the absorption band of carbonate ions (1354 cm−1), as proved by PXRD.28,47 When the reaction time exceeds 48 h, the ν2 and ν3 modes of nitrate disappear and the ν3 mode of carbonate is centred at 1354 cm−1.51
In ZAN/1 (Fig. 3(b)) the nitrate bands are always more intense (the ν3 mode at 1390 cm−1 and the ν2 mode at 827 cm−1). These signals may be due to either LDH in nitrate form or zinc hydroxide nitrate (Zn5(OH)8(NO3)2·2H2O); the latter can precipitate in the first stage of the synthesis. The bands at 1353 cm−1 and 1440 cm−1 are related to the traces of carbonate most likely adsorbed on the surface up to 48 h; after that, as observed by PXRD, the incorporation of carbonates also involves the interlayer region.
As regards the ZACl/1 sample, Fig. 3(c) shows that the solids contains mainly chloride anions with a very low amount of carbonate anions (bands at 1384, 1357 and 1398 cm−1). The bending mode of water (ν2) at 1620 cm−1 attests to the presence of weakly bonded water due to the formation of less hydrogen bonds.
In the 4000–1500 cm−1 spectral region of the ZnAlCO3 and ZAN/1 samples, reported in Fig. S4,† some bands with very low intensity are detected; these signals are ascribable to the intermediate products formed during the urea hydrolysis like cyanate, isocyanate, biuret and carbamate.52,53 In particular the band at 2212 cm−1 in the ZnAlCO3 until 24 h can be assigned to isocyanate N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO,54 which decomposes to carbonate after 48 h. Bands in the region 2200–2500 cm−1 can be assigned to physisorbed CO2.55
CO,54 which decomposes to carbonate after 48 h. Bands in the region 2200–2500 cm−1 can be assigned to physisorbed CO2.55
From FT-IR it can be inferred that after 48 h of reaction all the samples are constituted by pure LDH in carbonate, nitrate or chloride form depending on the synthetic parameters selected. An in-depth analysis on the composition of the samples coupled with the PXRD and FT-IR data allowed further insights into the mechanism of LDH formation.
Fig. 4 shows the trend of the pH of the mother liquor and of Al molar fraction in the solid during the different syntheses. In each case, the sample at 0 h contained mainly Al, and by comparing this result with the PXRD and IR spectra, it can be deduced that the solids were mainly constituted by amorphous aluminum hydroxide,29,36 the percentage of which increased as follows: ZAN/1 < ZnAlCO3 < ZACl/1. The higher is the amount of aluminum hydroxide precipitated at 0 h, the lower is the pH of the solution due to the higher consumption of OH− groups. However, while the pH of ZnAlCO3 and ZACl/1 increased very quickly with a change of 1.9 and 2.4 pH units in 48 h, respectively, ZAN/1 showed a slow pH increase with a change of only 0.9 units.
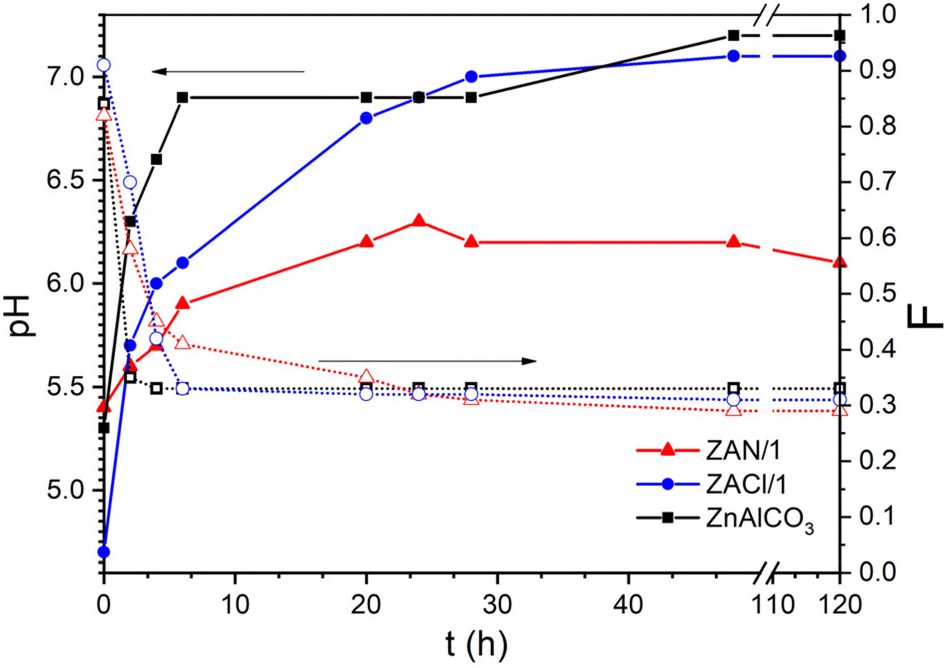 | ||
| Fig. 4 pH of the reaction solution (solid lines) and F (Al/(Zn + Al) molar fraction in the solid, dashed lines) collected during the synthesis for the indicated samples. | ||
Concerning the composition, after a few hours of reaction, the solids were clearly enriched in Zn ions, achieving the molar fraction typical of LDH. Note that an Al molar fraction, F, compatible with the existence of pure LDH (0.2 ≤ x ≤ 0.33), for all the samples, was obtained when the pH of the mother liquor was greater than 6.1. This value, in the case of ZnAlCO3 and ZACl/1, was reached in 4 and 6 h, respectively, from the beginning of the reaction; whereas ZAN/1 reached pH 6.3 in a longer time (20 h) and maintained this value for the whole duration of the synthesis. This value is one unit lower than the pH recorded in the ZACl/1 and ZnAlCO3 preparations. These findings suggest that the formation of LDH requires a pH higher than 6 and that the rate at which the system reaches this value depends on the concentration of urea and on the metal salt used: chloride or nitrate. In ZnAlCO3, the high urea concentration caused an increase in the urea hydrolysis rate with a consequent increase in pH. The ZAN/1 and ZACl/1 samples were prepared with identical urea concentration but different salts nitrate and chloride, respectively. The different trend of the pH value during the synthesis should be sought in the nature of the anion. Another important parameter to consider is the amount of precipitated metals. The content of Zn and Al was also determined in the mother liquor; these analyses confirmed the data shown in Fig. 4. Al3+ precipitated very quickly and, at time 0, was already absent in the solution. As a consequence, only the percentage of Zn in the mother liquor at different times was determined. These data indicate that after 6 h the Zn2+ precipitated in ZAN/1 is higher than that precipitated in ZACl/1: very likely, the formation of amorphous zinc hydroxide nitrate occurs and maintains the pH around 6. It was reported that freshly prepared Zn5(OH)8(NO3)2·2H2O has buffering properties, maintaining the pH of the solution at 6.1, in agreement with the pH value found during the synthesis of ZAN/1.56 During the time, both aluminum hydroxide and zinc hydroxide nitrate were converted into LDH. After 28 hours, the amount of Zn2+ in solution was very low and maintained a constant value. At this time, LDH had a stable composition, while the crystallinity, in terms of the size and order of crystals, was increased by prolonging the contact time with the mother liquors.
Fig. 5 displays the evolution of ZACl/1 crystals which resemble those found by Okamoto et al.29 Flat semi-circular crystals, covered by some smaller crystals, appeared after 6 h of reaction. Punctual EDX elemental analysis (shown in Fig. S6(a and b)†) showed that F of the larger crystals was about 0.43–0.45, which is a higher value than that of an ideal LDH indicating an excess of Al3+ in the structure. Conversely, in smaller crystals the F ranged from 0.25 to 0.33, suggesting, in some crystals, the presence of an excess of Zn2+. With the time, some semi-circular crystals grew to give single hexagonal platelets with 5–7 μm size (Fig. 5e′), while others fused together to form aggregates of hexagonal platelets (Fig. 5c′, d′ and e). According to Okamoto, the growth starts from seeds of aluminum hydroxides that act as LDH crystallization sites. The Zn2+ ions coming from the solution are adsorbed on aluminum hydroxides58 and deplete the seeds with the formation of the LDH structure having chloride anions in the interlayer region and leaving a hole at the site of the seeds. Very likely, when the crystallization process starts from the same seed, the development of geminated flat crystals occurs (Fig. 5b′–d′). The formation of geminated crystals is expected to be favoured by a high ratio between zinc ions and aluminum hydroxide seeds. This situation is verified after 20 h, since the amount of precipitated zinc is 90% of the initial value, the pH of the solution is close to the maximum value reached by this system and supplies enough OH− ions. By prolonging the reaction time, no more zinc ions precipitate and the growth of the crystals can be justified by a dissolution–reprecipitation mechanism. Indeed, F, determined by EDX (Fig. S6(c–e)†) at different points of the crystals collected after 48 and 120 h, is constant and ranges between 0.30 and 0.32.
 | ||
| Fig. 5 SEM images at different magnifications of ZACl/1 recovered after 6 (a and a′), 20 (b and b′), 28 (c and c′), 48 (d and d′), and 120 (e and e′) h of reaction. | ||
When the synthetic conditions of ZnAlCO3 are applied, sand rose aggregates of the hexagonal platelets are observed, and this morphology is retained throughout the synthesis, also regarding the size of the aggregates (Fig. 6). In this case, due to the high urea concentration, the pH of the solution increases to 6.9 in 6 h, leading to a massive precipitation of zinc ions that are adsorbed on the aluminum hydroxide and crystallize as LDH. The ratio between zinc ions and the aluminum hydroxide seeds is very high and several crystals can be formed from a single seed, yielding multiple geminated crystals. The F values determined in different zones of the crystals collected after 6, 48 and 120 h are always low and range from 0.26 to 0.29 (Fig. S7†). These crystals consist in small flat hexagonal specimens of 500 nm, arranged in aggregates of 2–3 μm. Over time (after 28 h), the small crystals that form the sand rose aggregates take on a more definite hexagonal shape due to a crystallization process.
 | ||
| Fig. 6 SEM images at different magnifications of ZnAlCO3 recovered after 6 (a and a′), 20 (b and b′), 28 (c and c′), 48 (d and d′), and 120 (e and e′) h of reaction. | ||
ZAN/1 shows a different behaviour: after 6 h, the formation of zinc hydroxide nitrate is assumed, besides the aluminum hydroxide. Fig. 7a and a′ show the presence of flower-like particles of about 5 μm of diameter covered by small particles. Punctual EDX analysis (see Fig. S8(a and b)†) performed on the small particles show that they are richer in zinc (F = 0.17–0.22) while in the flower-like particles F ranges between 0.3 and 0.33, in agreement with the ICP data. The corresponding TEM image (Fig. 7a′′) shows uniform particles. After 20 h the small particles are partially dissolved assuming a “C” shape as if they were consumed from the center (Fig. 7b and b′). The different texture of the small particles is highlighted by TEM in Fig. 7b′′ and reflects the different chemical composition. The “C” shaped particles appear fibrous and some voids are present near them. After 28 h the morphology is similar to the previous one, the number of voids increases and the “C” shaped particles are decrease (Fig. 7c, c′ and c′′). Over 48 h, flower-like particles with lacy edges and holes on the surface are obtained (Fig. 7d, d′ and d′′), and F is 0.29 (Fig. S8†). Finally, after 120 h, combined flat crystals are formed, but smaller (2.5 μm) and more irregular than those in the ZACl/1 case.
 | ||
| Fig. 7 SEM and TEM images at different magnifications of ZAN/1 recovered after 6 (a, a′ and a′′), 20 (b, b′ and b′′), 28 (c, c′ and c′′), 48 (d, d′ and d′′), and 120 (e, e′ and e′′) h of reaction. | ||
These findings suggest that both the aluminum hydroxide and zinc hydroxide nitrate phase are converted into LDH. The first acts as a crystallization seed, adsorbing the zinc ion derived from the solution and the zinc hydroxide nitrate that dissolves during the synthesis. The ability of zinc hydroxide nitrate to release Zn2+ or [Zn(OH)3]− under weakly acidic or basic conditions, respectively, was reported in a recent work.56 Upon the dissolution of the zinc hydroxide nitrate, particle holes are formed on the surface of LDH.
MgAl system
As far as MgAl LDH is concerned, the synthetic conditions used for ZnAl LDHs were not suitable to obtain LDHs in nitrate or chloride form; indeed, the low amount of urea used yielded quite amorphous materials after 24 h of reaction and the samples required longer reaction times to be converted into crystalline materials in carbonate form. Therefore, to prepare MgAlNO3 and MgAlCl from nitrate and chloride salt solutions, respectively, it was necessary to increase the R value up to 1.8, and the reaction time up to 48 h (sample MAN/2 and MACl/2 of Table S7†). This was probably due to the different solubility of zinc and magnesium hydroxides. Compared to the coprecipitation method, wherein NaOH is the alkaline agent and the pH of the solution is immediately very high (around 10), in homogeneous syntheses, the hydrolysis of urea produces a slow OH− release. Therefore, the precipitation of M(II) and Al(III) hydroxides is not simultaneous and the formation of the LDH structure occurs through an intermediate step that involves the precipitation of aluminum hydroxide species.59,60 Then, as the pH rises, divalent cations diffuse onto the aluminum hydroxide agglomerates and are adsorbed and incorporated on the surface, leading to the final LDH, either through surface growth or dissolution–reprecipitation mechanisms.31 In this second phase, the solubility of the M(II) hydroxide species is crucial and, since the suitable pH for the precipitation of Mg(OH)2 is higher than that of Zn(OH)2, more urea is required to assist the synthesis.61Preliminary experiments (see the ESI†) to elucidate the dependence of reaction time and salt concentration on this system have been carried out. From them, the best experimental conditions to obtain a crystalline MgAlNO3 phase were the following: ion concentration 1 M, R = 1.8, time 48 h, and under reflux, although the experimental analysis on the obtained solids showed the concomitant formation of a certain amount of the amorphous phase (see the ESI† for further details). A deeper investigation will be required on the MgAl system.
Conclusions
In this work, the reaction time, urea/(Al + Zn) molar ratio, and metal concentration were finely adjusted to obtain ZnAl LDH in a single step with the required anions in the interlayer region. In addition, the parameters affecting the morphology of the ZnAl in carbonate, nitrate and chloride forms have been studied. The micrographs obtained showed two different morphologies: sand rose aggregates, that are favoured by a high R value, and the presence of nitrate anions; hexagonal platelets that were obtained with low R values and the presence of chloride anions. In the future, similar studies will be applied to deeply investigate the MgAl LDH system.Author contributions
Conceptualization: M. N. and R. V.; data curation and methodology: M. N., R. V., A. D. M. and F. C; validation: M. N., R. V.; A. D. M. and F. C.; investigation: M. N., M. B., E. B. and A. D. M.; writing—original draft: M. N.; writing—review: M. N., R. V., A. D. M., E. B. and F. C.; editing: M. N. and R. V.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality. The authors also acknowledge MUR within the AMIS and DELPHI projects through the program “Dipartimenti di Eccellenza 2018-2022”.References
- Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova Science, New York, 2001 Search PubMed.
- U. Costantino, F. Leroux, M. Nocchetti and C. Mousty, LDH in physical, chemical, bio-chemical and life science, in Handbook of Clay Science, ed. G. L. E. Faiza Bergaya, Elsevier, Amsterdam, The Netherlands, 2013 Search PubMed.
- M. V. Bukhtiyarova, J. Solid State Chem., 2019, 269, 494–506 CAS.
- H. Du, D. Zhang, F. Peng, K. W. K. Yeung and X. Liu, Prog. Mater. Sci., 2024, 142, 101220 CAS.
- M. Laipan, J. Yu, R. Zhu, J. Zhu, A. T. Smith, H. He, D. O'Hare and L. Sun, Mater. Horiz., 2020, 7, 715–745 CAS.
- A. Donnadio, M. Bini, C. Centracchio, M. Mattarelli, S. Caponi, V. Ambrogi, D. Pietrella, A. Di Michele, R. Vivani and M. Nocchetti, ACS Biomater. Sci. Eng., 2021, 7, 1361–1373 CAS.
- P. R. Chowdhury, H. Medhi, K. G. Bhattacharyya and C. M. Hussain, Coord. Chem. Rev., 2024, 501, 215547 Search PubMed.
- V. Prevot and E. Bourgeat-Lami, Recent advances in layered double hydroxide/polymer latex nanocomposites: from assembly to in situ formation, in Woodhead Publishing Series in Composites Science and Engineering, Layered Double Hydroxide Polymer Nanocomposites, ed. S. Thomas and S. Daniel, Woodhead Publishing, 2020, pp. 461–495 Search PubMed.
- L. Ma, Q. Wang, S. M. Islam, Y. Liu, S. Ma and M. G. Kanatzidis, Am. Chem. Soc., 2016, 138, 2858–2866 CAS.
- Y.-D. Dong, Y. Shi, Y.-L. He, S.-R. Yang, S.-Y. Yu, Z. Xiong, H. Zhang, G. Yao, C.-S. He and B. Lai, Ind. Eng. Chem. Res., 2023, 62, 10828–10848 CAS.
- Progress in Layered Double Hydroxides From Synthesis to New Applications, in Series on Chemistry, Energy and the Environment, ed. M. Nocchetti, U. Costantino, K. M. Kadish and R. Guilard, World Scientific Publishing, Singapore, 2022, vol. 8 Search PubMed.
- E. Boccalon, G. Gorrasi and M. Nocchetti, Adv. Colloid Interface Sci., 2020, 285, 102284 CAS.
- S. Miyata and T. Kumura, Chem. Lett., 1973, 2, 843–848 Search PubMed.
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286–4291 CAS.
- Z. Gu, X. Xiang, G. Fan and F. Li, J. Phys. Chem. C, 2008, 112, 18459–18466 CAS.
- S. L. Xu, Z. Chen, B. Zhang, J. Yu, F. Z. Zhang and D. G. Evans, Chem. Eng. J., 2009, 155, 881–885 CAS.
- G. Hu, N. Wang, D. O'Hare and J. Davis, Chem. Commun., 2006, 42, 287–289 Search PubMed.
- G. Hu, N. Wang, D. O'Hare and A. Davis, J. Mater. Chem., 2007, 17, 2257–2266 CAS.
- F. Bellezza, A. Cipiciani, U. Costantino, M. Nocchetti and T. Posati, Eur. J. Inorg. Chem., 2009, 2603–2611 CAS.
- F. Bellezza, M. Nocchetti, T. Posati, S. Giovagnoli and A. Cipiciani, J. Colloid Interface Sci., 2012, 376, 20–27 CAS.
- L. Fagiolari, M. Bini, F. Costantino, G. Gatto, A. J. Kropf, F. Marmottini, M. Nocchetti, E. C. Wegener, F. Zaccaria, M. Delferro, R. Vivani and A. Macchioni, ACS Appl. Mater. Interfaces, 2020, 12, 32736–32745 CAS.
- T. Posati, F. Bellezza, A. Cipiciani, F. Costantino, M. Nocchetti, L. Tarpani and L. Latterini, Cryst. Growth Des., 2010, 10, 2847–2850 CAS.
- T. Posati, F. Costantino, L. Latterini, M. Nocchetti, M. Paolantoni and L. Tarpani, Inorg. Chem., 2012, 51, 13229–13236 CAS.
- J. Prince, A. Montoya, G. Ferrat and J. S. Valente, Chem. Mater., 2009, 21, 5826–5835 CAS.
- J. S. Valente, E. Lima, J. A. Toledo-Antonio, M. A. Cortes-Jacome, L. Lartundo-Rojas, R. Montiel and J. Prince, J. Phys. Chem. C, 2010, 114, 2089–2099 CAS.
- D. Sokol, D. E. L. Vieira, A. Zarkov, M. G. S. Ferreira, A. Beganskiene, V. V. Rubanik, A. D. Shilin, A. Kareiva and A. N. Salak, Sci. Rep., 2019, 9, 10419 Search PubMed.
- P. Benito, F. M. Labajos, J. Rocha and V. Rives, Microporous Mesoporous Mater., 2006, 94, 148–158 CAS.
- N. Iyi, T. Matsumoto, Y. Kaneko and K. Kitamura, Chem. Lett., 2004, 33, 1122–1123 CAS.
- K. Okamoto, N. Iyi and T. Sasaki, Appl. Clay Sci., 2007, 37, 23–31 CAS.
- U. Costantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1439–1446 CAS.
- M. Adachi-Pagano, C. Forano and J. P. Besse, J. Mater. Chem., 2003, 13, 1988–1993 CAS.
- T. Hibino and H. Ohya, Appl. Clay Sci., 2009, 45, 123–132 CAS.
- K. Tamura, R. Kawashiri, N. Iyi, Y. Watanabe, H. Sakuma and M. Kamon, ACS Appl. Mater. Interfaces, 2019, 11, 27954–27963 CAS.
- X. Y. Yu, T. Luo, Y. Jia, R. X. Xu, C. Gao, Y. X. Zhang, J. H. Liu and X. J. Huang, Nanoscale, 2012, 4, 3466–3474 CAS.
- A. Inayat, M. Klumpp and W. Schwieger, Appl. Clay Sci., 2011, 51, 452 CAS.
- Y. Yang, X. Zhao, Y. Zhu and F. Zhang, Chem. Mater., 2012, 24, 81–87 CAS.
- J. Liu, J. Song, H. Xiao, L. Zhang, Y. Qin, D. Liu, W. Hou and N. Du, Powder Technol., 2014, 253, 41–45 CAS.
- T. Hibino and H. Ohya, Appl. Clay Sci., 2009, 45, 123–132 CAS.
- P. Yang, J. Yu, Z. Wang, Q. Liu and T. Wu, React. Kinet. Catal. Lett., 2004, 83, 275–282 CAS.
- M. R. Berber, I. H. Hafez, K. Minagawa, M. Katoh, T. Mori and M. Tanaka, J. Mol. Struct., 2013, 1033, 104–112 CAS.
- X. Cheng, Y. Wang, Z. Sun, D. Sun and A. Wang, Water Sci. Technol., 2013, 67, 1757–1763 CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CAS.
- S. Marappa and P. V. Kamath, Ind. Eng. Chem. Res., 2015, 54, 11075–11079 CAS.
- I. C. Madsen, N. V. Y. Scarlett and A. Kern, Z. Kristallogr., 2011, 226, 944–955 CAS.
- H. D. Ruan, R. L. Frost and J. T. J. Kloprogge, Raman Spectrosc., 2001, 32, 745–750 CAS.
- S. J. Palmer, R. L. Frost and L.-M. Grand, J. Raman Spectrosc., 2011, 42, 1168–1173 CAS.
- J. K. Kloprogge, L. Hickey and R. L. Frost, J. Raman Spectrosc., 2004, 35, 967 CAS.
- R. L. Frost, W. Martens, Z. Ding, J. T. Kloprogge and T. E. Johnson, Spectrochim. Acta, Part A, 2003, 59, 291–302 Search PubMed.
- J. T. Kloprogge, L. R. Hickey and L. Frost, Mater. Chem. Phys., 2005, 89, 99–109 CAS.
- J. T. Kloprogge, D. Wharton, L. Hickey and R. L. Frost, Am. Mineral., 2002, 87, 623–629 CAS.
- S. J. Palmer, R. L. Frost, G. Ayoko and T. Nguyen, J. Raman Spectrosc., 2008, 39, 395–401 CAS.
- X. Ge, C. D. Gu, X. L. Wang and J. P. Tu, J. Mater. Chem. A, 2014, 2, 17066–17076 CAS.
- J. T. Kloprogge, L. Hickey, R. Trujillano, M. J. Holgado, M. S. San Román, V. Rives, W. N. Martens and R. L. Forst, Cryst. Growth Des., 2006, 6, 1533–1536 CAS.
- S. Faramawya, T. Zakib, A. A.-E. Sakra, O. Saberc, A. K. Aboul-Gheitd and S. A. Hassane, J. Nat. Gas Sci. Eng., 2018, 54, 72–82 Search PubMed.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CAS.
- P. Li, Z. P. Xu, M. A. Hampton, D. T. Vu, L. Huang, V. Rudolph and A. V. Nguyen, J. Phys. Chem. C, 2012, 116, 10325–10332 CAS.
- Y. Sun, X. Gao, N. Yang, X. Tantai, X. Xiao, B. Jiang and L. Zhang, Ind. Eng. Chem. Res., 2019, 58, 7937–7947 CAS.
- C. Tokoro, T. Sakakibara and S. Suzuki, Chem. Eng. J., 2015, 279, 86–92 CAS.
- S. S. C. Pushparaj, C. Forano, V. Prevot, A. S. Lipton, G. J. Rees, J. V. Hanna and U. G. Nielsen, J. Phys. Chem. C, 2015, 119, 27695–27707 CAS.
- L. B. Staal, S. S. C. Pushparaj, C. Forano, V. Prevot, D. B. Ravnsbæk, M. Bjerring and U. G. Nielsen, J. Mater. Chem. A, 2017, 5, 21795–21806 CAS.
- M. Li, T. Chowdhury, A. N. Kraetz, H. Jing, A. Dopilka, L. M. Farmen, S. Sinha and C. K. Chan, ChemEngineering, 2019, 3, 20 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis parameters of ZnAl LDH and MgAl LDH samples (Tables S1 and S2). Crystallographic data and refinement details for ZnAlCO3, ZnAlNO3 and ZnAlCl with added corundum as the internal standard for the evaluation of the amount of the amorphous fraction (Tables S3–S6). Rietveld plot of the last refinement cycle for ZnAlCO3, ZnAlNO3 and ZnAlCl (Fig. S1–S3). FT-IR of ZnAlCO3 and ZAN/1 in the 4000–1500 cm−1 spectral region of samples recovered at increasing times (Fig. S4). SEM images of ZACl/1, ZnAlCO3 and ZAN/1 after 0, 2, and 4 h of reaction (Fig. S5). EDX analysis on different points of ZACl/1, ZnAlCO3 and ZAN/1 crystals collected after 6, 48 and 120 h of reaction (Fig. S6–S8). Details on the synthesis parameters of MgAl LDH samples (Tables S7 and S8). PXRD patterns of MgAl LDH samples. See DOI: https://doi.org/10.1039/d4dt01529k |
| This journal is © The Royal Society of Chemistry 2024 |