
Oxadiazole-integrated heterocoronene discotics as ambipolar organic semiconductors†
Ritobrata
De‡
 a,
Joydip
De‡
a,
Santosh Prasad
Gupta
b,
Indu
Bala
a,
Ankita
c,
Tarun
c,
Upendra Kumar
Pandey
*c and
Santanu Kumar
Pal
*a
a,
Joydip
De‡
a,
Santosh Prasad
Gupta
b,
Indu
Bala
a,
Ankita
c,
Tarun
c,
Upendra Kumar
Pandey
*c and
Santanu Kumar
Pal
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector-81, SAS Nagar, Knowledge City, Manauli-140306, India. E-mail: skpal@iisermohali.ac.in; santanupal.20@gmail.com
bDepartment of Physics, Patna University, Patna-800005, India
cDepartment of Electrical Engineering, School of Engineering, Shiv Nadar Institution of Eminence, Gautam Buddha Nagar, Uttar Pradesh-201314, India. E-mail: upendra.pandey@snu.edu.in
First published on 13th December 2022
Abstract
The development of modern technologies has driven a quest for new semiconducting materials in optoelectronics, where self-assembled liquid crystal (LC) materials can play a potential role. The molecular engineering of disc-shaped LCs (DLCs) with suitable organic moieties, especially heterocyclic units, can lead to control over their columnar architecture in the nano-scale regime, which holds the key to tuning the charge-transport properties of the system. Here, we have successfully designed and synthesized room-temperature DLCs (1.1, 1.2 and 1.3) with 1,3,4-oxadiazole functional units acting as electron-deficient linkers between a central heterocoronene core and the peripheral alkoxy phenyl units. All the derivatives exhibited a broad columnar hexagonal mesophase range with high isotropic temperatures. When employed in space-charge limited current (SCLC) devices, they showed ambipolar charge transport behaviour in thin films, with maximum hole and electron mobilities of the order of 10−3 and 10−5 cm2 V−1 s−1, respectively.
Introduction
In recent years, both materials and synthetic chemists have focused on the strategy of molecular engineering to attain hierarchical superstructures at the nanoscale for modulating the performance of optoelectronic devices.1,2 In this aspect, liquid crystal (LC) materials have demonstrated their ability as a class of self-assembled soft materials, gaining increasing scope in organic electronics.3–5 True to their epithet, ‘liquid crystals’ maintain intermediary order and mobility with respect to both crystalline solids and isotropic liquids, thus striking the perfect balance for the practical fabrication of low-cost optoelectronic devices. While there are numerous supramolecular interactions at play in determining the functional behaviour of any such mesophase, the generic design of a discotic liquid crystal (DLC) with its rigid aromatic core and flexible peripheral alkyl units provides ample opportunities for exploring a wide range of organic moieties as building blocks.6 DLCs combine a high degree of order with the potential formation of large and uniform domains of well-aligned molecules during their self-assembly into columnar mesophases.7–11 Hence the dynamics of the system can be tuned at the molecular level through the inclusion of organic chromophores with varying optical and electronic properties to develop synthetically adjustable self-healing anisotropic DLC materials that are coveted for solution-processable optoelectronic devices. In accordance with this idea, the incorporation of heterocyclic units into the design of a DLC can manifest rewarding outcomes in terms of its thermal and physical properties. The presence of nitrogen, oxygen or sulphur atoms in a carbocyclic skeleton induces polarizability in the system.12 The ability of these electronegative heteroatoms to impart lateral or longitudinal dipoles, in combination with a reduced overall symmetry, is reflected in the mesophase behaviour.13–17 Especially in a DLC, if such electron-deficient heterocycles are attached to aromatic rings, the heterocycles behave as acceptors to the aromatic donors, and often a donor–acceptor interaction greatly influences the electronic nature of the mesophase.18 Following this train of thought, aromatic substituted oxadiazoles have made their way into the area of liquid crystalline chromophores.19,20 These moieties affluently enrich nematic, smectic and columnar mesophases, and even yield the elusive thermotropic nematic phase with biaxiality.21,22 The electron-accepting behaviour of 1,3,4-oxadiazole has also been established in the realm of DLCs, providing a rich area of research that requires exploration.23–29On the other hand, heterocoronene is a redox-active chromophore that shows high thermal stability up to 515 °C, and its photostability is comparable to the well-known dye perylene orange, i.e., N,N′-bis(2,6-diisopropylphenyl)-3,4,9,10-perylenetetracarboxylic diimide.30–32 Although identified as a promising outdoor colourant, the immense potential of this organic core in the field of electronics is yet to be fully realized. In 2019, De et al. reported the first examples of heterocoronene-based DLCs exhibiting efficient ambipolar charge transport behaviour in space charge limited current (SCLC) devices.33 These reported DLCs can retain their mesophases up to a maximum temperature of 95 °C. In the quest for DLCs with much wider mesophase ranges, we have reconstructed the design by decorating the heterocoronene core with four 1,3,4-oxadiazole (OXD) functional units (as illustrated in Fig. 1). The motivation of the study arose from two preliminary goals. The first was to stabilize the columnar order of the heterocoronene LC system over a broader range of temperatures. The second was to tune the electronic properties of the system via the introduction of an electron-withdrawing bridging unit between the core and the alkyl periphery. In the present work, with the alliance of the electron-deficient OXD unit, the designed discotic molecules (1.1–1.3, Fig. 1) retained their room-temperature columnar self-assembly with enormous mesophase widths. In addition, the electronic properties of the materials were studied using the SCLC technique in solution-processed thin films. The resultant fabricated hole-only and electron-only SCLC devices of compounds 1.1–1.3 exhibited ambipolar semiconducting behaviour with maximum hole and electron mobilities of the order of 10−3 and 10−5 cm2 V−1 s−1, respectively.
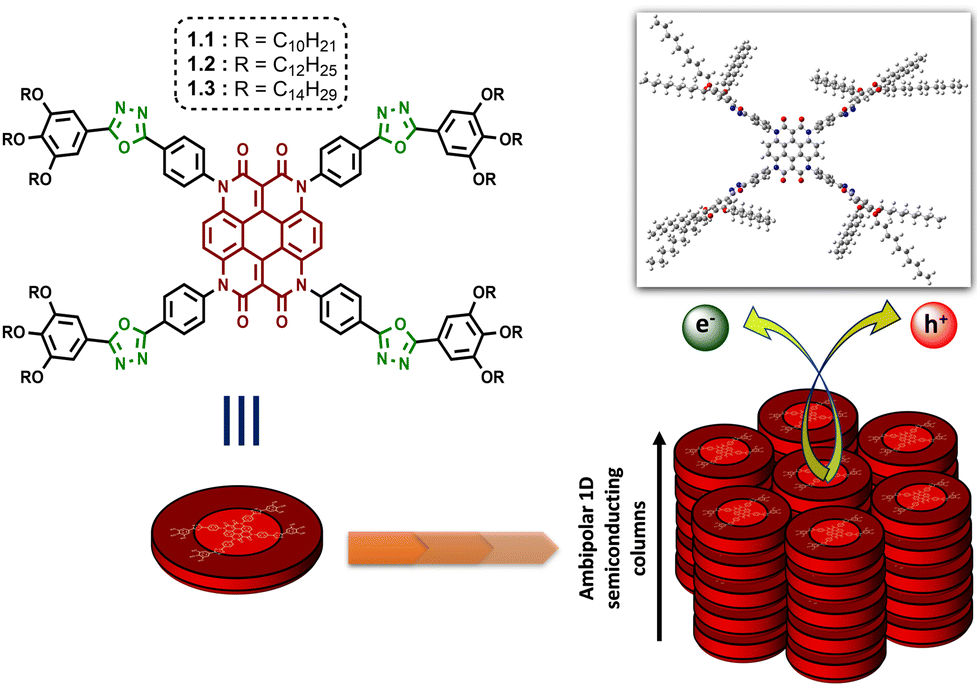 | ||
| Fig. 1 Molecular structure of oxadiazole integrated heterocoronene-based columnar DLCs for ambipolar charge transport (optimized geometry of 1.1 is shown in top-right corner). | ||
Results and discussion
Synthesis and characterization
The synthesis of the oxadiazole-modified heterocoronene-based DLCs (1.1–1.3) is illustrated in Scheme 1 and detailed in the ESI.† The synthetic procedure started with the esterification of commercially available gallic acid. Its corresponding methyl ester (3) underwent three-fold alkylation using an excess of alkyl bromide and potassium carbonate in anhydrous DMF with potassium iodide as a catalyst. The tri-alkylated methyl ester (4) was then converted back to the acid (5), the derivatization of which to the acyl chloride (6) was carried out using thionyl chloride and a catalytic amount of DMF. The highly reactive acyl chloride (6) thus generated was converted in situ to the oxadiazole precursor (7) by reacting with 4-nitrobenzohydrazide and pyridine in a dry THF medium. Subsequently, compound 7 was cyclized to give the 1,3,4-oxadiazole heterocyclic compound 8 by heating with POCl3. The nitro functionality of 8 was reduced to its corresponding amine via hydrogenation in the presence of palladium on activated charcoal. The oxadiazolyl aniline 9 underwent four-fold Buchwald–Hartwig amination to provide intermediate 2, which subsequently yielded a dark red solid as the targeted heterocoronene-oxadiazole (Het-OXD) compound through a microwave-assisted Knoevenagel reaction with diethyl malonate (yield: 66%, 63% and 56% for 1.1, 1.2 and 1.3, respectively). The final materials were characterized through 1H NMR, 13C NMR, IR and HRMS analyses (Fig. S1–S9, ESI†). All characterization results were well in accordance with the proposed molecular structure of the targeted compounds.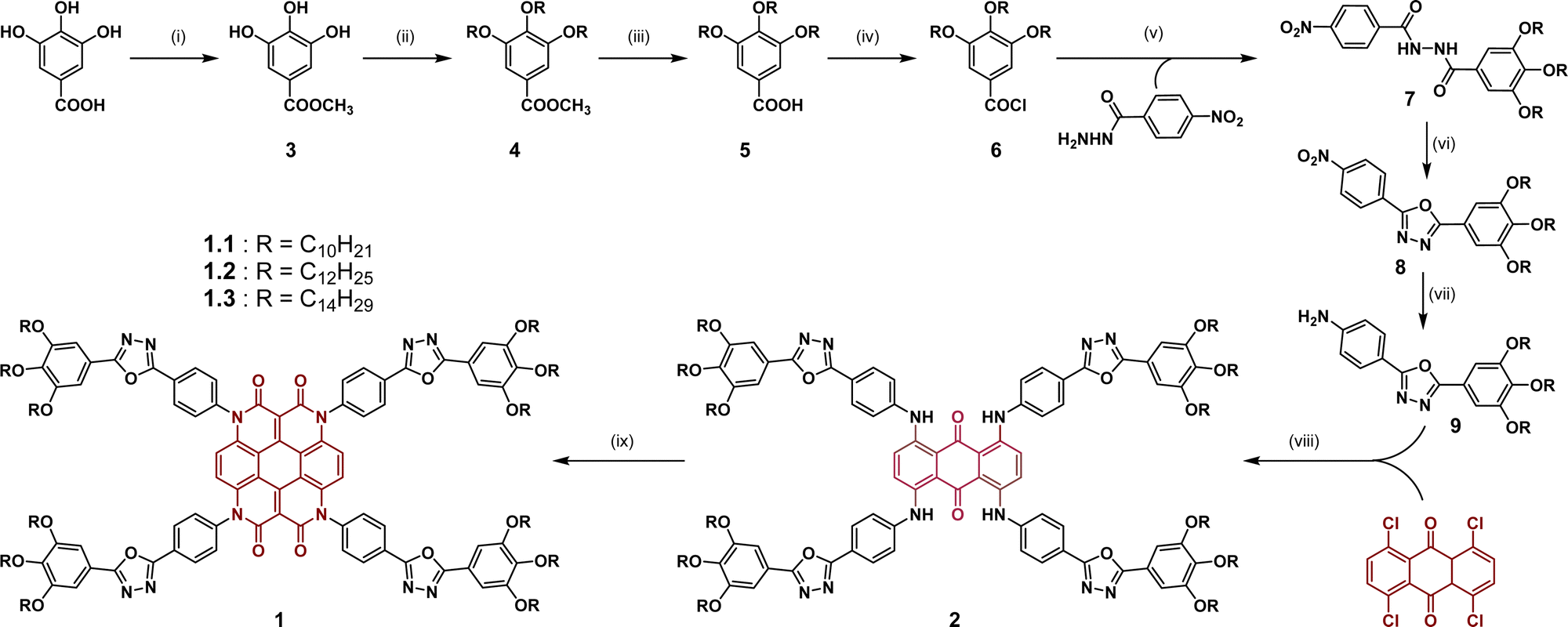 | ||
| Scheme 1 Synthesis of heterocoronene-oxadiazole derivatives 1.1–1.3. Reagents and conditions: (i) CH3OH, H2SO4, 60 °C, 18 h; (ii) K2CO3, KI, alkyl bromide, dry DMF, 90 °C, 24 h; (iii) KOH, EtOH, reflux, 24 h, conc. HCl, H2O; (iv) SOCl2, dry DCM, DMF (cat.), 18 h; (v) pyridine, dry THF, 18 h; (vi) POCl3, 100 °C, 18 h; (vii) Pd–C, H2, dry THF, 12 h; (viii) Cs2CO3, Pd2(dba)3, BINAP, dry toluene, reflux, 72 h; (ix) CH3COOK, diethyl malonate, dry DMF, microwave (172 °C), 90 min (yield: 1.1 = 66%, 1.2 = 63%, 1.3 = 56%). | ||
Thermal behaviour
A study of the mesophase behaviour of the final compounds was initiated using polarising optical microscopy (POM). Placed between a coverslip and an untreated glass slide, and upon cooling from their isotropic liquid states, all three Het-OXD derivatives (1.1–1.3) showed birefringent textures under cross-polarizers, indicating their anisotropic nature. The confocal fan-shaped textures as shown in Fig. 2b and Fig. S10 (ESI†) persisted at room temperature and exhibited shearability as well as fluidity upon pressing the glass slide, thus validating a columnar LC state for compounds 1.1–1.3 at ambient temperature.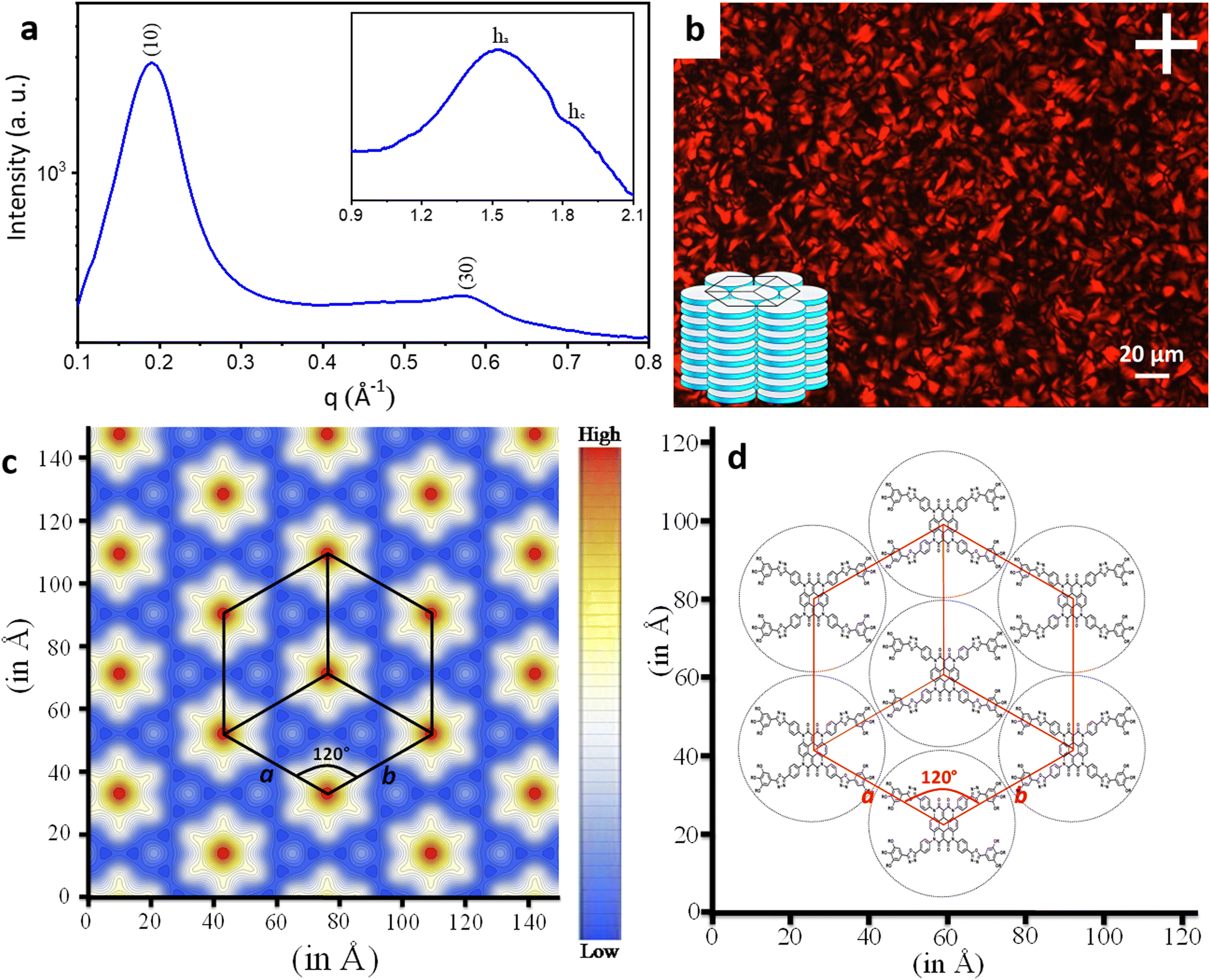 | ||
| Fig. 2 (a) Small- and mid-angle X-ray diffraction pattern of compound 1.1 at 25 °C (where the inset shows the wide-angle pattern). (b) Polarized optical micrograph of compound 1.1 at 27.1 °C under crossed polarizers (where the inset shows the Colh assembly). (c) Electron density map corresponding to the columnar hexagonal (Colh) phase of compound 1.1 at a temperature of 25 °C. Hexagons in the map showed the conventional unit cell of the Colh lattice, and there are three primitive unit cells within this conventional unit cell; a and b are the lattice parameters with a = b, and the angle between them is 120°. Deep red represents the highest electron density and deep blue denotes the lowest. (d) Two dimensional projection of the arrangement of Het-OXD molecules corresponding to the Colh phase. | ||
The LC phase transitions were further characterized via their differential scanning calorimetry (DSC) thermograms (Fig. S11, ESI†), which were well corroborated with the POM observations. The mesophase to isotropic transition temperatures of the Het-OXD DLCs were found to be very high (Table S1, ESI†). Enantiotropic LC behaviour holds for each of them (1.1–1.3), and they exhibit unusually wide mesophase windows. For example, compound 1.2, upon heating, shows an endothermic peak at 258.0 °C (ΔH = 5.24 kJ mol−1) corresponding to the clearing temperature. On cooling from the isotropic state, the exothermic peak for compound 1.2 is seen at 204.3 °C (ΔH = 4.47 kJ mol−1) for the isotropic to mesophase transition (Fig. S11b, ESI†). When compared with earlier reported heterocoronene DLCs without the oxadiazole heterocycles,33 the mesophase range is seen to be increased by almost 150 °C with the mere presence of the electron-deficient OXD. Thus, the OXD units are capable of stabilizing the columnar architecture at high temperatures, preventing disorder in the assembly and enabling efficient stacking of the discs through additional dipolar interactions.34,35 Also, the phase transitions for 1.1–1.3 were reproducible across several heating and cooling cycles (under POM), advocating the sustainable nature and thermal stability of the columnar assembly of the mesophase. Furthermore, thermogravimetric analysis (TGA) was performed to determine the thermal stability. This TGA study revealed that all three Het-OXD derivatives retain their stability with 5% weight loss at 361 °C, 353 °C and 351 °C for 1.1, 1.2 and 1.3, respectively (Fig. S12, ESI†). It can thus be inferred that the Het-OXD DLCs are thermally stable up to a range that extends well above their clearing temperature.
To gain further insight into the exact nature of the columnar mesophase, X-ray diffraction (XRD) studies were carried out. Fig. 2a shows the XRD pattern of compound 1.1. It exhibits two narrow peaks in the small-angle region with d-spacings in the ratio of 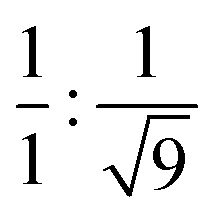 , which can be indexed as the (10) and (30) planes, respectively, of the two-dimensional hexagonal lattice. The calculated value of the lattice parameter, a, is equal to 38.20 Å (Table S2, ESI†). Also, a broad peak termed ha is visible in the wide-angle region. With a d-spacing of 4.16 Å, it indicates the reflection from the correlation of the fluidic alkyl chains. A broader peak hc, with a spacing of about 3.53 Å, in the same region is assigned to the π–π interaction between the discotic cores. The observation of the two-dimensional hexagonal lattices along with the hc peak is a confirmatory attribute of the columnar hexagonal (Colh) phase. Similarly, compounds 1.2 and 1.3 both show the Colh phase in their mesophase region (Fig. S13 and Table S2, ESI†). Furthermore, to gain a better understanding of the molecular arrangement in the mesophase, electron density maps (EDMs) were constructed (Fig. 2c shows that of compound 1.1, and Fig. S14 (ESI†) shows 1.2 and 1.3), using the information of the assigned indexes and intensities of the peaks. Thus, detailed analysis of the X-ray diffraction patterns and the derivation of their corresponding EDMs affirm that each molecule behaves as a disc that stacks itself into columns, and the columns in turn are further arranged on the two-dimensional lattices (2D-projection illustrated in Fig. 2d) leading to the Colh mesophase as discussed.
, which can be indexed as the (10) and (30) planes, respectively, of the two-dimensional hexagonal lattice. The calculated value of the lattice parameter, a, is equal to 38.20 Å (Table S2, ESI†). Also, a broad peak termed ha is visible in the wide-angle region. With a d-spacing of 4.16 Å, it indicates the reflection from the correlation of the fluidic alkyl chains. A broader peak hc, with a spacing of about 3.53 Å, in the same region is assigned to the π–π interaction between the discotic cores. The observation of the two-dimensional hexagonal lattices along with the hc peak is a confirmatory attribute of the columnar hexagonal (Colh) phase. Similarly, compounds 1.2 and 1.3 both show the Colh phase in their mesophase region (Fig. S13 and Table S2, ESI†). Furthermore, to gain a better understanding of the molecular arrangement in the mesophase, electron density maps (EDMs) were constructed (Fig. 2c shows that of compound 1.1, and Fig. S14 (ESI†) shows 1.2 and 1.3), using the information of the assigned indexes and intensities of the peaks. Thus, detailed analysis of the X-ray diffraction patterns and the derivation of their corresponding EDMs affirm that each molecule behaves as a disc that stacks itself into columns, and the columns in turn are further arranged on the two-dimensional lattices (2D-projection illustrated in Fig. 2d) leading to the Colh mesophase as discussed.
Photophysical, electrochemical and theoretical studies
The absorption and emission spectra of the Het-OXD compounds were recorded in chloroform solution to explore their photophysical properties. The three prominent absorption bands for each of the derivatives (Fig. S15, ESI†) at 308, 393 and 539 nm correspond to the β, p and α bands, respectively, for the heterocoronene unit.32 The emission spectra of 1.1–1.3 in chloroform solution at micromolar concentrations also showed similar peaks (Fig. S16, ESI†), with an emission maximum at 546 nm, which perfectly tallies with the visually perceivable orange fluorescence of the Het-OXD solutions under 365 nm UV light.Prior recognition of their redox activity is required to ascertain the charge transport behaviour of these Het-OXD derivatives. Moreover, a sound knowledge of the electrochemical behaviour of the compounds enables us to select appropriate metal electrodes with compatible work functions during device fabrication so that electron- and hole-only devices can be fabricated. With this aim, cyclic voltammetry experiments were carried out to determine the HOMO–LUMO energy levels of the molecule (Fig. S17, ESI†). The energy levels of the HOMO and LUMO were −5.86 and −3.46 eV, −5.76 and −3.58 eV, and −5.78 and −3.50 eV, for 1.1, 1.2 and 1.3, respectively, based on the oxidation and reduction curves of the cyclic voltammogram. The electrochemical band gaps were calculated as 2.40 eV for 1.1, 2.18 eV for 1.2 and 2.28 eV for 1.3 (Table 1).
| Compound | E ox,onset | E red,onset | E HOMO | E LUMO | ΔEg,CVe |
|---|---|---|---|---|---|
| a Onset potential of the oxidation peak, in V. b Onset potential of the reduction peak, in V. c Estimated from the formula EHOMO = −(4.8 − E1/2,Fc,Fc+ + Eoxd,onset) eV. d Estimated from the onset reduction peak values using ELUMO = −(4.8 − E1/2,Fc,Fc+ + Ered,onset) eV. E1/2,Fc,Fc+ = 0.51 eV. e Estimated from the formula ΔEg,CV = ELUMO − EHOMO. | |||||
| 1.1 | 1.53 | −0.87 | −5.86 | −3.46 | 2.40 |
| 1.2 | 1.43 | −0.75 | −5.76 | −3.58 | 2.18 |
| 1.3 | 1.45 | −0.83 | −5.78 | −3.50 | 2.28 |
DFT studies helped us to gain more information on the molecular orbitals and the electronic constituency of compounds 1.1–1.3. The optimized molecular structure and the contours of the frontier molecular orbitals for compounds 1.1–1.3 are shown in Fig. S18 (ESI†) (where the optimized geometry of 1.1 is also shown in Fig. 1). It is observed that, for all the derivatives, the core resides in a planar geometry, while the N-substituted phenyl rings along with the peripheral groups are tilted at an angle of 76–90°. The HOMO and LUMO are both distributed over the central heterocoronene unit, with a small portion of the HOMO localized on only one of the four radiating arms in the case of each Het-OXD derivative. The predominant delocalization of the electron density over the π-conjugated core leads the frontier orbitals having an increased time-average overlap, which is expected to favour charge transport in the present system.
Charge transport measurements
Both the electron and hole mobilities of the Het-OXD derivatives 1.1–1.3 extracted from SCLC measurements are summarized in Table 2 and in Tables S3 and S4 (ESI†). In brief, to extract the hole and electron mobility of 1.1–1.3, hole-only and electron-only devices were fabricated using the ITO/PEDOT:PSS/1.1–1.3/MoO3/Ag and ITO/ZnO/1.1–1.3/Ag device configurations, respectively. A combination of ITO/PEDOT:PSS and ITO/ZnO electrodes was used to establish near ohmic contact with the HOMO and LUMO of 1.1–1.3 for injecting holes and electrons in the SCLC devices. The thin film SCLC mobility was extracted by fitting the characteristic current density–voltage data (as shown in Fig. 3) on the semi-log plot using the modified Mott–Gurney equation, where electric field-dependent correction is also included in its original equation.36 It is represented aswhere εr is the relative dielectric constant of the material and ε0 is the permittivity of free space, J is the current density (the area of the sample was 6.6 mm2), V is the applied voltage and d is the thickness of the active layer of material. Here, γ is the fitting parameter that represents the strength of the field dependence on the mobility.
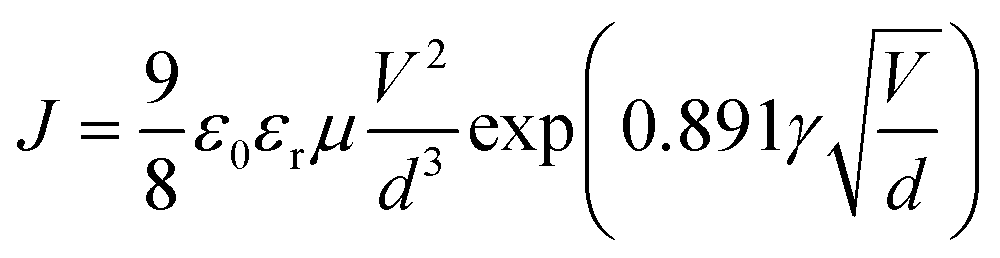
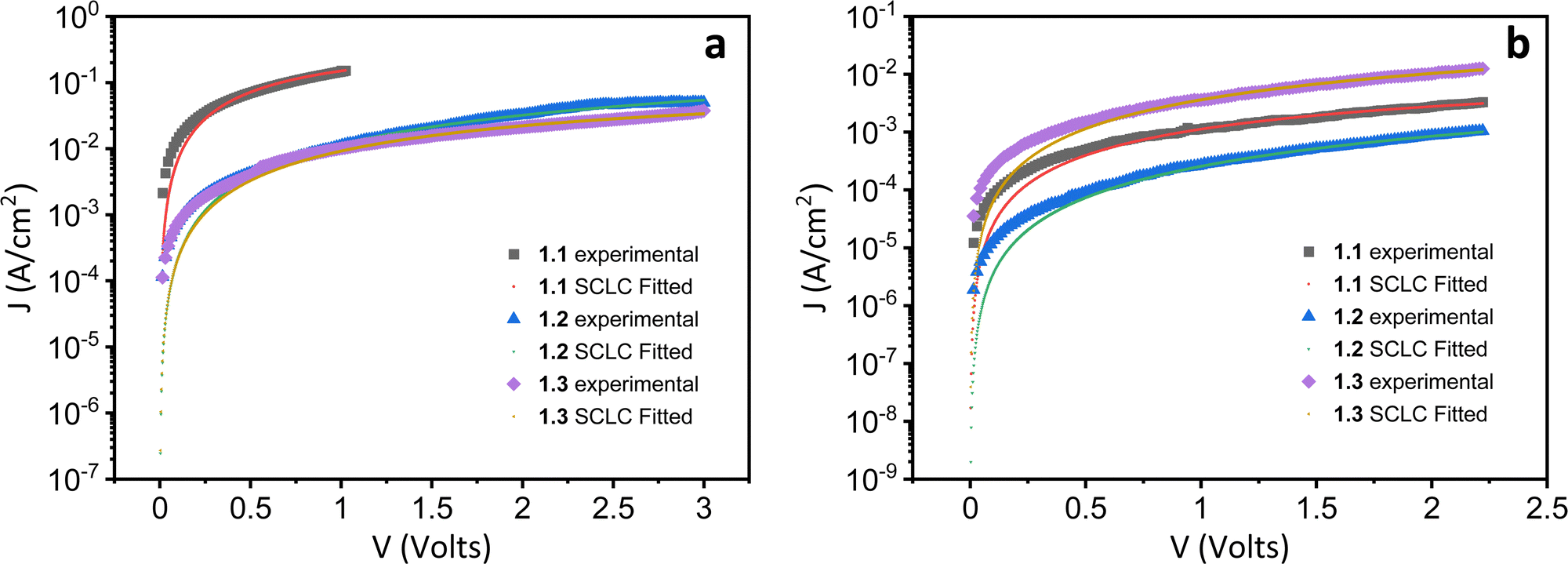 | ||
| Fig. 3 Current density–voltage characteristics as a semi-log plot obtained for (a) hole and (b) electron mobilities extracted from SCLC devices in the thin film of compounds 1.1–1.3 at room temperature. | ||
Generally, in sandwich cell structures, where the cell thickness is at the sub-micron level, the measured SCLC mobility is of the bulk.33,37 However, in this case, it is to be noted that the mobility values reported are those of thin films with a thickness of ∼100 nm, which are amorphous in nature. Hence the orientation of the DLCs cannot be ascertained here, unlike in sandwich cells where one can observe the alignment in their columnar direction. In the present case, the SCLC studies of reported DLCs were carried out in solution-processable thin-films because of their high isotropic temperatures that hindered the fabrication of melt-processable sandwich cells.
From the statistical data (Table 2 and Tables S3 and S4, ESI†), the average hole mobility values of Het-OXD derivatives 1.1–1.3 were 1.74 × 10−3, 1.46 × 10−4 and 2.01 × 10−4 cm2 V−1 s−1, respectively. The electron mobility values for Het-OXD derivatives 1.1–1.3 are 1.58 × 10−5 and 3.07 × 10−6 and 4.63 × 10−5 cm2 V−1 s−1, respectively.
The observed hole mobility exceeded that of the electrons by two orders of magnitude in 1.1, 1.2 and 1.3, which can be attributed to the small portion of the HOMO localized over one of the four arms that is additional to the central distribution, whereas the LUMO is only located on the central part for all derivatives (Fig. S18, ESI†).
It is to be noted here that a major motivation for differing the length of peripheral alkyl chains in 1.1, 1.2 and 1.3 was to study the change in the self-assembly and SCLC device performance of their respective mesophases. Although we observed a Colh assembly for all three derivatives throughout the mesophase range (as confirmed via POM and XRD), the effect of varying the alkyl chains is manifested in their isotropic temperatures. With the increase in alkyl chain length, the isotropic temperature decreases (Fig. S11 and Table S1, ESI†). Also, the thermal decomposition temperature is seen to decrease upon increasing the length of the alkyl tails (as evident from TGA). However, we did not observe any significant change in the electronic properties upon varying the alkyl chain length during cyclic voltammetry studies. In the case of SCLC experiments, the peripheral alkyl chains dictate the molecular alignment mainly in bulk measurements. Here in the amorphous thin films, the proper alignment of the director is not expected over a long range, as the orientation in columnar mesophases is highly surface dependent.38–40 Of course, correlations exist even in such a state, but deviations become prominent, and it is thus difficult in this case to draw definite conclusions based solely on the alkyl chain lengths.
Conclusions
In conclusion, we have reported a series of room-temperature DLCs based on oxadiazole-incorporated heterocoronene systems (1.1–1.3) where Colh self-assembly persists over a broad temperature range. In the present case, stabilization of the mesophase at higher temperatures was facilitated by the additional dipolar interactions of heterocyclic oxadiazole units with the π-stacked central heterocoronene fragment. All the compounds displayed ambipolar charge transport in thin film SCLC devices, with maximum hole and electron mobilities of the order of 10−3 and 10−5 cm2 V−1 s−1, respectively, which substantiates them as effective organic semiconductors for optoelectronic device applications. Intrigued by both the higher thin film hole mobility and the suitable HOMO energy level of 1.1–1.3, their application as a potential replacement for hole transporting materials in perovskite solar cells is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKP acknowledges the SERB Project (CRG/2019/000901/OC). RD acknowledges IISER Mohali for her fellowship. UKP is thankful to the Shiv Nadar Institution of Eminence for constant support as well as the I–V characterization facility of Dr R. Singh's lab. We thank NMR, SAXS/WAXS, LN2, and all other departmental/central facilities at IISER Mohali.References
- F. J. Martin-Martinez, K. Jin, D. López Barreiro and M. J. Buehler, ACS Nano, 2018, 12, 7425–7433 CrossRef CAS
.
- T. Q. Nguyen, R. Martel, M. Bushey, P. Avouris, A. Carlsen, C. Nuckolls and L. Brus, Phys. Chem. Chem. Phys., 2007, 9, 1515–1532 RSC
-
I. Bala, J. De and S. K. Pal, Molecular Architectonics and Nanoarchitectonics, Nanostructure Science and Technology, Springer Nature, 2022, 89–130 Search PubMed
- R. De, S. Sharma, S. Sengupta and S. K. Pal, Chem. Rec., 2022, 22, e202200056, DOI:10.1002/tcr.202200056
- Y. Wang, J. Shi, J. Chen, W. Zhu and E. Baranoff, J. Mater. Chem. C, 2015, 3, 7993–8005 RSC
- S. M. Said, M. S. Mahmood, M. N. Daud, M. F. Mohd Sabri and N. A. Sairi, Liq. Cryst., 2016, 43, 2092–2113 CrossRef CAS
- Q. Li and L. Li, Thermotropic Liq. Cryst. Recent Adv., 2007, 4991, 297–322 Search PubMed
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661–2670 RSC
- T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
- S. Kumar, Chem. Soc. Rev., 2006, 35, 83–109 RSC
- B. Roy, N. De and K. C. Majumdar, Chem. – Eur. J., 2012, 18, 14560–14588 CrossRef CAS PubMed
- A. Seed, Chem. Soc. Rev., 2007, 36, 2046–2069 RSC
- M. P. Aldred, R. Hudson, S. P. Kitney, P. Vlachos, A. Liedtke, K. L. Woon, M. O’Neill and S. M. Kelly, Liq. Cryst., 2008, 35, 413–427 CrossRef CAS
- M. Lehmann, G. Kestemont, R. G. Aspe, C. Buess-Herman, M. H. J. Koch, M. G. Debije, J. Piris, M. P. de Haas, J. M. Warman, M. D. Watson, V. Lemaur, J. Cornil, Y. H. Geerts, R. Gearba and D. A. Ivanov, Chem. – Eur. J., 2005, 11, 3349–3362 CrossRef CAS PubMed
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS
- K. Górski, K. Noworyta and J. Mech-Piskorz, RSC Adv., 2020, 10, 42363–42377 RSC
- H. H. G. Tsai, L. C. Chou, S. C. Lin, H. S. Sheu and C. K. Lai, Tetrahedron Lett., 2009, 50, 1906–1910 CrossRef CAS
- J. Han, J. Mater. Chem. C, 2013, 1, 7779–7797 RSC
- B. Schulz, M. Bruma and L. Brehmer, Adv. Mater., 1997, 9, 601–613 CrossRef CAS
- B. R. Acharya, A. Primak, T. J. Dingemans, E. T. Samulski and S. Kumar, Pramana, 2003, 61, 231–237 CrossRef CAS
- O. Francescangeli and E. T. Samulski, Soft Matter, 2010, 6, 2413–2420 RSC
- Y. D. Zhang, K. G. Jespersen, M. Kempe, J. A. Kornfield, S. Barlow, B. Kippelen and S. R. Marder, Langmuir, 2003, 19, 6534–6536 CrossRef CAS
- P. Zhang, B. Bai, H. Wang, S. Qu, Z. Yu, X. Ran and M. Li, Liq. Cryst., 2009, 36, 7–12 CrossRef CAS
- S. Varghese, N. S. S. Kumar, A. Krishna, D. S. S. Rao, S. K. Prasad and S. Das, Adv. Funct. Mater., 2009, 19, 2064–2073 CrossRef
- T. E. Frizon, A. G. Dal-Bó, G. Lopez, M. M. da Silva Paula and L. da Silva, Liq. Cryst., 2014, 41, 1162–1172 CrossRef CAS
- C. V. Yelamaggad, A. S. Achalkumar, D. S. S. Rao and S. K. Prasad, J. Org. Chem., 2009, 74, 3168–3171 CrossRef CAS PubMed
- A. A. Vieira, H. Gallardo, J. Barberá, P. Romero, J. L. Serrano and T. Sierra, J. Mater. Chem., 2011, 21, 5916–5922 RSC
- C. K. Lai, Y. C. Ke, J. C. Su, C. S. Li and W. R. Li, Liq. Cryst., 2002, 29, 915–920 CrossRef CAS
- J. Ivri, Z. Burshtein, E. Miron, R. Reisfeld and M. Eyal, IEEE J. Quantum Electron., 1990, 26, 1516–1520 CrossRef CAS
- M. D. Rahn, T. A. King, A. A. Gorman and I. Hamblett, Appl. Opt., 1997, 36, 5862 CrossRef CAS PubMed
- R. Duan, D. Schollmeyer, K. Müllen and C. Li, J. Mater. Chem. C, 2018, 6, 1334–1337 RSC
- J. De, I. Bala, S. P. Gupta, U. K. Pandey and S. K. Pal, J. Am. Chem. Soc., 2019, 141, 18799–18805 CrossRef CAS PubMed
- S. K. Pathak, S. Nath, J. De, S. K. Pal and A. S. Achalkumar, New J. Chem., 2017, 41, 9908–9917 RSC
- N. Tober, J. Winter, M. Jochem, M. Lehmann and H. Detert, Eur. J. Org. Chem., 2021, 798–809 CrossRef CAS
- P. N. Murgatroyd, J. Phys. D: Appl. Phys., 1970, 3, 151–156 CrossRef
- I. Bala, J. De, S. P. Gupta, H. Singh, U. K. Pandey and S. K. Pal, Chem. Commun., 2020, 56, 5629–5632 RSC
- C. Ruiz, U. K. Pandey, R. Termine, E. M. G. Frutos, G. L. Espejo, R. P. Ortiz, W. Huang, T. J. Marks, A. Facchetti, M. C. R. Delgado, A. Golemme and B. G. Lor, ACS Appl. Mater. Interfaces, 2016, 8, 26964–26971 CrossRef CAS PubMed
- T. S. Perova and J. K. Vij, Adv. Mater., 1995, 7, 919–922 CrossRef CAS
- T. S. Perova, J. K. Vij and A. Kocot, Europhys. Lett., 1998, 44, 198 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04144h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |